

Abstract
Epigenetic changes such as DNA methylation and histone methylation and acetylation alter gene expression at the level of transcription by upregulating, downregulating, or silencing genes completely. Dysregulation of epigenetic events can be pathological, leading to cardiovascular disease, neurological disorders, metabolic disorders, and cancer development. Therefore, identifying drugs that inhibit these epigenetic changes are of great clinical interest. In this review, we summarize the epigenetic events associated with different disorders and diseases including cardiovascular, neurological, and metabolic disorders, and cancer. Knowledge of the specific epigenetic changes associated with these types of diseases facilitates the development of specific inhibitors, which can be used as epigenetic drugs. In this review, we discuss the major classes of epigenetic drugs currently in use, such as DNA methylation inhibiting drugs, bromodomain inhibitors, histone acetyl transferase inhibitors, histone deacetylase inhibitors, protein methyltransferase inhibitors, and histone methylation inhibitors and their role in reversing epigenetic changes and treating disease.
Keywords: epigenetics, histone methylation, histone acetylation, DNA methylation, drugs, cardiovascular, neurological, metabolic, cancer, tumor suppressor genes, gene expression, gene silencing
What is Epigenetics?
Epigenetics is the study of heritable, reversible forms of gene regulation that are not dependent on the DNA sequence. This regulation includes DNA methylation and histone methylation, acetylation, ubiquitination, and phosphorylation. Recent studies have shown that epigenetics plays a central role in many types of diseases, including cardiovascular diseases, neurological diseases, metabolic disorders, and cancer.1 Because of the reversible nature of epigenetic events, researchers postulate that inhibition of epigenetic changes could be of valuable therapeutic potential. In fact, in some neurological diseases and cancers, epigenetic drugs are already in use.2,3 This review will discuss some of the important diseases that involve epigenetic changes and the drugs that are being developed to treat them.
In What Types of Cellular Events are Epigenetics Involved?
DNA methylation
Epigenetic events are involved throughout the entire human lifecycle, from embryogenesis to adulthood. Germ cells begin with a level of DNA methylation, inherited from their respective parents. Upon fertilization, a massive methylation overhaul occurs, in which most methylation is lost. As the zygote divides and development progresses, the embryo undergoes de novo methylation, reestablishing its original level of DNA methylation.4 Considering that all organisms begin as a single egg and that all cells in the final complicated organism contain the same genes, epigenetics must play an essential role.
DNA methylation exists primarily within the context of the cytosine–phosphate–guanine (CpG) dinucleotide.5 All changes in methylation are modulated by specific enzymes. DNA methyltransferase (DNMT) 3a, DNMT3b, and DNMT1 are three catalytically active enzymes required for formation and maintenance of DNA methylation patterns.6 DNMT1 acts as a maintenance methyltransferase, adding methyl groups to the unmethylated daughter strands of newly replicated DNA (Fig. 1). When CpG residues are methylated by DNMT1, methyl-binding domain protein (MBDP) and histone deacetylases (HDACs) are recruited, inhibiting transcription and silencing the gene. Deletion of DNMT1 results in global demethylation and embryonic lethality. DNMT3a and DNMT3b are responsible for de novo methylation after implantation and are highly expressed in developing embryos. DNMT3a and DNMT3b establish DNA methylation during embryonic stages and add new methyl groups to previously unmethylated DNA (Fig. 2).7
Figure 1.

After replication, the methylation (shown in red) on the specific CpG residues (not shown) of the mother strand is copied over onto the daughter strand by DNA methyltransferase 1 (DNMT1), maintaining the methylation pattern of the mother strand.
Figure 2.

RNA polymerase II (RNA Pol II) and transcription factors bind to unmethylated (shown in green) upstream promoter region of a gene and transcription proceeds. Methylation (shown in red) of specific CpG sites near the promoter regions allow binding of histone deacetylases (HDACs) and other methyl domain-binding proteins that prevents RNA Pol II binding and inhibits transcription. DNA methyl transferases (DNMTs) methylate CpG sites (not shown) within the promoter region. Methylation is also observed in the intragenic region, the function of which is still not well understood.
Chromatin packaging (histones)
In addition to methylation at CpG islands in DNA, other forms of expression-altering chemical modification exist at histones. Genomic DNA is packaged around histones to form a complex called chromatin. A unit of chromatin, known as a nucleosome, is composed of 146 base pairs of DNA wrapped around an octamer of four core histones (H2A, H2B, H3, and H4). With amino-terminal tails extending from the globular region of the histones, they become accessible to modifications, such as acetylation, methylation, phosphorylation, and ubiquitination.8 Histone modification can subsequently affect DNA processes, such as transcription, DNA repair and replication, and chromosomal organization. Acetylation and methylation of lysines and arginines at the histone tails are commonly analyzed; however, in contrast to DNA methylation, histone modification can be either activating or inhibiting. For example, H3K4 methylation activates gene expression, whereas H3K9 methylation, H3K27 trimethylation, and H3K20 trimethylation silence gene expression.9,10
Histone methylation is involved in several biological processes, including DNA repair, cell cycle, stress responses, development, differentiation, and aging. Any change in histone methylation can alter any one of these biological functions, resulting in the development of disease. For example, in many types of cancers including prostate, lung, kidney, breast, and pancreatic cancer, H3K4me2 is downregulated, which is often associated with high recurrence or poor survival rates. H3K27me3 is downregulated in gastric adenocarcinoma and ovarian cancer, and H4K20me3 downregulation is associated with lymphomas and colon cancer.11 Histone methylation changes are also associated with aging. For example, H4K20me3 levels increase with age in rat livers; however, H3K27me3 levels decrease in somatic tissues in aging Caenorhabditis elegans.12,13 These examples demonstrate a scenario where small molecule inhibitors of histone methylases could be used to demethylate the specific sites mentioned to develop anticancer and antiaging drugs.
Another important histone modification is the acetylation of lysine residues at the N-terminal region of histones (mainly H3), which regulates whether chromatin is in the open or closed formation (Fig. 3). Acetylated lysines provide an open chromatin conformation, whereas deacetylated lysines provide a closed chromatin conformation and inhibit transcription.14 The acetylation and deacetylation of histones are strictly regulated by enzymes called histone acetyl transferases (HATs) and histone deacetylases (HDACs). There are many forms of HATs and HDACs, which have a vast literature, but are out of the scope of this review.15 HATs such as HAT p300, CREB-binding protein (CBP), P300/CBP-associated factor (PCAF), and general control of amino acid synthesis protein 5 (GCN5) play a potential pathological role in asthma, chronic obstructive pulmonary disorder (COPD), and different types of cancers as well as learning and memory deficits.16–18 It is thought that HAT p300 and CBP regulate expression of tumor suppressor and promoter genes, and PCAF and GCNF perform global acetylations and nonhistone acetylations. GCNF is also crucial in cell-cycle progression, which is significant in cancer therapies.19,20 HATs p300 and PCAF often induce increased gene transcription in airway inflammation disorders such as asthma and COPD, leading to excessive expression of inflammatory genes. Initial studies in mice have shown that learning and memory impairments are a result of deficits in HATs, such as PCAF and CBP.21
Figure 3.
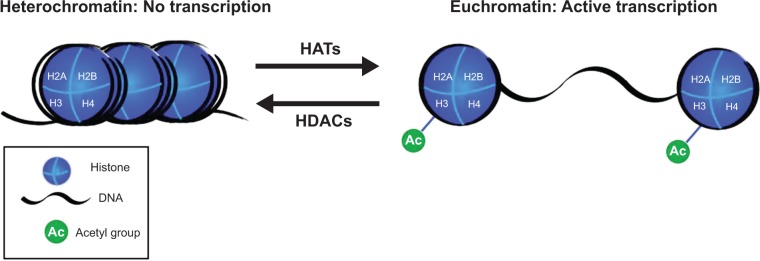
Histone acetyl transferases (HATs) add acetyl groups to lysine residues on the N-terminal end of the histone proteins of the nucleosome, causing the chromatin to de-condense, resulting in euchromatin, which is available for transcription. Histone deacetylases (HDACs) remove the acetyl groups, the chromatin condenses into heterochromatin, and is not transcribed.
HDACs
To achieve proper development and function, it is essential not only to acetylate and open certain regions of chromatin, but also to deacetylate and close other regions of the genome. HDACs are involved in the regulation of integrated cellular functions in the nucleus and cytoplasm. The general mechanism of these enzymes is to deacetylate specific lysine residues of histones and other proteins inducing formation of transcriptionally inactive heterochromatin. These modifications of HDACs and HATs are both reversible reactions and play a significant role in the regulation of transcription (Fig. 3).15
There are four general classes of HDACs: I, II, III, and IV. Classes I and II are involved in cellular growth and development, Class III HDACs are mainly involved in metabolic processes, and22 Class IV is comparatively new in the literature and is less studied. Class I HDACs remove acetyl groups from specific lysine residues on histones in the nucleosome, the genetic material coiled around histone subunits. Nucleosome modification affects how tightly DNA is wound around the histone structure, thus HDAC-mediated modifications can lead to induction of gene silencing.23 Class I HDACs are implicated in regulation by a process involving the methylation of CpG residues in upstream promoter regions of silenced genes. When these CpG residues are methylated, methyl domain-binding proteins (MDBP) actively recruit HDACs (mainly HDAC I, II, and III) to the histone, further inhibiting gene transcription (Fig. 2).24,25 Class II HDACs are able to shuttle back and forth between the nucleus and cytoplasm and are able to remove acetyl groups from proteins other than histones.26 For Class III HDAC (also known as sirtuins) mechanisms to proceed, nicotinamide adenine dinucleotide (NAD+) is required as an essential cofactor to remove the acetyl groups. Additionally, several sirtuins are known to regulate ATP production while others are known to protect against tumorigenesis during oxidative stress. Because of the wide range of target genes for HDACs, a variety of HDAC inhibitors (HDACi) are being developed, with hopes of treating a number of diseases including cancer, neurodegenerative disorders, cardiovascular diseases, and obesity.27,28
What Types of Diseases Involve Epigenetic Changes?
Cardiovascular diseases
The association of epigenetic changes with cardiovascular diseases is an emerging area of research. Although the studies are not expatiated on, there are many examples that demonstrate that histone and CpG residue modifications regulate important cardiovascular functions. Although the processes are not readily understood mechanistically, many studies have shown that alteration can lead to the development of atherosclerosis and cardiovascular disease.29,30 In particular, atheroprotective estrogen receptor genes ESR1 and ESR2 are often hypermethylated in human atherosclerosis.31 These genes are usually expressed in normal vascular smooth muscle cells; however, the expression levels decrease with age, resulting in vascular damage. Folic acid deficiency also portrays an epigenetic link to endothelial dysfunction, which is related to several cardiovascular diseases.32,33 Associations of elevated serum homocysteine levels and decreased nitric oxide production may be the cause of angiogenesis suppression.34 The most well-known endothelial gene regulated by the histone code is nitric oxide synthase 3 (NOS3), which codes for the endothelial nitric oxide synthase (eNOS) protein that catalyzes the formation of nitric oxide (NO), a vasodilator factor.35 In endothelial cells, eNOS is enriched; however, when exposed to short-term hypoxia, eNOS expression drastically decreases. Activating histone modifications, such as H3K9, H4K12, and H3K4, are observed in the NOS3 promoter region in endothelial cells, whereas these modifications are absent in vascular smooth muscle cells. In vascular cells, the promoter region is hypermethylated with methyl-binding proteins, rendering transcription inactive.36,37 Another example is cytosine methylation in the insulin-like growth factor 2 (IGF2) gene, which causes dysregulation of imprinting and is associated with a higher risk of coronary heart disease.38
A recent discovery links heritable coronary artery disease to the chromosome 9p21 locus.39,40 This locus does not transcribe any proteins but contains a lncRNA is called antisense noncoding RNA in the INK4 locus (ANRIL), which is known to regulate epigenetic function.41 Increased levels of ANRIL are associated with atherosclerotic plaque. Another study shows dysregulation of DNA methylation during embryogenesis, which may lead to congenital heart disease and increased risk of cardiovascular disease in adulthood.42,43 Hypomethylation of some genes has also been implicated in cardiovascular diseases. For example, global loss of genomic methylation content has been found in patients with hypertension.44 Ischemic heart disease and stroke are associated with reduced blood DNA methylation of Long Interspersed Nucleotide Element 1 (LINE-1). Additionally, hypomethylation of IGF2 and insulin/insulin-like growth factor 2 (INSIGF) genes, which are normally critical to metabolism of glucose and lipids, have been shown to increase risk of cardiovascular disease.45
Neurological disorders
Considering the vast potential of epigenetic modifications, it is not surprising that epigenetics plays a key role in development of the nervous system. In order to retain a multipotent state, many developmental and differentiation genes in neuronal precursor cells are silenced by CpG methylation. During neuronal differentiation, CpG methylation is lost and H3K4 dimethylation (H3K4me2) is gained.46,47 Regulation of methyl CpG-binding proteins is thus very important. Mutations, duplications, and insertions of the methyl CpG-binding protein 2 (MeCP2) gene are known to cause Rett syndrome, an X-linked neurological disorder that eventually causes severe mental retardation.48 Once MeCP2 binds to DNA at CpG islands, it interacts with other proteins, including HDACs, facilitating chromosome condensation and gene silencing. Interestingly, a recent study showed that neurological defects could be partially or completely reversed in mice with Rett syndrome, suggesting that early damage caused by dysregulation of MeCP2 is not necessarily permanent.49 Because females with Rett syndrome have one wild-type copy of the MeCP2 gene, possible epigenetic therapies could target the inactivated and methylation-silenced wild-type X-chromosomes.
The importance of epigenetic modifications in the nervous system continues throughout the lifespan, with many epigenetic changes associated with aging, long-term synaptic plasticity, and memory retention.50,51 Studies have shown that several neuronal genes display a gradual but continuous increase in CpG methylation from birth to old age.52,53 Several neurodegenerative disorders have been associated with dysregulation of these epigenetic changes. Parkinson’s disease, characterized by a decline in coordination and motor skills caused by neuronal death, is a late-onset progressive neurodegenerative disease. Recent studies have shown that the sequestration of the alpha-synuclein protein into Lewy bodies, a key feature of the disease, also involves the sequestration of DNMT1 into the cytoplasm. The decrease in nuclear DNMT1 leads to global neuronal hypomethylation.54,55 Huntington’s disease is another late-onset progressive neurodegenerative disorder. The disease is caused by an expanded polyglutamine repeat sequence in the huntingtin protein.56 The mutant huntingtin protein, unlike wild-type huntingtin, is able to enter the nucleus, where it binds and inhibits HATs, leading to a decrease in H3 and H4 histone acetylation, and thus to a global silencing of neuronal genes.57,58 Drugs able to correct the epigenetic imbalances in these disorders could be a very promising treatment for such diseases in the future.
Metabolic disorders
The epigenome is also subject to changes caused by environmental factors. For example, although monozygotic twins are born with nearly indistinguishable epigenetic patterns, as they age, the epigenomes diverge, explaining why one twin may be more susceptible to certain diseases than the other despite having identical DNA sequences.59 A recent study found that mice fed a diet deficient in folic acid, L-methionine, and choline exhibited deregulation of hepatic DNMT1 and methyl CpG-binding proteins.60 The changes were reversible if returned to a normal diet; however, prolonged changes in DNMT1 and methyl CpG-binding protein expression led to the development of hepatic carcinoma. Because even temporary changes in diet can lead to widespread epigenetic changes, the role of epigenetics in metabolic disorders is of great importance and could serve as an invaluable tool in the treatment and prevention of metabolically linked disorders.
Although not a direct cause, several epigenetic mechanisms have been implicated in the development of type II diabetes and obesity, with differential expression of certain genes leading to differential risks of disease.61 This differential expression can begin in utero, with maternal nutrition status having a large impact on the epigenetic status of the fetus. For example, one study linked in utero growth retardation to a progressive silencing (by methylation) of pancreatic islet factor 1 (Pdx1) in adulthood.62 Another study found that transient hyperglycemia led to the recruitment of the histone methyltransferase, Set7, and induced long-lasting changes in the histone methylation pattern of NFkB-65, a pro-inflammatory gene often associated with diabetic complications.63 Because epigenetic changes occur at many genes and stages of diabetes, there are many opportunities to utilize epigenetic therapy in the treatment of the disease.
Like diabetes, epigenetic changes that increase susceptibility to obesity can begin in utero. One study showed that there was a direct correlation between methylation status of the eNOS, RXRa, and SOD1 genes at birth with the level of adiposity at the age of 9 years.64 Another study implicated the H3K9 histone demethylase, Jhdm2a; a Jhdm2a knockout caused obesity and widespread downregulation of metabolically active genes in mouse models.65 Leptin, an adipose-derived hormone that regulates hunger and metabolism, is also subject to epigenetic control. Although the leptin promoter is heavily methylated at CpG islands in preadiposcytes, it becomes demethylated when maturing into a mature adipocyte cell. Interestingly, the leptin gene shows higher CpG methylation levels in mice with diet-induced obesity than in normal mice.66 There is clearly potential for epigenetic treatment of obesity, and this should be investigated further.
Cancer
Cancer is defined as an uncontrolled growth of cells with metastatic potential. It is caused by several mechanisms involving a combination of genetic and environmental sources. This has been well summarized by Hanahan and Weinberg who identified six potential ways in which cancer is produced, and deemed them the hallmarks of cancer.67 The way in which cancer progresses from a cancer stem/progenitor cell to various metastatic stages, however, is still poorly understood. Vogelstein and Kinzler postulated a stepwise progression of lung cancer.68 Recently, Sarkar et al postulated that epigenetic changes could influence cancer progenitor cell formation, cancer progression, and formation of stage-specific metastatic cancer.69 Epigenetic changes involve both histone modifications and DNA methylation at specific CpG residues. During carcinogenesis, many oncogenes, such as ras and src, become activated by mutations.70–73 Conversely, many tumor suppressor genes, which include both cell-cycle inhibitors and pro-apoptotic genes, are silenced by methylation of CpG islands in their promoter sites. In many cancers, there are several examples of silenced tumor suppressor genes, such as p21, p16, p27, differentiation marker RARβ2, and imprinted pro-apoptotic gene ARHI (in breast and ovarian cancer).74 The methylation of the upstream regions is caused by the upregulation of DNMT1 in cancer cells. The expression level of DNMT1 is observed to be high in almost all cancer cells compared with the normal tissue. DNMT1 expression alters with the cell-cycle progression of a normal cell, although it is always upregulated in cancer cells and maintains a higher methylation level. Once the DNA is methylated, methyl CpG-binding proteins and HDACs are recruited, preventing RNA polII-mediated transcription and inducing the closed chromatin state (Fig. 2). Unlike the genetic mutations that are also associated with cancer, the epigenetic changes that occur during this disease are, by nature, reversible and are thus a good target for epigenetic therapy. Sarkar et al proposed that targeting silenced tumor suppressor genes with epigenetic drugs could be an effective method to treat cancer. The group hypothesizes that re-expression of tumor suppressor genes weakens the cancer cells’ ability to withstand cytotoxic treatment, allowing for what would normally be a suboptimal dose of a cytotoxic drug to become an effective cancer killer.69,73,75
What are Epigenetic Drugs and What are their Uses?
Methylation inhibiting drugs
Nucleoside-like compounds are the oldest form of methylation inhibitors, and several of these compounds have been FDA approved for the treatment of certain cancers. 5-Azacytidine (Aza; market name Vidaza) has been known to have cytotoxic effects on cancer cells since 1968; however, its mechanism of action was discovered more recently.76 The drug is a cytidine analog, with a nitrogen atom in the place of Carbon 5. Once observed into the cell, it is phosphorylated and then incorporated into DNA during replication. The analog is recognized by DNMT1, and the normal reaction involving the transfer of a methyl group begins to take place. The nitrogen group in the fifth position, however, causes the formation of an irreversible DNMT1–aza linkage, which triggers the degradation of the enzyme and leads to widespread reductions in methylation.77,78 Because aza integrates into DNA during replication, rapidly dividing cancer cells are more susceptible to its effects. Although the drug has been FDA approved, there is still potential for improvement, as it is relatively unstable, can have toxic side effects, and is not available to be taken orally.
Zebularine is another cytidine analog that has a mechanism similar to aza, integrating into DNA and forming a covalent bond with DNMT1. Although the drug is not yet FDA approved, this isoform has had good results in mouse models. One study showed that Zebularine can inhibit DNA methylation and induce re-expression of methylation-silenced genes, even when given orally. This was the first drug of its kind to have successful inhibition of methylation without intravenous administration. Because Zebularine is more stable than aza and can be taken orally, it could be an effective anticancer drug in the future.79
Antisense oligonucleotides are also used to inhibit methylation. MG98 is a 20-base pair antisense oligonucleotide that binds the 3′ untranslated region of DNMT1, preventing transcription of the gene.80 Studies in mouse models of bladder and colon cancer showed that administration of MG98 led to re-expression of the tumor suppressor gene, p16. Clinical trials utilizing the drug have shown mixed results, but MG98 appears to have been successful in the treatment of advanced renal cell carcinoma. The most promising results came from a study in which MG98 was given in combination with Roferon-A, a known chemotherapeutic drug. Decreased levels of DNMT1 were observed and tumor progression was slowed, with minimal toxicity from MG98.81,82 Additional clinical trials examining the most effective dosages and dosing schedules for MG98 are currently in progress.
RG108 is a relatively new small molecule DNA methylation inhibitor that is currently being investigated. This drug does not intercalate into target DNA or bind to DNMT1 gene, but rather binds to and directly inhibits the DNMT1 enzyme active site. The in vitro use of RG108 in human cancer cell lines showed significant demethylation and re-expression of the p16 tumor suppressor gene and led to slowed cancer cell growth. Because the RG108 mechanism of action does not involve enzyme trapping, similar to nucleoside analogs, the toxicity of the drug is reduced. In addition, RG108 did not affect the methylation status of centromeric satellite repeats, an unexpected but advantageous trait that is likely to increase stability of hypomethylated chromatin.83 Because of RG108’s high specificity and low toxicity, this drug shows promise as an effective anticancer drug in the future.
Bromodomain and inhibitors
Bromodomains are conserved structural motifs associated with chromatin modifying proteins, such as HATs.84 Bromodomains, of which there are 61 different types, are considered epigenetic reader domains and are the only protein structure known to recognize acetylated lysine residues. This recognition is often a prerequisite for chromatin modification.85 Bromodomain and extra- terminal (BET) proteins are also known to mediate transcriptional elongation of acetylated chromatin, binding to the acetyl groups and interacting with RNAPII. Many studies have investigated the clinical importance of BRD4, a specific BET protein known to play an important role in mitosis.86,87 NUT-midline carcinoma is characterized by a BRD4 fusion protein that leads to global hypoacetylation and transcriptional repression.88 The same protein has been found to be overexpressed and associated with disease progression in plasma cell leukemia.89 Another study identified BRD4 as a regulator of human papillomavirus oncogene expression.90
Because of BET proteins’ unique function and specificity, they are a promising target for therapeutic interventions. Bromodomain inhibitors have been shown to be effective in the downregulation of c-Myc, a gene often implicated in cancer, by disruption of the interaction between BET proteins and acetylated histones. One study examined the effect of JQ1, a selective bromodomain inhibitor in mouse models of multiple myeloma, a myc-dependent cancer. JQ1 competitively binds acetylated lysine residues, displacing BET proteins from the histone, leading to widespread downregulation of c-Myc and its target proteins.91,92 Inhibitor of BET726 (I-BET726) is a selective small molecule inhibitor of BET proteins that binds to the acetyl-lysine recognition pocket of BET family proteins. It binds with high affinity to BRD2, BRD3, and BRD4 and competes with tetra-acetylated histone H4 peptides (K5ac, K8ac, K12ac, and K16ac) for binding to the bromodomains of these proteins. I-BET726 is therefore highly selective for BET family proteins. I-BET726 also directly regulates expression of BCL2, an antiapoptotic gene that is highly expressed in a number to tumor types. For example, I-BET726 has been found to inhibit neuroblastoma tumor growth.93
HAT inhibitors
Histone acetylase (HAT) inhibitor compounds have been identified to inhibit the catalytic activity of HATs in many cancers and diseases. Although they are not very selective and bind multiple classes of proteins, HAT inhibitors seem promising for treatment of various diseases. Bisubstrate inhibitors were the first discovered to selectively inhibit HATs p300 and PCAF and re-express tumor suppressor genes in cancers.94,95 Naturally occurring HAT inhibitors, such as curcumin, inhibit histone H3 and histone H4 acetylation by p300 and CBP.96 This results in inhibition of cell proliferation and induction of apoptosis as reported in tumor cells, and more recently studied in prevention of heart failure in rats.97,98 Garcinol and anacardiac acid also inhibit acetylation by p300 and PCAF, as well as induce apoptosis and downregulate global gene expression. These two natural compounds however exhibit relatively low potency, which limits the authenticity of their activity.99,100 Other inhibitors of HATs have been explored, such as isothiazolones that inhibit PCAF and p300. The inhibition is due to the structural differences in the isothiazolone family at the enzyme active site, where they are modified with diverse substitutions.101
Recent studies have progressed to yield increased selectivity of HATs. Lys-CoA, a large bisubstrate inhibitor, is seen to be a submicromolar inhibitor of p300 with high selectivity; however, it is generally inactive in mammalian cell systems, exhibiting nondrug-like properties unless modified with the co-administration of moderately cytotoxic detergents or administered via microinjection. C646, another p300 inhibitor, may be the only potent and selective HAT inhibitor discovered so far. The compound binds at the predicted druggable pocket of p300 and acts as a cofactor competitor.102,103 C646 can mimic the proapoptotic effect of RNA-mediated p300 knockdown, which involves both extrinsic and intrinsic cell death pathways, as seen in prostate cancer cells.104 Since HATs function in cells as part of large multiprotein complexes, the formation of these specific complexes may be necessary for the discovery of further inhibitors.
Protein methyltransferase inhibitors
Methylation of lysine and arginine residues plays an important role in gene transcription. These modifications are catalyzed by protein methyltransferases (PMTs). The enzymatic activities have suggested pathological roles in cancer, neurodegenerative diseases, and inflammatory diseases. Inhibition of PMTs has been shown to stop these enzymatic alterations.105 BIX-01294 was the first selective inhibitor of a protein lysine methyl transferase. Although the drug has good potency in terms of blocking protein–protein interactions, it is toxic in cellular assays at high concentrations. Second-generation inhibitors include E72 and UNC321, which incorporate 7-alkoxyamine tethered to the quinazoline core as a structural modification.106,107 Another drug, UNC0646, has a remarkable toxicity/function ratio in certain cell lines, such as MCF7, 22RV1, and IMR90 cells. UNC0638 is also potent, selective, and has low cell toxicity making it an excellent inhibitor of PMT.108
Histone methylation inhibitors
New inhibitors synthesized to inhibit trimethylation have been shown to re-activate developmentally regulated genes. Recently, 3- deazaneplanocin A (DZNep) was reposted to selectively inhibit trimethylation of lysine 27 on histone H3 (H3K27me3) and lysine 20 on histone H4 (H4K20me3), as well as reactivate silenced genes in cancer cells. A recent study observed that these inhibitors reactivate developmental genes that are not silenced by DNA methylation.109
HDAC inhibitors
Similar to HDAC proteins, HDACi are also structurally diverse. HDACi can be categorized into several groups according to structure, including short-chain fatty acids, hydroxamic acids, epoxyketones, and benzamides.110 Although HDACs appear widespread on DNA as chromatin structure remodelers, HDACi only affect 2–10% of expressed genes.111 Accordingly, this demonstrates the potential for specificity in using HDACi as a therapeutic drug, particularly in cancers, the current treatments for which often come with extreme side effects.
Hydroxamic acid inhibitors target Class I and II HDACs and have emerged as promising and potent treatments for cancers. In fact, aminosuberoyl hydroxamic acids, including suberanilohydroxamic acid (SAHA; market name Vorinostat), have been shown to inhibit HDACs and cell proliferation in nanomolar concentrations. In 2006, the US FDA approved Vorinostat as a treatment for progressive, persistent, or recurring cutaneous T-cell lymphoma, or for patients following two systemic chemotherapies.2 Vorinostat leads to hyperacetylation of histones as well as nonhistone proteins such as p53 and heat shock protein 90, inducing apoptosis and sensitizing tumors to cell death processes and other drugs. The ability of Vorinostat to sensitize cancerous cells to other drugs makes it an interesting candidate for combination epigenetic and non-epigenetic therapies. However, as Vorinostat has multiple targets, it also induces many side effects such as diarrhea, fatigue, nausea, and anorexia.112 Thus, the potential of HDACi to reduce these effects has not yet been realized.
A new approach to cancer drug development involves a combination of epigenetic drugs with other cytotoxic drugs, which is expected to increase cancer cell drug specificity and reduce toxic side effects. A recent study showed that HDAC inhibition sensitized breast and ovarian cancer cell lines to a variety of cytotoxic drugs, including calpeptin, TRAIL, and telomere homolog oligonucleotides.73 Interestingly, HDACi have also been shown to induce demethylation of silenced tumor-suppressor genes through the downregulation of DNMT1.113 The combination of HDACi and aza, a known methylation inhibitor, produced synergistic-type demethylation as compared to treatment with either alone.75 Clinical trials taking advantage of combination therapy involving HDACi have shown promising results. One trial administered aza in combination with entinostat (a class 1 HDACi) to patients with recurrent metastatic NSCLC. The combination was well tolerated and the majority of patients treated showed a decrease in methylation in at least two hypermethylated promoters. Eight of the 10 patients enrolled in the trial had either stable disease or objective responses to the therapy, with four of them showing major objective responses.72 ACY-1215 is an HDACi that specifically targets HDAC6. Preclinical trials involving administration of ACY-125 with bortezumib, a proteasome inhibitor, showed positive results, with the combination treatment inducing significant antimultiple myeloma effects in mouse models.114 Clinical trials in patients are in progress, with early results suggesting that the combination therapy is well tolerated and at least maintains a stable state of disease.115
In addition to treating cancers, HDACi also show great potential for the treatment of neurodegenerative and psychiatric diseases. HDACi have been shown to reduce memory loss and have neuroprotective effects in many studies. For example, one group showed that the administration of valproic acid after cerebral ischemia significantly reduced infarct size and neurological deficit scores, suggesting that HDACi could serve as a drug to prevent permanent brain damage following stroke.116 Another study showed that increased histone acetylation was associated with the recovery of long-term memories and learned behavior.117 HDAC2 was later found to negatively impact memory formation and synaptic plasticity, with mice overexpressing HDAC2 found to have decreased expression of several key neuronal genes, including BDNF, a gene known to be implicated in Alzheimer’s disease. HDAC2 knockout mice and mice given HDACi showed increased expression of the neuronal genes and increased memory formation and retention.118 Many studies have since investigated the effect of HDACi on memory retention in mouse models of Alzheimer’s disease, with promising results shown.119 The development of specific HDACi for use in human could be a considerable step in finding a cure to Alzheimer’s and related neurodengerative diseases, especially considering the recent shift of focus on drugs with neuroprotective rather than neuro-reparative effects for the treatment and prevention of the disease.120
In addition to minimizing brain damage after stroke, inhibition of HDACs in cultured embryonic cells favors myogenesis and angiogenesis and improves functional myocardial recovery after myocardial infarction (MI). Another use of HDACi is in stem cell therapy for heart muscle regeneration. Reprogramming differentiated cells, such as fibroblasts, into induced pluripotent stem (iPs) cells is accomplished through vector transduction of Yamanka factors. iPs cells can then be employed in therapies to be differentiated into patient-specific cells, such as vascular endothelium. Although current approaches for making iPS are very inefficient, HDACi such as Trichostatin A (TSA) have greatly improved the effectiveness of iPs cell induction, increasing cardiac-specific transcription factors, which play an important role in differentiation of stem cells into endothelial cells and cardiomyoctes.121
Future Direction of Epigenetic Drugs
Epigenetic changes involve histone and DNA modifications, which can result in drastic phenotypic changes—phenomena that are particularly interesting because these epigenetic events are inherently reversible. In response to circumstantial and environmental changes, epigenetic modifications can proceed forward or backward, even removing the modification completely and reverting the substrate back to its original state. Current research has shown that histone modifications are linked to CpG nucleotide methylation in DNA, thus connecting multiple forms of epigenetic modifications and regulations.73 This link poses a new scenario where an “epigenetic code” could dictate the expression of a particular set of genes, in essence serving as an “on/off” switch for many cellular events.122 As epigenetic drugs continue to be developed and increased sensitivity and specificity are obtained, greater control over this epigenetic switch is possible. Once the correct combinations of treatments are developed, it may be possible to utilize the switch to reverse the disease phenotype, particularly if drugs are administered during early disease progression. For example, it has been proposed that epigenetic drugs may prevent the formation of cancer progenitor cells while also killing drug-resistant cancer cells.70,72,122,123 As described in this review, there are many more examples of diseases for which epigenetic treatment holds great promise. Future studies will demonstrate how much exploitation of epigenetic events could be useful for preventing and treating different diseases.
Glossary
Abbreviations
- CpG
cytosine–phosphate–guanine dinucleotide
- DNMT3A
DNA methyltransferase 3A
- DNMT3B
DNA methyltransferase 3B
- DNMT1
DNA methyltransferase 1
- MBDP
methyl-binding domain protein
- H2A
histone 2A
- H2B
histone 2B
- H3
histone 3
- H4
histone 4
- H3K4
lysine 4 of histone 3
- H3K27
lysine 27 of histone 3
- H3K20
lysine 20 of histone 3
- H3K4me2
dimethylation of histone 3 lysine 4
- H3K27me3
trimethylation of histone 3 lysine 27
- H3K20me3
trimethylation of histone 3 lysine 20
- HAT
histone acetyl transferase
- HDAC
histone deacetylase
- CBP
CREB-binding protein
- PCAF
P300/CBP-associated factor
- GCN5
general control of amino acid synthesis protein 5
- COPD
chronic obstructive pulmonary disease
- GCNF
germ cell nuclear factor
- NAD+
nicotinamide-adenine-dinucleotide
- ATP
adenosine- triphosphate
- HDACi
histone deacetylase inhibitor
- ESR1
estrogen receptor 1
- ESR2
estrogen receptor 2
- NOS3
nitric oxide synthase 3
- eNOS
endothelial nitric oxide synthase
- H4K12
lysine 12 of histone 4
- NO
nitric oxide
- IGF2
insulin-like growth factor 2
- ANRIL
antisense noncoding RNA in the INK4 locus
- LINE-1
long interspersed nucleotide element 1
- INSIGF
insulin/insulin-like growth factor 2
- MeCP2
methyl CpG-binding protein 2
- Pdx1
pancreatic and duodenal homeobox 1
- Set7
Histone H3-K4 methyltransferase
- NFkB-65
NF-kappa-B p65 subunit
- RXRa
retinoid X receptor alpha
- SOD1
superoxide dismutase 1
- Jhdm2a
JmjC domain-containing histone demethylation protein 2A
- p21
cyclin-dependent kinase inhibitor 1
- p16
cyclin-dependent kinase inhibitor 2A
- p27
Cyclin-dependent kinase inhibitor 1B
- RARβ2
retinoic acid receptor β2
- ARHI
DIRAS family, GTP-binding RAS-like 3
- POLIII
RNA polymerase III
- Aza
5-aza-2′-deoxycytidine
- BET
bromodomain and extra-terminal
- RNAPII
RNA polymerase II holoenzyme
- BRD4
bromodomain containing 4
- NUT
nuclear protein in testis
- c-Myc
v-myc avian myelocytomatosis viral oncogene homolog
- JQ1, (S)-JQ1
bromodomain inhibitor (for BRD2, 3, 4, and T)
- BET726
bromodomain and extra-terminal 726
- I-BET726
inhibitor of bromodomain and extra-terminal 726
- BRD2
bromodomain containing protein 2
- BRD3
bromodomain containing protein 3
- K5ac
acetylation at lysine 5
- K8ac
acetylation at lysine 8
- K12ac
acetylation at lysine 12
- K16ac
acetylation at lysine 16
- BCL2
b-cell lymphoma 2
- Lys-CoA
conjugate of lysine and coenzyme A
- C646
histone acetyltransferase p300 inhibitor
- PMT
protein methyltransferase
- BIX-01294
diazepinquinazolin-amine derivative
- MCF7
human breast carcinoma cell line
- 22RV1
human prostate carcinoma cell line
- IMR90
human fetal lung cell line
- DZNep
3-deazaneplanocin A
- SAHA
suberoylanilide hydroxamic acid
- TRAIL
TNF-related apoptosis-inducing ligand
- NSCLC
non-small-cell lung cancer
- HDAC2
histone deacetylase 2
- BDNF
brain-derived neurotrophic factor
- MI
myocardial infarction
- iPs
induced pluripotent stem (cells)
- TSA
Trichostatin A
Footnotes
Author Contributions
All authors contributed to the writing, editing, and review of this article. SS conceived ideas and commentary presented here.
ACADEMIC EDITOR: Christian Bronner, Editor in Chief
FUNDING: The research work of SS was partially supported by a grant from ACS. SH, KL, and NS were supported by UROP, BU.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
This paper was subject to independent, expert peer review by a minimum of two blind peer reviewers. All editorial decisions were made by the independent academic editor. All authors have provided signed confirmation of their compliance with ethical and legal obligations including (but not limited to) use of any copyrighted material, compliance with ICMJE authorship and competing interests disclosure guidelines and, where applicable, compliance with legal and ethical guidelines on human and animal research participants. Provenance: the authors were invited to submit this paper.
REFERENCES
- 1.Ptak C, Petronis A. Epigenetics and complex disease: from etiology to new therapeutics. Annu Rev Pharmacol Toxicol. 2008;48:257–276. doi: 10.1146/annurev.pharmtox.48.113006.094731. [DOI] [PubMed] [Google Scholar]
- 2.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur RFDA. Approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 3.Mikaelsson MA, Miller CA. The path to epigenetic treatment of memory disorders. Neurobiol Learn Mem. 2011;96:13–18. doi: 10.1016/j.nlm.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 5.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 6.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 7.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 8.Marino-Ramirez L, Kann MG, Shoemaker BA, Landsman D. Histone structure and nucleosome stability. Expert Rev Proteomics. 2005;2:719–729. doi: 10.1586/14789450.2.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng JC, Karpen GH. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat Cell Biol. 2007;9:25–35. doi: 10.1038/ncb1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol. 2008;20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawakami K, Nakamura A, Ishigami A, Goto S, Takahashi R. Age-related difference of site-specific histone modifications in rat liver. Biogerontology. 2009;10:415–421. doi: 10.1007/s10522-008-9176-0. [DOI] [PubMed] [Google Scholar]
- 13.Maures TJ, Greer EL, Hauswirth AG, Brunet A. The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin- dependent manner. Aging Cell. 2011;10:980–990. doi: 10.1111/j.1474-9726.2011.00738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 15.Sarkar S, Longacre M, Tatur N, Heerboth S, Lapinska K. Encyclopedia of Analytical Chemistry. John Wiley & Sons, Ltd; 2006. Available at http://onlinelibrary.wiley.com/doi/10.1002/9780470027318.a9365/abstract. [Google Scholar]
- 16.Barnes PJ. Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol. 2009;71:451–464. doi: 10.1146/annurev.physiol.010908.163257. [DOI] [PubMed] [Google Scholar]
- 17.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 2009;8:1056–1072. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 18.Banerjee HN, Verma M. Epigenetic mechanisms in cancer. Biomark Med. 2009;3:397. doi: 10.2217/bmm.09.26. [DOI] [PubMed] [Google Scholar]
- 19.Iyer NG, Özdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225–4231. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- 20.Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert Rev Anticancer Ther. 2010;10:935–954. doi: 10.1586/era.10.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maurice T, Duclot F, Meunier J, et al. Altered memory capacities and response to stress in p300/CBP-associated factor (PCAF) histone acetylase knockout mice. Neuropsychopharmacology. 2007;33:1584–1602. doi: 10.1038/sj.npp.1301551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Ruijter AJM, van Gennip AH, Caron HN, Kemp S, van Kuilenburg ABP. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hildmann C, Wegener D, Riester D, et al. Substrate and inhibitor specificity of class 1 and class 2 histone deacetylases. J Biotechnol. 2006;124:258–270. doi: 10.1016/j.jbiotec.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 24.Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 25.Ng HH, Zhang Y, Hendrich B, et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet. 1999;23:58–61. doi: 10.1038/12659. [DOI] [PubMed] [Google Scholar]
- 26.Yang X-J, Grégoire S. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol Cell Biol. 2005;25:2873–2884. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu T, Liu PY, Marshall GM. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009;69:1702–1705. doi: 10.1158/0008-5472.CAN-08-3365. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto H, Schoonjans K, Auwerx J. Sirtuin functions in health and disease. Mol Endocrinol. 2007;21:1745–1755. doi: 10.1210/me.2007-0079. [DOI] [PubMed] [Google Scholar]
- 29.Ordovás JM, Smith CE. Epigenetics and cardiovascular disease. Nat Rev Cardiol. 2010;7:510–519. doi: 10.1038/nrcardio.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lund G, Zaina S. Atherosclerosis: an epigenetic balancing act that goes wrong. Curr Atheroscler Rep. 2011;13:208–214. doi: 10.1007/s11883-011-0174-3. [DOI] [PubMed] [Google Scholar]
- 31.Post WS, Goldschmidt-Clermont PJ, Wilhide CC, et al. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res. 1999;43:985–991. doi: 10.1016/s0008-6363(99)00153-4. [DOI] [PubMed] [Google Scholar]
- 32.Beedle AS, Buklijas T, Gluckman PD, Hanson MA, Low FM. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol. 2009;5:401. doi: 10.1038/nrendo.2009.102. [DOI] [PubMed] [Google Scholar]
- 33.Mattson MP, Kruman II, Duan W. Folic acid and homocysteine in age-related disease. Ageing Res Rev. 2002;1:95–111. doi: 10.1016/s0047-6374(01)00365-7. [DOI] [PubMed] [Google Scholar]
- 34.Rodríguez-Nieto S, Chavarría T, Martínez-Poveda B, Sánchez-Jiménez F, Rodríguez Quesada A, Medina MA. Anti-angiogenic effects of homocysteine on cultured endothelial cells. Biochem Biophys Res Commun. 2002;293:497–500. doi: 10.1016/S0006-291X(02)00232-2. [DOI] [PubMed] [Google Scholar]
- 35.Fish JE, Matouk CC, Rachlis A, et al. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem. 2005;280:24824–24838. doi: 10.1074/jbc.M502115200. [DOI] [PubMed] [Google Scholar]
- 36.Fish JE, Yan MS, Matouk CC, et al. Hypoxic repression of endothelial nitricoxide synthase transcription is coupled with eviction of promoter histones. J Biol Chem. 2010;285:810–826. doi: 10.1074/jbc.M109.067868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McQuillan LP, Leung GK, Marsden PA, Kostyk SK, Kourembanas S. Hypoxia inhibits expression of eNOS via transcriptional and posttranscriptional mechanisms. Am J Physiol. 1994;267:H1921–H1927. doi: 10.1152/ajpheart.1994.267.5.H1921. [DOI] [PubMed] [Google Scholar]
- 38.Sharma P, Kumar J, Garg G, et al. Detection of altered global DNA methylation in coronary artery disease patients. DNA Cell Biol. 2008;27:357–365. doi: 10.1089/dna.2007.0694. [DOI] [PubMed] [Google Scholar]
- 39.McPherson R, Pertsemlidis A, Kavaslar N, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holdt LM, Beutner F, Scholz M, et al. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol. 2010;30:620–627. doi: 10.1161/ATVBAHA.109.196832. [DOI] [PubMed] [Google Scholar]
- 41.Yap KL, Li S, Muñoz-Cabello AM, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38:662–674. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singhal A. Early nutrition and long-term cardiovascular health. Nutr Rev. 2006;64:S44–S49. doi: 10.1301/nr.2006.may.s44-s49. [DOI] [PubMed] [Google Scholar]
- 43.Dolinoy DC, Weidman JR, Jirtle RL. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol. 2007;23:297–307. doi: 10.1016/j.reprotox.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 44.Zhang D, Yu Z, Cruz P, Kong Q, Li S, Kone BC. Epigenetics and the control of epithelial sodium channel expression in collecting duct. Kidney Int. 2009;75:260–267. doi: 10.1038/ki.2008.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baccarelli A, Wright R, Bollati V, et al. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology. 2010;21:819–828. doi: 10.1097/EDE.0b013e3181f20457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirabayashi Y, Gotoh Y. Epigenetic control of neural precursor cell fate during development. Nat Rev Neurosci. 2010;11:377–388. doi: 10.1038/nrn2810. [DOI] [PubMed] [Google Scholar]
- 47.Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 49.Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of Neurological Defects in a Mouse Model of Rett Syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurodevelopmental and neurodegenerative disease. Nat Med. 2012;18:1194–1204. doi: 10.1038/nm.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hernandez DG, Nalls MA, Raphael GJ, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20:1164–1172. doi: 10.1093/hmg/ddq561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siegmund KD, Connor CM, Campan M, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One. 2007;2:e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Desplats P, Spencer B, Coffee E, et al. α-Synuclein sequesters Dnmt1 from the nucleus. J Biol Chem. 2011;286:9031–9037. doi: 10.1074/jbc.C110.212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kontopoulos E, Parvin JD, Feany MB. α-Synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- 56.MacDonald ME, Ambrose CM, Duyao MP, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 57.Cong S-Y, Pepers BA, Evert BO, et al. Mutant huntingtin represses CBP, but not p300, by binding and protein degradation. Mol Cell Neurosci. 2005;30:12–23. doi: 10.1016/j.mcn.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 58.Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2005;13:539–550. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghoshal K, Li X, Datta J, Bai S, Al E. A folate- and methyl-deficient diet alters the expression of DNA methyltransferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats 1, 2. J Nutr. 2006. Available at http://www.highbeam.com/doc/1P3-1046731851.html. [DOI] [PMC free article] [PubMed]
- 61.Wren JD, Garner HR. Data-mining analysis suggests an epigenetic pathogenesis for type 2 diabetes. J Biomed Biotechnol. 2005;2005:104–112. doi: 10.1155/JBB.2005.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pinney SE, Santos LJJ, Han Y, Stoffers DA, Simmons RA. Exendin-4 increases histone acetylase activity and reverses epigenetic modifications that silence Pdx1 in the intrauterine growth retarded rat. Diabetologia. 2011;54:2606–2614. doi: 10.1007/s00125-011-2250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brasacchio D, Okabe J, Tikellis C, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes. 2009;58:1229–1236. doi: 10.2337/db08-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Godfrey KM, Sheppard A, Gluckman PD, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011;60:1528–1534. doi: 10.2337/db10-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tateishi K, Okada Y, Kallin EM, Zhang Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature. 2009;458:757–761. doi: 10.1038/nature07777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Milagro FI, Campión J, García-Díaz DF, Goyenechea E, Paternain L, Martínez JA. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J Physiol Biochem. 2009;65:1–9. doi: 10.1007/BF03165964. [DOI] [PubMed] [Google Scholar]
- 67.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 68.Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9:138–141. doi: 10.1016/0168-9525(93)90209-z. [DOI] [PubMed] [Google Scholar]
- 69.Byler S, Goldgar S, Heerboth S, et al. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. 2014;34:1071–1077. [PubMed] [Google Scholar]
- 70.Mataga M, Rosenthal S, Heerboth S, et al. Anti-breast cancer effects of histone deacetylase inhibitors and calpain inhibitor. Anticancer Res. 2012;32:2523–2530. [PubMed] [Google Scholar]
- 71.Frew A, Lindemann R, Martin B, et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc Natl Acad Sci U S A. 2008;105(32):11317–11322. doi: 10.1073/pnas.0801868105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sarkar S, Faller DV. T-oligos inhibit growth and induce apoptosis in human ovarian cancer cells. Oligonucleotides. 2011;21(1):47–53. doi: 10.1089/oli.2010.0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sarkar S, Horn G, Moulton K, et al. Cancer development, progression, and therapy: an epigenetic overview. Int J Mol Sci. 2013;14:21087–21113. doi: 10.3390/ijms141021087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Monier R. Oncogenes and anti-oncogenes in tumorigenesis. Reprod Nutr Dev. 1990;30:445–454. doi: 10.1051/rnd:19900319. [DOI] [PubMed] [Google Scholar]
- 75.Sarkar S, Goldgar S, Byler S, Rosenthal S, Heerboth S. Demethylation and re-expression of epigenetically silenced tumor suppressor genes: sensitization of cancer cells by combination therapy. Epigenomics. 2013;5:87–94. doi: 10.2217/epi.12.68. [DOI] [PubMed] [Google Scholar]
- 76.Kaminskas E, Farrell AT, Wang Y-C, Sridhara R, Pazdur R. FDA drug approval summary: azacitidine (5-azacytidine, Vidaza™) for injectable suspension. Oncologist. 2005;10:176–182. doi: 10.1634/theoncologist.10-3-176. [DOI] [PubMed] [Google Scholar]
- 77.Momparler RL. Pharmacology of 5-Aza-2′-deoxycytidine (decitabine) Semin Hematol. 2005;42(suppl 2):S9–S16. doi: 10.1053/j.seminhematol.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 78.Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci U S A. 1984;81:6993–6997. doi: 10.1073/pnas.81.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cheng JC, Matsen CB, Gonzales FA, et al. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst. 2003;95:399–409. doi: 10.1093/jnci/95.5.399. [DOI] [PubMed] [Google Scholar]
- 80.Amato RJ. Inhibition of DNA methylation by antisense oligonucleotide MG98 as cancer therapy. Clin Genitourin Cancer. 2007;5:422–426. doi: 10.3816/CGC.2007.n.029. [DOI] [PubMed] [Google Scholar]
- 81.Davis AJ, Gelmon KA, Siu LL, et al. Phase I and pharmacologic study of the human DNA methyltransferase antisense oligodeoxynucleotide MG98 given as a 21-day continuous infusion every 4 weeks. Invest New Drugs. 2003;21:85–97. doi: 10.1023/a:1022976528441. [DOI] [PubMed] [Google Scholar]
- 82.Winquist E, Knox J, Ayoub J-P, et al. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: a National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Invest New Drugs. 2006;24:159–167. doi: 10.1007/s10637-006-5938-1. [DOI] [PubMed] [Google Scholar]
- 83.Brueckner B, Boy RG, Siedlecki P, et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005;65:6305–6311. doi: 10.1158/0008-5472.CAN-04-2957. [DOI] [PubMed] [Google Scholar]
- 84.Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002;513:124–128. doi: 10.1016/s0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- 85.Dawson MA, Kouzarides T, Huntly BJP. Targeting epigenetic readers in cancer. N Engl J Med. 2012;367:647–657. doi: 10.1056/NEJMra1112635. [DOI] [PubMed] [Google Scholar]
- 86.Jang MK, Mochizuki K, Zhou M, Jeong H-S, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 87.Maruyama T, Farina A, Dey A, et al. Mammalian bromodomain protein, Brd4, interacts with replication factor C and inhibits progression to S phase. Mol Cell Biol. 2002;22:6509–6520. doi: 10.1128/MCB.22.18.6509-6520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.French CA, Kutok JL, Faquin WC, et al. Midline carcinoma of children and young adults with NUT rearrangement. J Clin Oncol. 2004;22:4135–4139. doi: 10.1200/JCO.2004.02.107. [DOI] [PubMed] [Google Scholar]
- 89.Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Smith JA, White EA, Sowa ME, et al. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc Natl Acad Sci U S A. 2010;107:3752–3757. doi: 10.1073/pnas.0914818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mertz JA, Conery AR, Bryant BM, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wyce A, Ganji G, Smitheman KN, et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS One. 2013;8:e72967. doi: 10.1371/journal.pone.0072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dekker FJ, Haisma HJ. Histone acetyl transferases as emerging drug targets. Drug Discov Today. 2009;14:942–948. doi: 10.1016/j.drudis.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 95.Zheng Y, Thompson PR, Cebrat M, et al. Selective HAT inhibitors as mechanistic tools for protein acetylation. Methods Enzymol. 2003;376:188–199. doi: 10.1016/S0076-6879(03)76012-1. [DOI] [PubMed] [Google Scholar]
- 96.Balasubramanyam K, Varier RA, Altaf M, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279:51163–51171. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- 97.Bachmeier B, Nerlich A, Iancu C, et al. The chemopreventive polyphenol curcumin prevents hematogenous breast cancer metastases in immunodeficient mice. Cell Physiol Biochem. 2007;19:137–152. doi: 10.1159/000099202. [DOI] [PubMed] [Google Scholar]
- 98.Morimoto T, Sunagawa Y, Kawamura T, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest. 2008;118:868–878. doi: 10.1172/JCI33160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Balasubramanyam K, Altaf M, Varier RA, et al. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem. 2004;279:33716–33726. doi: 10.1074/jbc.M402839200. [DOI] [PubMed] [Google Scholar]
- 100.Hemshekhar M, Sebastin Santhosh M, Kemparaju K, Girish KS. Emerging roles of anacardic acid and its derivatives: a pharmacological overview. Basic Clin Pharmacol Toxicol. 2012;110:122–132. doi: 10.1111/j.1742-7843.2011.00833.x. [DOI] [PubMed] [Google Scholar]
- 101.Stimson L, Rowlands MG, Newbatt YM, et al. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol Cancer Ther. 2005;4:1521–1532. doi: 10.1158/1535-7163.MCT-05-0135. [DOI] [PubMed] [Google Scholar]
- 102.Bowers EM, Yan G, Mukherjee C, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003;278:19134–19140. doi: 10.1074/jbc.M301580200. [DOI] [PubMed] [Google Scholar]
- 104.Santer FR, Höschele PPS, Oh SJ, et al. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Ther. 2011;10:1644–1655. doi: 10.1158/1535-7163.MCT-11-0182. [DOI] [PubMed] [Google Scholar]
- 105.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 106.Chang Y, Ganesh T, Horton JR, et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J Mol Biol. 2010;400:1–7. doi: 10.1016/j.jmb.2010.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu F, Chen X, Allali-Hassani A, et al. Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J Med Chem. 2010;53:5844–5857. doi: 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fu L, Yan F-X, An X-R, Hou J. Effects of the histone methyltransferase inhibitor UNC0638 on histone H3K9 dimethylation of cultured ovine somatic cells and development of resulting early cloned embryos. Reprod Domest Anim. 2014;49(2):21–25. doi: 10.1111/rda.12277. [DOI] [PubMed] [Google Scholar]
- 109.Miranda TB, Cortez CC, Yoo CB, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8:1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 111.Belvedere S, Witter DJ, Yan J, Secrist JP, Richon V, Miller TA. Aminosuberoyl hydroxamic acids (ASHAs): a potent new class of HDAC inhibitors. Bioorg Med Chem Lett. 2007;17:3969–3971. doi: 10.1016/j.bmcl.2007.04.089. [DOI] [PubMed] [Google Scholar]
- 112.Richon VM. Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br J Cancer. 2006;95:S2–S6. [Google Scholar]
- 113.Sarkar S, Abujamra AL, Leow JE, Forman LW, Perrine SP, Faller DV. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 2011;31:2723–2732. [PubMed] [Google Scholar]
- 114.Santo L, Hideshima T, Kung A, et al. Preclinical activity, pharmacodynamic and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119(11):2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Raje N, Vogl D, Parameswaran H, et al. ACY-1215, a selective histone deacetylase 6 inhibitor: interim results of combination therapy with bortezomib in patients with multiple myeloma. American Society of Hematology Annual Meeting. Ernest N. Morial Convention Center, New Orleans. Oral Presentation. 2013 [Google Scholar]
- 116.Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89:1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- 117.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 118.Guan J-S, Haggarty SJ, Giacometti E, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kilgore M, Miller CA, Fass DM, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2010;35:870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Salomone S, Caraci F, Leggio GM, Fedotova J, Drago F. New pharmacological strategies for treatment of Alzheimer’s disease: focus on disease modifying drugs. Br J Clin Pharmacol. 2012;73:504–517. doi: 10.1111/j.1365-2125.2011.04134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Webster AL, Yan MS, Marsden PA. Epigenetics and cardiovascular disease. Can J Cardiol. 2013;29:46–57. doi: 10.1016/j.cjca.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 122.Byler S, Sarkar S. Do epigenetic drug treatments hold the key to killing cancer progenitor cells? Epigenomics. 2014;6:161–165. doi: 10.2217/epi.14.4. [DOI] [PubMed] [Google Scholar]
- 123.Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2:S4–S11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]