

Summary
A deletion on human chromosome 16p11.2 is associated with autism spectrum disorders. We deleted the syntenic region on mouse chromosome 7F3. MRI and high-throughput single-cell transcriptomics revealed anatomical and cellular abnormalities, particularly in cortex and striatum of juvenile mutant mice (16p11+/−). We found elevated numbers of striatal medium spiny neurons (MSNs) expressing the dopamine D2 receptor (Drd2+) and fewer dopamine-sensitive (Drd1+) neurons in deep layers of cortex. Electrophysiological recordings of Drd2+ MSN revealed synaptic defects, suggesting abnormal basal ganglia circuitry function in 16p11+/− mice. This is further supported by behavioral experiments showing hyperactivity, circling, and deficits in movement control. Strikingly, 16p11+/− mice showed a complete lack of habituation reminiscent of what is observed in some autistic individuals. Our findings unveil a fundamental role of genes affected by the 16p11.2 deletion in establishing the basal ganglia circuitry and provide insights in the pathophysiology of autism.
Introduction
Autism spectrum disorders (ASD) are characterized by social deficits, language impairments, and stereotyped behaviors manifested in early childhood (Geschwind and Levitt, 2007). Epidemiological studies have reported a dramatic increase in the prevalence of ASD (Fombonne, 2003), now estimated to affect more than 1 in 100 children (Baron-Cohen et al., 2009). A number of genomic loci have been associated with increased risk for ASD (Abrahams and Geschwind, 2008 and Persico and Bourgeron, 2006). A copy number variation (CNV) on human chromosome 16p11.2 is among the most common genetic variations found in ASD (Weiss et al., 2008). Patients with this deletion display motor deficits, speech/language delay, and cognitive impairments, accompanied by ASD, attention deficit hyperactivity disorder (ADHD), seizures, and hearing disorders (Bijlsma et al., 2009, Fernandez et al., 2010 and Shinawi et al., 2010). Conversely, a duplication of 16p11.2 is associated with schizophrenia (McCarthy et al., 2009).
The most common deletion in the 16p11.2 locus associated with ASD causes loss of 550 kb of genomic DNA and haploinsufficiency of 26 genes. Knockdown and overexpression studies have attempted to model these gene dosage changes, implicating two genes, Kctd13 and Taok2, in altered brain size and neurite morphogenesis, respectively (de Anda et al., 2012 and Golzio et al., 2012). However, it is not known whether knockdown-mediated dosage changes accurately model loss of a single allele for each of these genes. A 16p11.2 CNV adult mouse model was recently reported to display activity-related behavioral deficits and subtle morphological changes in the ventral midbrain (Horev et al., 2011). Nevertheless, the defects in brain development in the context of the 16p11.2 deletion that may underlie behavioral abnormalities in patients remain unclear.
Neural circuits modulated by the neurotransmitter dopamine (DA) play an important role in motor, cognitive, and emotional control (for review, see DeLong and Wichmann, 2009). DA neurons in the ventral midbrain send projections to the striatum and cortex. The striatum contains DA-sensitive medium spiny neurons (MSNs) and is the entry point of the basal ganglia (BG) circuitry, which plays a major role in motor control, motivation, and attention. MSNs that express either dopamine D1 (Drd1+) or D2 (Drd2+) receptors act antagonistically through the direct (striatonigral) and indirect (striatopallidal) pathways, respectively (Kravitz et al., 2012). DA also modulates the activity of Drd1+ neurons in deeper layers of cortex. The role of these cells in regulating behavior has not been studied extensively. Some cortical Drd1+ cells project back to striatal MSNs, providing important top-down control of movements, motivation, and attention. These cells have also been proposed to play a role in gain control of cortical inputs, as well as in mediating the effects of DA on learning and memory (Olsen et al., 2012, Seong and Carter, 2012 and Thurley et al., 2008). The circuits modulated by DA play an important role in the pathophysiology of several neurologic and psychiatric diseases. ADHD is clinically treated with drugs altering DA levels, like dexamphetamine and methylphenidate, suggesting DA misregulation as a key element in the etiology of this disorder. In contrast, agents like risperidone that block D2 receptors (D2Rs) are used both to control irritability in ASD and as antipsychotics in schizophrenia, implicating these circuits in the biogenesis of these disorders. Although DA-modulated circuits are strongly implicated in schizophrenia, ADHD, and ASD, the underlying anatomical or molecular defects in patients are largely unknown.
We generated a mouse model for the 16p11.2 deletion. Using high-throughput multiplex single-cell gene expression analysis (sc-qPCR) to identify cell-type-specific deficits across the developing mouse brain, we found that 16p11.2 heterozygous (16p11+/−) mice have increased numbers of Drd2+ striatal MSNs, as well as fewer Drd1+ neurons in cortex. MRI revealed anatomical defects of BG nuclei, direct targets of BG output structures, and several cortical regions. Electrophysiological recordings suggested synaptic alterations in Drd2+ MSNs. Finally, extensive behavioral analyses carried out by two independent laboratories revealed that 16p11+/− mice exhibit normal social behavior but show hyperactivity and deficits in movement control, hearing, and habituation to familiarity. Taken together, our findings suggest that BG circuitry and DA signaling play a critical role in the defects of the 16p11.2 deletion, and more generally, in the pathophysiology of ADHD and ASD.
Results
A Mouse Model for the Human Chromosome 16p11.2 Microdeletion
The chromosome 16p11.2 CNV encompasses 26 genes (the 16p11 genes) that are highly conserved on mouse chromosome 7F3 (Figure 1A). To generate a mouse model of the 16p11.2 deletion, we introduced LoxP sites flanking the genes deleted in human patients (Figures 1B, S1A, and S1B). The targeting strategy also included an mCherry reporter gene coupled to deletion of the region. Successful targeting of mouse embryonic stem cells (mESCs) was verified by Southern blotting and PCR (Figures S1C–S1I; Table S1). The modified mESCs were injected into blastocysts, implanted into pseudopregnant female mice, and chimeric offspring were subsequently bred to (1) C57BL/6N females to produce heterozygous floxed (16p11flx/+) mice and (2) HPRT-Cre transgenic females (Tang et al., 2002) to produce mice lacking one copy of the 16p11 genes (16p11+/−). Heterozygous deletion was confirmed by PCR genotyping, as well as mCherry fluorescence (Figures 1B, inset, and 1C). Quantitative real-time RT-PCR (qPCR) analysis of neonate brain RNA showed a global reduction in transcription of 40%–60% for the 16p11 genes upon loss of one copy (Figure 1D). F1 and further generations of 16p11+/− male mice were consequently backbred to C57BL/6N females.
Figure 1. A Mouse Model for the Human 16p11.2 Microdeletion.
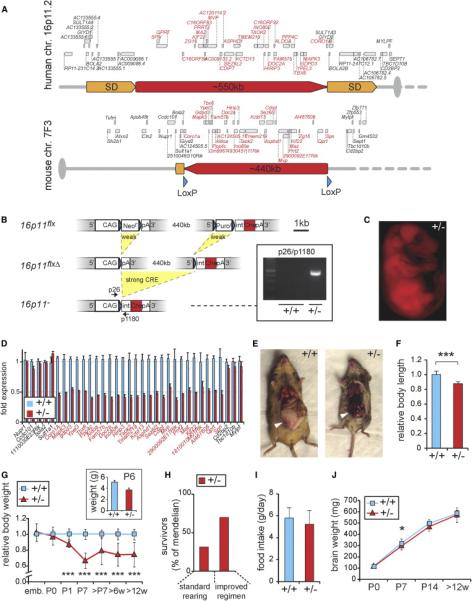
(A) Top: the region on human chromosome 16p11.2 is flanked by segmental duplications (SD), which likely mediate CNVs of the locus by nonhomologous recombination. Bottom: the syntenic region on mouse chromosome 7F3, in which LoxP sites (blue arrowheads) were inserted at positions indicated.
(B) Sequential recombination steps yield deletion of 440 kb containing the mouse 16p11 genes. pA, poly A; int, intron; CAG, chicken β-actin enhanced CMV promoter; Neor and Puror, neomycin and puromycin resistance cassettes; STOP, translational stop codon; Che, mCherry. Scale bar, 1 kb. Inset, Genotyping of F1 offspring from floxed (16p11flx/+) chimeras and HPRT-Cretg/+ females shows germline transmission. Arrows in scheme show PCR primer positions.
(C) Red fluorescence in E16.5 16p11+/− embryo confirms mCherry expression and deletion of 16p11 genes.
(D) At birth (P0): qPCR analysis shows global downregulation of 16p11 genes (red) in the brain upon deletion of one allele, whereas neighboring genes are unaffected.
(E) Adult animals (4 months): 16p11+/− females lack abdominal fat pads (white arrowheads) typical for this age.
(F) Adult animals (3 months): relative body length (n = 12).
(G) Relative body weight (n ≥ 6 per time point) across development normalized to the average of gender-matched wild-type littermates. Inset: independent analysis of body weight performed at NIMH at P6.
(H) At 6 weeks of age: increased juvenile mortality of 16p11+/− mice is rescued by improved nutritional regimen and alleviated sibling competition.
(I) Adult animals (3 months): food intake is comparable between 16p11+/− and wild-type animals.
(J) Postnatal developmental trajectory of brain weight as a measure for global brain growth (n ≥ 6).
Data are mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. Inset in (G), NIMH.
16p11+/− mice were born at Mendelian ratios. As adults, they showed a 12.2% reduction in average body length (p = 5.09 × 10−7; Figures 1E and 1F), a reduction in the accumulation of abdominal fat pads, and significantly reduced body weight starting at early postnatal age (Figure 1G). A small number of 16p11+/− animals recovered to nearly normal body weight in adulthood. Severely affected mice were runty and died within the first postnatal weeks (Figure 1H), resulting in lower-than-Mendelian ratios of survivors past the age of tagging (typically 6 weeks). An improved nutritional regimen and alleviated competition by wild-type siblings rescued 16p11+/− pup viability to 60% of Mendelian expectation (Figure 1H; Supplemental Experimental Procedures). Food intake in adult 16p11+/− animals was normal (Figure 1I). 16p11+/ − pups appeared hyperactive and displayed severe deficits in motor coordination including tumbling and tremor (Movie S1). These findings were replicated and quantified in adult animals as described below. In contrast to the decreased body size, the brain weight of 16p11+/− mice was indistinguishable from controls (Figure 1J) throughout development, with the exception of 1-week-old (P7) pups, in which the brain weight was mildly reduced (p < 0.05).
MRI and Diffusion Tensor Image Analyses of Juvenile Brains
MRI in P7 mice revealed decreased brain volume throughout the 16p11+/− brain (total −13.0%; Figures 2A–2C and 2E), consistent with decreased brain weight at this age. The relative volume (normalized to total brain volume) of several structures revealed more complex abnormalities, particularly in the BG, direct targets of BG output structures (thalamus and superior colliculus), and major afferent regions to the BG (cortex and thalamus). In the BG, the dorsal striatum showed significant expansion in the dorsofrontal direction and reduction at ventrocaudal areas, although the relative volume was unchanged (Figure 2A; +0.42%, n.s.). The relative volume of the nucleus accumbens (NAc) was increased by 4.83% in 16p11+/− pups compared to wild-type (Figure 2B; q < 0.01). The globus pallidus (GP), a major target of striatal afferents, showed a 2.79% increase in relative volume in 16p11+/− pups (Figure 2C; q < 0.01), suggesting a potential link between striatal and pallidal abnormalities. These changes in the BG were accompanied by major structural defects in brain regions projecting to striatum. Abnormalities in the 16p11+/− cortex encompassed increased thickness in medial areas including motor cortices (Figure 2D). In contrast, lateral, and ventral areas, including sensory (e.g., primary auditory cortex) and insular cortices, showed significant reduction in thickness. Mesodiencephalic structures were increased in size, including the thalamus (+9.6%, q < 0.01), hypothalamus (+4.2%, q < 0.01), and superior and inferior colliculi (+ 5.9% and +11.7% respectively, both with q < 0.01). In conclusion, MRI data suggest major structural abnormalities in the early postnatal 16p11+/− brain, affecting primarily the BG, cortex, and regions of the mesodiencephalon, which receive projections from BG output structures.
Figure 2. Anatomical Abnormalities in Juvenile 16p11+/− Mice.
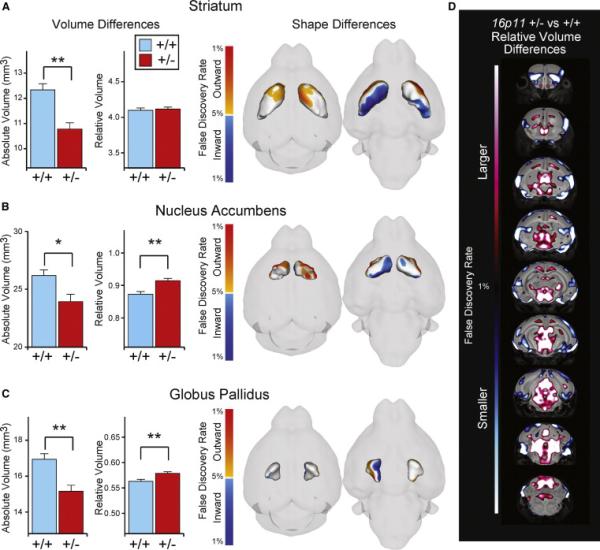
MRI analysis of mouse brains at P7 (n = 26 for each genotype).
(A–C) Volume and shape difference are displayed for the BG regions, namely, the striatum (A), GP (B), and NAc (C). Volumes in (A)–(C) are shown as both absolute (in mm3) and relative volumes. Shape differences in (A)–(C) show 3D surface renderings of the given region of interest. Highlighted on that surface are significant shape differences (q < 0.05) between the 16p11+/− mouse and control. Orange, outward movement; blue, inward movement.
(D) Coronal flythrough highlighting significant differences in the relative volume of the 16p11+/− mouse and control (red: larger, blue: smaller); only highly significant areas are shown (q < 0.01). Error bars represent SEM. *q < 0.05, **q < 0.01.
Single-Cell Gene Expression Analysis
The complex anatomical defects in the brain of juvenile 16p11+/− mice suggested defects in specific cell populations. We previously established multiplex sc-qPCR using nanofluidic array technology as a powerful tool to study the cellular composition of heterogeneous cell populations in vitro (Pasca et al., 2011 and Yoo et al., 2011). Here, we adapted the method to study the composition of cells in defined anatomical regions of the brain: cerebral cortex, hippocampus, subpallium (including striatum, NAc, GP, and amygdala), and mesodiencephalon (Figure 3A). To ensure that our findings would not reflect secondary consequences of the early postnatal weight loss in 16p11+/− pups, we performed this analysis in neonate mice (P0), which were morphologically indistinguishable from wild-type. After tissue dissociation, single cells were sorted into 96-well PCR plates using fluorescence activated cell sorting (FACS). Expression of 190 genes (Table S2) was measured in each of more than 2,000 single cells. We selected genes that by virtue of their expression pattern best distinguished specific neural cell types, encoding transcription factors, neurotransmitter receptors, synthetic enzymes, and guidance molecules. In addition, we included the 16p11 genes.
Figure 3. Single-Cell Gene Expression Profiling of Cell Types in the Neonate Brain.

(A) Experimental workflow: brain dissection, single-cell sorting by FACS, reverse transcription (RT), and preamplification of cDNAs of interest. A total of 190 genes were profiled by multiplex qPCR using two 96.96 dynamic arrays per sample plate. ctx, cortex; hc, hippocampus; spa, subpallium; ms-dienc, mesodiencephalon.
(B and C) Identification of cell-type-specific gene clusters for the subpallium (B) and cortex (C) by coexpression mapping (n > 500 cells). The dendrogram shows the proximity of genes based on their coexpression with all other genes. Listed genes are known to be expressed in a cell-type- and/or developmental-stage-specific manner and used accordingly for identification of cell-type-specific gene clusters. (D and E) Separate clustering for 16p11+/− and wild-type samples (n ≈ 250 cells each) of subpallium (D) and cortex (E).
Color code matches (C) and (D), respectively. LVW, lateral ventricular wall; LMS, lateral migratory stream; SVZ, subventricular zone; VZ, ventricular zone.
sc-qPCR data showed a lognormal distribution (Figure S2A) and high variability across cells (average SD: 1.73 PCR cycles, average range: 7.93 PCR cycles; n = 172 genes; n > 2,000 cells, Figure S2B) consistent with previous studies suggesting that mRNA levels in a single cell are largely a function of the bursting kinetics of gene transcription (,Bengtsson et al., 2005 and Suter et al., 2011). To eliminate this source of variability data were binarized and a Fisher's exact test p value (pFET) was calculated for the observed coexpression of a gene with every other gene based on the expected coexpression frequency if they were randomly assigned to cells. The logarithm of these p values (log[pFET]) was supplied with an algebraic sign, in order to recover information about increased coexpression or mutual exclusiveness that was lost in the process of assessing the significance of the deviation from the expected coexpression of genes (Experimental Procedures; Supplemental Experimental Procedures). The diagonal symmetric coexpression matrix was then subjected to unsupervised clustering (Figures 3 and S2I). The resulting gene clusters identified major neuronal subtypes in the examined brain regions. By example, the subpallium (Figure 3B) included GABAergic neurons, striatal MSNs, striatopallidal MSNs, striatal interneurons, neurons of the amygdala, lateral migratory stream, GP, and lateral ventricular wall progenitors (Arlotta et al., 2008, Ding et al., 2012, Marin et al., 2000 and Nóbrega-Pereira et al., 2010). In the cortex (Figure 3C), we found gene clusters that distinguish progenitor populations, cortical interneurons, and distinct groups of cortical excitatory neurons. Among these are deeper layer excitatory neurons that are born earliest during corticogenesis and already express mature markers indicating synaptogenesis, upper layer excitatory neurons generated late in embryogenesis, and ventricular zone (VZ) progenitors, which in the neonate also start to express glial progenitor markers (Arlotta et al., 2005, Chen et al., 2008, Lai et al., 2008 Molnár and Cheung, 2006 Sasaki et al., 2008 and Takemoto et al., 2011). This analysis strategy therefore identified gene clusters correlating with specific cell types previously described using other methods in vivo.
Altered DA Signaling in the Neonate 16p11+/− Brain
Using these tools, we found that 16p11+/− and wild-type mice have highly similar cell-type-specific gene clusters, suggesting that the mutation does not lead to the generation of ectopic cell types or to complete loss of existing ones (Figures 3D and 3E). However, when we quantified the cellular composition we found striking differences between 16p11+/− and wild-type mice (Figures 4 and S3). There was a significant increase in the number of MSNs expressing Drd2 in 16p11+/− mice (+47.3%, p = 0.003; Figure 4A). This population of cells also expresses Adora2a (+43.0%, p = 0.005), as well as other markers of GABAergic and MSN identity, such as Penk (+83.4%, p = 0.002), Nfh (+111.7%, p = 0.005), Plxnd1 (+58.4%, p = 0.001), Reelin (+71.1%, p = 1 × 10−4), Robo1 (+29.5%, p = 3 × 10−4), Foxp1 (+24.1%, p = 3 × 10−7), and Ctip2 (+20.2%, p = 3 × 10−6). The number of cells coexpressing all of these genes was significantly elevated, providing evidence for a general increase in the number of Drd2+ MSNs and not just an increase in Drd2 expression resulting in higher chance of detection. Furthermore, the increase in Drd2+ MSNs was not at the expense of the Drd1+ MSN population, because we found no significant change Drd1+ cell numbers in the 16p11+/− subpallium (p = 0.086) but observed an overall increase in the total number of cells expressing either Drd2 and/or Drd1 (p = 0.017, Figure 4B), suggesting an increased MSN pool in the striatum. Interestingly, we also observed an increased number of cells that coexpress both Drd1 and Drd2 in 16p11+/− mice (p = 4 × 10−4). These cells were rarely observed in wild-type mice suggesting that the 16p11+/− mutation alters not only the number of Drd2+ MSNs, but also the process of MSN specification.
Figure 4. Altered DA Signaling in the Neonate 16p11+/− Brain.
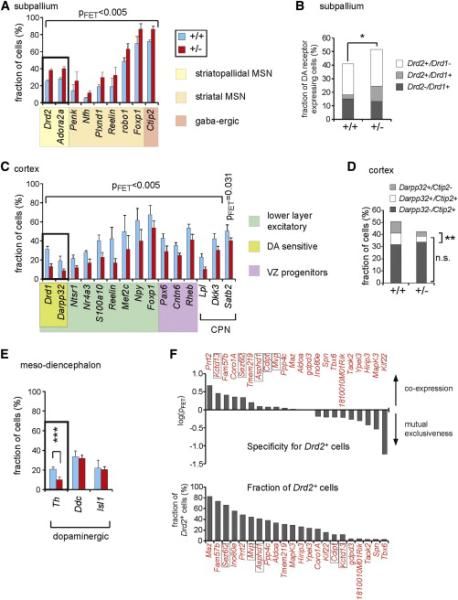
(A) Numbers of subpallial cells expressing genes specific to GABAergic neurons, striatal MSNs, and striatopallidal MSNs suggest an increase in the number of indirect pathway MSNs and total MSNs in 16p11+/− mice.
(B) Striatal MSNs as defined by DA receptor gene expression only. Note that the increase in Drd2+ MSNs is not at the expense of Drd1+ cells, but is likely due to a total increase in the number of MSNs consistent with (A).
(C) Abnormalities in deeper layer cortical excitatory neurons and Pax6+ VZ progenitors as well as callosal projection neurons (CPN). Reduction of Darpp32 and Drd1 expression indicate a loss of dopamine-sensitive cells.
(D) Combined consideration of Ctip2 and Darpp32 expression suggests a lower number of dopamine-sensitive cells in the deeper cortical layers but no major reduction in deeper layer corticofugal cell types. n.s., not significant.
(E) Decreased expression of Th in ventral midbrain DA cells.
(F) Top: coexpression of the 16p11 genes in striatopallidal MSNs as defined by Drd2 expression. Framed 16p11 genes have been reported in ASD individuals with a smaller deletion in the 16p11.2 locus. Bottom: fraction of Drd2+ cells expressing each of the 16p11 genes.
Error bars represent SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 where not otherwise indicated.
Although Drd1+ cell numbers were not reduced in the striatum, we observed a significant decrease in Drd1+ cells in the cortex (−58.2%, p = 1 × 10−6; Figure 4C). This reduction was accompanied by reduction of another marker for DA-sensitive deeper layer excitatory neurons, Darpp32 (−55.6%, p = 9 × 10−5), as well as a battery of other genes specific to deeper layer neurons, including Ntsr1 (−44.1%, p = 0.001), Nr4a3 (−39.4, p = 9 × 10−4), S100a10 (− 44.1%, p = 2 × 10−6), Reelin (−60.0%, p = 4 × 10−12), Mef2c (−37.6%, p = 1 × 10−5), Npy (−35.5%, p = 8 × 10−8), and Foxp1 (−20.4%, p = 9 × 10−4). Importantly, based on the expression of Ctip2, a marker specific for corticofugal projecting cell types such as corticospinal motor neurons, not all deeper layer neurons appeared equally affected. We found no major reduction in Ctip2+ cells, but fewer Ctip2+ cells coexpressing Drd1 and Darpp32 (Figure 4D), again pointing toward a reduction in DA sensitivity in these cells. Furthermore, we observed a downregulation of genes in the cluster specific to ventricular zone (VZ) progenitors, particularly Pax6 (− 32.9%, p = 2 × 10−4), and three genes reported to be specific to callosal projection neurons (CPN), Lpl (−56.9%, p = 7 × 10−6), Dkk3 (−28.2%, p0.002), and Satb2 (−54.0%, p = 0.031) (Alcamo et al., 2008 and Molyneaux et al., 2009) (Figure 4C). The latter result was corroborated by immunohistochemical staining in P7 mice showing reduced numbers of SATB2+ neurons (−18.4% p = 0.0014, Figures S4A and S4B) in 16p11+/− brains compared to wild-type. Furthermore, the results are consistent with data from diffusion tensor imaging indicating morphological abnormalities in the corpus callosum of 16p11+/− pups (Figures S4C and S4D).
sc-qPCR also revealed changes in the expression of specific genes in 16p11+/ − mice independent of gene clusters. For example, tyrosinehydroxylase (Th), which encodes a rate-limiting enzyme in the DA synthesis pathway, was decreased in mesodiencephalic DA cells (−52%, p = 0.001), although other genes expressed in this cell lineage, such as dopadecarboxylase (Ddc) and Isl1, respectively, were unaffected (Figure 4E). Together, our results from multiplex sc-qPCR indicate a major imbalance in the DA signaling system of 16p11+/− mice.
Finally, we examined which of the 16p11 genes are expressed in the Drd2+ MSNs and therefore might be important for the defect observed in the mice (Figure 4F). We found that Kctd13 (p = 0.407), Prrt2 (p = 0.227), Fam57b (p = 0.401), Sez6l2 (p = 0.456), and Coro1a (p = 0.350) are enriched in Drd2+ cells, whereas Kif22 (p = 0.058), Hirip3 (p = 0.394), Mapk3 (p Ypel3 (p = 0.516) or Taok2 (p = 0.293), and = 0.542) show increased specificity for other cell types at this developmental stage. Interestingly, Kctd13 and Sez6L2 are located within a smaller 16p11.2 deletion of a patient diagnosed with ASD and Kctd13 was identified as a gene that controls neuronal progenitor proliferation in zebrafish (Golzio et al., 2012).
Neuroanatomical Analysis of Drd2+ Cells
To independently determine whether the 16p11.2 deletion caused increased numbers of Drd2+ cells in the striatum, we crossed the 16p11+/− mice with Drd2-EGFP BAC transgenic (Gong et al., 2003) mice that express GFP under control of the Drd2 gene-regulatory region, specifically in striatopallidal MSNs. We examined brains of Drd2-EGFPtg/+;16p11+/− mice at P7, when most structures of the brain have reached relatively definitive morphology and major migratory streams and projections have arrived at their target regions. We found a significant increase in the fraction of GFP+ cells in mutant Drd2-EGFPtg/+;16p11+/− mice in both the ventral and dorsal striatum (+60.6%, p = 1.7 × 10−5 and +56.4%, p = 2.3 × 10−7 respectively) compared to controls (Drd2-EGFPtg/+;16p11+/+, Figures 5A–5C). In addition, we observed severe enlargement of the GP (Figure 5D), which receives input from Drd2+ MSNs, as well as ectopic GFP+ projections to GPm, which normally receives input from striatal Drd1+ cells (Figures 5E and 5F). This suggests that heterozygous deletion of the 16p11 genes results in aberrant expression of Drd2 in cells projecting along the direct pathway to the GPm. Furthermore, antibody staining against DARPP32 to label DA-sensitive neurons in cortex of 16p11+/− mice revealed a significant loss of DARPP32 expression in deep layers (−23.6%, p = 0.0149, Figures 5G–5I), consistent with our sc-qPCR results. Finally, we tested whether above described cellular phenotypes could be validated in an independent, previously described 16p11.2 deletion mouse model (16p11.2df/+) (Horev et al., 2011). 16p11.2df/+ mice were bred with Drd1a-TdTomatotg/+ and subsequently with Drd2-EGFPtg/+ mice. Juvenile (P7) Drd1a-TdTomatotg/+;Drd2-EGFPtg/+;16p11.2df/+ animals displayed increased numbers of GFP+ MSNs and decreased numbers of TdTomato-expressing deep layer cortical neurons as compared to controls (Drd1a-TdTomatotg/+; Drd2-EGFPtg/+;16p11.2+/+, Figures S5A–S5E). Taken together, these findings confirm our single-cell study and support the idea that the 16p11.2 deletion affects DA-sensitive neuronal circuits.
Figure 5. Excess Numbers of Striatopallidal MSNs and Hypodopaminergia in Cortex of Juvenile 16p11+/− Mice.
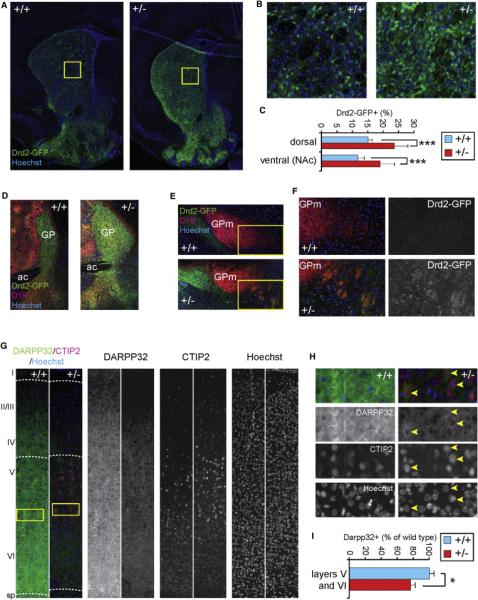
(A) Coronal cryosections show the expression of a Drd2-GFP BAC transgene in the mouse striatum at P7.
(B) Magnification of boxed regions of the dorsal striatum from (A).
(C) Quantification of GFP+ cells (n = 3 animals per genotype).
(D) The GP, the output structure of striatopallidal projecting (Drd2+) MSNs, is enlarged in 16p11+/− brains.
(E) GPm, the striatonigral (Drd1+ MSNs) output structure contains Drd2-GFP+ fibers in 16p11+/− not found in wild-type.
(F) Magnification of boxed regions in (E).
(G) Somatosensory cortex in sagittal sections of P7 brains shows downregulation of DARRP32 expression. CTIP2 was used for visualization of layer V (large, bright CTIP2+ pyramidal neurons), and layer VI (smaller, less bright CTIP+ neurons). Blue: Hoechst nuclear stain.
(H) Magnification of boxed area in (G). Although DARPP32 expression is much weaker in 16p11+/− cortex, some DARPP32+ cells can still be identified (yellow arrowheads).
(I) Quantification of DARPP32+ cells in cortical layers V and VI (irrespective of expression level).
Error bars represent SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
Electrophysiology of Striatal MSNs
We next examined whether changes in the DA-sensitive circuitry of the developing 16p11+/− brain affected the function of striatal MSNs by performing electrophysiological recordings from MSNs in the NAc while stimulating striatal afferents. AMPA receptor-mediated excitatory postsynaptic currents (AMPAR EPSCs) revealed comparable I/V relationships in 16p11+/− and wild-type mice (Figure 6A) and thus no significant change in the AMPAR EPSC rectification index (defined as current amplitude at +40 mV over the current amplitude at −70 mV) (Figure 6B). This suggests that the stoichiometry of synaptic AMPARs in ventral striatal MSNs is unaffected in 16p11+/− mice and that the vast majority of these AMPARs contain the GluA2 subunit. However, a clear increase in the ratio of the AMPAR EPSC to NMDA receptor-mediated EPSC (AMPAR/NMDAR ratio) was observed in 16p11+/− MSNs (Figure 6C), in addition to a significant decrease in the paired-pulse ratios (PPRs) across multiple interstimulus intervals (ISIs) (Figure 6D). Miniature AMPAR EPSC (mEPSC) recordings further showed an increase in the mean mEPSC frequency (wild-type: 1.82 ± 0.19, 16p11+/−: 2.90 ± 0.45, p = 0.039) (Figure 6E). The mean mEPSC amplitude was not significantly changed in 16p11+/− MSNs (wild-type: 18.64 ± 2.14, 16p11+/−: 19.76 ± 1.27, p = 0.540) (Figure 6F). In summary, the decrease in PPRs and increase in mEPSC frequency in 16p11+/− MSNs strongly suggest that the release probability of excitatory synapses on MSNs is increased. The increased AMPAR/NMDAR ratio suggests that excitatory synapses on 16p11+/− MSNs also exhibit postsynaptic alterations. These may include increase in quantal size preferentially at synapses with mEPSCs below our detection threshold. Although these physiological measurements demonstrate that 16p11+/− MSNs exhibit changes in excitatory synaptic function, they do not address the possibility of additional morphological changes in striatal MSNs. However, analysis of MSN primary dendrite number and spine density indicated no significant differences between wild-type and 16p11+/− (Figures 6G–6I).
Figure 6. Deficits at Excitatory Synapses onto NAc MSNs in 16p11+/− Mice.
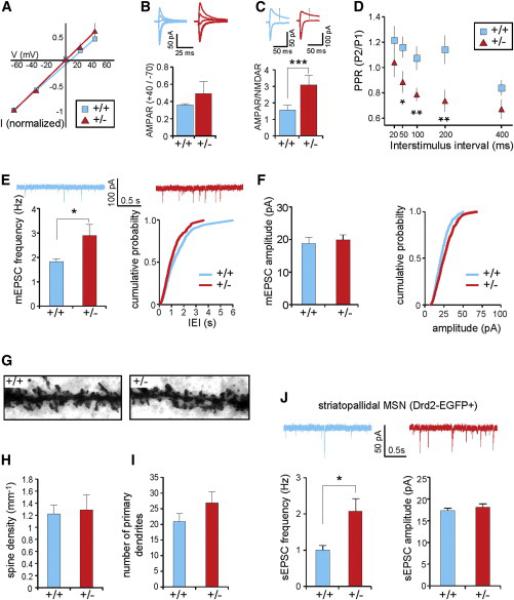
Electrophysiological recordings in NAc MSNs at 4–8 weeks.
(A and B) Comparable I/V relationships (A) AMPAR rectification index (B) in 16p11+/− and wild-type mice (n = 9 cells for each genotype).
(C) Increased AMPAR/NMDAR ratio in 16p11+/− mice.
(D) Decreased paired-pulse ratios (PPRs) across multiple interstimulus intervals (ISIs) in 16p11+/− mice.
(E) Consistent with a higher presynaptic release probability, significant increase in mEPSC frequency in 16p11+/− mice (n = 12 wild-type cells, n = 14 16p11+/− cells).
(F) Comparable mEPSC amplitude between genotypes.
(G–I) Morphological analysis (8 weeks of age) of MSN dendrites (n = 10 cells per genotype). (G) Representative Golgi-stained MSN dendrites covered with dendritic spines. (H and I) No change in spine density (H) or number of primary dendrites (I) was detected.
(J) Recording of synaptic events in Drd2-EGFP+ MSNs of the 16p11+/− NAc (4–6 weeks) show a significant increase in sEPSC frequency, whereas the amplitude remained unchanged (n = 9 wild-type cells, n = 10 16p11+/− cells).
Error bars represent SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
Previous studies have suggested that release probability at excitatory synapses terminating on indirect-pathway MSNs is higher than at synapses terminating on direct-pathway MSNs (Grueter et al., 2010 and Kreitzer and Malenka, 2007). The decreased PPR and increased mEPSC frequency could thus be a result of oversampling indirect-pathway MSNs in 16p11+/− mice, due to the increased number of D2R-expressing cells. To test this, we again used the Drd2-EGFP BAC transgene to specifically target indirect-pathway MSNs by recording from GFP+ neurons in the NAc. We observed a significantly increased EPSC frequency (sEPSCs) recorded in GFP+ MSNs of mutants compared to wild-type (Drd2-EGFPtg/+;16p11.2+/−: 2.07 ± 0.35; Drd2-EGFPtg/+;16p11.2+/+: 1.0 ± 0.13; p < 0.02) (Figure 6J). These data argue against oversampling of Drd2+ neurons as an explanation for the observed electrophysiological phenotype in 16p11+/− striatum and instead argue for a fundamental alteration in ventral striatal circuitry.
Behavioral Analysis
DA pathways in the BG are important for motor, emotional, and cognitive function. The changes in DA signaling and striatal circuitry observed in 16p11+/− mice suggest possible mechanisms relevant for deficits observed in 16p11.2 deletion syndrome. We therefore tested 16p11+/− mice in a battery of behaviors to assay defects in locomotor activity, social interaction, working memory, and sensory processing. To control for variability across testing sites, these studies were conducted in parallel by the Stanford Behavioral and Functional Neuroscience Laboratory (SBFNL) and by the Laboratory of Behavioral Neuroscience at the National Institute of Mental Health (NIMH). Tests were performed on increasing C57BL/6N background after hybrid founder mice had been backcrossed into C57BL/6N for at least five to seven times.
Eight- to 12-week-old 16p11+/− mice did not show gross defects in a battery of tests for general health and neurological reflexes (Tables S3 and S4; Figure S6A). The mice also had normal olfactory abilities, as assessed in the olfactory habituation/dishabituation test (Figure S6B), and normal vision, as assessed by the forepaw reaching test (Table S3). However, 16p11+/− mice lacked a startle response even to sounds at 120 dB (NIMH: at 100 dB, F1,30 = 8.04, p < 0.01; at 110 dB, F1,30 = 29.1, p < 0.001; at 120 dB, F1,30 = 28.2, p < 0.001, Figure 7A). This change was likely due to a defect in auditory perception or processing because the mice had normal startle responses to air puffs but had defects in evoked electrical responses (not shown).
Figure 7. Behavioral Deficits of Adult 16p11+/− Animals.

(A) Adult 16p11+/− mice (2–3 months) display a significantly reduced startle response at increasing decibels (NIMH: wild-type n = 17, 16p11+/− n = 15) and repeated 20-stimulus presentations (inset, SBFNL).
(B and C) Movement control: 16p11+/− mice show lack of gait fluidity (B) and frequent tremor (C).
(D) Hyperactivity of 16p11+/− mice in a home-cage environment.
(E) In a novel empty-cage environment, 16p11+/− mice exhibited significantly more hanging, less self-grooming, and less resting than wild-type littermates. A fraction of 16p11+/− mice showed continuous circling.
(F) Follow-up quantification of circling behavior in a rotational assay in a cylindrical cage (inset, total number of rotations over the 30 min period).
(G) Adult 16p11+/− mice exhibited initial hypoactivity and abnormal dishabituation to novel environment in the open field. Inset, similar behaviors of the 16p11+/− mice in the open-field test independently reproduced at NIMH.
(H and I) Adult 16p11+/− mice display altered performance in a novel object recognition test (H) and a six-trial novel object recognition assay (I) compared to wild-type littermates. 16p11+/− mice did not show a significant preference for the novel object, compared to wild-type mice (H). This effect was due to a lack of habituation to the familiar object in the 16p11+/− mice. This was further corroborated in a six-trial novel object recognition test, where the 16p11+/− mice spent longer time in trials 2–4 and 6 sniffing the first object that had been presented as a novel object in trial 1 and also longer time sniffing the second novel object in trial 5 than the control mice (I). (J) Adult 16p11+/− mice showed hyperactivity in the activity chamber during a 10 min period.
(K) Acute administration of risperidone had no significant effect on the activity level of 16p11+/− mice in the activity chamber, whereas wild-type littermates exhibited a dramatic decrease in activity level after risperidone administration.
For all panels, mean ± SEM is presented for each data point; *p < 0.05, – **p < 0.01, and ***p < 0.001. (A), (E), (G) inset, and (H): NIMH; (A) inset, (B) (D), (F), (G), and (I–K): SBFNL.
Motor Behavior
Although the analysis of gait and locomotor activity in adult 16p11+/− mice showed normal performance in a rotarod test (Figures S6C and S6D), a significant fraction of mice displayed tremor (16p11+/−: 19.4%, p < 0.001) and a decrease in fluid gait (wild-type: 19.4%, 16p11+/−: 52.8, p < 0.001, Figures 7B and 7C). These findings are consistent with tremor and severe motor coordination defects observed in 10-day-old pups (Movie S1) and suggest an underlying developmental defect rather than a neurodegenerative process.
We next investigated activity patterns in a home-cage environment in the dark using the PhenoTyper system for 4 hr. The 16p11+/− mice moved approximately 2.5 times the distance of wild-type littermates (Figure 7D; effect of genotype: F1,11 = 26.57, p = 0.003; effect of time: F3,33 = 12.78, p < 0.0001; effect of genotype × time interaction F3,33 = 0.659, NS; overall distance moved: t11 = 5.15, p = 0.003). 16p11+/ − mice also exhibited more bouts of hanging (U = 380.5, p < 0.05), less self-grooming (U = 385.0, p < 0.02), and less resting (U = 369.5, p < 0.05) compared to wild-type in the home cage (Figures 7E and S6E). In addition, we observed a significant increase in circling behavior in a fraction (18.75%) of 16p11+/ − mice (Figure 7F, t131 = 3.404, p < 0.0001). These behaviors are indications of significant hyperactivity in 16p11+/− mice in an otherwise dark, low-stress, and familiar environment.
In contrast, when observed in an open-field test, under bright light in a novel environment, the 16p11+/− mice showed initial hypoactivity that gradually disappeared over the course of the first 10 min as well as a lack of habituation to the novel environment (Figure 7G, effect of genotype: F1,86 = 17.72, p < 0.0001). Based on analysis of the time spent in the center versus in the periphery of the chamber, we found no evidence that this initial hypoactivity reflected increased anxiety (Figure S6F). The initial hypoactivity of 16p11+/− mice in the open-field test might therefore reflect deficits related to motor initiation rather than anxiety.
Social Behavior
In humans, activity in the striatal circuits has been correlated with social deficits relevant to autism (Cascio et al., 2012 and Insel, 2003). Normal sociability was detected in multiple cohorts of adult 16p11+/− mice in a three-chamber social interaction test (Figure S7). Both sexes exhibited significant sociability as well as preference for social novelty spending significantly more time in a chamber containing a mouse than in one containing an object (sociability test) and more time in a chamber containing an unfamiliar mouse than one containing a familiar mouse (social novelty test). Quantification of sniffing time in these two tests corroborated chamber time data. Similar results were obtained on reciprocal social interactions with two cohorts of juvenile mice (stage P21–P25, Figure S6J). Overall, these results indicate largely normal social interactions in 16p11+/− mice.
Novelty-Seeking Behavior
Autism is also associated with a preference for sameness and an aversion to novelty. Wild-type mice seek novel objects and spend more time with novel objects than familiar ones. In addition, they gradually lose interest in familiar objects over time (habituation). In a conventional novel object recognition test to determine if mice learn and remember familiar versus unfamiliar objects (Figure 7H), we exposed control and 16p11+/− mice first to two identical objects and then 1 hr later to one of the now-familiar identical objects and one new object and quantified the time that each genotype spent sniffing each object. Wild-type mice spent significantly more time sniffing the novel object than the familiar object (F1,23 = 16.16, p < 0.001), indicating normal learning and memory, whereas 16p11+/− mice did not display any preference for the novel object over the familiar object (F1,25 = 1.07, NS), indicating a cognitive deficit (Figure 7H). Two standard cognitive tasks could not be conducted, the Morris water-maze task because the 16p11+/− mice frequently sank rather than swimming when placed in the pool, and fear conditioning because of the hearing deficit (Figures S6G and 7A, respectively). In other tests performed to evaluate memory, the 16p11+/ − mice were indistinguishable from control mice in their performance (Y maze and a modified Barnes Maze, Figures S6H and S6I). We then tested whether 16p11+/− mice become desensitized to novel objects by presenting control and 16p11+/− mice with the same object four times with 10 min intertrial interval (ITI), followed by presentation of a novel object, and then reintroduction of the familiar object (Figure 7I). Although control mice showed normal habituation to the novel object, 16p11+/ − mice showed increased interest for the familiar object that did not decline over the course of presentations (effect of genotype F1,68 = 5.99, p × trial interaction F3,204 = 0.017; effect of genotype = 3.92, p < 0.01). This result suggests that 16p11+/− mice lack the habituation normally observed in response to novel objects.
Effects of Risperidone on Activity Levels
The importance of BG function in motor control suggests a correlation of the increased numbers of Drd2+ cells in the striatum with the hyperactivity of 16p11+/− mice. We therefore treated 16p11+/− mice with risperidone, a D2R antagonist used clinically in patients with ASD. Risperidone has multiple effects in mice but primarily acts as a sedative. We introduced 16p11+/− and control mice into an activity chamber in a standard, low-stress assay for base-line activity similar to home cage performed in the dark. The 16p11+/− mice moved significantly more than control mice over the course of 10 min (Figure 7J). We then injected the mice with either saline vehicle or 0.2 mg/kg of risperidone and again observed their activity over the course of 10 min. Injection of risperidone significantly decreased the activity levels of wild-type mice (Figure 7K, t40 = 7.41, p < 0.0001) but had no sifnificant effect on those of 16p11+/− mice (t39 = 1.44, NS). Consistent with this, acute administration of risperidone had no overt effect on circling behavior in a subgroup of the 16p11+/− mice (not shown). This suggests that 16p11+/− mice are less susceptible to sedation by this D2R antagonist.
Discussion
We have generated a mouse model for the 16p11.2 microdeletion syndrome and identified a set of anatomical, behavioral, and electrophysiological phenotypes that provide insights into the developmental consequences of this mutation. In agreement with a previous paper describing a different mouse with the same deletion, we found that the 16p11+/− mice are born at Mendelian ratios, have impaired early-postnatal survival, are smaller than wild-type mice and are hyperactive. However, by using a comprehensive set of tools at ASD-relevant developmental stages we uncovered cellular and behavioral defects that significantly expand our knowledge of the phenotypic consequences of this deletion.
Anatomically we found abnormalities in the BG circuitry, namely, striatum and GP, as well as direct BG input (cortex) and output (thalamus and superior colliculus) structures. Using in vivo high-throughput single-cell gene expression profiling, we identified developmental changes in the BG- and DA-regulated circuitry of 16p11+/− mice, including increased numbers of Drd2+ MSNs in the striatum and downregulation of DA signaling components (Drd1 and Darpp32) in deeper cortical layers. Furthermore, expression of Th in cells of the mesodiencephalon was decreased. These abnormalities were accompanied by both pre- and postsynaptic defects at NAc MSNs as assessed by electrophysiology. Overall, these changes likely contribute to severe motor deficits observed in juvenile and adult 16p11+/− mice, including hyperactivity in a home-cage environment, tremor, lack of gait fluidity, and circling. Furthermore, a behavioral correlate of the increased numbers of striatopallidal MSNs is provided by the significantly reduced sensitivity of 16p11+/− mice to the D2R antagonist risperidone. This differential effect of risperidone suggests involvement of indirect pathway deficits in the abnormal activity pattern of 16p11+/− mice and thus provides evidence for another link between cellular and behavioral deficits in these mice. The 16p11+/− mice also showed a lack of habituation to familiarity in experiments testing novelty recognition, in the absence of overt memory defects. Taken together, these findings strongly indicate that the 16p11.2 deletion alters the gross developmental trajectory of the brain, including anatomy, connectivity, and information processing by the BG as well as circuit modulation by DA. Nevertheless, despite compelling evidence for the involvement of disrupted BG function in the phenotypes of 16p11+/ − mice, other brain regions, including—but not limited to—input and output structures of the BG, may also contribute. Our study provides evidence that BG function is altered by the 16p11.2 deletion and may provide important clues about the underlying basis of ASD and other psychiatric disorders.
Comparison of 16p11.2 Deletion in Mice and Humans
There are several similarities and notable differences between our mouse model and patients with 16p11.2 deletions. MRI data suggest a relative increase in size of various brain regions, including nuclei of the BG and mesodiencephalon. This could be seen as a parallel to the frequently described macrocephaly in human 16p11.2 deletion patients and other types of ASD (Courchesne et al., 2007). There is also an apparent decrease in the size of the corpus callosum, which has been repeatedly observed in ASD and ADHD (Giedd et al., 1994 and Gilliam et al., 2011), as well as mouse models for ASD (Ellegood et al., 2013 and Wahlsten et al., 2003). The 16p11+/− mice show hyperactivity, a potential correlate for ADHD observed in 16p11.2 patients (Shinawi et al., 2010). The mice also show defects in smooth motor movements that may be analogous to the highly prevalent motor delay described for 16p11.2 patients. Finally, defects in habituation to familiar objects are reminiscent of behavioral inflexibility reported in ASD. Together, these phenotypes suggest that the mouse model may provide some insights into the underlying circuits that lead to developmental defects in individuals with 16p11.2 deletions.
There are also a number of differences between the 16p11+/− mouse and individuals with the 16p11.2 deletion. Patients have a tendency to be obese, whereas the 16p11+/− mice are smaller and leaner than wild-type. Our early observations suggest reduced weight loss and improved growth performance of 16p11+/− pups when littermate competition is decreased, making it unlikely that the abnormal weight of 16p11+/− mice is related to mechanisms acting in humans, in which social and cultural factors eliminate a similar early postnatal selection pressure. Nevertheless, we cannot exclude the possibility that nutritional status of the pups could have affected postnatal brain development that may, in turn, have contributed to our observed behavioral phenotypes. In conclusion, our mouse model for the 16p11.2 deletion exhibits many phenotypes suggestive of abnormal function in the BG- and DA-regulated circuits, with behavioral and anatomical parallels in human patients. Interpretation of the human deficits in light of abnormal BG function may be important for understanding the underlying molecular mechanisms and developing therapeutic approaches targeting cellular defects in 16p11.2 deletion patients.
Experimental Procedures
A detailed description of all methods is provided in the Supplemental Experimental Procedures available online. All animal experiments were in accordance with the National Institutes of Health and Stanford guidelines for care and use of laboratory animals and approved by the National Institute of Mental Health Animal Care and Use Committee.
ESC Targeting
Sequential mouse ESC targeting was done according to standard protocols including LoxP sites (chr7:133842117 and chr7:134285222) and a fluorescent mCherry reporter transgene (Figures 1A–1D and S1A–S1K). Sequential targeting of DNA constructs resulted in both cis and trans arrangements of the LoxP sites in different mESC clones, as verified by PCR analysis of Cre-transfected ESC clones (Figures S1L and S1M). Germline transmission was successful only for the cis arrangement.
MRI Study
Diffusion tensor images of 52 brains (16p11+/ − and 26 wild-type) were acquired on a 7 Tesla MRI scanner (Varian) using a custom built three coil solenoid array to acquire images from three brains in parallel (Nieman et al., 2007). Image averaging and registration, was used to provide measures of local differences in anatomy between mice. Multiple comparisons were controlled for by using the false discovery rate (FDR) (Genovese et al., 2002).
96.96 Dynamic Arrays, Data analysis, and Coexpression Mapping
sc-qPCR experiments and FACS sorting by the Stanford Shared FACS Facility were performed as previously described (Pasca et al., 2011 and Yoo et al., 2011). A major challenge of single-cell gene expression analysis is the fluctuation of mRNA pools caused by transcriptional kinetics of individual alleles (Suter et al., 2011). A thorough assessment of the basic properties of the sc-qPCR data, which addressed (1) variability of gene expression level between single cells, (2) housekeeping genes and their value for normalization, (3) reflection of gene expression changes in the data, and (4) false-positives/negatives (Figures S2C – S2H; Supplemental Experimental Procedures), is provided in the Supplemental Experimental Procedures. Across-chip normalization was done on the mean expression of the ubiquitous Rps18 and Gapdh genes.
It remains unclear, given the biological difficulties we addressed earlier, what the value of the expression level of a cell-type-specific gene within a single cell is for classifying cell types. We therefore restricted our analysis to coexpression of genes. Data were clustered using the R-based heatmap tool (heatmap.2, R package: gplots) provided by Los Alamos National Laboratory (http://www.hiv.lanl.gov) and the manhattan distance between coexpression profiles of genes combined with Ward’s clustering method (Ward, 1963). Gene clusters were associated with known cell types reported in the literature and publicly available gene expression repositories (including Allen Brain Atlas for developing mouse brain, at e18 and P4). An example for coexpression mapping is provided in Supplemental Experimental Procedures.
Electrophysiology
Parasagittal slices (250 μm) containing the NAc core were prepared from wild-type and 16p11+/− mice on a C57BL/6N background (age P28–56), as described previously (Dölen et al., 2013).
Behavioral Assays at SBFNL
Age-matched 16p11+/− and wild-type mice were maintained on a C57BL/6N background, after backcrossing with C57BL/6N wild-type mice for five to seven generations. Four cohorts of male and female mice (aged 2–9 months) were blindly tested in behavioral paradigms. SHIRPA, home-cage behavioral monitoring, activity chamber, open field, grip strength, rotarod, startle response, Y maze spontaneous alternation task, modified Barnes maze, novel object recognition, three-chamber sociability and social novelty tasks, and associated statistical analyses were performed according to previously established protocols at SBFNL (Bader et al., 2011, Coutellier et al., 2012, Faizi et al., 2011, Faizi et al., 2012 and Ishizaki et al., 2010). For risperidone treatment, mice of both genotypes and genders were semirandomly divided into vehicle or risperidone treatment groups based on their activity levels in an activity chamber. One hour after acute, intraperitoneal risperidone administration (conc.: 0.2 mg/kg, vol.: 10 ml/kg) locomotor activity levels were measured for 10 min in the activity chamber.
Behavioral Assays at the NIMH
Twelve 16p11+/+ females and six 16p11+/− males were transferred from Stanford University to the National Institute of Mental Health in Bethesda, MD, and bred as described in Supplemental Experimental Procedures to generate two cohorts for behavioral testing. Pup body weight, juvenile reciprocal social interactions, three-chambered social approach, general health and neurological reflexes, open-field activity, novel empty cage, acoustic startle response, olfactory habituation/dishabituation, and novel object recognition are fully described in the Supplemental Experimental Procedures and previous publications (Brielmaier et al., 2012, Chadman et al., 2008, Ey et al., 2012, Silverman et al., 2011, Yang et al., 2011, Yang et al., 2012 and Yang and Crawley, 2009).
Supplementary Material
Acknowledgments
We would like to thank Ulrich Elling and Josef Penninger (IMBA Vienna, Austria) for help and advice on mouse ESC targeting, Marty Bigos of the Stanford shared FACS facility for assistance with clone sorting, Renee Reijo-Pera for generously providing the Biomark Instruments, Kristin L. Sainani for advice in statistical analysis of mouse behavioral data, and Yishan Sun for validating and providing primer pairs for the single-cell gene expression analysis. This research was funded by the Simons foundation SFARI grant no. 204340 (R.E.D. and J.N.C.), Nina Jauw (R.E.D.), the Swiss National Science Foundation (nos. PBSKP3-123434 and PA00P3_134196, T.P.), the NIMH Intramural Research Program and the University of California Davis MIND Institute (M.Y., D.K., D.L., and J.N.C.), an F31 NRSA from the NIMH (no. MH090648-02, G.P.), a K99 from the NIH/NIMH (no. MH091160, R.M.), and the Institute of Neurological Disorders and Stroke P30 center core grant no. NS069375-01A1 (M.S.).
Footnotes
Author Contributions
T.P. made the mice. M.Y., R.M., P.L.B., D.K., D.L., N.L.S., Z.Z., and M.A.M. performed and analyzed behavioral experiments; T.P. and G.P. performed sc-qPCR experiments and data analysis; J.E. and J.P.L. performed MRI experiments; T.P., G.P., E.F., K.C., and P.R. performed IHC stainings; G.D., B.A.G., and C.G. performed electrophysiological characterization; and R.E.D., J.N.C., M.S., R.C.M, T.P., M.Y., R.M., G.P., J.E., G.D., P.L.B., and J.P.L. planned the experiments and wrote the paper.
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Fariñas I, Grosschedl R, McConnell SK. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron. 2008;57:364–377. doi: 10.1016/j.neuron.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Jabaudon D, Yoshida Y, Macklis JD. Ctip2 controls the differentiation of medium spiny neurons and the establishment of the cellular architecture of the striatum. J. Neurosci. 2008;28:622–632. doi: 10.1523/JNEUROSCI.2986-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader PL, Faizi M, Kim LH, Owen SF, Tadross MR, Alfa RW, Bett GC, Tsien RW, Rasmusson RL, Shamloo M. Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proc. Natl. Acad. Sci. USA. 2011;108:15432–15437. doi: 10.1073/pnas.1112667108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron-Cohen S, Scott FJ, Allison C, Williams J, Bolton P, Matthews FE, Brayne C. Prevalence of autism-spectrum conditions: UK schoolbased population study. Br. J. Psychiatry. 2009;194:500–509. doi: 10.1192/bjp.bp.108.059345. [DOI] [PubMed] [Google Scholar]
- Bengtsson M, Ståhlberg A, Rorsman P, Kubista M. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res. 2005;15:1388–1392. doi: 10.1101/gr.3820805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, van Haeringen A, Fransen van de Putte DE, Anderlid BM, Lundin J, Lapunzina P, Pérez Jurado LA, Delle Chiaie B, et al. Extending the phenotype of recurrent rearrangements of 16p11.2: deletions in mentally retarded patients without autism and in normal individuals. Eur. J. Med. Genet. 2009;52:77–87. doi: 10.1016/j.ejmg.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Brielmaier J, Matteson PG, Silverman JL, Senerth JM, Kelly S, Genestine M, Millonig JH, DiCicco-Bloom E, Crawley JN. Autism-relevant social abnormalities and cognitive deficits in engrailed-2 knockout mice. PLoS ONE. 2012;7:e40914. doi: 10.1371/journal.pone.0040914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascio CJ, Foss-Feig JH, Heacock JL, Newsom CR, Cowan RL, Benningfield MM, Rogers BP, Cao A. Response of neural reward regions to food cues in autism spectrum disorders. J. Neurodev. Disord. 2012;4:9. doi: 10.1186/1866-1955-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadman KK, Gong S, Scattoni ML, Boltuck SE, Gandhy SU, Heintz N, Crawley JN. Minimal aberrant behavioral phenotypes of neuroligin-3 R451C knockin mice. Autism Res. 2008;1:147–158. doi: 10.1002/aur.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Wang SS, Hattox AM, Rayburn H, Nelson SB, McConnell SK. The Fezf2-Ctip2 genetic pathway regulates the fate choice of subcortical projection neurons in the developing cerebral cortex. Proc. Natl. Acad. Sci. USA. 2008;105:11382–11387. doi: 10.1073/pnas.0804918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, Morgan J. Mapping early brain development in autism. Neuron. 2007;56:399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Coutellier L, Beraki S, Ardestani PM, Saw NL, Shamloo M. Npas4: a neuronal transcription factor with a key role in social and cognitive functions relevant to developmental disorders. PLoS ONE. 2012;7:e46604. doi: 10.1371/journal.pone.0046604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Anda FC, Rosario AL, Durak O, Tran T, Gräff J, Meletis K, Rei D, Soda T, Madabhushi R, Ginty DD, et al. Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat. Neurosci. 2012;15:1022–1031. doi: 10.1038/nn.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong M, Wichmann T. Update on models of basal ganglia function and dysfunction Parkinsonism Relat. Disord. 2009;15(Suppl 3):S237–S240. doi: 10.1016/S1353-8020(09)70822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JB, Oh WJ, Sabatini BL, Gu Semaphorin C. 3E-Plexin-D1 signaling controls pathwayspecific synapse formation in the striatum. Nat. Neurosci. 2012;15:215–223. doi: 10.1038/nn.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen G, Darvishzadeh A, Huang KW, Malenka RC. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature. 2013;501:179–184. doi: 10.1038/nature12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegood J, Babineau BA, Henkelman RM, Lerch JP, Crawley JN. Neuroanatomical analysis of the BTBR mouse model of autism using magnetic resonance imaging and diffusion tensor imaging. Neuroimage. 2013;70:288–300. doi: 10.1016/j.neuroimage.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ey E, Yang M, Katz AM, Woldeyohannes L, Silverman JL, Leblond CS, Faure P, Torquet N, Le Sourd AM, Bourgeron T, et al. Absence of deficits in social behaviors and ultrasonic vocalizations in later generations of mice lacking neuroligin4. Genes Brain Behav. 2012;11:928–941. doi: 10.1111/j.1601-183X.2012.00849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faizi M, Bader PL, Tun C, Encarnacion A, Kleschevnikov A, Belichenko P, Saw N, Priestley M, Tsien RW, Mobley WC, Shamloo M. Comprehensive behavioral phenotyping of Ts65Dn mouse model of Down syndrome: activation of β1-adrenergic receptor by xamoterol as a potential cognitive enhancer. Neurobiol. Dis. 2011;43:397–413. doi: 10.1016/j.nbd.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faizi M, Bader PL, Saw N, Nguyen TV, Beraki S, Wyss-Coray T, Longo FM, Shamloo M. Thy1-hAPP(Lond/Swe+) mouse model of Alzheimer's disease displays broad behavioral deficits in sensorimotor, cognitive and social function. Brain Behav. 2012;2:142–154. doi: 10.1002/brb3.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, Szatmari P, Joseph-George AM, Mackay S, Whitten K, Noble B, et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J. Med. Genet. 2010;47:195–203. doi: 10.1136/jmg.2009.069369. [DOI] [PubMed] [Google Scholar]
- Fombonne E. The prevalence of autism. JAMA. 2003;289:87–89. doi: 10.1001/jama.289.1.87. [DOI] [PubMed] [Google Scholar]
- Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage. 2002;15:870–878. doi: 10.1006/nimg.2001.1037. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr. Opin. Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Giedd JN, Castellanos FX, Casey BJ, Kozuch P, King AC, Hamburger SD, Rapoport JL. Quantitative morphology of the corpus callosum in attention deficit hyperactivity disorder. Am. J. Psychiatry. 1994;151:665–669. doi: 10.1176/ajp.151.5.665. [DOI] [PubMed] [Google Scholar]
- Gilliam M, Stockman M, Malek M, Sharp W, Greenstein D, Lalonde F, Clasen L, Giedd J, Rapoport J, Shaw P. Developmental trajectories of the corpus callosum in attention-deficit/hyperactivity disorder. Biol. Psychiatry. 2011;69:839–846. doi: 10.1016/j.biopsych.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat. Neurosci. 2010;13:1519–1525. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horev G, Ellegood J, Lerch JP, Son YE, Muthuswamy L, Vogel H, Krieger AM, Buja A, Henkelman RM, Wigler M, Mills AA. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism Proc. Natl. Acad. Sci. USA. 2011;108:17076–17081. doi: 10.1073/pnas.1114042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR. Is social attachment an addictive disorder? Physiol. Behav. 2003;79:351–357. doi: 10.1016/s0031-9384(03)00148-3. [DOI] [PubMed] [Google Scholar]
- Ishizaki T, Erickson A, Kuric E, Shamloo M, HaraNishimura I, Inácio AR, Wieloch T, Ruscher K. The asparaginyl endopeptidase legumain after experimental stroke. J. Cereb. Blood Flow Metab. 2010;30:1756–1766. doi: 10.1038/jcbfm.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat. Neurosci. 2012;15:816–818. doi: 10.1038/nn.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lai T, Jabaudon D, Molyneaux BJ, Azim E, Arlotta P, Menezes JR, Macklis JD. SOX5 controls the sequential generation of distinct corticofugal neuron subtypes. Neuron. 2008;57:232–247. doi: 10.1016/j.neuron.2007.12.023. [DOI] [PubMed] [Google Scholar]
- Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J. Neurosci. 2000;20:6063–6076. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, Wellcome Trust Case Control Consortium et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár Z, Cheung AF. Towards the classification of subpopulations of layer V pyramidal projection neurons. Neurosci. Res. 2006;55:105–115. doi: 10.1016/j.neures.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Fame RM, MacDonald JL, MacQuarrie KL, Macklis JD. Novel subtype-specific genes identify distinct subpopulations of callosal projection neurons. J. Neurosci. 2009;29:12343–12354. doi: 10.1523/JNEUROSCI.6108-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman BJ, Lerch JP, Bock NA, Chen XJ, Sled JG, Henkelman RM. Mouse behavioral mutants have neuroimaging abnormalities. Hum. Brain Mapp. 2007;28:567–575. doi: 10.1002/hbm.20408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nóbrega-Pereira S, Gelman D, Bartolini G, Pla R, Pierani A, Marín O. Origin and molecular specification of globus pallidus neurons. J. Neurosci. 2010;30:2824–2834. doi: 10.1523/JNEUROSCI.4023-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen SR, Bortone DS, Adesnik H, Scanziani M. Gain control by layer six in cortical circuits of vision. Nature. 2012;483:47–52. doi: 10.1038/nature10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Paşca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 2011;17:1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 2006;29:349–358. doi: 10.1016/j.tins.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Tabata H, Tachikawa K, Nakajima K. The cortical subventricular zone-specific molecule Svet1 is part of the nuclear RNA coded by the putative netrin receptor gene Unc5d and is expressed in multipolar migrating cells. Mol. Cell. Neurosci. 2008;38:474–483. doi: 10.1016/j.mcn.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Seong HJ, Carter AG. D1 receptor modulation of action potential firing in a subpopulation of layer 5 pyramidal neurons in the prefrontal cortex. J. Neurosci. 2012;32:10516–10521. doi: 10.1523/JNEUROSCI.1367-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 2010;47:332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Turner SM, Barkan CL, Tolu SS, Saxena R, Hung AY, Sheng M, Crawley JN. Sociability and motor functions in Shank1 mutant mice. Brain Res. 2011;1380:120–137. doi: 10.1016/j.brainres.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter DM, Molina N, Gatfield D, Schneider K, Schibler U, Naef F. Mammalian genes are transcribed with widely different bursting kinetics. Science. 2011;332:472–474. doi: 10.1126/science.1198817. [DOI] [PubMed] [Google Scholar]
- Takemoto M, Hattori Y, Zhao H, Sato H, Tamada A, Sasaki S, Nakajima K, Yamamoto N. Laminar and areal expression of unc5d and its role in cortical cell survival Cereb. Cortex. 2011;21:1925–1934. doi: 10.1093/cercor/bhq265. [DOI] [PubMed] [Google Scholar]
- Tang SH, Silva FJ, Tsark WM, Mann JR. A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis. 2002;32:199–202. doi: 10.1002/gene.10030. [DOI] [PubMed] [Google Scholar]
- Thurley K, Senn W, Lüscher HR. Dopamine increases the gain of the input-output response of rat prefrontal pyramidal neurons. J. Neurophysiol. 2008;99:2985–2997. doi: 10.1152/jn.01098.2007. [DOI] [PubMed] [Google Scholar]
- Wahlsten D, Metten P, Crabbe JC. Survey of 21 inbred mouse strains in two laboratories reveals that BTBR T/+ tf/tf has severely reduced hippocampal commissure and absent corpus callosum. Brain Res. 2003;971:47–54. doi: 10.1016/s0006-8993(03)02354-0. [DOI] [PubMed] [Google Scholar]
- Ward JH. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963;58:236. [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, Autism Consortium et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Yang M, Crawley JN. Simple behavioral assessment of mouse olfaction. Curr. Protoc. Neurosci. 2009;8:8.24. doi: 10.1002/0471142301.ns0824s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Silverman JL, Crawley JN. Automated three-chambered social approach task for mice. Curr. Protoc. Neurosci. 2011;8:8.26. doi: 10.1002/0471142301.ns0826s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Bozdagi O, Scattoni ML, Wöhr M, Roullet FI, Katz AM, Abrams DN, Kalikhman D, Simon H, Woldeyohannes L, et al. Reduced excitatory neurotransmission and mild autismrelevant phenotypes in adolescent Shank3 null mutant mice. J. Neurosci. 2012;32:6525–6541. doi: 10.1523/JNEUROSCI.6107-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, Lee-Messer C, Dolmetsch RE, Tsien RW, Crabtree GR. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature. 2011;476:228–231. doi: 10.1038/nature10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.