

Abstract
In the search for peroxisome proliferator-activated receptor gamma (PPARγ) active constituents from the roots and rhizomes of Notopterygium incisum, 11 new polyacetylene derivatives (1–11) were isolated. Their structures were elucidated by NMR and HRESIMS as new polyyne hybrid molecules of falcarindiol with sesquiterpenoid or phenylpropanoid moieties, named notoethers A–H (1–8) and notoincisols A–C (9–11), respectively. Notoincisol B (10) and notoincisol C (11) represent two new carbon skeletons. When tested for PPARγ activation in a luciferase reporter assay with HEK-293 cells, notoethers A–C (1–3), notoincisol A (9), and notoincisol B (10) showed promising agonistic activity (EC50 values of 1.7 to 2.3 μM). In addition, notoincisol A (9) exhibited inhibitory activity on NO production of stimulated RAW 264.7 macrophages.
Qiang Huo, the dried roots and rhizomes of Notopterygium incisium and N. forbesii (Umbelliferae), is used in traditional Chinese medicine for treating the common cold and inflammatory diseases such as rheumatoid arthritis and as diaphoretics, antifebrile agents, and analgesics.1 Its potential anti-inflammatory constituents include coumarins, phenethyl ferulate, falcarindiol, and (−)-bornyl ferulate.2−4 Among them, falcarindiol is also known for its antimicrobial5−7 and cytotoxic8,9 activities.
Peroxisome proliferator-activated receptor gamma (PPARγ) is involved in the regulation of various metabolic and inflammatory processes. Moreover, some PPARγ activators are used as drugs.10 Many PPARγ-active compounds have already been identified from natural sources.11 On the other hand, the high concentration of NO released by aberrant expression of inducible nitric oxide synthases (iNOS) plays an important role in inflammatory processes and in the pathophysiology of many diseases.12,13
Recently, we have identified a series of falcarindiol derivatives from rhizome and roots of Notopterygium incisum Ting ex H.T. Chang with significant PPARγ agonistic activity, which also inhibited NO production in LPS/IFN-gamma-activated RAW264.7 macrophages.4,14 In this work, 11 further polyyne derivatives with new hybrid structures, notoethers A–H (1–8) and notoincisols A–C (9–11), were purified from the roots and rhizomes of N. incisum. Their structures were elucidated by NMR and HRESIMS as new polyyne hybrid molecules with unusual structural features: eight of them are ethers comprising a falcarindiol and a sesquiterpenoid subunit (notoethers A–H, 1–8), while three compounds comprise a falcarindiol and a phenylpropanoid subunit (notoincisols A–C, 9–11). Notoincisol B (10) and notoincisol C (11) represent two novel skeletons. The compounds were examined for their potential to transactivate a PPARγ-driven luciferase reporter gene in HEK-293 cells and to inhibit LPS/IFNγ-induced NO production in RAW264.7 macrophages.

Results and Discussion
Novel Polyyne Hybrid Compounds from Notopterygium incisum
The dichloromethane-soluble extract from roots and rhizomes of N. incisum exerted significant PPARγ activation in a PPARγ-driven luciferase reporter gene assay (2.5 ± 0.28-fold activation, p < 0.001).14 Fractionation of the extract by several chromatographic separation steps on normal- and reversed- phase silica gel yielded 11 new polyyne derivatives (1–11). Notoethers A–H (1–8) are four pairs of isomeric ethers, each consisting of a falcarindiol unit and a sesquiterpene unit. This is the first report of polyynes fused with sesquiterpenoids, and also the second type of polyacetylene adducts connected through an ether bond, besides reported polyacetylene coumarin adducts.15,16 Notoincisols A–C (9–11) are adducts of a polyacetylene and a phenylpropanoid unit, with 10 and 11 representing two new carbon skeletons.
Notoether A (1) was obtained as a colorless oil. The HRESIMS, 13C NMR, and HSQC data indicated a molecular formula of C32H50O3. Four acetylene carbon signals, at δ 80.0, 79.9, 69.6, and 69.3 ppm, and four alkene carbons, at δ 116.1, 136.3, 128.1, and 134.4, together with a characteristic alkene proton at δ 5.79 (ddd, J = 16.9, 10.2, 4.7 Hz) and a pair of terminal alkene protons at δ 5.58 and 5.50 suggested the presence of a falcarindiol unit.16 The remaining 15 carbon resonances including four tertiary methyl groups indicated an additional sesquiterpene unit. Complete assignments of the 1D and 2D NMR signals and comparison with literature data revealed the second part of the molecule to be a 4,11-eudesmane diol.17 The HMBC correlation between C-4′ (δ 80.0) of the eudesmane and H-3 (δ 4.84) of the falcarindiol unit indicated an ether linkage between these two parts, which was also supported by a NOESY correlation between Me-15′ (δ 1.11) of the eudesmane and H-3 (δ 4.84) of the falcarindiol unit. The relative configuration of the eudesmane diol was determined by analyzing NOESY correlations, together with coupling constants from the 1D proton spectrum and cross-peak intensities in the DQF-COSY spectrum. The observed NOEs, typical for a trans-fused decalin system in a chair conformation, indicated the β-orientations of Me-14′ and Me-15′. The quadruplet signal of the axial H-6′ (δ 1.01) in combination with the large J coupling (12.3 Hz) required axial orientations of both the H-7′ and H-5′ methine protons. Therefore, the relative configuration at the stereogenic centers was determined as depicted. The unsubstituted sesquiterpene, cryptomeridol, has been previously obtained by chemical modification of β-eudesmane.17
Notoether B (2) was obtained as a colorless oil. The HRESIMS, 13C NMR, and HSQC data indicated a molecular formula of C32H50O3. Four acetylene and four alkene carbons with chemical shifts like those of compound 1, as well as the corresponding alkene protons, revealed the presence of a falcarindiol unit. Correlations from the 2D NMR spectra confirmed a cryptomeridiol structure. A comparison with reference spectroscopic data obtained for falcarindiol showed different chemical shifts of the carbons centered on C-8, including upfield shifts of C-8 (−1.3 ppm), C-6 (−0.9 ppm), and C-10 (−3.1 ppm), as well as downfield shifts of C-7 (1.8 ppm) and C-9 (0.9 ppm). Further evidence came from the HMBC correlation between C-4′ (δ 79.5) and H-8 (δ 5.01) and the NOESY correlation between Me-15′ and H-8. On the basis of these results, the structure of 2 was elucidated as a falcarindiol unit connected at C-8 via an ether bond to C-4′ of cryptomeridiol.
Notoether C (3) was obtained as a colorless gum. The HRESIMS, 13C NMR, and HSQC data indicated a molecular formula of C32H50O4. A falcarindiol subunit was identified from four acetylene signals and four alkene carbon signals, as well as from three alkene protons and two oxygenated methine proton (H-3, H-8) signals. Their chemical shift values closely resembled those of compound 1. Complete NMR resonance assignments revealed that compound 3 consists of a falcarindiol unit attached to a trihydroxyeudesmane moiety. An HMBC correlation between H-3 (δ 4.86) and C-4′ (δ 79.3) as well as a NOESY correlation between H-3 and Me-14′ indicated that these two parts are connected via an ether bond between C-3 and C-4′. The relative configuration of the sesquiterpene unit was determined by analyzing NOE and coupling constant data. A NOE between Me-14 and Me-15 indicated their β-orientations. In contrast, the H-1′, H-5′, and H-7′ protons were assigned with an α-orientation, which corresponded to these protons with an axial orientation in a trans-decalin structure. The H-5′ singlet and the missing cross-peak in the DQF-COSY spectrum for H-6′ indicated an equatorial orientation of H-6′. Therefore, an axial orientation of the hydroxy group at C-6′ could be suggested. Based on these data, the sesquiterpene unit was identified as 1′β,4′α,6′β-trihydroxyeudesmane. An acetylated derivative of this sesquiterpene has been isolated but not fully characterized by Onorato and co-workers.18
Notoether D (4) was obtained as a colorless gum. The HRESIMS, 13C NMR, and HSQC data indicated a molecular formula of C32H50O4. A falcarindiol unit was identified by its specific carbon and proton shifts, for which the chemical shift values correlated closely with those of compound 2 and therefore indicated C-8 substitution. Assignment of the 2D NMR spectra confirmed the presence of a 1′β,4′α,6′β-trihydroxyeudesmane residue substituted at position C-4′. An HMBC correlation between H-8 and C-4′, as well as a NOESY correlation between H-8 and Me-14′, indicated these two parts to be connected through an ether bond between C-8 and C-4′.
Notoether E (5) was obtained as colorless needles. The HRESIMS, 13C NMR, and HSQC data indicated a molecular formula of C32H50O4. The falcarindiol unit was recognized again by its characteristic set of carbon and proton signals, for which the chemical shift values correlated closely with those of compounds 1 and 3, and hence indicated a C-3 substitution of the falcarindiol moiety. Assignments of the 1D and 2D NMR spectra were used to further identify a 1′,4′,6′-trioxygenated eudesmane unit substituted at C-4′. The absence of a NOESY correlation between H-5′ and the angular Me-14′ supported the occurrence of a trans-fused A/B ring system. Coupling of H-1′ (dd, J = 10.9, 4.1 Hz) and the NOESY correlation between H-5′ and H-1′ suggested the β-orientation of the C-1′ hydroxy group. Coupling of the bridgehead H-5′ (d, J = 10.9 Hz) with the H-6′ of hydroxy methine (dt, J = 10.4, 3.3 Hz) revealed their vicinal diaxial relationship and, hence, an equatorial arrangement for the C-6′ hydroxy group, which was also confirmed by the NOESY correlation observed between Me-14′ and H-6′. A NOESY correlation between H-6′ and the angular Me-15′, as well as between Me-14′ and Me-15′, suggested the C-4′ oxy group to be also α-oriented. It is noteworthy that the two free hydroxy group proton signals of the sesquiterpenoid moiety showed different patterns. The more low-field signal showed a doublet (δ 4.76, J = 2.6 Hz, OH-6′), while a singlet could be found at high field (δ 2.36, OH-1′). The low-field pattern of 6′-OH supported a syn-equatorial configuration for -O-4′ and OH-6′, which allows an intramolecular hydrogen bonding that may resist hydrogen exchange with the residual solvent water indicated by a flat OH-1′.19 Thus, with one of the two small couplings of H-6′ (2.6 Hz) assigned to the OH-6′ hydroxy group proton, the remaining small coupling of H-6′ (3.7 Hz) requires a cis relationship to the adjacent H-7′, suggesting the axial orientation of the isopropyl group. The axial α-orientation of the isopropyl group was supported also by strong NOEs between H-11′, H-5′, and H-9′, respectively. When comparing its C-7′ chemical shift (δ 46.2) with the counterpart carbon of zingibertriol (δ 51.5), the difference (Δδ = −5.3 ppm) was close to that between equatorial and axial isopropylcyclohexane (Δδ = −3.4 ppm, −150 °C).20−22 The sesquiterpenoid moiety of compound 5 was therefore identified as the C-7 epimer of zingibertriol.21,22 Accordingly, compound 5 was elucidated as a falcarindiol unit substituted at C-3 through an ether bond by 7-epizingibertriol (C-4′).
Notoether F (6) was obtained as a colorless gum. The HRESIMS, 13C NMR, and HSQC data indicated a molecular formula of C32H50O4. Through assignments of the 1D and 2D NMR spectra, a C-8-substituted falcarindiol moiety was identified in the same way as in the case of compounds 2 and 4. The other unit of this molecule was also found to be a C-4′-substituted 7S isomer of zingibertriol. Therefore, compound 6 was established as a falcarindiol unit substituted at C-8 through an ether bond by the 7S isomer of zingibertriol (C-4′).
Figure 1.

NOESY correlations and intramolecular hydrogen bond (dashed) of the sesquiterpenoid moiety of 5 and 6.
Notoether G (7) was obtained as a colorless gum. The HRESIMS, 13C NMR, and HSQC data indicated a molecular formula of C32H50O4. The falcarindiol moiety and its substitution at C-3 were apparent as described for earlier compounds. Unlike for compounds 1–6, NMR experiments revealed the presence of a different type of sesquiterpenoid. The most obvious difference was that both tertiary methyl groups of compound 7 showed HMBC correlations with three carbons, including one methylene, one methane, and one oxygenated tertiary carbon. Assignment of the 1D and 2D NMR data observed and comparison with literature data revealed that the 13C NMR data were consistent with those previously reported for teuclatriol (guaiane-4,6,10-triol), except for chemical shift changes of C-10′ (Δδ = 7.4 ppm) and its vicinal C-1′ (Δδ = −2.1 ppm), C-9′ (Δδ = −5.3 ppm), and C-14′ (Δδ = −2.9 ppm), which suggested substitution at C-10′.23 Two broad singlets (δ 2.38, 2.24), each integrating for one proton, assigned to OH-4′ and OH-6′, were observed, with their shapes due to proton exchange. These data, together with HMBC correlation between C-4′ and H-3, as well as the NOESY correlation between H-3 and Me-14′, were used to establish compound 7 as a falcarindiol unit substituted at C-3 by teuclatriol (C-10′) via an ether bond.
Notoether H (8) was obtained as a colorless gum. The HRESIMS, 13C NMR, and HSQC data indicated a molecular formula of C32H50O4. 1D and 2D NMR spectroscopic data analysis revealed the presence of a falcarindiol moiety substituted at the C-8 position. The remaining 15 carbons could be associated with a teuclatriol residue. The two hydroxy group singlets (δ 2.37, OH-4′; δ 2.23, OH-6′) exhibited similar chemical shifts to those of 7. Furthermore, their sharp signal shape indicated the absence of any proton exchange. For the same reason, the OH-3 (δ 1.91) proton of falcarindiol, which was absent in the other compounds isolated, was also observed. In contrast to OH-6′ of compounds 5 and 6, no downfield shift of the hydroxy group proton caused by intramolecular hydrogen bonding was observed, although OH-4′ and OH-6′ are both syn-equatorial in 7 and 8. This is due to the distance between the two hydroxy group protons, which is larger in the guaiane skeleton when compared to the eudesmane skeleton. These arguments together with an HMBC correlation between C-4′ and H-8, as well as a NOESY correlation between Me-14′ and H-8, confirmed 8 as a falcarindiol unit substituted by teuclatriol via a (C-8)–O–(C-10′) ether bond.
Notoincisol A (9) was obtained as a colorless oil. The HRESIMS, 13C NMR, and HSQC data indicated a molecular formula of C27H32O5. The falcarindiol moiety and its substitution at C-8 were identified through assignments of 1D and 2D NMR signals. The proton signals observed included three ABX coupled benzene protons at δ 7.03 (s), 6.92 (d, J = 8.2 Hz), and 7.07 (br d, J = 8.3 Hz), two trans vinyl protons at δ 7.65 (d, J = 15.8 Hz) and 6.28 (d, J = 15.4 Hz), and a methoxy singlet at δ 3.93. These 1H NMR data and 10 remaining carbon signals suggested the presence of a ferulic acid ester unit.24 Further evidence came from an HMBC correlation between C-9′ and H-8. Therefore, compound 9 was elucidated as a falcarindiol moiety esterified at C-8 with ferulic acid. A biogenic pathway to form compound 9 from falcarindiol and ferulic acid is proposed (Figure 2). Compound 9 was found to be unstable, as it visibly changed color when exposed to elevated temperatures (45 °C) or was kept under direct sunlight at ambient temperature for an extended time period.
Figure 2.

Possible biogenetic pathway for notoincisols A (9) and C (11).
Notoincisol B (10) was obtained as a colorless gum. The HRESIMS, 13C NMR, and HSQC data indicated a molecular formula of C27H32O4. The 1H NMR spectrum showed three alkene signals at δ 6.11 (m), 5.70 (t, J = 9.4 Hz), and 5.65 (m), with similar chemical shifts and identical splitting patterns to that of H-2, H-9, and H-10 of a falcarindiol unit. The 2D NMR data further revealed two terminal alkene protons at δ 5.49 (d, J = 18.0 Hz) and 5.24 (d, J = 10.7 Hz), as well as protons of two oxygenated methines at δ 5.80 (d, J = 6.1 Hz) and 5.49 (d, J = 8.7 Hz), which corresponded to H-1cis, H-1trans, H-3, and H-8, respectively. Furthermore, a seven-membered aliphatic chain connected to the alkene C-10 was identified. A downfield shift occurred for C-3. Two alkylene carbons at C-6 (δ 80.5) and C-7 (δ 99.0), both showing an HMBC correlation with H-8, showed significant downfield shifts when compared to model compounds. Instead of two alkyne carbons at the C-4 and C-5 positions, two quaternary aromatic carbons were observed, with HMBC correlations to H-3 and weak HMBC correlations to H-8. This suggested that C-4 and C-5 are aromatic carbons. The H-3 proton showed HMBC correlations with six carbons, including a pair of olefinic carbons (C-1, C-2) and a pair of aromatic carbons (C-4, C-5), originating from falcarindiol. The other two HMBC correlations were between H-3 and the oxygenated methylene group C-9′ and a quaternary aromatic carbon (C-8′). The H-9′ proton showed HMBC correlations with the C-7′, C-4, and C-3, indicating a five-membered ring. In turn, H-7′ showed HMBC correlations with the aromatic carbons C-4 and C-6′. The remaining two aromatic proton resonances of H-2′ and H-5′ were observed as singlets and were both correlated in the HMBC spectrum with the two quaternary aryl carbons C-4′ (δ 146.5) and C-3′ (δ 147.7), for which the 13C NMR chemical shift values indicated an ortho-relationship of the phenolic carbons. In addition, H-5′ correlated with C-1′ and C-3′, while H-2′ correlated with C-6′ and C-7′. The position of a methoxy group was determined by the HMBC correlation of its protons with C-3′. Analysis of all the aforementioned correlations led to the assignment of compound 10 as depicted, which is a cyclization and oxidation product of a falcarindiol and a hydroxy-methoxy phenyl propane unit. In this molecule, a second aromatic ring is formed, comprising carbons C-4 and C-5 of the falcarindiol and carbons C-7′ and C-8′ of the phenyl propane unit. This is the first time that this type of carbon skeleton has been described.
Notoincisol C (11) was obtained as a colorless gum. The HRESIMS, 13C NMR, and HSQC data indicated a molecular formula of C27H32O4. The 1H NMR spectrum showed three alkene signals at δ 6.10 (m), 5.53 (t, J = 10.2 Hz), and 5.74 (m), with similar chemical shifts and identical splitting patterns to that of H-2, H-9, and H-10 of a falcarindiol unit. A 2D NMR experiment further revealed two terminal alkene protons at δ 5.57 (d, J = 17.5 Hz) and 5.30 (d, J = 10.1 Hz), as well as protons of two oxygenated methines at δ 5.18 (br s) and 6.13 (d, J = 9.2 Hz), which correspond to H-1cis, H-1trans, H-3, and H-8, respectively. Also identified was a seven-membered aliphatic chain connected to alkene C-10. A major downfield shift occurred for C-8, and two alkylene carbons at C-4 (δ 97.2) and C-5 (δ 81.9), which both showed HMBC correlations with H-3, experienced considerable downfield shift when compared to model compounds. Instead of two alkyne carbons, which are supposed to be C-6 and C-7, two quaternary aromatic carbons were apparent, which both showed HMBC correlations with H-8 and weak HMBC correlations with H-3. As in the case of compound 10, these data suggested a conversion of the alkyne carbons C-6 and C-7 into aromatic ones. C-7 showed HMBC correlations with three protons, including one aryl proton (H-7′), one oxygenated methylene (H-9′), and one oxygenated methane (H-8). The H-9′ signal gave HMBC correlations with four carbons, including one now aromatic carbon (C-7) of falcarindiol origin, an oxygenated methine (C-8), a quaternary aromatic carbon (C-8′), and an unsubstituted aromatic carbon (C-7′), indicating a five-membered ring, which resembled that of compound 10. In turn, H-7′ showed HMBC correlations with the aromatic carbons C-2′, C-6′, C-7, and oxygenated methylene C-9′. The remaining two aromatic proton resonances, H-2′ and H-5′, gave clear singlet structures, and both showed HMBC correlations with the two quaternary carbons C-4′ (δ 146.5) and C-3′ (δ 147.7), for which the 13C NMR chemical shift values indicated an ortho-position of the phenolic carbons. In addition, H-5′ correlated with C-1′ and C-3′, while H-2′ correlated with C-6′ and C-7′. The position of a methoxy group was determined by the HMBC correlation of its protons with C-3′. The aforementioned correlations led to the structure of compound 11 as depicted. Like compound 10, this is also a cyclization and oxidation product of a falcarindiol and a hydroxy-methoxy phenyl propane unit. A biogenic pathway to form compound 11 from falcarindiol and ferulic acid via compound 9 is proposed (Figure 2).
PPARγ Agonistic Effects
The isolated compounds were assessed for their PPARγ activation effects. Notoethers A–C (1–3), notoincisol A (9), and notoincisol B (10) were the most potent partial PPARγ agonists among the tested compounds, with EC50 values ranging from 1.7 to 2.3 μM and a maximal fold activation ranging from 1.6 to 2.8 (see Table 5 for comparison of the EC50 values and maximal fold activation induced by 1–3, 9, and 10, as well as Figure 3 for comparison of the effectiveness of all tested compounds at 10 μM). For comparison, the full PPARγ agonist pioglitazone used as positive control activated 6.6-fold at 5 μM (Figure 3) with an EC50 value of 0.21 μM (not shown).
Table 5. PPARγ Agonistic Effects Determined in a PPARγ-Driven Luciferase Reporter Assay.
| compound | EC50 (μM) | maximal fold activation |
|---|---|---|
| 1 | 1.9 | 1.6 |
| 2 | 1.7 | 2.0 |
| 3 | 2.0 | 1.6 |
| 9 | 2.3 | 2.8 |
| 10 | 1.7 | 2.3 |
Figure 3.

PPARγ agonistic effects of polyacetylene adducts from N. incisum. (***p ≤ 0.001, n.s.: not significant; n = 3, ANOVA).
Molecular Modeling of the Investigated Compounds with the PPARγ Ligand Binding Domain
In a previous study, molecular details for the PPARγ binding mode of falcarindiol were investigated by molecular docking studies.14 Falcarindiol was observed to occupy parts of the entrance region of the PPARγ ligand binding domain and established interactions with the mainly hydrophobic binding site arms I and II. Since the active compounds from this study have some structural features in common with falcarindiol but are significantly larger, it was investigated how the proposed binding mode would shift within the binding site and if the docking could distinguish between active and inactive polyyne hybrid compounds.
In general, the PPARγ ligand binding domain is Y-shaped and is divided into three parts: the entrance domain, arm I, and arm II (Figure 4).25 While the PPAR ligand binding site entrance is lined by several polar residues (e.g., Arg288, Glu291, Glu295, Glu343), the two branches of the binding pocket—arm I and arm II—are formed by mainly hydrophobic amino acids. Arm I, however, accommodates some moderately hydrophobic residues (e.g., Cys285, Ser289, His323, His449, and Tyr473). Falcarindiol-type polyacetylenes were supposed to form hydrophobic contacts with residues of arm I (e.g., Ile326, Tyr327, Phe363), arm II (e.g., Ile281), and the entrance region (e.g., Ala292, Met329, Leu330, and Leu333). The hydroxy groups formed hydrogen bonds with the backbone amide of Cys285 in arm I and the carboxy group of Glu295 at the entrance.
Figure 4.
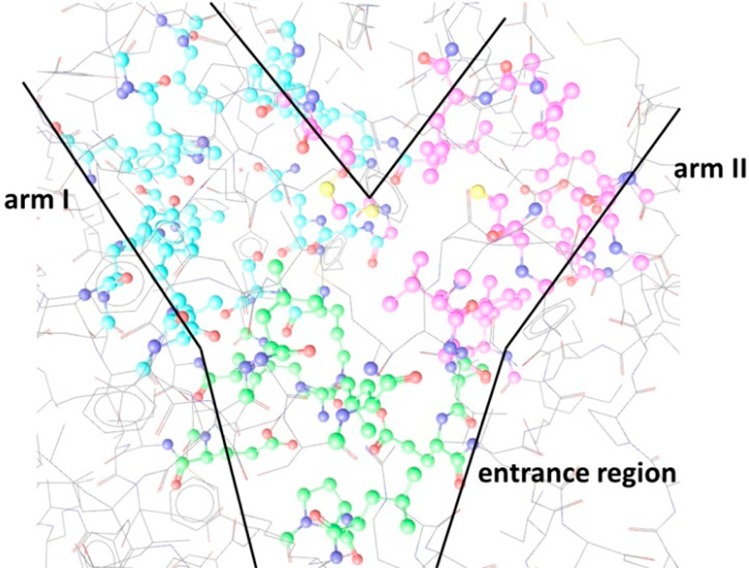
Overall topology of the PPARγ ligand binding site. Amino acids from the entrance region are marked in green, from arm I in blue, and from arm II in pink.
Analyzing the protein–ligand interactions of all active compounds and comparing them to the ones of the inactive molecules revealed that hydrogen bonding with Ser289 was associated with the activity (Figure 5). While all active compounds formed this hydrogen bond in their best-ranked docking pose, none of the inactive structures did so. Ser289 is one amino acid in the core of the ligand binding site, where it is involved in the molecular recognition of many PPARγ agonists, as observed in X-ray crystal structures.26 The docking results rationalized the observed in vitro PPARγ activation by compounds 1, 2, 3, 9, and 10 and could serve as an inspiration for synthetic optimization to develop more potent partial agonists.
Figure 5.
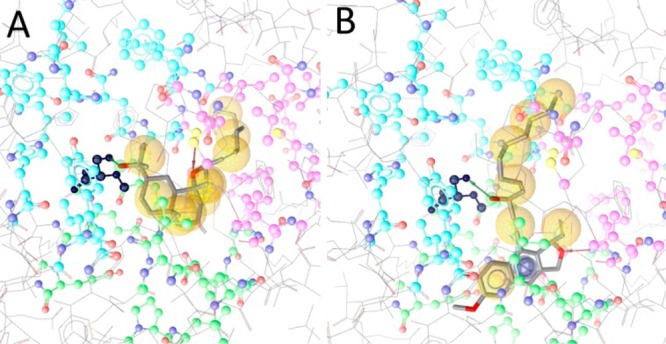
Representative docking poses of compounds 2 (A) and 10 (B) fitted into the PPARγ ligand binding site. Both compounds occupied parts of the entrance region (green) and extend into one of the hydrophobic arms (blue and pink) with their long alkyl chains. One of the smaller hydrophobic parts reached into the other hydrophobic arm. The hydrogen bond with Ser289, which was only observed with the active compounds 1, 2, 3, 9, and 10, is depicted as a green arrow. Ser289 is highlighted in black. Hydrophobic contacts with the binding site are shown as yellow spheres; those with the hydrogen bonds, as arrows.
Inhibitory Effects on the NO Production by iNOS
The new polyacetylene derivatives were tested for inhibition of NO production in stimulated RAW 264.7 macrophages. While most of the compounds were inactive at the concentrations tested (data not shown), notoincisol A (9) inhibited NO production with an IC50 value (14.6 ± 0.7 μM) comparable to those of polyacetylenes previously isolated from N. incisum (around 10 to 30 μM).4
Conclusion
The search for constituents with PPARγ agonistic activity of N. incisum led to the identification of active novel polyacetylene adducts (1, 2, 3, 9, and 10). Their preliminary structure–activity relationship was rationalized by molecular docking experiments. Notoincisol A (9) was also able to inhibit NO production in LPS-induced RAW 264.7 macrophages. These compounds can serve as starting points for chemical modifications in order to optimize potency, selectivity, safety, and pharmacokinetic parameters, thereby offering new scaffolds for the development of compounds to treat inflammatory processes and possibly the related metabolic syndrome.
Experimental Section
General Experimental Procedures
Melting points were determined with a Reichert melting point apparatus and are uncorrected. Optical rotations were taken with a PerkinElmer 341 polarimeter. 1H, 13C, and 2D NMR spectra were recorded in CDCl3 on Unity 600 (Varian, Palo Alto, CA, USA) and Avance 700 (Bruker, Billerica, MA, USA) spectrometers. Chemical shifts are expressed in δ (ppm) with reference to CDCl3. All spectra were recorded at 25 °C. LC-ESIMS were carried out using a Thermo Finnigan LCQ Deca XP Plus mass spectrometer connected to a Surveyor HPLC system (Thermo Fisher, Waltham, MA, USA), with a Zorbax SB-C18 narrow bore (3.5 μm) 2.1 × 150 mm column (Agilent, Santa Clara, CA, USA). Open column chromatography was carried out using MCI CH-P 20P resin (Mitsubishi Chemical, Tokyo, Japan), octadecyl silica gel (25–40 μm, Fuji Silysia, Kasugai, Japan), Sephadex LH-20 (GE Healthcare, Little Chalfont, UK), and silica gel (15–40 μM, Merck KGaA, Darmstadt, Germany) as stationary phase. TLC was conducted on silica gel 60 F254 and silica gel 60 RP-18 F254s plates (Merck KGaA). All chemical reagents (AR grade) were purchased from Carl Roth GmbH + Co. KG (Karlsruhe, Germany).
Accurate mass determinations were performed using an LC/FTMS system consisting of an Exactive Orbitrap mass spectrometer, equipped with a heated ESI II source (Thermo Fisher) and operated in ultra-high-resolution mode (100.000) coupled to a U-HPLC system (Thermo Fisher). Operating conditions for the ESI source used in the positive ionization mode were as follows: 3.5 kV spray voltage, 325 °C capillary temperature, 300 °C heater temperature, sheath gas flow rate 45 units, and auxiliary gas flow 10 units (units refer to arbitrary values set by the Exactive software). Nitrogen was used for sample nebulization. U-HPLC separations were performed on a Hypersil Gold C18 (Thermo Fisher), 1.9 μm, 2.1 × 50 mm i.d. HPLC column, operated at 30 °C. Each 10 min chromatographic run was carried out at a flow rate of 0.3 mL/min with a binary mobile phase consisting of acetonitrile (A) and 0.1% formic acid (B) using a step gradient profile of 50% A for 0.5 min and increased up to 100% A in 5 min, kept isocratic at 100% for 0.5 min, then decreased down to 50% A in 0.1 min. After re-equilibration at 50% A for 3.9 min, the next sample was injected.
Plant Material
Rhizoma et Radix Notopterygii (2 kg) were purchased in 2008 from Plantasia, Oberndorf, Austria, and authenticated as Notopterygium incisum via DNA-based identification.4 A voucher specimen (no. 650107) has been deposited at the Department of Pharmacognosy, Institute of Pharmaceutical Sciences, Karl-Franzens-University Graz.
Extraction and Isolation
The plant material (2 kg) was ground and percolated with CH2Cl2 (17.5 L), to produce 250 g of an extract. Then, 145 g of the extract was partitioned twice between n-hexane and MeOH (1.5:1), to obtain n-hexane- (39 g) and MeOH-soluble (104 g) portions. The MeOH layer was further partitioned twice between CH2Cl2 and a 60% MeOH–water solution (1:1) to obtain a CH2Cl2 layer (94 g) and an aqueous MeOH layer (1.5 g).
The dried CH2Cl2 layer (94 g) was fractionated using MCI CHP-20P resin, with a MeOH–H2O gradient (40% to 100% MeOH) as mobile phase, to afford 313 fractions. To facilitate subsequent biological testing, 0.3% of each of these fractions was sampled and recombined according to their TLC profile to afford 10 pooled fractions (fraction pools first to 10th), which were assayed together with the CH2Cl2 layer for PPARγ activation. The most active fraction pools, 8 and 9 (eluting with 65–90% MeOH), were subjected to successive column chromatography, including RP-18 (MeOH–H2O, 85:15), Sephadex LH-20 (MeOH–H2O, 60:40), silica gel (n-hexane–ethyl acetate, 3:1 or 2.2:1), and preparative HPLC (acetonitrile–H2O, 68:32, 80:20, or 95:5), to afford 11 compounds: 1 (6.3 mg), 2 (11.2 mg), 3 (1.5 mg), 4 (1.1 mg), 5 (10.9 mg), 6 (9.6 mg), 7 (3.6 mg), 8 (2.3 mg), 9 (4.7 mg), 10 (3.1 mg), and 11 (0.68 mg).
Notoether A (1):
C32H50O3, colorless oil; [α]D20 +155.1 (c 0.2, MeOH); UV (MeCN–H2O) λmax 246, 260 nm; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Tables 1 and 2; positive ESIMS m/z 505 [M + Na]+, 987 [2 M + Na]+; HRESIMS m/z 505.3652 [M + Na]+ (calcd for C32H50O3Na, 505.3658).
Table 1. 13C NMR Spectroscopic Data (150 MHz, CDCl3) for Compounds 1–11a.
| δC |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
| 1 | 116.1 | 117.3 | 116.2 | 117.4 | 117.3 | 117.3 | 116.3 | 117.4 | 117.5 | 116.3 | 117.0 |
| 2 | 136.3 | 136.2 | 136.2 | 136.1 | 135.2 | 136.1 | 136.3 | 136.1 | 135.9 | 136.2 | 137.0 |
| 3 | 61.0 | 63.7 | 61.5 | 63.7 | 61.9 | 63.6 | 62.1 | 63.7 | 63.7 | 85.0 | 64.1 |
| 4 | 80.0 | 77.5 | 79.9 | 77.7 | 78.0 | 78.5 | 79.4 | 77.7 | 78.5 | 141.2 | 97.2 |
| 5 | 69.3 | 71.0 | 69.4 | 71.0 | 70.9 | 70.5 | 69.8 | 70.9 | 70.5 | 112.2 | 81.9 |
| 6 | 69.6 | 67.8 | 69.7 | 68.0 | 68.9 | 69.0 | 69.3 | 68.3 | 69.4 | 80.5 | 111.8 |
| 7 | 79.9 | 81.7 | 79.3 | 81.4 | 80.1 | 79.4 | 79.2 | 81.3 | 77.0 | 99.0 | 142.2 |
| 8 | 58.6 | 57.2 | 58.8 | 57.7 | 58.6 | 57.3 | 58.8 | 58.1 | 60.2 | 59.1 | 79.5 |
| 9 | 128.1 | 128.6 | 127.9 | 128.5 | 127.9 | 126.8 | 128.0 | 128.2 | 124.1 | 129.1 | 127.8 |
| 10 | 134.4 | 131.4 | 134.7 | 131.8 | 134.5 | 133.5 | 134.8 | 131.7 | 136.7 | 134.0 | 135.0 |
| 11 | 27.8 | 28.1 | 27.8 | 28.2 | 27.8 | 28.1 | 27.8 | 28.1 | 28.1 | 27.9 | 28.1 |
| 12 | 29.5 | 29.5 | 29.5 | 29.4 | 29.4 | 29.2 | 29.4 | 29.4 | 29.3 | 29.6 | 30.0 |
| 13b | 29.3 | 29.4 | 29.3 | 29.4 | 29.3 | 29.4 | 29.3 | 29.5 | 29.3 | 29.4 | 29.6 |
| 14b | 29.3 | 29.3 | 29.3 | 29.3 | 29.3 | 29.3 | 29.3 | 29.3 | 29.3 | 29.3 | 29.4 |
| 15 | 31.9 | 32.0 | 31.9 | 32.0 | 31.9 | 31.9 | 31.9 | 32.0 | 32.0 | 31.9 | 32.0 |
| 16 | 22.8 | 22.8 | 22.8 | 22.8 | 22.8 | 22.8 | 22.8 | 22.8 | 22.8 | 22.8 | 22.8 |
| 17 | 14.3 | 14.3 | 14.3 | 14.3 | 14.2 | 14.2 | 14.3 | 14.3 | 14.3 | 14.2 | 14.3 |
| 1′ | 40.8 | 40.9 | 80.4 | 80.5 | 79.4 | 79.4 | 43.5 | 43.2 | 127.0 | 128.9 | 128.8 |
| 2′ | 22.5 | 22.4 | 28.4 | 28.4 | 27.6 | 27.8 | 24.0 | 23.7 | 109.5 | 106.2 | 106.2 |
| 3′ | 37.4 | 38.5 | 38.2 | 38.2 | 36.1 | 37.3 | 41.4 | 41.3 | 146.9 | 147.8 | 147.7 |
| 4′ | 80.0 | 79.5 | 79.3 | 79.2 | 82.3 | 82.2 | 81.3 | 81.3 | 148.3 | 146.6 | 146.6 |
| 5′ | 50.9 | 51.2 | 54.1 | 53.5 | 50.3 | 50.7 | 55.0 | 54.8 | 114.9 | 108.2 | 108.1 |
| 6′ | 22.0 | 21.8 | 67.1 | 67.0 | 71.8 | 71.8 | 71.5 | 71.5 | 123.5 | 129.9 | 130.0 |
| 7′ | 49.3 | 49.8 | 50.1 | 50.5 | 46.2 | 46.4 | 52.0 | 52.1 | 146.1 | 119.0 | 119.1 |
| 8′ | 19.9 | 20.0 | 20.5 | 20.5 | 22.3 | 22.4 | 20.5 | 20.5 | 114.7 | 135.5 | 136.2 |
| 9′ | 44.9 | 45.2 | 41.6 | 41.8 | 36.8 | 36.8 | 42.8 | 43.0 | 165.8 | 72.3 | 72.0 |
| 10′ | 34.8 | 34.8 | 39.4 | 39.4 | 40.9 | 41.0 | 82.9 | 82.5 | |||
| 11′ | 73.5 | 73.1 | 29.1 | 29.0 | 25.9 | 25.8 | 29.7 | 29.7 | |||
| 12′b | 27.4 | 27.6 | 20.7 | 20.9 | 22.5 | 22.5 | 21.7 | 21.7 | |||
| 13′b | 27.0 | 27.1 | 21.2 | 21.2 | 24.1 | 24.1 | 21.4 | 21.3 | |||
| 14′ | 19.2 | 19.3 | 15.1 | 15.1 | 14.9 | 15.0 | 19.3 | 20.0 | |||
| 15′ | 20.6 | 20.7 | 21.4 | 22.8 | 19.7 | 18.5 | 23.3 | 23.3 | |||
| 3′-OMe | 56.1 | 56.1 | 56.1 | ||||||||
13C NMR of 11 was taken at 175 MHz, CDCl3.
Signals are interchangeable.
Table 2. 1H NMR Spectroscopic Data (600 MHz, CDCl3) for Compounds 1–4a.
| δH (J in Hz) |
||||
|---|---|---|---|---|
| H | 1 | 2 | 3 | 4 |
| 1 | 5.41 d (17.5) | 5.46 d (18.5) | 5.42 d (17.0) | 5.47 |
| 5.16 | 5.24 d (10.7) | 5.18 d (10.2) | 5.25 d (10.6) | |
| 2 | 5.79 ddd (16.9, 10.2, 4.7) | 5.94 ddd (17.1, 10.1, 5.4) | 5.83 ddd (17.1, 10.3, 4.8) | 5.94 ddd (16.5, 10.2, 5.5) |
| 3 | 4.84 br d (4.1) | 4.93 br s | 4.85 br t (4.6) | 4.93 br t (4.7) |
| 8 | 5.16 | 5.01 d (6.7) | 5.20 | 5.03 br d (6.7) |
| 9 | 5.50 dd (10.7, 8.4) | 5.45 | 5.51 m | 5.45 m |
| 10 | 5.58 dt (10.5, 7.5) | 5.44 | 5.60 m | 5.45 m |
| 11 | 2.10 q (7.3) | 2.08 m | 2.10 q (7.3) | 2.07 m |
| 12 | 1.38 m | 1.39 m | 1.38 p (6.8) | 1.39 m |
| 13b | 1.28 | 1.30 | 1.28 | 1.29 |
| 14b | 1.28 | 1.30 | 1.28 | 1.29 |
| 15 | 1.27 | 1.27 | 1.27 | 1.27 |
| 16 | 1.29 | 1.29 | 1.29 | 1.30 |
| 17 | 0.88 | 0.89 | 0.88 t (6.8) | 0.89 |
| 1′ | 1.38 m | 1.38 d (13.4) | 3.23 br d (10.4) | 3.19 br d 9.6) |
| 1.06 m | 1.04 dt (3.5, 12.7) | |||
| 2′ | 1.60 m | 1.59 m | 1.72 m | 1.72 m |
| 1.30 m | 1.30 m | 1.59 m | 1.59 m | |
| 3′ | 1.66 m | 1.83 d (10.9) | 1.84 dt (12.8, 3.4) | 1.82 m |
| 1.63 m | 1.48 m | 1.74 m | 1.55 m | |
| 4′ | ||||
| 5′ | 1.45 d (12.3) | 1.26 d (12.3) | 1.29 s | 1.14 s |
| 6′ | 2.03 d (12.5) | 1.87 d (11.8) | 4.63 s | 4.54 s |
| 1.01 q (12.3) | 0.96 q (12.0) | |||
| 7′ | 1.46 m | 1.28 m | 0.91 m | 0.81 br t (10.5) |
| 8′ | 1.60 m | 1.58 m | 1.63 m | 1.61 m |
| 1.54 m | 1.51 m | 1.44 m | 1.46 m | |
| 9′ | 1.45 d (13.0) | 1.44 d (12.3) | 1.90 dt (12.7,3.3) | 1.89 br d (13.6 |
| 1.21 m | 1.15 m | 1.10 td (12.2,3.4) | 1.06 br t (13.1) | |
| 10′ | ||||
| 11′ | 1.51 m | 1.53 m | ||
| 12′ | 1.21 s | 1.19 s | 0.95 d (6.7) | 0.95 d (6.8) |
| 13′ | 1.22 s | 1.19 s | 0.96 d (6.8) | 0.93 d (6.8) |
| 14′ | 0.89 s | 0.88 s | 1.20 s | 1.20 s |
| 15′ | 1.11 s | 1.18 s | 1.51 s | 1.58 s |
Multiplicity of obscured signals is not labeled.
Signals are interchangeable.
Notoether B (2):
C32H50O3, colorless oil; [α]D20 +68.4 (c 0.4, MeOH); UV (MeCN–H2O) λmax 246, 260 nm; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Tables 1 and 2; positive ESIMS m/z 505 [M + Na]+; HRESIMS m/z 505.3652 [M + Na]+ (calcd for C32H50O3Na, 505.3658).
Notoether C (3):
C32H50O4, colorless gum; [α]D20 +155.3 (c 0.05, MeOH); UV (MeCN–H2O) λmax 246, 260 nm; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Tables 1 and 2; positive ESIMS m/z 499 [M + H]+, 997 [2 M + H]+; HRESIMS m/z 521.3607 [M + Na]+ (calcd for C32H50O4Na, 521.3607).
Notoether D (4):
C32H50O4, colorless gum; [α]D20 +8.6 (c 0.04, MeOH); UV (MeCN–H2O) λmax 246, 260 nm; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Tables 1 and 2; positive ESIMS m/z 521 [M + Na]+; HRESIMS m/z 521.3602 [M + Na]+ (calcd for C32H50O4Na, 521.3607).
Notoether E (5):
C32H50O4, colorless needles, mp: 95–98 °C; [α]D20 +144.9 (c 0.4, MeOH); UV (MeCN–H2O) λmax 246, 260 nm; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Tables 1 and 3; positive ESIMS m/z 521 [M + Na]+, 1019 [2 M + Na]+; HRESIMS m/z 521.3600 [M + Na]+ (calcd for C32H50O4Na, 521.3607).
Table 3. 1H NMR Spectroscopic Data (600 MHz, CDCl3) for Compounds 5–8a.
| δH (J in
Hz) |
||||
|---|---|---|---|---|
| H | 5 | 6 | 7 | 8 |
| 1 | 5.41 d (17.0) | 5.47 d (17.5) | 5.42 d (17.2) | 5.47 d (17.2) |
| 5.21 d (10.1) | 5.25 d (10.1) | 5.18 d (10.4) | 5.25 d (10.1) | |
| 2 | 5.80 ddd (16.9, 10.1, 5.3) | 5.94 ddd (16.4, 10.2, 5.4) | 5.81 ddd (17.0, 10.2, 4.8) | 5.94 ddd (16.1, 10.2, 5.4) |
| 3 | 4.82 d (4.4) | 4.93 d (4.6) | 4.79 br d (4.8) | 4.93 br t (5.8) |
| 8 | 5.17 d (8.4) | 5.08 d (8.2) | 5.20 d (8.5) | 4.97 d (7.1) |
| 9 | 5.50 dd (10.6, 8.5) | 5.41 t (9.5) | 5.51 dd (10.7, 8.4) | 5.41 |
| 10 | 5.59 dt (10.8, 7.5) | 5.51 m | 5.60 dt (10.7, 7.5) | 5.44 |
| 11 | 2.09 q (7.2) | 2.08 | 2.11 q (7.7) | 2.07 m |
| 12 | 1.38 p (6.9) | 1.38 | 1.38 | 1.38 |
| 13b | 1.28 | 1.28 | 1.28 | 1.29 |
| 14b | 1.28 | 1.28 | 1.28 | 1.29 |
| 15 | 1.26 | 1.26 | 1.26 | 1.27 |
| 16 | 1.29 | 1.29 | 1.29 | 1.29 |
| 17 | 0.88 t (6.9) | 0.88 t (6.8) | 0.88 t (6.8) | 0.89 t (6.8) |
| OH-3 | 1.91 d (6.5) | |||
| OH-8 | 1.85 | |||
| 1′ | 3.32 dd (10.9, 4.1) | 3.36 dd (11.5, 3.8) | 2.04 m | 1.94 td (10.5,2.2) |
| 2′ | 1.72 m | 1.75 dq (12.5, 4.0) | 1.85 m | 1.82 m |
| 1.57 | 1.57 dt (15.0, 3.4) | 1.61 m | 1.48 m | |
| 3′ | 2.00 m | 2.08 | 1.66 | 1.66 |
| 1.54 | 1.89 td (13.6, 3.9) | |||
| 5′ | 1.78 d (10.9) | 1.83 d (10.8) | 1.80 t (9.7) | 1.80 t (9.6) |
| 6′ | 4.27 ddd (10.4, 3.7, 2.6) | 4.19 ddd (10.4, 4.3, 3.2) | 4.14 dd (8.7, 4.5) | 4.13 |
| 7′ | 1.66 | 1.62 m | 1.07 m | 1.06 m |
| 8′ | 1.67 | 1.66 m | 1.40 m | 1.40 |
| 1.54 | 1.52 | |||
| 9′ | 1.52 | 1.51 | 1.95 dd (13.2, 5.3) | 2.01 dd (12.6, 5.8) |
| 1.28 | 1.29 | 1.54 m | 1.50 | |
| 11′ | 1.98 m | 1.99 dq (13.9,6.6) | 1.68 | 1.67 |
| 12′b | 0.93 d (6.7) | 0.93 d (6.7) | 0.99 d (6.5) | 0.98 d (6.6) |
| 13′b | 1.10 d (6.6) | 1.09 d (6.5) | 1.04 d (6.6) | 1.03 d (6.6) |
| 14′ | 0.99 s | 0.98 s | 1.25 s | 1.32 s |
| 15′ | 1.53 s | 1.39 s | 1.28 s | 1.26 s |
| OH-1′ | 2.36 br s | 2.39 br s | ||
| OH-4′ | 2.37 s | |||
| OH-6′ | 4.76 d (2.6) | 4.66 d (3.1) | 2.23 s | |
Multiplicity of obscured signals is not labeled.
Signals are interchangeable.
Notoether F (6):
C32H50O4, colorless gum; [α]D20 +108.9 (c 0.3, MeOH); UV (MeCN–H2O) λmax 246, 260 nm; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Tables 1 and 3; positive ESIMS m/z 499 [M + H]+, 997 [2 M + H]+, 1019 [2 M + Na]+; HRESIMS m/z 521.3600 [M + Na]+ (calcd for C32H50O4Na, 521.3607).
Notoether G (7):
C32H50O4, colorless gum; [α]D20 +148.5 (c 0.07, MeOH); UV (MeCN/H2O) λmax 246, 260 nm; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Tables 1 and 3; positive ESIMS m/z 521 [M + Na]+; HRESIMS m/z 521.3600 [M + Na]+ (calcd for C32H50O4Na, 521.3607).
Notoether H (8):
C32H50O4, colorless gum; [α]D20 +34.8 (c 0.08, MeOH); UV (MeCN–H2O) λmax 246, 260 nm; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Tables 1 and 3; positive ESIMS m/z 521 [M + Na]+; HRESIMS m/z 521.3600 [M + Na]+ (calcd for C32H50O4Na, 521.3607).
Notoincisol A (9):
C27H32O5, light green oil; [α]D20 +85.5 (c 0.09, MeOH); UV (MeCN–H2O) λmax 237, 328 nm; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Tables 1 and 4; positive ESIMS m/z 437 [M + H]+; HRESIMS m/z 437.2353 [M + H]+ (calcd for C27H33O5, 436.2328).
Table 4. 1H NMR Spectroscopic Data (600 MHz, CDCl3) for Compounds 9–11a.
| δH (J in
Hz) |
|||
|---|---|---|---|
| H | 9 | 10 | 11 |
| 1 | 5.47 d (16.6) | 5.49 d (18.0) | 5.57 (17.5) |
| 5.26 d (10.2) | 5.24 d (10.7) | 5.30 (10.1) | |
| 2 | 5.93 ddd (16.1, 10.2, 5.3) | 6.11 ddd (16.8, 10.3, 6.2) | 6.10 m |
| 3 | 4.93 br s | 5.80 d (6.1) | 5.18 br s |
| 8 | 6.28 d (9.1) | 5.49 d (8.7) | 6.13 d (9.2) |
| 9 | 5.56 t (9.7) | 5.70 t (10.7, 8.4) | 5.53 t (10.2) |
| 10 | 5.70 dt (10.9, 7.6) | 5.65 dt (10.7, 7.3) | 5.74 m |
| 11 | 2.19 m | 2.21 q (7.3) | 2.46 m, 2.25 m |
| 12 | 1.39 m | 1.43 p (7.3) | 1.49 |
| 13b | 1.29 | 1.31 | 1.33 |
| 14b | 1.29 | 1.31 | 1.31 |
| 15 | 1.25 | 1.25 | 1.28 |
| 16 | 1.27 | 1.28 | 1.30 |
| 17 | 0.87 t | 0.85 t (6.8) | 0.89 |
| OH-3 | |||
| OH-8 | |||
| 2′ | 7.03 s | 7.08 s | 7.10 s |
| 5′ | 6.92 d (8.2) | 7.72 s | 7.72 s |
| 6′ | 7.07 br d (8.3) | ||
| 7′ | 7.65 d (15.8) | 7.46 s | 7.48 s |
| 8′ | 6.28 d (15.4) | ||
| 9′ | 5.27d (12.2) | 5.26 d (11.9) | |
| 5.17d (12.1) | 5.13 d (12.4) | ||
| OH-4′ | 5.96 s | ||
| OMe-3′ | 3.93 s | 4.02 s | 4.03 s |
Multiplicity of obscured signals is not labeled.
Signals are interchangeable.
Notoincisol B (10):
C27H32O4, colorless gum; [α]D20 +268.9 (c 0.1, MeOH); UV (MeCN–H2O) λmax 244, 315, 346; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Tables 1 and 4; positive ESIMS m/z 421 [M + H]+; HRESIMS m/z 421.2370 [M + H]+ (calcd for C27H33O4, 421.2379).
Notoincisol C (11):
C27H32O4, colorless gum; [α]D20 +25.8 (c 0.03, MeOH); UV (MeCN–H2O) λmax 244, 316, 346; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Tables 1 and 4; positive ESIMS m/z 421 [M + H]+; HRESIMS m/z 421.2370 [M + H]+ (calcd for C27H33O4, 421.2379).
Cell Culture Reagents, Chemicals, and Plasmids
Dulbecco’s modified Eagle’s medium (DMEM) and fetal calf serum (FCS) were supplied by Lonza (Basel, Switzerland). Pioglitazone, a full PPARγ agonist, was used as positive control and was purchased from Molekula Ltd. (Shaftesbury, UK). For evaluation of PPARγ activation the test compounds were first dissolved in DMSO, divided into aliquots, and frozen at −20 °C until used. In all experiments, DMSO was applied as solvent control, and the final concentration of DMSO was always kept at ≤0.1%. The PPAR luciferase reporter plasmid (tk-PPREx3-luc) was a kind gift from Prof. Ronald M. Evans (Howard Hughes Medical Institute, La Jolla, CA, USA), the plasmid encoding enhanced green fluorescent protein (pEGFP-N1) was supplied by Clontech (Mountain View, CA, USA), and the plasmid encoding human PPARγ (pSG5-PL-hPPAR-γ1) was kindly supplied by Prof. Walter Wahli and Prof. Beatrice Desvergne (Center for Integrative Genomics, University of Lausanne, Switzerland).
PPARγ Luciferase Reporter Gene Transactivation
The PPARγ luciferase reporter gene assay was performed as previously described.27 Briefly, HEK-293 cells (ATCC, Manassas, VA, USA) were grown in DMEM with 2 mM glutamine, 10% FBS, 100 U/mL benzylpenicillin, and 100 μg/mL streptomycin. Cells were grown in 10 cm dishes (6 × 106 cells/dish) for 18 h and then transfected by the calcium phosphate precipitation method with 4 μg of plasmid encoding human PPARγ (pSG5-PL-hPPAR-γ1), 4 μg of reporter plasmid (tk-PPREx3-luc), and 2 μg of pEGFP-N1 as control for internal normalization.28 After 6 h, the cells were transferred to 96-well plates (5 × 104 cells/well) in DMEM without phenol red supplemented with charcoal-stripped FBS, glutamine, and antibiotics. The cells were treated with different concentrations of the indicated compounds and incubated for 18 h. Cells were then lysed, and the luminescence of the firefly luciferase and the fluorescence of EGFP were quantified with a GeniosPro microplate reader (Tecan, Grödig, Austria). The luminescence was finally normalized to the EGFP-derived fluorescence from each well to account for differences in transfection efficiency or cell number.
Inhibition of NO Production in Stimulated RAW 264.7 Macrophages
RAW 264.7 macrophages were stimulated with lipopolysaccharides (LPS) and interferon-γ (IFNγ) for induction of iNOS gene expression. The effects on NO production were determined by photometric quantification of nitrite accumulation in cell culture supernatants using the Griess assay compared with a sodium nitrite standard curve after 16 h of incubation with the respective sample as described by Baer et al. with slight modifications.29,30 Activity is referred to nitrite accumulation of cells treated with LPS/IFN-γ/DMSO (final concentration of 0.1% DMSO serves as solvent control). Test compounds were first dissolved in DMSO and then diluted with PBS to obtain respective concentrations. l-NMMA (N-monomethyl-l-arginine), a known nonselective inhibitor of all NOS isoforms, was used as a positive control. IC50 determination was performed in eight concentrations, each in at least three independent experiments, every time in duplicate.
Statistical Methods and Data Analysis
Statistical analyses for effects on PPARγ were performed with the GraphPad Prism software version 4.03. Nonlinear regression (with sigmoidal dose response) was used to calculate the EC50 values and maximal fold activation.
IC50 values for effects on NO production were calculated with the SigmaPlot program package employing the four-parameter logistic regression model. Statistical differences were evaluated using one-way ANOVA. Differences with a p value < 0.05 were considered significant.
Molecular Docking
The prediction of binding modes for the investigated compounds was accomplished in a docking study. A quantum mechanics-polarized ligand docking (QPLD) workflow available within Maestro version 9.2.112 (Schrödinger, LLC, New York, NY, 2011,www.schrodinger.com) was applied as described previously.14 For each molecule, up to 10 docking poses were calculated. For further investigations, the highest ranked docking solution as determined by the Emodel scoring function was used. Visual inspection and analysis of the retrieved binding poses were performed with LigandScout 3.1 (Inte:Ligand GmbH, Maria Enzersdorf, Austria, 2012, www.inteligand.com).31
Acknowledgments
We gratefully acknowledge the funding provided by the Austrian Science Fund (FWF): S 10705, S 10704, and S 10711 (NFN Drugs from Nature Targeting Inflammation (DNTI)). We also gratefully acknowledge the skillful technical assistance of J. Benedics and M. Gössinger. We would like to thank Dr. Zi-Yu Wang for discussions on the biogenetic pathway.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Sun Y.; Xu Q. Pharmacol. Res. 2002, 46, 333–337. [DOI] [PubMed] [Google Scholar]
- Okuyama E.; Nishimura S.; Ohmori S.; Ozaki Y.; Satake M.; Yamazaki M. Chem. Pharm. Bull. 1993, 41, 926–929. [DOI] [PubMed] [Google Scholar]
- Zschocke S.; Lehner M.; Bauer R. Planta Med. 1997, 63, 203–206. [DOI] [PubMed] [Google Scholar]
- Blunder M.; Liu X.; Kunert O.; Winkler N. A.; Schinkovitz A.; Schmiderer C.; Novak J.; Bauer R. Planta Med. 2014, 80, 415–418. [DOI] [PubMed] [Google Scholar]
- Rollinger J. M.; Zidorn C.; Dobner M. J.; Ellmerer E. P.; Stuppner H. Z. Naturforsch., C J. Biosci. 2003, 58, 553–557. [DOI] [PubMed] [Google Scholar]
- Deng S.; Wang Y.; Inui T.; Chen S.-N.; Farnsworth N. R.; Cho S.; Franzblau S. G.; Pauli G. F. Phyther. Res. 2008, 22, 878–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman J. D.; Evangelopoulos D.; Gupta A.; Prieto J. M.; Gibbons S.; Bhakta S. Phyther. Res. 2013, 27, 993–998. [DOI] [PubMed] [Google Scholar]
- Dall’Acqua S.; Viola G.; Piacente S.; Cappelletti E. M.; Innocenti G. J. Nat. Prod. 2004, 67, 1588–1590. [DOI] [PubMed] [Google Scholar]
- Meot-Duros L.; Cerantola S.; Talarmin H.; Le Meur C.; Le Floch G.; Magne C. Food Chem. Toxicol. 2010, 48, 553–557. [DOI] [PubMed] [Google Scholar]
- Cariou B.; Charbonnel B.; Staels B. Trends Endocrinol. Metab. 2012, 23, 205–215. [DOI] [PubMed] [Google Scholar]
- Wang L.; Waltenberger B.; Pferschy-Wenzig E.-M.; Blunder M.; Liu X.; Malainer C.; Blazevic T.; Schwaiger S.; Rollinger J. M.; Heiss E. H.; Schuster D.; Kopp B.; Bauer R.; Stuppner H.; Dirsch V. M.; Atanasov A. G.. Biochem. Pharmacol. 2014, Jul 30. pii: S0006-2952(14)00424-9. doi: 10.1016/j.bcp.2014.07.018. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy R. M.; Amin A. R.; Abramson S. B. Arthrit. Rheumatol. 1998, 41, 1141–1151. [DOI] [PubMed] [Google Scholar]
- Kleinert H.; Pautz A.; Linker K.; Schwarz P. M. Eur. J. Pharmacol. 2004, 500, 255–266. [DOI] [PubMed] [Google Scholar]
- Atanasov A. G.; Blunder M.; Fakhrudin N.; Liu X.; Noha S. M.; Malainer C.; Kramer M. P.; Cocic A.; Kunert O.; Schinkovitz A.; Heiss E. H.; Schuster D.; Dirsch V. M.; Bauer R. PLoS One 2013, 8, e61755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furumi K.; Fujioka T.; Fujii H.; Okabe H.; Nakano Y.; Matsunaga H.; Katano M.; Mori M.; Mihashi K. Bioorg. Med. Chem. Lett. 1998, 8, 93–96. [DOI] [PubMed] [Google Scholar]
- Fujioka T.; Furumi K.; Fujii H.; Okabe H.; Mihashi K.; Nakano Y.; Matsunaga H.; Katano M.; Mori M. Chem. Pharm. Bull. 1999, 47, 96–100. [DOI] [PubMed] [Google Scholar]
- Ando M.; Arai K.; Kikuchi K.; Isogai K. J. Nat. Prod. 1994, 57, 1189–1199. [DOI] [PubMed] [Google Scholar]
- Garcia-Granados A.; Martinez A.; Molina A.; Onorato M. E. Phytochemistry 1986, 25, 2171–2173. [Google Scholar]
- Irwin M. A.; Geissman T. A. Phytochemistry 1973, 12, 849–852. [Google Scholar]
- Squillacote M. E.; Neth J. M. Magn. Reson. Chem. 1987, 25, 53–56. [Google Scholar]
- Nukina M.; Otuki T.; Kurebayashi T.; Hosokawa K.; Sekine M.; Ito S.; Suenaga M.; Sato A.; Sassa T. Tennen Yuki Kagobutsu Toronkai Koen Yoshishu 1996, 38th, 391–396. [Google Scholar]
- Takahashi H.; Yoshioka S.; Kawano S.; Azuma H.; Fukuyama Y. Chem. Pharm. Bull. 2002, 50, 541–543. [DOI] [PubMed] [Google Scholar]
- Bruno M.; de la Torre M. C.; Rodriguez B.; Omar A. A. Phytochemistry 1993, 34, 245–247. [Google Scholar]
- Balde A. M.; Claeys M.; Pieters L. A.; Wray V.; Vlietinck A. J. Phytochemistry 1991, 30, 1024–1026. [Google Scholar]
- Zoete V.; Grosdidier A.; Michielin O. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2007, 1771, 915–925. [DOI] [PubMed] [Google Scholar]
- Dixit V. A.; Bharatam P. V. J. Comput. Med. 2013, 2013, 38. [Google Scholar]
- Rozema E.; Atanasov A. G.; Fakhrudin N.; Singhuber J.; Namduang U.; Heiss E. H.; Reznicek G.; Huck C. W.; Bonn G. K.; Dirsch V. M.; Kopp B. Evid.-Based Complementary Altern. Med. 2012, 2012, 983023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham F. L.; Van der Eb A. J. Virology 1973, 52, 456–467. [DOI] [PubMed] [Google Scholar]
- Baer H. P.; Schmidt K.; Mayer B.; Kukovetz W. R. Life Sci. 1995, 57, 1973–1980. [DOI] [PubMed] [Google Scholar]
- Konkimalla V. B.; Blunder M.; Bauer R.; Efferth T. Biochem. Pharmacol. 2010, 79, 1573–1580. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Langer T. J. Chem. Inf. Model. 2005, 45, 160–169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.