

Abstract
Disruption of cell-cell junctions and the concomitant loss of polarity, downregulation of tumor-suppressive adherens junctions and desmosomes represent hallmark phenotypes for several different cancer cells. Moreover, a variety of evidence supports the argument that these two common phenotypes of cancer cells directly contribute to tumorigenesis. In this study, we aimed to determine the status of intercellular junction proteins expression in JJ012 human malignant chondrosarcoma cells and investigate the effect of the antitumorigenic cytokine, proline-rich polypeptide-1 (PRP-1) on their expression. The cell junction pathway array data indicated downregulation of desmosomal proteins, such as desmoglein (1,428-fold), desmoplakin (620-fold) and plakoglobin (442-fold). The tight junction proteins claudin 11 and E-cadherin were also downregulated (399- and 52-fold, respectively). Among the upregulated proteins were the characteristic for tumors gap junction β-5 protein (connexin 31.1) and the pro-inflammatory pathway protein intercellular adhesion molecule (upregulated 129- and 43-fold, respectively). We demonstrated that PRP-1 restored the expression of the abovementioned downregulated in chondrosarcoma desmosomal proteins. PRP-1 inhibited H3K9-specific histone demethylase activity in chondrosarcoma cells in a dose-dependent manner (0.5 µg/ml PRP, 63%; 1 µg/ml PRP, 74%; and 10 µg/ml PRP, 91% inhibition). Members of the H3K9 family were shown to transcriptionally repress tumor suppressor genes and contribute to cancer progression. Our experimental data indicated that PRP-1 restores tumor suppressor desmosomal protein expression in JJ012 human chondrosarcoma cells and inhibits H3K9 demethylase activity, contributing to the suppression of tumorigenic potential in chondrosarcoma cells.
Keywords: chondrosarcoma, proline-rich polypeptide-1, desmosomal proteins, H3K9-specific histone demethylase, cell-cell adhesion
Introduction
Chondrosarcoma is a highly aggressive bone cancer of mesenchymal origin, which is resistant to chemo- and radiation therapies, leaving surgical resection as the only option. Metastatic spread of malignant cells eventually develops following surgical resection. The malignant progression to chondrosarcoma has been suggested to involve a degree of mesenchymal-to-epithelial transition (MET) and it has been demonstrated in an in vitro model that chondrosarcoma cells may transition to a more epithelial-like phenotype under certain conditions, with the acquisition of epithelial markers, such as E-cadherin, desmocollin 3, maspin and 14-3-3σ due to the epigenetic switch (1). The epithelial-to-mesenchymal transition (EMT) and its reverse process, MET, play central roles in embryogenesis. EMT is an important process that permits epithelial cells to dissolve intercellular connections and acquire mesenchymal properties, exhibiting augmented migratory ability. The hallmarks of EMT include loss of structural adhesion components, such as E-cadherin, cell-cell adhesion glycoprotein, and gain of mesenchymal cell markers, such as vimentin. E-cadherin expression is lost in carcinomas during EMT and restored during MET, providing a survival advantage for these cells in a challenging ectopic environment. Aberrant activation of EMT has been reported to contribute to tumour progression/metastasis. During metastasis-associated MET, the epithelial tumour cells, which have undergone EMT and are present in the blood circulation, reverse the EMT process to grow at distant sites as epithelial tumour cells (2,3). Sarcoma metastasis is not well understood (1). Sarcomas are considered to arise from mesenchymal cells and, thus, do not have a baseline epithelial differentiation state that is seen in several carcinomas. This fact excludes sarcomas from the EMT-MET metastasis paradigm, whereby tumor cells in carcinomas must lose their epithelial characteristics to escape the primary tumor, but regain them to colonize the secondary site. Undifferentiated Ewing's sarcoma cells, for example, may innately have the right balance of epithelial and mesenchymal characteristics that allow for successful growth at the primary and secondary sites, as well as a high propensity for metastasis. Providing support for this intrinsic ability, micrometastatic spread is ubiquitous in Ewing's sarcoma patients and occurs during the early stages of the disease, suggesting a parallel progression of tumor and metastases. This is in contrast to certain carcinomas, such as colorectal cancer, where a slow progression and accumulation of mutations eventually leads to an invasive malignancy. A ‘passive metastasis’ model was recently proposed for Ewing's sarcoma dissemination to contrast the deliberate steps required in carcinoma metastasis (4). MET in sarcoma is represented by expression of epithelial-like markers, such as E-cadherin, whereas mesenchymal markers predominate in tumor cells. Disruption of cell-cell junctions and the concomitant loss of polarity represent hallmark phenotypes for a number of different cancer cells. Moreover, a variety of evidence supports the argument that these two common phenotypes of cancer cells directly contribute to tumorigenesis, rather than simply representing indirect consequences of the transformed state of these cells (5). Intercellular junctions are of paramount importance in EMT and MET. A disruption of this cell-cell communication, through multiple mechanisms, may in the short term be a protective mechanism to limit the spread of toxicity in a tissue following chemical or radiation damage. However, sustained downregulation confers a loss of tumor-suppressive action (6).
Adherens junctions, also referred to as belle desmosomes, are protein complexes that are found in cell-cell junctions and their cytoplasmic face is linked to the actin cytoskeleton. Three main protein families constitute traditional adherens junction complexes: Transmembrane cadherins, armadillo proteins and cytoskeletal adaptors. The cytoplasmic tails of cadherins bind members of the armadillo protein family, such as β-catenin. Cadherins communicate with the actin cytoskeleton through contacts with β-catenin (7).
Similar to adherens junctions, desmosomes comprise three main protein families, namely cadherins, armadillo proteins and plakins. The two types of desmosomal cadherins, namely the desmogleins (DSG) 1–4 and the desmocollins (DSC) 1–3, mediate adhesion between apposing cells through interactions of their ectodomains. Intracellularly, desmosomal cadherins bind to the armadillo proteins junction plakoglobin (JUP, γ-catenin) and plakophilins (PKP) 1–3, which help bridge the cadherins to the intermediate filament cytoskeleton. The loss or reduction of one or more desmosome components, including DSG 1–3, DSC 2, DSC 3, JUP, PKP 1–3 and DSP, is observed during development and/or the progression of various human cancers. Adherens junctions and desmosomes are known to be downregulated in cancer, as they exert tumor-suppressive effects (8–11).
The other main types of vertebrate intercellular junctions may be broadly categorized as ‘communicating’ and ‘occluding’ junctions. The latter, also known as tight junctions, seal adjacent membranes through multiple spanning transmembrane proteins, such as occludins and claudin (5).
Finally, there are also gap junctions, some of which may be downregulated in cancer (12), or may not depend on cellular context. Gap junctions are intercellular membrane channels that allow the direct exchange of small molecules (<1.2 kD) between adjacent cells and are composed of two hemichannels (connexons), which are in turn formed by the oligomerization of six protein subunits, termed connexins.
The focus of the present study was the investigation of intercellular junction protein expression in human malignant chondrosarcoma and the effect of the antiproliferative cytostatic, proline-rich polypeptide-1 (PRP-1) (13–18) on their expression. PRP-1 was originally isolated from the neurosecretory granules of bovine neurohypophysis. This compound is a cytokine immunomodulator (17), with a wide spectrum of properties (18).
Materials and methods
Cell lines and tissue culture
JJ012 chondrosarcoma cells were cultured in a monolayer, as previously described (13–16), in a medium consisting of Dulbecco's modified Eagle's medium, supplemented with F12, 10% fetal bovine serum, 25 µg/ml ascorbic acid, 100 ng/ml insulin, 100 nM hydrocortisone and 1% penicillin/streptomycin. Upon confluency, the cells were trypsinized and seeded in 6-well cluster dishes at a concentration of 1×106 cells/ml and incubated for 24 h in a 5% CO2 incubator. The experiments were performed in biological triplicates. The cells were treated with PRP-1 peptide in the experimental samples, while no peptide was added to the control wells.
Gel electrophoresis and western blotting
Upon confluency, the cells were trypsinized and seeded in 6-well cluster dishes at a concentration of 1×106 cells/ml. The experimental samples were treated with PRP-1 in corresponding concentrations, whereas control samples were not treated with the peptide. The cells were incubated for 24 h in a 5% CO2 incubator at 37°C. On the following day, the cells were washed with ice-cold phosphate-buffered saline. A protease inhibitor was added to the cell lysis buffer (C2978; Sigma-Aldrich, St. Louis, MO, USA) in a 1:100 ratio. The cells were collected with a scraper and centrifuged at 15,000 × g at 4°C. The supernatant was collected and the protein concentration was measured. The pellets were frozen at −80°C until loading on the gel (20 µg/lane). Polyacrylamide gel electrophoresis and western blotting reagents were supplied by Lonza, Inc., (Allendale, NJ, USA) and all the related procedures followed the company's protocol. The catalog numbers for the reagents and the suppliers are listed below.
Pager Gold Precast Gels (59502; 10% Tris-Glycine; Lonza, Inc.); ECL reagent (RPN2109; GE Healthcare, Little Chalfont, UK); Western Blocker solution (W0138; Sigma-Aldrich); ProSieve Quad Color Protein marker (4.6–300 kD, 00193837; Lonza, Inc.); 20X reducing agent for ProSieve ProTrack Dual Color Loading buffer (00193861; Lonza, Inc.); ProTrack Loading buffer (00193861; Lonza, Inc.); ProSieve ProTrack Dual Color Loading buffer EX running buffer (00200307; Lonza, Inc.); ProSieve EX Western Blot Transfer buffer (00200309; Lonza, Inc.); Immobilon®-P Polyvinylidene difluoride membranes (P4188; Sigma-Aldrich).
Antibodies
Primary
Rabbit anti-desmoplakin antibody (ab71690; Abcam, Cambridge, MA, USA); rabbit anti-JUP antibody (2309S; Cell Signaling Technology, Inc., Beverly, MA, USA); mouse anti-DSG 2 antibody (ab14415; Abcam); rabbit anti-claudin 11 antibody (LS-C6168; LifeSpan Biosciences, Inc., Seattle, WA, USA); mouse anti-E-cadherin (32A8) (5296; Cell Signaling Technology, Inc.); and mouse monoclonal anti-α tubulin antibody (T5168; Sigma-Aldrich).
Secondary
Anti-mouse IgG (A9044; Sigma-Aldrich); and goat anti-rabbit IgG peroxidase conjugate (A0545; Sigma-Aldrich).
RNA isolation
The cells were spun down, the medium was aspirated, the pellets were snap-frozen and shipped to the Qiagen Service Core for Genomics and Gene Expression.
RNA was isolated using RNEasy Mini kit (74104; Qiagen, Hilden, Germany) following the manufacturer's protocol for cells. RNA quality was determined using the Agilent BioAnalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA) with RNA 6000 Nano kits (5067-1511; Agilent Technologies). The total RNA yield, 260/280 and 260/230 ratios were measured using a NanoDrop spectrophotometer 2000 (Thermo Scientific, Waltham, MA, USA).
Reverse transcription-polymerase chain reaction (RT-PCR)
The following methods were implemented to assess gene expression. RNA was isolated using the Qiagen RNEasy Mini kit (74101; Qiagen). RNA concentration was determined with the NanoDrop 2000 spectrophotometer. An RNA quality check was performed using BioAnalyzer 2100 (Agilent Technologies). The RT reaction was performed using RT2 First Strand kit (330401; Qiagen) with 100 ng RNA input per sample. RT-PCR was performed using the ABI 7900 PCR system (Applied Biosystems, Foster City, CA, USA) in a 384-well format. Ct values were normalized using the Ct value of glyceraldehyde 3-phosphate dehydrogenase. Data analysis was performed using the data analysis tool at http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php.
Cell junction pathway
This array analysis was performed by Qiagen (SABiosciences) using PAHS-213Z cell junction finder arrays.
Dispase assay
Dispase assay was performed as previously described (19). Dispase II was supplied by Roche Life Science, Indianapolis, IN (04942078001).
Nuclear extraction
The extraction procedure was performed with the EpiSeeker Nuclear Extraction kit (ab113474; Abcam).
H3K9 demethylase activity assay
The quantification of H3K9-specific histone demethylase activity was performed using the EpiSeeker Histone H3 (K9) Demethylase Activity Quantification Assay kit (113458; Abcam).
Results
Downregulation of desmosomal and tight junction proteins in the JJ012 human chondrosarcoma cell line
We investigated the status of intercellular junction protein expression in JJ012 human malignant chondrosarcoma cells and investigated the effect of the antitumorigenic cytokine, PRP-1, on their expression. The cell junction pathway array data indicated downregulation of desmosomal proteins, such as DSG (1,428-fold), desmoplakin (620-fold) and JUP (442-fold). The tight junction proteins claudin 11, claudin I and E-cadherin were also downregulated (399-, 206- and 52-fold, respectively) (Table I, Fig.1).
Table I.
Downregulated proteins in the JJ012 human chondrosarcoma cell line detected by cell junction pathway arrays.
| Cellular localization | Gene symbol | Gene name | Fold downregulationa |
|---|---|---|---|
| Desmosomes | DSG2 | Desmoglein-2 | 1,428 |
| Desmosomes | DSP | Desmoplakin | 620 |
| Desmosomes | JUP | Junction plakoglobin | 442 |
| Tight junctions | CLDN11 | Claudin 11 | 399 |
| Tight junctions | CLDN1 | Claudin 1 | 206 |
| Tight junctions | CDH1 | E-cadherin | 52 |
| Gap junctions | GJB2 | Connexin 26; gap junction β-2 protein | 49 |
| Desmosomes | DSC3 | Desmocollin 3 | 49 |
| Focal adhesions | ITGB3 | Integrin, β 3; platelet glycoprotein IIIa; antigen CD61 | 22 |
| Focal adhesions | ITGA7 | α 7 integrin gene | 20 |
| Tight junctions | JAM2 | Junctional adhesion molecule B | 12 |
| Gap junctions | GJA3 | GJA3 gap junction protein, α 3 | 15 |
| Tight junctions | CLDN9 | Claudin 9 | 10 |
| Focal adhesions | ITGA9 | Integrin, α 9 | 8 |
| Tight junctions | OCLN | Occludin | 5 |
| Tight junctions | CLDN6 | Claudin 6 | 4 |
P<0.05. Fold downregulation of intercellular junction proteins in chondrosarcoma COMPARED TO control chondrocytes. THE most SIGNIFICANTLY downregulated were desmosomal proteins desmoglein 2, desmoplakin and junction plakoglobin. Among THE downregulated proteins were ALSO Claudin 11, Claudin 1 and E-cadherin, representing adherens and tight junctions. These proteins were SELECTED FOR western blot ANALYSIS FOLOWING treatment with proline-rich polypeptide-1.
Figure 1.

Heat map of JJ012 human chondrosarcoma gene expression in cell junction pathway profile array. Downregulation of desmosomal proteins, including plakoglobin (JUP), desmoglein 2 (DSG2) and desmoplakin (DSP), was observed in the JJ012 human chondrosarcoma cell line. Experimental samples (JJ012 chondrosarcoma cells), were labeled as Sarc 1, 2 and 3 and controls (C-28 chondrocytes) were labeled as Sarc 4, 5 and 6. Dark color, upregulated genes; light color, downregulated genes.
By contrast, the cell junction pathway array indicated the upregulation of gap junction β-5 protein (GJB5, connexin 31.1) and the pro-inflammatory pathway protein intercellular adhesion molecule (ICAM), which are both common in tumors and pro-inflammatory pathways (Table II, Fig. 1).
Table II.
Upregulated proteins in the JJ012 human chondrosarcoma cell line detected by cell junction pathway arrays.
| Cellular localization | Gene symbol | Gene name | Fold upregulationa |
|---|---|---|---|
| Gap junctions | GJB5 | Gap junction β-5 protein | 129 |
| Tight junctions | ICAM1 | Intercellular adhesion molecule 1; CD54; cluster of differentiation 54 | 43 |
| Gap junctions | GJE1 | Gap junction protein, ε 1 | 34 |
| Tight junctions | ICAM2 | Intercellular adhesion molecule 2 | 23 |
| Focal adhesions | CAV1 | Caveolin-1 | 15 |
| Adherens junctions | PVRL1 | Poliovirus receptor-related 1 herpes virus entry mediator C; nectin-1; CD111 | 8 |
| Focal adhesions | ITGA2 | Integrin, α 2 (CD49B, α 2 subunit of VLA-2 receptor) | 8 |
| Tight junctions | ESAM | Endothelial cell-selective adhesion molecule | 7 |
| Hemidesmosomes | DST | Dystonin | 5 |
| Focal adhesions | ITGA6 | Integrin, α 6 | 6 |
| Focal adhesions | ITGA5 | Integrin, α 5 | 6 |
| Focal adhesions | ITGA4 | Integrin, α 4 | 4 |
| Focal adhesions | ITGA3 | Integrin, α 3 | 3 |
| Tight junctions | CLDN4 | Claudin 4 | 3 |
| Tight junctions | CLDN12 | Claudin 12 | 3 |
P<0.05. Gap junction β-5 protein and ICAM1 were among THE most SIGNIFICANTLY upregulated proteins in human chondroSARcoma cellS. Both proteins are known to be involved in tumorigenic and pro-inflammatory pathways.
PRP-1 restores desmosomal protein expression in the JJ012 human chondrosarcoma cell line
The selection of the proteins for western blot analysis was based on the cell junction pathway arrays data. The majority of the downregulated desmosomal and tight junction proteins were included in these series. The western blot experiments indicated the restoration of desmosomal proteins JUP, DSG and desmoplakin following PRP-1 treatment (10 µg/ml) in a dose-dependent manner. PRP-1 did not exert any effect on tight junction protein expression, including E-cadherin (Fig. 2).
Figure 2.
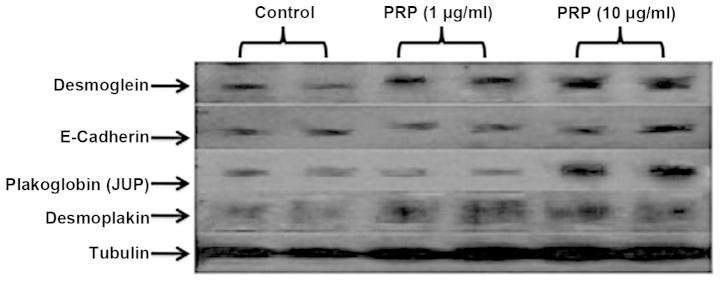
Restoration of desmosomal protein gene expression following treatment with proline-rich polypeptide-1 (PRP-1). PRP-1 restored desmosomal protein expression, including desmoglein 2, plakoglobin (JUP), desmoplakin expression levels, in a dose-dependent manner. For JUP, the restorative effect of PRP-1 appeared at 10 µg/ml, whereas for desmoglein 2, it gradually increased upon treatment with 1–10 µg/ml PRP-1. For desmoplakin, a strong effect was already present at 1 µg/ml PRP-1 and continued at 10 µg/ml. However, PRP-1 did not affect E-cadherin protein levels, which, despite downregulation in chondrosarcoma, was weakly expressed due to mesenchymal-to-epithelial transition, characteristic to sarcomas. PRP-1 did not exert any effect on the expression of claudins (data not shown). α tubulin was used as a control.
Restoration of tumor suppressor proteins with PRP-1 was cell-cell adhesion-independent
To determine whether the restoration of the desmosomal proteins with PRP-1 was adhesion-dependent, we performed a cell-cell adhesion dispase assay. By dividing the number of cells disassociated by the dispase by the total number of cells in the trypsin, we obtained a percent disassociation. The higher the number, the less adhesive the cells. As seen in Fig. 3, PRP-1 in different concentrations did not exert any effect on dispase activity.
Figure 3.

Proline-rich polypeptide-1 (PRP-1) did not affect cell-cell adhesion in the dispase assay. PRP-1 did not exert any effect in the dispase assay, which was used to determine cell-cell adhesion. The restoration of desmosomal proteins as tumor suppressors was independent from their adhesion properties.
PRP-1 inhibited H3K9 demethylase activity in the JJ012 human chondrosarcoma cell line
We conducted experiments to determine H3K9-specific histone demethylase activity in JJ012 human chondrosarcoma cells and investigated the effect of PRP-1 on the enzymes activity, which is known to be cellular context-dependent. Lysine histone methylation is one of the most important epigenetic marks and is essential for the regulation of multiple cellular processes. The methylation of H3K9 is of particular importance, as it is associated with repression regions of the genome. H3K9 demethylase (including JMJD1 and JMJD2) families have been found to be involved in cancer progression. The total H3K9 demethylase activity in chondrosarcoma cells was detected as 421 ng/h/µg and was inhibited by PRP-1 in a dose-dependent manner: 0.5 µg/ml, 63%f; 1 µg/ml, 74%; and 10 µg/ml, 91% inhibition (Table III).
Table III.
H3K9-specific histone demethylase (HDM) activity in the JJ012 cell line and inhibition with different concentrations of proline-rich polypeptide-1 (PRP-1).
| H3K9 HDM activity (RFU/h/μg) | H3K9 HDM activity (ng/h/μg) | PRP-1 (μg/ml) | Inhibition (%) |
|---|---|---|---|
| 843 | 421 | 0.5 | 63 |
| 1 | 74 | ||
| 10 | 91 |
Total H3K9 HDM activity was estimated in JJ012 human chondrosarcoma cellS. H3K9 HDM activity was inhibited by PRP-1 in A dose-DEPENDENT manner, WITH 91% inhibition upon treatment with 10 μg/ml PRP-1. RFU, RELATIVE FLUORESCENCE UNITS.
Discussion
We previously described the antitumorigenic effect of cytostatic PRP-1 in the JJ012 human chondrosarcoma cell line (13–16). In our attempt to further elucidate the antiproliferative effect of this peptide, we conducted experiments to determine its possible role in the regulation of intercellular junctions. Intercellular junctions are crucial for cell function and enable cell interactions and contact between the plasma membranes of adjacent cells. Most importantly, the intercellular junction desmosomal proteins, unlike the majority of gap junction proteins, manifest tumor suppressor functions, which are lost due to the downregulation of these proteins in tumors. To the best of our knowledge, this is the first attempt to characterize the status of intercellular adhesion junction proteins in the JJ012 human chondrosarcoma cell line and investigate the effect of the cytostatic PRP-1 peptide on the expression of these proteins. Cell junction pathway array profiling was performed to compare the expression of intercellular junction proteins in JJ012 human chondrosarcoma cells to that in control C-28 chondrocytes.
The array data indicated the upregulation of GJB5 (connexin 31.1) and the pro-inflammatory pathway protein ICAM, which are both common in tumors (Table II, Fig. 1). Upregulated GJB5 protein was found in colon cancer, which regulates cell adhesion, motility, proliferation and metastasis. Gap junction proteins promote stromal-epithelial interactions in tumors, play a role in tumor progression and regulate the survival of certain stem cells (20). ICAM was reported to play an important role in cancer invasion (21). Strong downregulation of desmosomal proteins, such as DSG, desmoplakin and JUP was observed. The tight junction proteins claudin 11 and E-cadherin were also downregulated (Table I, Fig. 1). Currently available evidence indicates that desmosomes play an important role in the regulation of cell proliferation, differentiation and signal transduction. A recent study using mouse genetic approaches uncovered tumor suppressor properties and demonstrated that desmosome downregulation occurs prior to that of adherens junctions (22). Desmosome loss promotes cancer as a specific mechanism, rather than by a general change in the differentiation status, such as EMT (23,24). The fact that some experiments support a tumour-suppressive role for desmosomes in cancer and others provide evidence for an oncogenic function, may reflect real context-dependent differences in the contribution of desmosomes to cancer (25,26). The selection of the proteins to be subjected to western blotting analysis was based on the level of downregulation of the proteins in JJ012 human chondrosarcoma cells compared to control C-28 chondrocytes in cell junction pathway arrays data. We did not investigate the effect of PRP-1 on the upregulated proteins, as we focused on underexpressed junctional proteins in tumorigenesis and the effect of antitumorigenic PRP-1 on their restoration in chondrosarcoma. In our western blot experiments, we were able to demonstrate the restoration of the expression of the desmosomal proteins JUP, desmoplakin and DSG following treatment with PRP-1, compared to untreated controls, in chondrosarcoma cells (Fig. 2). JUP encodes for the cell-adhesion protein γ-catenin. In conjunction with β-catenin, γ-catenin links E-cadherin to the actin cytoskeleton, forming intercellular cadherin-catenin complexes, a key part of the extracellular matrix. Loss of JUP as a tumor suppressor is associated with increased cancer invasion. JUP may efficiently suppress the tumorigenicity of cells in the presence of, or independently of the cadherin-catenin complex (27,28). Several groups have observed the loss of JUP expression in a wide range of tumors. The majority of those studies investigated JUP in conjunction with other adhesive junctional proteins and demonstrated that loss of JUP expression, which is an early event in tumorigenesis, in conjunction with the lack of expression of other cell-cell adhesion proteins, such as E-cadherin, α-catenin, β-catenin, DSG, or desmoplakin, resulted in increased tumor formation and size and was correlated with advanced tumor stage, poor patient survival and increased metastasis (29). Decreased expression/loss of desmoplakin and DSG as tumor suppressors was reportedly attributed to invasive tumorigenic potential (9,11). PRP-1 did not exert any effect on occluding/tight junction proteins, such as claudins (data not shown) and E-cadherin (Fig. 2). The latter was weakly expressed in chondrosarcoma cells due to MET, as was previously discussed. Of note, the dispase assay results did not indicate any change in cell-cell adhesion in human chondrosarcoma cells following PRP-1 treatment (Fig. 3). Thus, PRP-1 restored cell adhesion intercellular junction desmosomal protein expression without restoring cell-cell adhesion. This fact supports the hypothesis that the tumor-suppressive function of desmosomal proteins is independent from their cell-cell adhesive properties in certain cases and is possibly tissue-specific. JUP, for example, suppressed keratinocyte motility through both cell-cell adhesion-dependent and -independent mechanisms (30). An analysis of the JUP promoter described CpG islands within the promoter and it was observed that inhibition of DNA methylation may result in increased JUP expression (29). The formation of the H3K9me3 mark in promoter-associated chromatin has important consequences on long-term gene silencing, as this modification is considered to be a prelude to the recruitment of DNA methyltransferases that catalyze DNA methylation. Methylation at the CpG dinucleotides near gene promoters is associated with highly stable gene silencing that may be inherited with high fidelity over the course of multiple successive cell divisions (31–33).
Based on this information, one may expect activation of H3K9 demethylase following PRP-1 treatment, since PRP-1 restored the expression of desmosomal proteins. The subsequent set of experiments was focused on H3K9 demethylase activity estimation in chondrosarcoma cells and the effect of PRP-1 on this epigenetic enzymes activity.
Our experimental results, however, revealed a strong dose-dependent inhibition of H3K9 demethylase activity in JJ012 cells upon PRP-1 treatment (Table III). This fact may be explained by other literature results, demonstrating that JMJD2A, a H3K9me3 demethylase, transcriptionally repressed tumor suppressor genes and contributed to cancer progression (34). Moreover, a previous study reported the G1/S arrest of cancer cells following knockdown of JMJD2A (35). This is in accord with our previous observations, where PRP-1 exerts its cytostatic effect by causing G1/S arrest (15). To the best of our knowledge, this is the first report on intercellular junction protein expression in JJ012 human chondrosarcoma cells, providing new experimental evidence on desmosomal protein expression restoration by PRP-1 in a cell-cell adhesion-independent manner, accompanied by inhibition of H3K9-specific histone demethylase activity, leading to suppression of the tumorigenic potential in chondrosarcoma cells.
Acknowledgements
We would like to thank the scientific team of SABiosciences for their help with the cell junction pathway array data. This study was supported by the University of Miami Tissue Bank Research account.
References
- 1.Fitzgerald MP, Gourronc F, Teoh ML, et al. Human chondrosarcoma cells acquire an epithelial-like gene expression pattern via an epigenetic switch: evidence for mesenchymal-epithelial transition during sarcomagenesis. Sarcoma. 2011;2011(598218) doi: 10.1155/2011/598218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang J, Du X, Wang G, et al. Mesenchymal to epithelial transition in sarcomas. Eur J Cancer. 2014;50:593–601. doi: 10.1016/j.ejca.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Wells A, Yates C, Shepard CR. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin Exp Metastasis. 2008;25:621–628. doi: 10.1007/s10585-008-9167-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiles ET, Bell R, Thomas D, Beckerle M, Lessnick SL. ZEB2 represses the epithelial phenotype and facilitates metastasis in Ewing sarcoma. Genes Cancer. 2013;4:486–500. doi: 10.1177/1947601913506115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Latorre IJ, Frese KK, Javier RT. Tigh. junction proteins and cancer. In: Gonzalez-Mariscal L, editor. Tight Junctions. Springer US; New York, NY: 2006. pp. 116–134. [Google Scholar]
- 6.Chipman JK, Mally A, Edwards GO. Disruption of gap junctions in toxicity and carcinogenicity. Toxicol Sci. 2003;71:146–153. doi: 10.1093/toxsci/71.2.146. [DOI] [PubMed] [Google Scholar]
- 7.Gill GA, Buda A, Moorghen M, et al. Characterisation of adherens and tight junctional molecules in normal animal larynx; determining a suitable model for studying molecular abnormalities in human laryngopharyngeal reflux. J Clin Pathol. 2005;58:1265–1270. doi: 10.1136/jcp.2004.016972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolegraff K, Nava P, Helms MN, et al. Loss of desmocollin-2 confers a tumorigenic phenotype to colonic epithelial cells through activation of Akt/β-catenin signaling. Mol Biol Cell. 2011;22:1121–1134. doi: 10.1091/mbc.E10-10-0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, Chen Y, Cui T, et al. Desmoplakin acts as a tumor suppressor by inhibition of the Wnt/β-catenin signaling pathway in human lung cancer. Carcinogenesis. 2012;33:1863–1870. doi: 10.1093/carcin/bgs226. [DOI] [PubMed] [Google Scholar]
- 10.Holen I, Whitworth J, Nutter F, et al. Loss of plakoglobin promotes decreased cell-cell contact, increased invasion and breast cancer cell dissemination in vivo. Breast Cancer Res. 2012;14:R86. doi: 10.1186/bcr3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yashiro M, Nishioka N, Hirakawa K. Decreased expression of the adhesion molecule desmoglein-2 is associated with diffuse-type gastric carcinoma. Eur J Cancer. 2006;42:2397–2403. doi: 10.1016/j.ejca.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 12.Leithe E, Sirnes S, Omori Y, et al. Downregulation of gap junctions in cancer cells. Crit Rev Oncog. 2006;12:225–256. doi: 10.1615/critrevoncog.v12.i3-4.30. [DOI] [PubMed] [Google Scholar]
- 13.Galoian K, Scully S, McNamara G, et al. Antitumorigenic effect of brain proline rich polypeptide-1 in human chondrosarcoma. Neurochem Res. 2009;34:2117–2121. doi: 10.1007/s11064-009-0009-6. [DOI] [PubMed] [Google Scholar]
- 14.Galoian K, Temple TH, Galoyan A. Cytostatic effect of the hypothalamic cytokine PRP-1 is mediated by mTOR and cMyc inhibition in high grade chondrosarcoma. Neurochem Res. 2011;36:812–818. doi: 10.1007/s11064-011-0406-5. [DOI] [PubMed] [Google Scholar]
- 15.Galoian KA, Temple TH, Galoyan A. Cytostatic effect of novel mTOR inhibitor, PRP-1 (galarmin) in MDA 231 (ER-) breast carcinoma cell line. PRP-1 inhibits mesenchymal tumors. Tumour Biol. 2011;32:745–751. doi: 10.1007/s13277-011-0176-3. [DOI] [PubMed] [Google Scholar]
- 16.Galoian K, Temple TH, Galoyan A. mTORC1 inhibition and ECM-cell adhesion-independent drug resistance via PI3K-AKT and PI3K-RAS-MAPK feedback loops. Tumour Biol. 2012;33:885–890. doi: 10.1007/s13277-011-0315-x. [DOI] [PubMed] [Google Scholar]
- 17.Galoyan AA, Aprikyan VS. A new hypothalamic polypeptide is a regulator of myelopoiesis. Neurochem Res. 2002;27:305–312. doi: 10.1023/a:1014959212651. [DOI] [PubMed] [Google Scholar]
- 18.Galoyan A. Neurochemistry of brain neuroendocrine immune system: signal molecules. Neurochem Res. 2000;25:1343–1355. doi: 10.1023/a:1007656431612. [DOI] [PubMed] [Google Scholar]
- 19.Silberberg M, Charron AJ, Bacallao R, et al. Mispolarization of desmosomal proteins and altered intercellular adhesion in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol. 2005;288:F1153–F1163. doi: 10.1152/ajprenal.00008.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delker DA, McGettigan BM, Kanth P, Pop S, Neklason DW, Bronner MP, Burt RW, Hagedorn CH. RN. sequencing of sessile serrated colon polyps identifies differentially expressed genes and immunohistochemical markers. PLoS One. 2014;9:e88367. doi: 10.1371/journal.pone.0088367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosette C, Roth RB, Oeth P, et al. Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis. 2005;26:943–950. doi: 10.1093/carcin/bgi070. [DOI] [PubMed] [Google Scholar]
- 22.Chun MG, Hanahan D. Genetic deletion of the desmosomal component desmoplakin promotes tumor microinvasion in a mouse model of pancreatic neuroendocrine carcinogenesis. PLoS Genet. 2010;6:e1001120. doi: 10.1371/journal.pgen.1001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dusek RL, Attardi LD. Desmosomes: new perpetrators in tumour suppression. Nat Rev Cancer. 2011;11:317–323. doi: 10.1038/nrc3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chidgey M, Dawson C. Desmosomes: a role in cancer? Br J Cancer. 2007;96:1783–1787. doi: 10.1038/sj.bjc.6603808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delva E, Tucker DK, Kowalczyk AP. The desmosome. Cold Spring Harb Perspect Biol. 2009;1:a002543. doi: 10.1101/cshperspect.a002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green KJ, Gaudry CA. Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol. 2000;1:208–216. doi: 10.1038/35043032. [DOI] [PubMed] [Google Scholar]
- 27.Rieger-Christ KM, Ng L, Hanley RS, et al. Restoration of plakoglobin expression in bladder carcinoma cell lines suppresses cell migration and tumorigenic potential. Br J Cancer. 2005;92:2153–2159. doi: 10.1038/sj.bjc.6602651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simcha I, Geiger B, Yehuda-Levenberg S, et al. Suppression of tumorigenicity by plakoglobin: an augmenting effect of N-cadherin. J Cell Biol. 1996;133:199–209. doi: 10.1083/jcb.133.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aktary Z, Pasdar M. Plakoglobin: role in tumorigenesis and metastasis. Int J Cell Biol. 2012;2012(189521) doi: 10.1155/2012/189521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin T, Getsios S, Caldelari R, et al. Plakoglobin supresses keratinocyte motility through both cell-cell adhesion-dependent and -independent mechanisms. Proc Natl Acad Sci USA. 2005;102:5420–5425. doi: 10.1073/pnas.0501676102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tam LW, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19:1438–1449. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi L, Sun L, Li Q, et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc Natl Acad Sci USA. 2011;108:7541–7546. doi: 10.1073/pnas.1017374108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirmizis A, Bartley SM, Kuzmichev A, Margueron R, et al. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 2004;18:1592–1605. doi: 10.1101/gad.1200204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li LL, Xue AM, Li BX, et al. JMJD2A contributes to breast cancer progression through transcriptional repression of the tumor suppressor ARHI. Breast Cancer Res. 2014;16:R56. doi: 10.1186/bcr3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kogure M, Takawa M, Cho HS, et al. Deregulation of the histone demethylase JMJD2A is involved in human carcinogenesis through regulation of the G(1)/S transition. Cancer Lett. 2013;336:76–84. doi: 10.1016/j.canlet.2013.04.009. [DOI] [PubMed] [Google Scholar]