

Abstract

Three new decalin-type tetramic acid analogues, pyrrolocins A (1), B (2), and C (3), were defined as products of a metabolic pathway from a fern endophyte, NRRL 50135, from Papua New Guinea. NRRL 50135 initially produced 1 but ceased its production before chemical or biological evaluation could be completed. Upon transfer of the biosynthetic pathway to a model host, 1–3 were produced. All three compounds are structurally related to equisetin-type compounds, with 1 and 3 having a trans-decalin ring system, while 2 has a cis-fused decalin. All were active against Mycobacterium tuberculosis, with the trans-decalin analogues 1 and 3 exhibiting lower MICs than the cis-decalin analogue 2. Here we report the isolation, structure elucidation, and antimycobacterial activities of 1–3 from the recombinant expression as well as the isolation of 1 from the wild-type fungus NRRL 50135.
Papua New Guinea is a hot spot for plant biodiversity, harboring an estimated 20 000 individual species of vascular plants. An estimated 60% of these are endemic to Papuasia, one of the highest rates of endemism in the world.1 Further, it is thought that most tropical plants contain multiple endophytic fungi,2 providing an immense unexplored reservoir of secondary metabolites.
As part of our International Cooperative Biodiversity Group (ICBG) program, we screened extracts of endophytic fungi isolated from Papua New Guinea terrestrial and marine sources for antibacterial activities. A methanol extract of a phylogenetically novel strain (NRRL accession number 50135) isolated from the stem of an Asplenium sp. fern growing in Watunou, Milne Bay Province, Papua New Guinea, was identified as selectively active against Gram-positive bacteria including Mycobacterium tuberculosis H37Ra (ATCC 25177), Staphylococcus aureus, Streptococcus pneumoniae, and Bacillus subtilis ATCC6633. We employed an antimycobacterial bioassay-guided isolation to obtain the TB-active metabolite pyrrolocin A (1), a trans-fused decalin-containing tetramic acid analogue related to equisetin.3 When NRRL 50135 stopped producing 1, we used a recombinant expression platform4 to induce its production. This heterologous expression platform successfully produced 1, matching the initial molecular weight, UV, NMR, and CD spectra, as well as two additional desmethyl analogues, pyrrolocin B (2) and pyrrolocin C (3). The three compounds have different configurations in the decalin ring system, with 2 having the cis-fused decalin ring and 1 and 3 having the trans-decalin configuration. To our knowledge, this is the first report of both cis and trans configurations being isolated from a single biological source and may be valuable in understanding the biosynthesis of this group of tetramic acid analogues. Furthermore, transplanting, or indeed rescuing, synthetic pathways from one organism into more tractable backgrounds unquestionably furthers drug discovery by magnifying the number and amount of rare natural chemicals with potential pharmacological activities, i.e., accessing dormant biosynthetic potential with a concomitant increase in production. Skillful manipulation of these pathways could theoretically also steer biosynthesis of undesirable intermediates or end products to pharmacologically desirable entities in quantities useful for experimentation or production.
Tetramic
acids are characterized by the presence of a 2,4-pyrrolidinedione
moiety in their structures. Tetramic acids that are related to equisetin
are particularly important because of their reported antimicrobial
activities,3,5−9 HIV-1 integrase inhibition,10,11 and microtubule assembly
inhibition.12 The mechanism of their antimicrobial
activities has been purported to be due to inhibition of SecA,13 inhibition of undecaprenyl pyrophosphate synthase,14 and inhibition of the histidine kinase WalK.15 Due to their biological activities, the biosynthesis
of this group of compounds and related polyketides was also the subject
of interest in several reviews.16−18
Results and Discussion
Initially, growth of the fungus on semisolid media led to the robust production of 1 at approximately 50 mg L–1. The compound was purified on the basis of promising activity against M. tuberculosis. The methanol extract from NRRL 50135 was purified using a preparative C18 column to isolate 1. High-resolution ESIMS determined its molecular formula to be C27H40NO5. The 13C NMR spectra (Table 1) showed 27 carbon signals, while its 1H NMR spectra presented five olefinic protons, five methyls, four methylenes, and six methines. The carbon connectivities in the decalin ring and the olefinic side chain were established by COSY, HMBC, and HSQC. Their association with equisetin-type tetramic acid was evident from a review of literature data that showed that 1 is related to phomasetin, with the addition of two carbons in the olefinic side chain and with the presence of an N-methyl group. In order to establish the relative and absolute configuration of 1 and evaluate its antimycobacterial activity, we fermented NRRL 50135 for additional material. Unfortunately, as is often observed in secondary metabolites isolated from cultivated microorganisms, NRRL 50135 ceased production of 1 before completion of this work. In order to achieve production of 1, we identified the biosynthetic locus for its production and moved it into the expression host, Fusarium heterosporum, as reported elsewhere.4 In this context, the major compounds produced were not those found in NRRL 50135, but instead comprised approximately 800 mg of desmethyl pyrrolocin analogues B (2) and C (3), along with a smaller amount of 1, per kilogram of fermentation.
Table 1. 1H and 13C NMR Data for Compound 1 (400 and 100 MHz, respectively, δ in ppm), 2, and 3 (500 and 125 MHz, respectively).
|
1a |
2b |
3a |
||||
|---|---|---|---|---|---|---|
| pos | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) |
| 1 | 195.7, C | 204.5, C | 198.6,c C | |||
| 2 | 48.8, C | 52.5, C | 49.0, C | |||
| 2-Me | 13.7, CH3 | 1.33, s | 19.2, CH3 | 1.30, s | 13.3, CH3 | 1.32, s |
| 3 | 39.3, CH | 1.57, br dd (10.2, 10.2) | 38.4, CH | 2.72, br d (10.2) | 40.4, CH | 1.57, m |
| 4 | 27.7, CH2 | 1.89, m | 24.6, CH2 | 1.38, m | 27.5, CH2 | 1.93, m |
| 1.00, m | 1.45, m | 1.00, m | ||||
| 5 | 35.4, CH2 | 1.70, m | 36.6, CH2 | 1.67, m | 35.4, CH2 | 1.71, m |
| 1.00, m | 0.87, dd (13.4, 12.6, 2.9) | 1.00, m | ||||
| 6 | 32.7, CH | 1.47, m | 29.8, CH | 1.40, d (5.3) | 32.9, CH | 1.49, m |
| 6-Me | 22.2, CH3 | 0.87, d (6.7) | 23.2, CH3 | 0.81, d (6.5) | 22.1, CH3 | 0.88, d (6.5) |
| 7 | 42.0, CH2 | 1.77, m | 38.4, CH2 | 1.56, d (14.0) | 42.0, CH2 | 1.77, m |
| 0.81, m | 1.11, dt (12.3, 5.3) | 0.82, dd (12.9, 11.7) | ||||
| 8 | 38.7, CH | 1.77, m | 36.7, CH | 2.09, br s | 38.7, CH | 1.79, m |
| 9 | 126.0, CH | 5.18, s | 126.8, CH | 5.09, s | 126.0, CH | 5.19, s |
| 10 | 130.9, C | 133.7, C | 131.6, C | |||
| 10-Me | 22.2, CH3 | 1.52, s | 23.3, CH3 | 1.63, s | 22.4, CH3 | 1.52, s |
| 11 | 48.8, CH | 3.14, m | 43.5, CH | 3.20, d (9.8) | 48.4, CH | 3.18, m |
| 12 | 130.7, CH | 5.21, dd (15.4, 9.8) | 134.1, CH | 5.50, dd (14.2, 10.5) | 131.0, CH | 5.21d |
| 13 | 131.3, CH | 5.69, dd (15.4, 10.4) | 135.9, CH | 6.04, dt (10, 3) | 132.1, CH | 5.72, dd (15.6, 10.0) |
| 14 | 130.9, CH | 5.86, dd, (15.4, 10.5) | 136.1, CH | 6.09, dt (10, 3) | 131.4, CH | 5.90, dd (15.0, 11.2) |
| 15 | 129.5, CH | 5.49, ddd, (15.4, 10, 7) | 130.3, CH | 5.63, pentet (7.5) | 130.3, CH | 5.51, dt (15.1, 7.0) |
| 16 | 42.1, CH2 | 2.09, ddd (13.8, 7, 7) | 42.1, CH2 | 2.24, pentet (6.4) | 42.4, CH2 | 2.09, m |
| 1.99, ddd (13.8, 7, 7) | 2.18, pentet (6.4) | 2.00, pentet (6.8) | ||||
| 17 | 65.6, CH | 3.56, pentet (6.5) | 68.7, CH | 3.78, m | 65.9, CH | 3.59, m |
| 18 | 22.7, CH3 | 0.98, d (6.5) | 23.1, CH3 | 1.14, d (6.20) | 23.1, CH3 | 0.98, d (6.1) |
| 2′ | 176.0,c C | 180.8, C | 179.2,c C | |||
| 3′ | 100.6,c C | 100.0,c C | 100.0,c C | |||
| 4′ | 190.6,c C | 192.9, C | 190.5, C | |||
| 5′ | 67.6, CH | 3.77, m | 62.4, CH | 3.87, br s | 63.3, CH | 3.79, m |
| 6′ | 58.0, CH2 | 3.80, dd (12, 2) | 64.3, CH2 | 3.83, dd (11.4, 3) | 60.4, CH2 | 3.62, d (11.0) |
| 3.68, dd, (12, 1.8) | 3.77, m | 3.57, dd (12.2, 5.9) | ||||
| N-Me | 26.8, CH3 | 2.93, s | ||||
DMSO-d6.
CD3OD.
Broad signal.
Overlapped with H-9.
Compound 2, which was isolated as an off-white, amorphous solid, was the major metabolite found in the extract. High-resolution ESIMS determined its molecular formula to be C26H38NO5. Likewise 3, which was also isolated as an off-white, amorphous solid, has a molecular formula of C26H38NO5, as established by high-resolution ESIMS. Comparison of the UV spectra and 1H, 13C, COSY, HSQC, and HMBC data of 2 and 3 revealed that the two have the same carbon structure and that they differ only in their stereochemistry. Furthermore, the HRESIMS and NMR data showed that 2 and 3 are the N-desmethyl analogues of 1. However, the proton and carbon chemical shift values of 1 were more similar to 3 than to 2 (Table 1).
Relative Configuration of the Decalin Ring Moiety
NMR data showed that compound 2 has a cis-fused decalin structure. In the 1H NMR spectrum of 2, the decalin ring junction signals H-3 (δ 2.72 ppm) and H-8 (δ 2.09 ppm) were deshielded compared to the same protons in 1 and 3. This is consistent with other cis-decalin tetramic acid analogues5,11,15 when compared to their trans-fused counterparts.3,6−8,10,14 The NMR data also indicated that the fused cyclohexyl ring assumes a chair conformation where H-3 is axial while H-8 is equatorial (Figure 1). The absence of a diaxial coupling between these two protons caused H-8 to be seen as a broad singlet in the 1H NMR spectrum, while H-3 was seen as a broad doublet with J = 10.2 Hz due to its coupling with H-4ax (δ 1.38 ppm). The ROESY spectrum of 2 also showed a correlation between H-3 and H-8, which confirmed that the two are not in a diaxial configuration (Figure 1). H-3 was further observed to correlate with H-5ax (δ 0.87 ppm) and H-7ax (δ 1.11 ppm), while H-8 was also seen to correlate with both H-7ax and H-7eq (δ 1.56 ppm). This was also the case with the equatorial C-6 methyl (δ 0.81 ppm), which exhibited same intensity ROESY correlations with both H-7ax and H-7eq and with H-5ax and H-5eq (δ 1.67 ppm). On the other hand, H-6ax was seen to correlate with H-4ax (δ 1.45 ppm), H-7eq, and H-5eq The fused cyclohexenyl ring also seems to assume a pseudochair configuration with H-3, C-2 methyl, and H-8 in equatorial positions, which was deduced from their ROESY correlations. Correlations were also observed between H-4ax and H-12 since C-4 and C-12 are in pseudoaxial positions. Likewise, the pseudoaxial H-9 (δ 5.09 ppm) also correlated with H-6ax.
Figure 1.

Relevant ROESY correlations in 2.
The relative configuration of 3 differs from that of 2 at stereocenters C-8 and C-11. The first indication was the upfield shifts of the proton signals H-3 (δ 1.57 ppm) and H-8 (δ 1.79 ppm) as mentioned above. H-8 was also more deshielded than H-3 compared to cis-fused decalins, where H-3 is more deshielded.5,11,15 The ROESY spectrum established the relative configuration of 3 (Figure 2). The chair conformation of the cyclohexyl ring was also deduced from the following ROESY correlations: H-3 correlated with H-5ax (δ 1.71 ppm) and H-7ax (δ 0.82 ppm); H-8 correlated with H-4ax (δ 1.00 ppm) and H-6ax (δ 1.49 ppm). This therefore indicated that the C-6 methyl is equatorial. The equatorial C-6 methyl also correlated equally to H-5ax (δ 1.71 ppm) and H-5eq (δ 1.00 ppm) and to H-7eq (δ 1.77 ppm). The fused cyclohexenyl ring in 3 also seems to adopt a pseudochair configuration where H-3 and the C-2 methyl are anti to each other in pseudodiaxial positions. H-11 and C-10 methyl appeared to be in pseudoequatorial and pseudoaxial positions, respectively, due to the observed correlation between the C-10 methyl and C-2 methyl.
Figure 2.

Relevant ROESY correlations in 3.
Absolute Configuration: The Tetramic Acid Ring
The absolute configuration of C-5′ was determined by oxidative bond cleavage of the tetramic ring followed by acid hydrolysis (Figure 3).19 The crude extract containing both compounds 2 and 3 was reacted with sodium hypochlorite and sodium hydroxide at room temperature for 8 h.19 The oxidation products were extracted using ethyl acetate and evaporated in vacuo. The residue was hydrolyzed using 6 M HCl for 15 h at 110 °C to yield serine. Marfey derivatization20 of the hydrolytic product was performed using FDLA (1-fluoro-2,4-dinitrophenyl-5-l-alanine amide). The resulting hydrolytic product serine-FDLA (HPS-FDLA) was analyzed by analytical HPLC together with reference standards l- and d-serine, which were derivatized in the same way as the hydrolytic product serine. The HPS-FDLA eluted at 25.00 min, while d-serine-FDLA injected alone and l-serine-FDLA injected alone eluted at 24.83 and 24.63 min, respectively. The HPS-FLDA that was spiked with l-serine-FDLA showed two peaks, at 24.54 and 24.99 min, while the HPS-FDLA that was spiked with d-serine-FDLA showed one peak, at 24.96 min. The HPS-FDLA was also analyzed by LC-MS and showed the expected ion peak at m/z 339 [M + H]+.
Figure 3.

(a) Oxidative bond cleavage;17 (b) acid hydrolysis; (c) derivatization with Marfey’s reagent.

Absolute Configuration: The C-17 Hydroxy Group of the Olefinic Side Chain
Mosher derivatization was performed on 2 and 3 separately. Each compound was reacted with (R)- and (S)-MTPA chloride to form (S)-MTPA and (R)-MTPA esters, respectively. 1H NMR analysis of the Mosher esters was performed, and the ΔδS–R calculated (Figure 4).21 Both 2 and 3 were found to have the 17R configuration.
Figure 4.

ΔδS–R values obtained in pyridine-d5 of the MTPA esters of 1 and 2.
Absolute Configuration: The Decalin Ring of 2
Without any derivatization step, the exciton CD method22 was used to elucidate the absolute configuration of 2. This was possible because two existing chromophores can possibly interact and exhibit exciton split CD spectra, which can then be observed as a positive or negative Cotton effect. The positive Cotton effect that is predicted from the configuration of the two chromophores of 2 (Figure 5A) was observed in the experimental CD spectra of 2 (Figure 5B). The tetramic acid ring attached at C-2 and the conjugated diene attached at C-11 are two chromophores that coupled to cause the positive Cotton effect that was observed in the experimental CD spectrum of 2. The absolute configuration of 2 was therefore assigned as 2R, 3S, 6S, 8S, 17R, and 5′R. This method has been applied to establish the absolute configuration of natural products with pre-existing chromophores, including abscisic acid, quassin, dendryphiellin F, and arnottin II.22
Figure 5.

(A) Expected Cotton effect of 2 according to the exciton CD method of determining absolute configuration. (B) Experimental CD spectrum of 2.
Absolute Configuration: The Decalin Ring of 3
The optical rotation [α]D = +130 and the CD spectrum of 3 (Figure 6) were almost identical to that of phomasetin ([α]D = +93.9) and ent-equisetin (Table 2) and opposite of that of altersetin/coniosetin (Figure 7).6,8 The C-5′R configuration of the tetramic ring as determined above is also consistent with that of phomasetin. The absolute configuration of 3 was therefore assigned as 2R, 3S, 6S, 8R, 17S, and 5′R.
Figure 6.

Experimental CD spectrum of 3 in methanol.
Table 2. CD Data of 3 and Related Tetramic Acid Analogues.
| compound | CD λmax nm (Δε) | ||||
|---|---|---|---|---|---|
| 3 | 225 (+4.8) | 232 (+6.5) | 260 (+2.6) | 288 (+7.5) | 330 (0) |
| phomasetin10 | 225 (+3.2) | 232 (+4.4) | 260 (+1.0) | 290 (+5.2) | 330 (0) |
| ent-equisetin10 | 235 (+6.0) | 250 (+4.0) | 291 (+12.5) | 330 (0) | |
| equisetin10 | 227 (−5.5) | 235 (−7.5) | 260 (−3.0) | 290 (−8.9) | 330 (0) |
| altersetin6 | 212 (−2.9) | 232 (−18.1) | 253 (−5.0) | 282 (−15.3) | 330 (0) |
| coniosetin8 | 227 (−11.4) | 232 (−12.0) | 251 (−1.4) | 283 (−3.0) | 330 (0) |
Figure 7.
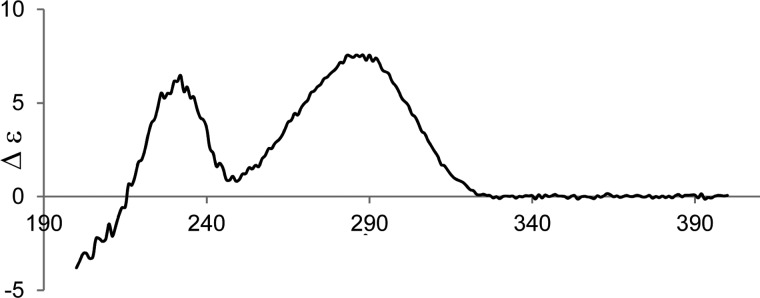
Experimental CD spectrum of coniosetin (reprinted with permission from ref (8)).
Absolute Configuration of 1
Compound 1, which was isolated as an off-white oil, was a minor compound in the recombinant expression. Its HPLC retention time (Figure 8A and B), high-resolution ESIMS, UV, 1H, 13C, COSY, HMBC, and HSQC NMR spectra, and CD data matched those of 1 from the wild-type fungus NRRL 50135. Determination of the absolute configuration of 1 was achieved by comparison of its ROESY and CD spectra with 3. The ROESY spectrum of 1 was very similar to that of 3, which indicates that 1 and 3 have the same relative configuration. The similarity of the CD spectra of these two compounds further revealed that 1 and 3 have the same absolute configuration.
Figure 8.

(A) HPLC chromatogram with PDA detection of the original extract of NRRL 50135. (B) HPLC chromatogram with PDA detector of the extract from the expression platform in mutated F. heterosporum.
Biosynthesis of Pyrrolocins and Related Compounds
The co-occurrence of 1, 2, and 3 seems to suggest that 2 and 3 are cis- and trans-adducts originating from an intramolecular Diels–Alder reaction in the biosynthetic process.16,17 Chemically, this reaction usually prefers to go through an endo transition state, which leads to the cis-decalin isomer, while the less favored exo transition state leads to the trans-decalin. In secondary metabolism, the favored stereochemical route is usually controlled by enzymes, rather than by solution chemistry. For example, the fungal polyketide solanapyrone is cyclized by a fairly novel enzyme after release of the intermediate from the polyketide synthase (PKS).16,17 In a more relevant example the fungal decalin lovastatin is produced by a PKS that is similar to the pyrrolocin PKS. In lovastatin biosynthesis, it is the PKS itself that controls decalin stereochemistry, leading to strict production of the trans-decalin. The relevant biosynthetic intermediate is proposed to contain an ene that is activated by conjugation to an enzyme-bound thioester. By contrast, the synthetic analogue of the proposed intermediate leads to a mixture of cis- and trans-decalin compounds, reflecting a mixture of endo- and exo-Diels–Alder reactions, respectively.
Equisetin and its related compounds are thought to be synthesized by a similar mechanism, wherein the PKS itself would dictate whether the Diels–Alder reaction follows the endo or exo pathway. In the present study, 1 was found as the major product by NRRL 50135, and only trace amounts of 3 and none of 2 were detected (Figure 8A), revealing that only trans-decalins were produced in the wild-type fungus. On the other hand, the heterologous expression yielded 2 and 3 in a ∼2:1 ratio, which is what one would expect for a nonenzymatic reaction (Figure 8B). Although there are many reasons for this, the result implies that a separate Diels–Alderase domain might be involved in the biosynthesis of equisetin-like compounds, which in the present study might not be present in the heterologous expression. As postulated by other authors,17 it seems that LovB (the lovastatin nonaketide synthase) and EqxS (equisetin PKS-NRPS hybrid), at the very least, contain a binding pocket to direct the stereochemical outcome of the reaction. On the other hand, the direct product of the pyrrolocin PKS still contains a carbonyl in the correct position to activate an ene for the Diels–Alder reaction. By contrast, in lovastatin biosynthesis, more advanced PKS intermediates no longer contain an activated ene. This result implies that perhaps pyrrolocins are enzymatically cyclized in the wild-type host, but not in the recombinant host.
Of additional interest, despite the structural similarities between pyrrolocins and compounds such as equisetin, these compounds are often enantiomers of each other. While this fact is very convenient in terms of assigning the absolute configuration of the compound series as we have done here, it is curious. Why is the d-amino acid always found with one decalin absolute configuration and the l-amino acid with the other? We propose two possibilities. First, the presence of the d- or l-amino acid may be controlled enzymatically; the configuration would then dictate the stereochemical course of the Diels–Alder reaction. Second, since the α position is relatively labile in the tetramic acid motif, the amino acid may begin as l-configured and equilibrate to the thermodynamically favored configuration based upon the stereochemistry of the decalin ring. These ideas provide a testable hypothesis that will enable understanding of the timing and biochemical basis of the Diels–Alder reaction in pyrrolocins and the many structural analogues.
Biological Activity
Compounds 1–3 were found to be active against M. tuberculosis, with 1 and 3 being more potent than 2 (Table 3). The data seem to indicate that a trans configuration and N-methylation enhance the potency of antimicrobial activity.
Table 3. Antimycobacterial Activities of 1–3.
| compound | anti-TB IC50 (μM) | cytotoxicity IC50 (μM) |
|---|---|---|
| 1 | 26.3 | 76.6 |
| 2 | 112.9 | 167.0 |
| 3 | 56.4 | 112.9 |
| rifampicin | 0.152 | not determined |
Experimental Section
General Experimental Procedures
UV spectra were measured on a Hewlett-Packard 8452A diode array spectrophotometer (Agilent, Santa Clara, CA, USA). IR spectra were recorded using a JASCO (Easton, MD, USA) FT/IR-400 spectrometer. Specific rotations were recorded on a PerkinElmer (Downers Grove, IL, USA) 343 digital polarimeter in MeOH at 22 °C. CD spectra were recorded on an Aviv model 420 CD spectrometer (Aviv Biomedical, Inc., Lakewood, NJ, USA) in methanol at 25 °C. NMR spectra were recorded on a Varian (Palo Alto, CA, USA) INOVA at 500 MHz for 1H and 125 MHz for 13C using vendor-supplied pulse sequences.
Accurate mass measurements were performed by HRESIMS on a Micromass Q-tof Micro (Waters, Milford, MA, USA) using positive ion mode and an FTMS (LTQ-FT, ThermoFisher, Waltham, MA, USA). HPLC was performed on an Agilent 1200 series equipped with a PDA detector (Agilent Technologies). A Luna 5 μm C18 column, 250 × 10 mm (Phenomenex, Torrance, CA, USA), was used for the isolation of the three compounds. Supelco Diaion HP20SS was purchased from Sigma-Aldrich (St. Louis, MO, USA). TLC was conducted using Kieselgel 60 F254 (Merck, Whitehouse Station, NJ, USA).
Cultivation and Biological Source
The fern Asplenium sp. was collected in Watunou, Milne Bay Province, Papua New Guinea, by a team from Wyeth (now Pfizer). A piece of the stem was rigorously surface-sterilized, and the sterile stem was placed on malt extract agar containing antibacterials and 1 μg/mL cyclosporin until fungal hyphae were observed to protrude from the stem. The strain was initially isolated with the name ENDO-0549, and after axenic cultivation, it was deposited in the NRRL culture collection as NRRL 50135. Phylogenetic analysis using molecular markers identified NRRL 50135 as a potentially novel genus in the order Diaporthales. The sequences were deposited in GenBank (KM107910).
Preparation of Inocolum of NRRL 50135
NRRL 50135 was cultured on Difco potato dextrose agar. A 250 mL Erlenmeyer flask containing 50 mL of seed medium, Difco potato dextrose broth, was inoculated with agar-grown culture. The seed was incubated at 22 °C with shaking at 200 rpm until sufficient cell density was achieved to inoculate the production fermentation.
Fermentation of NRRL 50135
The production fermentation medium consisted of 15 g of nongauze milk-filter paper (KenAG Animal Care Group, Ashland, OH, USA) cut into 8 mm wide by 60 mm long strips and 102 g of milled long grain white rice wetted with 125 mL of 0.1% yeast extract solution per 2.8 L Fernbach flask. The fermentation was inoculated with 20 mL of the seed and incubated stationary at 22 °C for 15 days.
Isolation of 1 from NRRL 50135
The cells were extracted three times with methanol (0.6 L). The combined extract was evaporated under reduced pressure to dryness. The methanol extract was then purified by reversed-phase HPLC using a C18 column (Phenomenex Luna, 12 μp, 50 × 250 mm) and a gradient solvent of 85–100% acetonitrile in water (40 mL/min, 0 min run), with both solvents containing 0.01% in volume of trifluoroacetic acid (TFA). The peak at 18 min, monitored at 293 nm, was concentrated to give partially purified pyrrolocin. The compound was further chromatographed by HPLC using a different column (YMC ODS-A, 10 μp, 30 × 250 mm) and a gradient solvent of 80–100% acetonitrile in water (20 mL/min, 23 min run), with both solvents containing 0.01% in volume of TFA. The peak at 17 min was collected and evaporated under reduced pressure to afford 1 as a colorless oil (7.2 mg).
Fermentation Procedure for F. heterosporum
Mutant strains of F. heterosporum carrying pyrrolocin biosynthetic genes were maintained and grown as described.4
Isolation of 1–3 from F. hereosporum
Three-week cultures of F. heterosporum grown on 500 g of corn grit agar at room temperature in 2.5 L Fernbach flasks were extracted twice with acetone for 12 h. Extracts were pooled and dried invacuo using a rotary evaporator (IKA Works, Inc., Wilmington, NC, USA) to afford the crude extract. A portion of the crude extract (6.7%) was dissolved in MeOH, mixed with Diaion HP20SS, and dried. The resin was loaded into a column (8.5 cm × 2.0 cm i.d.) and fractionated using 40 mL each of the following solvents: 100% water, 75% H2O/25% 2-propanol, 50% H2O/50% 2-propanol, 25% H2O/75% 2-propanol, and 100% MeOH to yield five fractions, designated FW, F1, F2, F3, and F4.23 The fractions were collected and solvents evaporated using a rotary evaporator. Bioassay-guided fractionation showed anti-TB activity for F3. F3 was purified by semipreparative HPLC using a reversed-phase C18 column at a 3.5 mL/min flow rate as follows: 60% ACN/20% 0.1% aqueous TFA from 0 to 5 min, linear gradient from 60% to 100% ACN from 5 to 20 min. Compound 1 (1.5 mg) eluted at 18.7 min, 2 (5.0 mg) at 16.0 min, and 3 (11.5 mg) at 13.0 min.
Preparation of (R)- and (S)-MTPA Ester Derivatives of 2 and 3(24)
2 and 3 (0.5 mg each) were separately dissolved in pyridine-d6 (150 μL) and transferred into clean NMR tubes. Under a N2 gas stream, (R)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (5 μL) was added into each sample, and the NMR tubes were then carefully shaken to mix the samples and the MTPA chloride evenly. The NMR tubes were allowed to stand at room temperature and monitored every 2 h by 1H NMR. The reaction was found to be complete after 6 h. 1H NMR data of the (S)-MTPA ester derivative of 2 (2a) (500 MHz, pyridine-d6, data were assigned on the basis of its 1H–1H COSY correlations): δ 0.81 (3H, d, J = 6.3 Hz, H3-6-Me), 1.21 (3H, s, H3-2-Me), 1.31 (3H, d, J = 6.3 Hz, H3-18), 2.44 (2H, m, H2-16), 5.12 (1H, s, H-9). 1.64 (3H, s, H3-10-Me), 5.58 (1H, p, J = 7.3 Hz, H-15), 5.68 (1H, dd, J = 14.7, 10.7 Hz, H-12), 6.11 (1H, dd, J = 14.7, 10.0 Hz, H-13), 6.20 (1H, dd, J = 10.0, 14.0 Hz, H-14). 1H NMR data of the (S)-MTPA ester derivative of 3 (3a) (500 MHz, pyridine-d6, data were assigned on the basis of its 1H–1H COSY correlations): δ 0.81 (2H, d, J = 6.6 Hz, H3-6-Me), 1.17 (3H, d, J = 6.3 Hz, H3-18), 1.48 (3H, s, H3-2-Me), 2.22 (2H, m, H2-16), 3.31 (1H, m, H-11), 5.40 (1H, m, H-15), 5.58 (1H, m, H-12), 5.91 (1H, m, H-14), 6.13 (1H, m, H-13).
In the manner described for 2a and 3a, 2 and 3 (0.5 mg each) were separately dissolved in pyridine-d6 and reacted with (S)-(−)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (5 μL) in NMR tubes at room temperature for 6 h to afford the (R)-MTPA esters of 2 (2b) and 3 (3b). 1H NMR data of the (R)-MTPA ester derivative of 2b (500 MHz, pyridine-d6): δ 0.82 (1H, d, J = 6.4 Hz, H3-6-Me), 1.21 (3H, d, J = 6.4 Hz, H3-18), 1.25 (3H, s, H3-2-Me), 1.64 (3H, s, H3-10-Me), 2.44 (2H, m, H2-16), 5.09 (1H, s, H-9), 5.60 (1H, p, J = 7.1 Hz, H-15), 5.69 (1H, m, H-12), 6.04 (1H, dd, J = 10.7, 14.7 Hz, H-13), 6.22 (1H, dd, J = 10.6, 14.9 Hz, H-14). 1H NMR data of the (R)-MTPA ester derivative of 3b (500 MHz, pyridine-d6, data were assigned on the basis of its 1H–1H COSY correlations): δ 0.80 (3H, d, J = 6.3 Hz, H3-6-Me), 1.10 (3H, d, J = 6.4 Hz, H3-18), 1.52 (3H, s, H3-2-Me), 1.80 (3H, s, H3-10-Me), 2.22 (2H, m, H2-16), 3.39 (1H, m, H-11), 5.45 (1H, m, H-15), 5.58 (1H, m, H-12), 5.96 (1H, m, H-14), 5.99 (1H, m, H-13).
Oxidative Bond Cleavage of the Tetramic Acid Ring19
One F. heterosporum sample lacked a methyltransferase and made only 2 and 3. The crude methanol extract was obtained and dried, and a portion (0.2 g) was dissolved in methanol (6 mL). Then 1 M NaOH (0.75 mL) was slowly added, followed by a NaOCl solution (available chlorine 8.5–13.5%, 3.5 mL). The mixture was stirred at rt for 8 h. Then, 1 M aqueous Na2SO2 (3 mL) was added, and the mixture was neutralized by addition of 1 M HCl. After removal of the solvent, the residue was diluted with H2O, and then the resulting mixture was extracted with ethyl acetate. The organic layer was dried in vacuo. The residue was dissolved in 6 N HCl (0.5 mL) and heated in a sealed ampule vial at 110 °C for 15 h. The solvent was removed in vacuo. The absolute stereochemistry of the resulting serine was determined by the advanced Marfey method.20
The acid hydrolysate was dissolved in 1 mL H2O. To a 100 μL aliquot were added 1% FDAA (1-fluoro-2,4-dinitrophenyl-5-l-alanine amide) in acetone (200 μL) and 40 μL of 1 N NaHCO3.The mixture was heated at 40 °C for 1 h in a water bath. The vial was then cooled, and 2 M HCl (20 μL) was added. This solution was analyzed by analytical HPLC using the following method: 0 min 5% ACN/95% aqueous TFA (0.1%), 0–2 min 5–25% ACN, 2–3 min 25% ACN, 3–48 min 25–70% ACN, 48–50 min 70% ACN.
Antimycobacterial Assay
M. tuberculosis H37Ra growth inhibition was quantified using the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay modified from previously published methods.25 Plant extracts and control drugs were dissolved in dimethyl sulfoxide (DMSO) to produce test stock solutions, and DMSO served as a negative control. M. tuberculosis cultures were dispensed in 200 μL of ADC (Remel, Lenexa, KS, USA)-enriched 7H9 medium into 96-well culture clusters at 100 000 cells per well. Then 1 μL of DMSO (control) or DMSO containing extract or drug dilution was added in quadruplicate wells. After incubation for 4 days at 37 °C, 11 μL of sterile MTT (5 mg/mL in PBS) was added and further incubated for 12 h. The insoluble formazan salt formed was solubilized by the addition of 50 μL of a solubilization solution (5% SDS w/v, 50% DMF v/v, 45% H2O v/v). A570 was measured using a Multiskan FC plate reader (Fisher Scientific, Waltham, MA, USA). All data were corrected against media-only blank wells by subtracting media-only A570. The percent inhibition was calculated as the fraction of the sum of the test wells over the sum of control wells subtracted from 1 and multiplied by 100. The IC50 was determined from the percent inhibition as the concentration of test material that inhibited at least 50% of growth of the test organism.
Cytotoxicity Assay
The cytotoxicity assay utilized CEM-TART cells obtained from the NIH AIDS Reagent Reference Program (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH).26 Cells were grown in complete RPMI 1640 in a jacketed incubator with 5% CO2 at saturated humidity and at 37 °C. The assay used round-bottom 96-well culture cluster plates. Compounds to be tested were added in quadruplicate wells, while controls were as follows: solvent only (DMSO) in 16 wells, of which 8 received media only (“media only” blank) and 8 received 20 000 cells/well CEM-TART cells (positive growth control). A further 8 wells received a decreasing amount of doxorubicin in duplicate wells (4 concentrations) and CEM-TART cells serving as positive cytotoxicity control. After 4 days of incubation 11 μL/well of 5 mg/mL MTT in PBS was added, followed by 2 h of further incubation. Cells were pelleted by centrifugation, and the medium in each cell was aspirated off using a vacuum aspirator. DMSO (150 μL/well) was added to dissolve the insoluble formazan salt. To completely dissolve the formazan precipitate, plates were usually agitated for 5 min using a Fisher Scientific MS1 S7 plate shaker. A570 was determined using a Scientific Multiskan FC (Fisher Scientific) plate reader. The resulting data were processed with a specially made Excel spreadsheet to calculate the percent inhibition as follows: the average of the media-only blank was subtracted from all wells. The average of the test wells for each compound was subtracted from the average of the positive control wells and divided by the average of the positive growth control, resulting in a fraction of control. To determine the percent fraction of inhibition, the fraction of control was subtracted from unity and multiplied by 100. The IC50 was determined from the percent inhibition as the concentration of test material that inhibited at least 50% of TART cell growth.
Pyrrolocin A (1):
brownish oil; [α]22D +56.1 (c 0.03, MeOH); UV (MeOH) λmax nm (log ε 244 (3.48), 295 (3.05); CD λmax nm (Δε) 239 (+5.82), 294 (+2.40); IR (NaCl disk) νmax 3317, 2946, 2832, 2161, 2012, 1977, 1679; 1H NMR (DMSO-d6, 400 MHz) and 13C NMR (DMSO-d6, 100 MHz) see Table 1; HRESIMS m/z 458.2906 [M + H]+ (calculated for C27H40NO5 458.2906).
Pyrrolocin B (2):
off-white solid; [α]22D −150 (c 0.025, MeOH); UV (MeOH) λmax nm (log ε) 238 (4.4156), 286 (4.0263); CD λmax nm (Δε 236 (−16.9), 283 (3.0); IR (NaCl disk) νmax 3325, 2945, 2834, 2362, 2340, 1683, 1653, 1456; 1H NMR (CD3OD, 500 MHz) and 13C NMR (CD3OD, 125 MHz) see Table 1; HRESIMS m/z 444.2751 [M + H]+ (calculated for C26H38NO5 444.2750; Δ +0.2 ppm).
Pyrrolocin C (3):
off-white solid; [α]22D +130 (c 0.025, MeOH); UV (MeOH) λmax nm (log ε) 247 (4.4776), 286 (4.1049); CD λmax nm (Δε) 232 (6.46), 288 (7.56); IR (NaCl disk) νmax 3300, 2945, 2916, 2868, 2843, 2155, 1678, 1575, 1453; 1H NMR (DMSO-d6, 500 MHz) and 13C NMR (DMSO-d6, 125 MHz) see Table 1; HRESIMS m/z 444.2762 [M + H]+ (calculated for C26H38NO5 444.2750; Δ +2.7 ppm).
Acknowledgments
The authors wish to gratefully acknowledge NIH support through the ICBG 5UO1TW006671-10 R and NSF 0957791. R.C.J. wishes to acknowledge the following persons for their technical assistance: M. Jacobsen for the measurement of the CD spectra, C. Serrano for the IR and OR, T. E. Smith for the LC-MS, and A. Fleming for the ECD calculations.
Supporting Information Available
1D and 2D NMR spectra for compounds 1–3 and HPLC chromatograms of the FDLA derivatives are available as Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# (R.C.J.) De La Salle Health Sciences Institute College of Pharmacy, Congressional Ave., Dasmariňas, Cavite 4114, Philippines.
Author Contributions
All authors have contributed to the manuscript and have given approval to the final version.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Beehler B. M., Swartzendruber J. F., Eds. In Papua New Guinea Conservation Needs Assessment, Vol. 2; Biodiversity Support Program: Boroko, Papua New Guinea, 1993. [Google Scholar]
- Arnold A. E.; Lutzoni F. Ecology 2007, 88, 541–549. [DOI] [PubMed] [Google Scholar]
- Vesonder R. F.; Tjarks L. W.; Rohwedder W. K.; Burmeister H. R.; Laugal J. A. J. Antibiot. 1979, 32, 759–761. [DOI] [PubMed] [Google Scholar]
- Kakule T. B.; Jadulco R. C.; Koch M.; Janso J. E.; Barrows L. R.; Schmidt E. W.. Native promoter strategy for high-yielding synthesis and engineering of fungal secondary metabolites. ACS Synth. Biol. DOI: 10.1021/sb500296p. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros C.; Dix A.; Katz B.; Vasina Y.; Pearce C. J. Antibiot. 2003, 56, 862–865. [DOI] [PubMed] [Google Scholar]
- Hellwig V.; Grothe T.; Mayer-Bartschmid A.; Endermann R.; Geshke F.; Henkel T.; Stadler M. J. Antibiot. 2002, 55, 881–891. [DOI] [PubMed] [Google Scholar]
- Ondeyka J. G.; Smith S. K.; Zink D. L.; Vicente F.; Basilio A.; Bills G. F.; Polishook J. D.; Garlisi C.; Mcguinness D.; Smith D.; Qiu H.; Gill C. J.; Donald R. G. K.; Phillips J. W.; Goetz M. A.; Singh S. B. J. Antibiot. 2014, 1–5. [DOI] [PubMed] [Google Scholar]
- Segeth M. P.; Bonnefoy A.; Brönstrup M.; Knauf M.; Schummer D.; Toti L.; Vertesy L.; Wetzel-Raynal M. C.; Wink J.; Seibert G. J. Antibiot. 2003, 56, 114–122. [DOI] [PubMed] [Google Scholar]
- Sugie Y.; Dekker K. A.; Inagaki T.; Kim Y.-J.; Salakibara T.; Sakemi S.; Sugiura A.; Brennan L.; Duignan J.; Sutcliffe J. A.; Kojima Y. J. Antibiot. 2002, 55, 19–24. [DOI] [PubMed] [Google Scholar]
- Signh S. B.; Zink D. L.; Goetz M. A.; Dombrowski A. W.; Polishook J. D.; Hazuda D. J. Tetrahedron Lett. 1998, 39, 2243–2246. [Google Scholar]
- Singh S. B.; Zink D. L.; Heimbach B.; Genilloud O.; Teran A.; Silverman K. C.; Lingham R. B.; Felock P.; Hazuda D. J. Org. Lett. 2002, 4, 1123–1126. [DOI] [PubMed] [Google Scholar]
- Namikoshi M.; Kobayashi H.; Yosimoto T.; Hosoya T. J. Antibiot. 1997, 50, 890–892. [DOI] [PubMed] [Google Scholar]
- Sugie Y.; Inagaki S.; Kato Y.; Nishida H.; Pang C.-H.; Saito T.; Sakemi S.; Dib-Hajj F.; Mueller J. P.; Sutcliffe J.; Kojima Y. J. Antibiot. 2002, 55, 25–29. [DOI] [PubMed] [Google Scholar]
- Inoshi J.; Shigeta N.; Fukuda T.; Uchida R.; Nonaka K.; Masuma R.; Tomoda H. J. Antibiot. 2013, 66, 549–554. [DOI] [PubMed] [Google Scholar]
- Watanabe T.; Igarashi M.; Okajima T.; Ishii E.; Kino H.; Hatano M.; Sawa R.; Umekita M.; Kimura T.; Okamoto S.; Eguchi Y.; Akamatsu Y.; Utsumi R. Antimicrob. Agents Chemother. 2012, 56, 3657–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C. D.; Vederas J. C. Biopolymers 2010, 93, 755–763. [DOI] [PubMed] [Google Scholar]
- Kelly W. L. Org. Biol. Chem. 2008, 6, 4483–4493. [DOI] [PubMed] [Google Scholar]
- Stocking E. M.; Williams R. M. Angew. Chem., Int. Ed. 2003, 42, 3078–3115. [DOI] [PubMed] [Google Scholar]
- Minowa N.; Kodam Y.; Harimaya K.; Mikawa T. Heterocycles 1998, 48, 1639–1642. [Google Scholar]
- Marfey P. Calsberg Res. Commun. 1984, 591–596. [Google Scholar]
- Ohtani I.; Kusumi T.; Kashman Y.; Kakisawa H. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar]
- Nobuyuki H.; Nakanishi K.; and Berova N. In Comprehensive Chiroptical Spectroscopy: Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products, And Biomolecules; Berova N.; Polavarapu P. L.; Nakanishi K.; Woody R. W., Eds.; John Wiley and Sons, Inc.: NJ, 2012; Vol. 2, Chapter 4, pp 154–155. [Google Scholar]
- Bugni T. S.; Harper M. K.; McCulloch M. W. B.; Reppart J.; Ireland C. M. Molecules 2008, 13, 1372–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B.-N.; Park E. J.; Mbwambo Z. H.; Santarsiero B. D.; Mesecar A. D.; Fong H. H. S.; Pezzuto J. M.; Kinghorn A. D. J. Nat. Prod. 2002, 65, 1278–1282. [DOI] [PubMed] [Google Scholar]
- Koch M.; Bugni T. S.; Sondossi M.; Ireland C. M.; Barrows L. R. Planta Med. 2010, 76, 1678–1682. [DOI] [PubMed] [Google Scholar]
- Chen H.; Boyle J. T.; Malim M. H.; Cullen B. R.; Lyerly H. K. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 7678–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.