

Abstract
Insulin resistance plays a central role in the development of the metabolic syndrome, but how it relates to cardiovascular disease remains controversial. Liver insulin receptor knockout (LIRKO) mice have pure hepatic insulin resistance. On a chow diet, LIRKO mice have a proatherogenic lipoprotein profile with reduced HDL cholesterol and VLDL particles that are markedly enriched in cholesterol. This is due to increased secretion and decreased clearance of apoB-containing lipoproteins, coupled with decreased triglyceride secretion secondary to increased expression of PGC-1β, which promotes VLDL secretion, but decreased expression of SREBP-1c, SREBP-2 and their targets, the lipogenic enzymes and the LDL receptor. Within twelve weeks on an atherogenic diet, LIRKO mice show marked hypercholesterolemia, and 100% of LIRKO mice, but 0% of controls, develop severe atherosclerosis. Thus, insulin resistance at the level of the liver is sufficient to produce the dyslipidemia and increased risk of atherosclerosis associated with the metabolic syndrome.
Introduction
Atherosclerotic cardiovascular disease is a major cause of morbidity and mortality in Western society, and much effort has been directed towards understanding its pathogenesis and identifying the risk factors associated with it. One of the most important predisposing factors for atherosclerosis is the metabolic syndrome. The metabolic syndrome is a constellation of abnormalities, including central obesity, glucose intolerance and type 2 diabetes, hypertension, and a dyslipidemia characterized by increased serum triglycerides, decreased high density lipoprotein (HDL) cholesterol, and increased small, dense low density lipoprotein (LDL) particles (Haffner et al., 1998; Grundy et al., 2004; Ford, 2005). The metabolic syndrome affects more than 27% of adults in the United States (Ford et al., 2004; Hedley et al., 2004) and increases the risk of cardiovascular disease two- to three-fold (Isomaa et al., 2001). Despite the alarming prevalence of the metabolic syndrome and the magnitude of risk it confers, however, defining the pathogenic links between the metabolic syndrome and cardiovascular disease has been difficult, and even the question of whether they are united by some common underlying pathophysiology remains a matter of intense debate (Kahn et al., 2005; Zimmet et al., 2005).
Insulin resistance has long been considered to be central to the pathophysiology of the metabolic syndrome (Reaven, 1988; Biddinger and Kahn, 2006). In the liver, insulin resistance is manifest by the blunted ability of insulin to activate its receptor kinase and its downstream targets (Saad et al., 1992; Kerouz et al., 1997), resulting in incomplete suppression of hepatic glucose production (Lewis et al., 1996). It is not clear, however, whether all metabolic pathways become resistant to insulin (Shimomura et al., 2000; Saad et al., 1993). For example, lipogenesis, which is positively regulated by insulin, is increased in the metabolic syndrome. This could be because lipogenesis remains sensitive to insulin, and is driven excessively by the compensatory hyperinsulinemia which occurs in insulin resistant states (Reaven, 1988; Reaven, 2004; Biddinger et al., 2005; Biddinger and Kahn, 2006). Alternatively, lipogenesis could also become resistant to insulin, but be driven by other factors in the metabolic syndrome, such as excessive carbohydrate intake (Schwarz et al., 2003).
The co-existence of hyperinsulinemia and other hormonal and metabolic changes has made it difficult to dissect the role of insulin resistance in the pathogenesis of the metabolic syndrome. This is particularly true of VLDL secretion, which is increased in the metabolic syndrome and thought to drive the other aspects of the dyslipidemia associated with this disorder (Sparks and Sparks, 1994; Zammit, 2002; Ginsberg, 1996; Ginsberg and Huang, 2000). Under normal conditions, insulin targets apoB, the principal protein component of VLDL, for intracellular degradation and thereby decreases VLDL secretion acutely (Sparks and Sparks, 1994; Ginsberg, 2006). However, insulin also stimulates lipogenesis, which can promote VLDL secretion (Shimomura et al 1999; Horton, 2002). Therefore, the increase in VLDL secretion associated with the metabolic syndrome could be due to either insulin resistance driving VLDL secretion by failing to degrade apoB or hyperinsulinemia driving VLDL secretion through excessive lipogenesis (Sparks and Sparks, 1994; Lewis et al., 1995; Malmstrom et al., 1997b; Reaven, 1997; Reaven and Laws, 1994). Determining the relative roles played by insulin resistance versus hyperinsulinemia or other factors in the development of the dyslipidemia associated with the metabolic syndrome remains a fundamental, but unanswered question, with important therapeutic implications.
In the present study, we have used the liver insulin receptor knockout (LIRKO) mouse as a model of pure hepatic insulin resistance. These mice develop hyperinsulinemia, but their livers are unable to respond to it. Therefore, the LIRKO model is a unique tool to dissect the effects of hepatic insulin resistance on the development of dyslipidemia and atherosclerosis (Michael et al., 2000; Biddinger et al., 2005). We find that triglyceride and cholesterol secretion are uncoupled in LIRKO mice, such that the secretion of triglycerides is decreased, but the secretion of apoB and cholesterol are robust. Moreover, LDL receptor expression and the clearance of apoB-containing particles are reduced. This results in a pro-atherogenic distribution of the serum cholesterol, characterized by decreased HDL cholesterol and an increase in cholesterol-enriched apoB containing lipoprotein particles. When challenged with an atherogenic diet, LIRKO mice rapidly develop severe hypercholesterolemia and extensive atherosclerosis. Therefore, hepatic insulin resistance alone can produce both dyslipidemia and the increased risk of atherosclerosis associated with the metabolic syndrome.
Results
LIRKO mice were created by breeding mice carrying insulin receptor alleles modified with loxP sites with mice carrying the Cre-recombinase driven by the albumin promoter, as previously described (Michael et al., 2000). On normal chow (9% fat with negligible amounts of cholesterol), LIRKO mice were non-obese, with normal serum free fatty acid levels and mild to moderately elevated serum glucose levels (Table I) relative to their littermate controls (Lox mice). LIRKO mice also developed marked hyperinsulinemia, due to the compensatory secretion of insulin from the β-cells of the pancreas coupled with a defect in insulin clearance due to the lack of insulin receptors in the liver (Michael et al., 2000). We have previously shown that the LIRKO liver fails to respond to this hyperinsulinemia, as insulin is unable to stimulate phosphorylation of its downstream targets, the insulin receptor substrates (IRS)-1 and –2, or normally suppress expression of phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (Michael et al., 2000). As previously observed, liver weights were decreased by approximately 30% due, at least in part, to decreased glycogen stores (Michael et al., 2000). Hepatic triglyceride content was unchanged, but there was a two-fold increase in hepatic cholesterol, a four-fold increase in hepatic cholesterol esters and a 40% increase in free hepatic cholesterol (Table I).
Table I. Phenotypic and Biochemical Characterization of LIRKO mice.
Data were obtained from two- to three-month old male LIRKO mice and their littermate Lox controls on a standard chow diet. Mice were sacrificed in the non-fasted state, at which time serum and liver samples were collected for further analysis. (n=5-8, p<0.05)
| LOX | LIRKO | |
|---|---|---|
| Weight (g) | 32.7 ± 0.8 | 30.9 ± 0.7 |
| Liver Weight (mg/ g BW) | 42.4 ±1.2 | 27.1 ± 2* |
| Serum glucose (mg/dL) | 132 ± 17 | 170 ± 32 |
| Serum Insulin (ng/mL) | 1.1 ± 0.1 | 12 ± 3* |
| FFA (mequiv/L) | 0.71 ± 0.60 | 0.70 ± 0.13 |
| Hepatic Lipids (mg/ g Liver) | ||
| Triglycerides | 3.58 ± 1.0 | 4.09 ± 0.8 |
| Total Cholesterol | 1.1 ± 0.1 | 2.1 ± 0.2** |
| Unesterified Cholesterol | 1.0 ± 0.1 | 1.4 ± 0.1** |
| Cholesteryl Esters | 0.3 ± 0.0 | 1.3 ± 0.2** |
Dyslipidemia in LIRKO mice
Despite the fact that serum cholesterol levels were normal (Figure 1A) and serum triglyceride levels were reduced by 50% (Figure 1B) on a normal chow diet, hepatic insulin resistance alone was sufficient to produce atherogenic changes in lipoprotein metabolism. Thus, FPLC analysis of serum from LIRKO and Lox control mice revealed that HDL cholesterol, the major form of circulating cholesterol in the mouse, was decreased by 50%, whereas VLDL cholesterol was increased three-fold (Figure 1C). Consistent with the decreased total serum triglyceride levels, FPLC analysis also showed that VLDL and LDL/IDL particles in the LIRKO mouse contained reduced levels of triglycerides (Figure 1D). Concomitant with these differences, the levels of apoB100 and apoB48, the major lipoproteins of VLDL, LDL and chylomicrons were increased in LIRKO mice, whereas the levels of apoA-I and apoE were not (Figure 1E). Interestingly, ApoE was present in the more buoyant lipoprotein fractions of LIRKO, but not control, serum. Therefore, even though insulin resistance did not alter total serum cholesterol levels, it produced a distinctly pro-atherogenic distribution of cholesterol with a shift towards apoB-containing lipoproteins.
Figure 1. FPLC profiles of plasma lipoproteins from Lox and LIRKO mice.
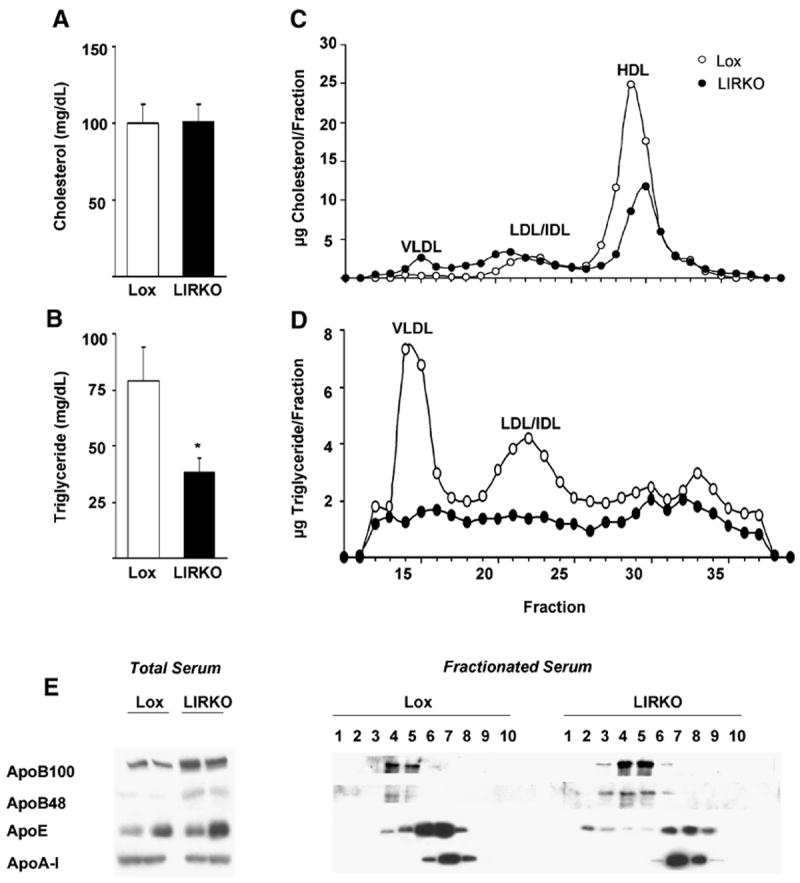
Serum was obtained from six month old mice on a normal chow diet, after a 4.5 hour fast, and (A) total cholesterol or (B) triglycerides was measured (n=5-6, p=0.04). Serum was subjected to FPLC analysis, and (C) cholesterol and (D) triglycerides were measured in each of the eluted fractions. Data are presented as the average of 2-4 mice per genotype. Similar results were obtained in two other experiments. (E) Apolipoprotein levels were examined by immunoblotting whole serum (left) or serum subjected to fractionation by FPLC. FPLC samples were prepared by pooling equal volumes of each fraction from three mice. In this and all other figures, error bars represent the SEM.
Lipid analysis of VLDL particles isolated from these mice by ultracentrifugation confirmed their abnormal lipid composition (Table II). In control mice, VLDL particles were composed largely of triglycerides, with an 8:1 weight ratio of triglycerides to total cholesterol. In contrast, triglycerides accounted for only 43% of the lipid in VLDL isolated from LIRKO serum, producing a 1:1 ratio of triglycerides to total cholesterol. Therefore, isolated hepatic insulin resistance was sufficient to produce VLDL particles enriched in cholesterol.
Table II. Lipoprotein Composition.
Six month old mice were fasted for 4.5 hours. Serum five mice of each genotype was pooled and subjected to sequential ultracentrifugation to obtain VLDL, IDL, LDL and HDL. The amounts of triglyceride, total cholesterol, free cholesterol and phospholipids were determined for each fraction. The relative lipid composition, by weight, of each particle was calculated.
| Lox | LIRKO | ||
|---|---|---|---|
| VLDL | |||
| TG | 80 | 43 | |
| FC | 5 | 3 | |
| CE | 5 | 46 | |
| PL | 11 | 8 | |
| IDL | |||
| TG | 50 | 14 | |
| FC | 13 | 6 | |
| CE | 15 | 25 | |
| PL | 23 | 55 | |
| LDL | |||
| TG | 21 | 10 | |
| FC | 15 | 19 | |
| CE | 27 | 44 | |
| PL | 37 | 27 | |
| HDL | |||
| TG | 2 | 3 | |
| FC | 14 | 16 | |
| CE | 40 | 39 | |
| PL | 45 | 42 | |
Dysregulation of PGC-1β, SREBP-1c and SREBP-2
Several transcription factors, including the sterol regulatory binding element proteins (SREBP) and their coactivators, especially peroxisome proliferator-activated receptor-γ coactivator (PGC)-1β, have been shown to regulate the synthesis and secretion of VLDL (Lin et al., 2005; Kalaany and Mangelsdorf, 2006; Wolfrum and Stoffel, 2006; Horton et al., 2002) In non-fasted mice on a chow diet, PGC-1β, as well as PGC-1α, a related co-activator involved in regulation of gluconeogenic and mitochondrial gene expression (Yoon et al., 2001), were increased two- to three-fold at the mRNA and protein levels (Figure 2A). In contrast, SREBP-1c, which regulates lipogenic gene expression, and SREBP-2, which regulates expression of the cholesterologenic enzymes and the LDL receptor, were decreased by 40-80% at the mRNA levels in LIRKO livers. After fasting and re-feeding, which normally leads to a dramatic induction of SREBP-1c (Matsuzaka et al., 2004), nuclear SREBP-1c, which represents the active form of SREBP-1c, was reduced >95% in LIRKO livers relative to controls (Figure 2A).
Figure 2. Insulin resistance alters gene expression.
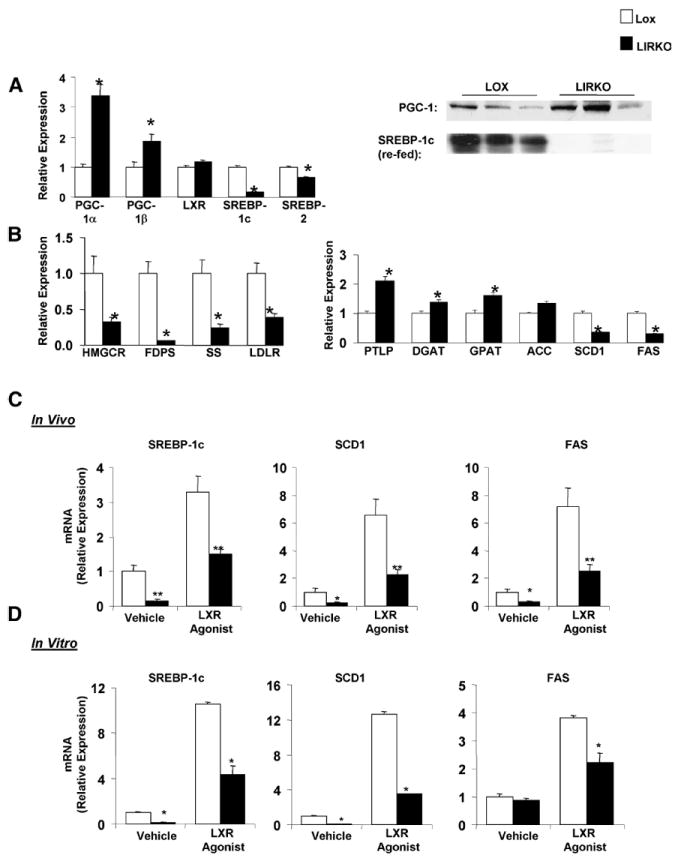
(A) Expression of the PGC and SREBP expression was measured in the livers of two-month old, non-fasted mice on a chow diet by real time PCR analysis or immunoblotting of liver extracts (PGC-1) or nuclear extracts (SREBP-1). SREBP-1 protein was also measured in two-month old mice after fasting and re-feeding (n=4-8, *p<0.05,). (B) Real time PCR analysis of cDNA prepared from the livers of two to four month old non-fasted chow fed mice. See text for gene names. (C) Two-month old Lox and LIRKO mice were gavaged with 40 mg/kg LXR agonist (T090137) or vehicle every 24 hours for two doses, and sacrificed four hours after the second dose, in the non-fasted state. Real time PCR was performed on cDNA prepared from these livers (n=5-8, *p<0.05, **p<0.005). (D) Hepatocytes were isolated from two to three month old Lox and LIRKO mice, and cultured overnight in the presence of 100 nM insulin, and either 5 μM LXR agonist or vehicle. RNA was extracted and subjected to real time PCR analysis. (n=4-6, *p<0.05)
Many of the targets of SREBP-2, including the cholesterologenic enzymes, HMG CoA reductase (HMGCR), squalene synthase (SS) and farnesyl diphosphate synthetase (FDPS), as well as the LDL receptor (LDLR), were decreased by 60-90% in LIRKO livers. However, the targets of SREBP-1c, the genes of fatty acid and triglyceride metabolism, showed a mixed pattern of change in the LIRKO liver. Some enzymes, like glycerol-3-phosphate acyltransferase (GPAT) and diacylglycerol acyltransferase (DGAT) which are involved in the packaging of fatty acids into triglycerides, and phospholipid transfer protein (PTLP), which also traffics lipids, were significantly increased in LIRKO livers. In contrast, stearoyl-CoA desaturase 1 (SCD1) and fatty acid synthase (FAS) were decreased by 40 to 90%, and acetyl-CoA carboxylase (ACC) was unchanged (Figure 2B).
SREBP-1c is regulated in part by insulin and in part by liver X receptor (LXR) (Repa et al., 2000; Tobin et al., 2002; Chen et al., 2004; Liang et al., 2002), but how these two factors interact in control of SREBP-1c remains debated (Hegarty et al., 2005; Dif et al., 2006; Cagen et al., 2005). LXR mRNA (Figure 2A) and protein levels were essentially unchanged (data not shown) in LIRKO mice. To determine whether activation of LXR could rescue lipogenic gene expression in the absence of insulin signaling, we treated LIRKO mice and controls with an LXR agonist, T090137, for two days (Figure 2C). The LXR agonist markedly increased expression of SREBP-1c and its lipogenic targets in both Lox and LIRKO livers. However, expression of these genes remained significantly lower in LIRKO livers even after pharmacological stimulation of LXR. Therefore, insulin action, in addition to LXR activation, is necessary for maximal induction of SREBP-1c and lipogenesis. Similar results were obtained in hepatocytes isolated from Lox and LIRKO mice and studied in vitro (Figure 2D), indicating that these effects were mediated directly by insulin resistance at the level of the liver.
Increased secretion and decreased clearance of apoB-lipoproteins
Given the changes in gene expression and the presence of abnormal, cholesterol-enriched VLDL particles in LIRKO serum, we measured VLDL secretion in chow-fed mice by inhibiting VLDL clearance with Triton WR1339. As shown in Figure 3A, the rate of triglyceride secretion was decreased by 66% in LIRKO mice. Three hours after injection of Triton WR1339, VLDL was isolated by ultracentrifugation, and consistent with the decreased rate of triglyceride secretion, VLDL from LIRKO mice contained 50% less triglyceride (Figure 3B). However, VLDL cholesterol levels were similar in control and LIRKO mice (Figure 3C). Thus, insulin resistance in the liver led to an uncoupling of cholesterol and triglyceride secretion, such that only triglyceride secretion was impaired, resulting in cholesterol-rich VLDL particles.
Figure 3. VLDL metabolism.
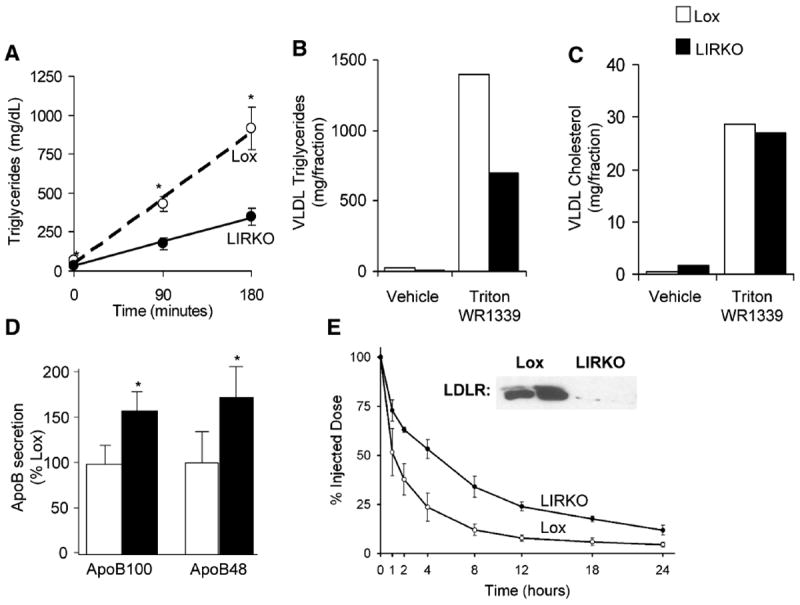
A-C) Two- to three- month old chow-fed Lox and LIRKO mice were injected with Triton WR1339 after a six hour fast. Serum triglycerides were measured at 0, 90 and 180 minutes (n=5-7, p ≤ 0.01 at each time point). After 180 minutes, serum was collected and equal volumes were pooled, and subjected to ultracentrifugation to obtain the VLDL fraction. VLDL triglycerides (A) and cholesterol (B) were measured. (D, E) apoB secretion and VLDL clearance were measured in four-month old mice after ten weeks on the Western diet. (D) The rate of apoB synthesis was measured as the amount of radiolabeled apoB100 and apoB48 present in the serum one hour after the injection of Triton WR1339 and [35S]methionine, normalized to the amount present in the Lox control (n=4-5, *p<0.01) (E) Fractional clearance of LDL apoB was determined by measuring plasma radioactivity over 24 hrs after injection of 125I –labeled mouse LDL. Results are expressed as the percentage of LDL radioactivity remaining in plasma at each time point (n=3). Inset shows LDL receptor immunoblot of liver extracts prepared from these mice.
Given the marked effects of hepatic insulin resistance on lipoprotein metabolism, we subjected LIRKO mice and their controls to the dietary stress of a Western diet, comprised of 40% fat (21% milk fat), 34.1% sucrose, 0.15% cholesterol. After ten weeks on the Western diet, LIRKO mice developed marked hypercholesterolemia relative to Lox mice (180 ± 19 mg/dl in Lox mice, versus 350 ±86 mg/dl LIRKO mice, n=5-11), demonstrating their inability to maintain cholesterol homeostasis in the presence of excess dietary fat and cholesterol.
Insulin normally inhibits secretion of apoB, the principal structural component of VLDL, and promotes its clearance (Sparks and Sparks, 1994). The hypercholesterolemia of LIRKO mice on the Western diet was due to defects in both of these processes. Secretion of both isoforms of apoB, apoB100 and apoB48, was increased 50-70% in LIRKO mice (Figure 3D). On the other hand, apoB-containing lipoprotein clearance, measured as the fraction removal rate of radio-iodinated mouse LDL, was markedly reduced (Figure 3E), with an increase in LDL half life from one hour in Lox mice, to more than four hours in LIRKO mice. Consistent with these changes, LDL receptor protein (Figure 3E, inset) was decreased 90% or more in the livers of LIRKO mice on a Western diet.
LIRKO mice develop severe hypercholesterolemia and atherosclerosis on an atherogenic diet
The low HDL cholesterol and increase in cholesterol rich VLDL suggested that LIRKO mice might be more susceptible to the development of atherosclerosis. To test this directly, we subjected Lox and LIRKO mice to an atherogenic Paigen diet (15% dairy fat, 1% cholesterol, 0.5% cholic acid) (Paigen et al., 1987). This diet is high in fat and cholesterol, like the Western diet, but it also contains cholic acid, which increases intestinal cholesterol absorption and produces an even greater degree of hypercholesterolemia. Interestingly, on this diet, although LDL receptor protein was markedly decreased, LDL receptor mRNA levels were similar (Supplemental Figure 1). After two months on the atherogenic Paigen diet, serum cholesterol levels rose to 310 ± 24 mg/dl in control mice, but to 781 ± 98 mg/dl in LIRKO mice (Figure 4A). FPLC fractionation revealed that the excess cholesterol was found exclusively in the apoB-containing atherogenic lipoproteins: LDL, IDL and VLDL (Figure 4B). Remarkably, despite this dramatic increase in total serum cholesterol, HDL cholesterol levels were still significantly lower in LIRKO mice than Lox mice. Similar changes were observed upon FPLC analysis of serum from mice placed on the Western diet (data not shown). In contrast, the amount and distribution of serum triglycerides were not different between Lox and LIRKO mice on either the atherogenic Paigen diet (Figures 4C and 4D) or the Western diet (data not shown).
Figure 4. Dyslipidemia and Atherosclerosis on the Atherogenic Diet.
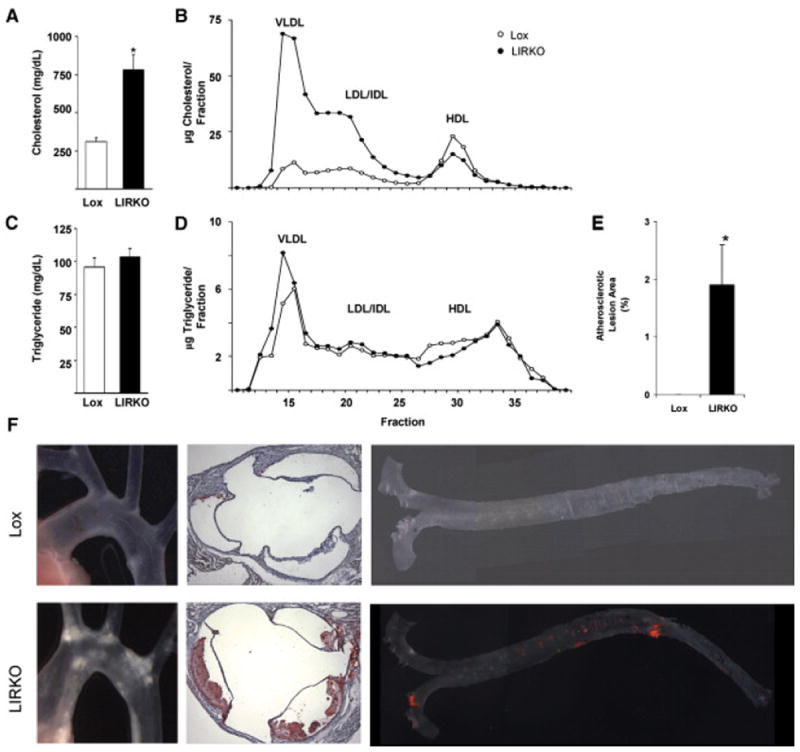
A-D) Serum was obtained from four month old mice fed the atherogenic diet for two months, after a six hour fast. Total cholesterol (A) and triglycerides (B) were measured in the serum. Equal amounts of serum were pooled from two mice of each genotype, and subjected to FPLC fractionation. Cholesterol (C) and triglycerides (D) were measured in each fraction. Data are presented as an average of three samples per genotype.
E-F) Lox and LIRKO mice were placed on an atherogenic diet at two months of age, and sacrificed three to four months later, at which time the aorta was examined for the presence of atherosclerosis (E) Quantitation of the plaque area in Lox and LIRKO mice (*p=0.01, using a Mann-Whitney U-test). (F) Gross dissected anatomy in situ (left); sections through aortic sinus and the aortic valve stained with hematoxylin and eosin (middle); and entire aorta with adventitial fat carefully removed, flat mounted “en face” and stained for fat with Sudan IV (right).
The effects of hepatic insulin resistance and the atherogenic diet on atherosclerosis were striking. After twelve weeks on this diet, all of the LIRKO mice, of both sexes, but not a single Lox mouse, developed overt atherosclerosis (n= 10-15 mice of each sex per genotype). Atherosclerotic plaques were easily visible on transillumination of the unstained aortic arch of LIRKO mice in situ (Figure 4F, left). Oil-Red-O staining of sections through the aortic sinus revealed intimal fat accumulation in LIRKO mice (Figure 4F, center). Staining of the entire aorta with Sudan IV demonstrated extensive disease throughout the aorta of LIRKO mice, but no visible atheroma in control mice subjected to the same dietary stress (Figure 4F, right). By quantitation, 1.7% of the surface of LIRKO aortas was stained by Sudan IV, even though no staining was detectable in Lox aortas (Figure 4E). Thus, by multiple criteria, LIRKO mice developed severe atherosclerosis whereas controls did not.
Discussion
Although the association between the metabolic syndrome, dyslipidemia and increased cardiovascular disease risk is generally accepted, how each of these relates to insulin resistance is controversial (Kahn et al., 2005). Our studies in LIRKO mice show that hepatic insulin resistance causes a pro-atherogenic distribution of serum cholesterol, with a 50% decrease in HDL cholesterol and an increase in non-HDL cholesterol. Furthermore, on both the Western diet and the atherogenic Paigen diet, HDL cholesterol levels remain low, yet LIRKO developed severe hypercholesterolemia with the excess cholesterol associated with apoB-containing lipoproteins, i.e. VLDL, LDL and IDL. As a consequence of these pro-atherogenic changes in lipid and lipoprotein metabolism, LIRKO mice, but not control mice, developed atherosclerosis within twelve weeks on an atherogenic diet. Therefore, hepatic insulin resistance alone is sufficient for the development of dyslipidemia and, when coupled with a permissive diet, the development of atherosclerosis.
Insulin resistance acts at several levels to produce dyslipidemia in the LIRKO mouse. First, it increases apoB secretion by decreasing intracellular degradation of apoB (Sparks and Sparks, 1994; Fisher et al., 2001). Second, it alters the expression of SREBP-1c, SREBP-2, PGC-1α and PGC-1β. SREBP-1c, which mediates the lipogenic response to insulin (Shimomura et al., 1999b), is decreased in LIRKO mice, even in the presence of an LXR agonist, indicating that insulin resistance suppresses SREBP-1c through mechanisms independent of LXR ligand generation. This decrease in SREBP-1c expression results in reduced VLDL triglyceride secretion and hypotriglyceridemia on a chow diet. Insulin resistance also promotes the accumulation of cholesterol, which inhibits SREBP-2 by preventing its maturation (Brown and Goldstein, 1997), and thereby decreases expression of its target, the LDL receptor, and apoB clearance. Insulin may also play a role in stabilizing LDL receptor protein, since the differences in LDL receptor expression were greater at the protein level than the mRNA level. In addition, PGC-1 expression is upregulated in LIRKO mice and this, along with other factors such as Foxa2, could further increase apoB secretion (Lin et al., 2005; Kalaany and Mangelsdorf, 2006; Wolfrum and Stoffel, 2006; Horton et al., 2002). Finally, hepatic insulin resistance could alter lipoprotein metabolism through secondary changes in the hormonal and metabolic milieu, such as hyperglycemia and hyperinsulinemia.
Dietary cholesterol and fat intake clearly interact with hepatic insulin resistance in production of the dyslipidemia of metabolic syndrome. On a chow diet, serum triglyceride levels are low, and total serum cholesterol is normal. Nonetheless, the distribution of this cholesterol is distinctly pro-atherogenic with reduced levels of HDL and increased levels of non-HDL cholesterol. When LIRKO mice are challenged with a high-fat, high-cholesterol diet, serum triglycerides normalize to the level of Lox controls, but cholesterol levels increase progressively to almost 800 mg/dl, while still retaining their atherogenic profile with low HDL cholesterol. Therefore, hepatic insulin resistance is able to produce a significant derangement of cholesterol metabolism, which progresses in the presence of dietary stress to marked hypercholesterolemia and atherosclerosis.
The distribution of serum cholesterol in LIRKO mice recapitulates the dyslipidemia of human metabolic syndrome, with increased VLDL cholesterol and decreased HDL cholesterol. In contrast, most commonly used mouse models of the metabolic syndrome, such as leptin deficient ob/ob mice and mice with diet induced obesity, show increased HDL cholesterol (Supplemental Table 1). Yet, LIRKO mice do not develop the hypertriglyceridemia that is characteristic of human metabolic disease. Serum triglyceride levels are under complex regulation (reviewed in (Ginsberg, 1996), but one important factor driving hypertriglyceridemia is insulin-stimulated hepatic de novo lipogenesis, and this is mediated largely by SREBP-1c (Shimomura et al., 1998; Shimomura et al., 1999b). Supplemental Figure 2 shows that SREBP-1c and its targets are reduced in mice deficient in hepatic insulin signaling, such as LIRKO mice and mice made insulinopenic by streptozotocin. In contrast, SREBP-1c is activated in ob/ob mice and mice with diet induced obesity, consistent with previous reports in the literature on these and other models of the metabolic syndrome (Shimomura et al., 2000; Shimomura et al., 1999a).
These data have led to the concept of partial insulin resistance, in which defects in one arm of the insulin signaling pathway lead to impaired control of glucose homeostasis, while lipogenesis continues to be activated by hyperinsulinemia or some alternative pathway. (Siri et al., 2001; Biddinger et al., 2005; Elam et al., 2001) In particular, it appears that the phosphotidylinositol 3-kinase/Akt arm of the insulin signaling pathway, which is thought to mediate many of insulin’s metabolic effects (Garg, 1996), becomes resistant to insulin, whereas the MAPK arm of the insulin signaling pathway remains intact. (Cusi et al., 2000; Maezono et al., 1998). Our data are entirely consistent with this hypothesis, as restoring Akt activity in LIRKO mice on the atherogenic diet, using a constitutively active form of Akt, normalizes serum glucose and the distribution of serum cholesterol by increasing HDL and decreasing non-HDL cholesterol (Supplemental Figure 3). These data further suggest that in the metabolic syndrome, insulin resistance drives changes in cholesterol and glucose homeostasis, whereas hyperinsulinemia or other factors drive hypertriglyceridemia, and together these produce the full complement of lipid abnormalities associated with the metabolic syndrome in humans.
Hypertriglyceridemia in humans is thought to decrease HDL cholesterol by stimulating cholesteryl ester transfer protein (CETP), an enzyme present in humans but not mice (Nagashima et al., 1988; de Grooth et al., 2004) to remove cholesteryl esters on HDL, by exchanging them for triglycerides on VLDL (Garg, 1996; Ginsberg, 1996; Rashid et al., 2002). The fact that LIRKO mice show a major reduction in HDL cholesterol levels even in the absence of hypertriglyceridemia and CETP indicates that insulin resistance alone can decrease HDL cholesterol independently of hypertriglyceridemia. Although the exact mechanism by which insulin resistance produces this effect remains to be determined, preliminary studies show that HDL clearance is normal in LIRKO mice. Therefore, hepatic insulin resistance reduces HDL cholesterol by either decreasing HDL production or promoting a shift of HDL cholesterol to non-HDL lipoproteins.
In relating the LIRKO model to the metabolic syndrome, it is important to remember that insulin resistance in human metabolic disease differs in several respects from the LIRKO model. First, the insulin resistance associated with the metabolic syndrome and type 2 diabetes involves many tissues of the body in addition to the liver, including muscle, fat, β-cells, the vasculature and other cells. Recent studies have shown that these other tissues may also participate in the dyslipidemia and accelerated atherosclerosis associated with these disorders. For example, Baumgartl et al. (Baumgartl et al., 2006) have shown that insulin resistance in cells of the myeloid lineage may protect against atherosclerosis in apoE deficient mice, whereas Han et al. (Han et al., 2006) found that insulin receptor deficiency in macrophages may increase ER stress-induced apoptosis and necrosis in advanced atherosclerotic lesions. Knockout of insulin receptors in endothelial cells has been shown to protect against retinal neovascularization and reduce expression of several vasoactive substances including eNOS (Kondo et al., 2003), which could contribute to the development of atherosclerosis. Therefore, the development of dyslipidemia and atherosclerosis in the metabolic syndrome is ultimately dependent upon the net effects of insulin resistance in both hepatic and extra-hepatic tissues. Moreover, as noted above, in human disease, the effects of the dietary and other genetic factors are superimposed upon the effects of insulin resistance in production of the metabolic syndrome. Despite these differences, however, many of the changes in cholesterol metabolism observed in the LIRKO model are also observed in mice and humans with the metabolic syndrome. First, obese, insulin resistant mice and humans show increased secretion of apoB and VLDL (Lewis et al., 1995; Lewis et al., 1993; Malmstrom et al., 1997a) (Bartels et al., 2002) (Cohen et al., 2002) (Siri et al., 2001). Second, several rodent models of obesity have also been shown to have decreased expression of the LDL receptor (Roberts et al., 2004) (Lundasen et al., 2003; Liao et al., 1997). Finally, humans with metabolic disease show a defect in the catabolism of apoB containing lipoproteins (Ouguerram et al., 2003) (Duvillard et al., 2000; Howard et al., 1987; Chan et al., 2003). Therefore, insulin resistance in the context of the metabolic syndrome may drive dyslipidemia and atherosclerosis by the same mechanisms as it does in the LIRKO model.
In summary, we have shown that insulin resistance plays a critical and central role in the development of dyslipidemia and atherosclerosis. This finding is of clinical importance because it suggests that the metabolic syndrome is not merely a collection of abnormalities that should be considered and treated independently, as some experts have advocated (Kahn et al., 2005). Rather, it appears that the metabolic syndrome is truly a syndrome, in which disturbances in glucose and cholesterol metabolism both stem from a defect in insulin signaling. These data further suggest that finding and reversing the molecular lesions that produce the insulin resistance associated with the metabolic syndrome will result in more effective treatment of this disorder.
Experimental Procedures
Mice, diets and treatments
Generation and genotyping of LIRKO (Cre+/-, IRflox/flox) mice and their littermate Lox controls (Cre-/-, IRflox/flox) has been described previously (Michael et al., 2000). LIRKO mice were generated on a mixed genetic background, including 129/sv, C57BL/6, FVB, and DBA, and inbred for more than ten generations.
In most studies, mice were male, chow-fed (Purina Lab Diet 9F Mouse Chow 5020, Pharmaserv, containing 9.0% fat and 0.221 ppm cholesterol), and sacrificed in the non-fasted state. Alternatively, mice were given ad libitum access to either the atherogenic Paigen diet (The Jackson Laboratories, Bar Harbor, ME), or a western diet (21% milk fat, 34.1% sucrose, 0.15% cholesterol). For refeeding studies, mice were fasted for 24 hours, and then re-fed for eight hours with a high-carbohydrate diet. LXR agonist (Sigma) was prepared in 1% carboxymethylcellulose and given at a dose of 40 mg/kg by gavage every 24 hours for two days to three month old mice, and then sacrificed four hours after the second dose in the non-fasted state. Adenovirus encoding myr-Akt or LacZ (Ono et al., 2003) were prepared as previously described (Taniguchi et al., 2006), and injected into the tail vein four days prior to sacrifice.
Phenotypic and Biochemical characterization
Plasma measurement were made using the following assays: insulin, ELISA (Crystal Chem Inc.); leptin, ELISA (Crystal Chem, Inc.); free fatty acids, colorimetric assay, (Wako); total cholesterol or triglycerides (measured as total triglycerides), colorimetric assay (Stanbio, or Sigma). Hepatic lipid analysis was performed by the Lipid, Lipoprotein and Atherosclerosis Core of the Vanderbilt Mouse Metabolic Phenotyping Centers as described previously (Biddinger et al., 2006).
Fast performance liquid chromatography analysis
Serum from mice fasted 4-6 hours was subjected to fast performance liquid chromatography as previously described (Babaev et al., 2000). Cholesterol and triglycerides were measured in the eluted fractions using Sigma kits 352 and 339, adapted for microtiter plates. For apolipoprotein analysis, equal aliquots of three FPLC fractions were pooled together and subjected to SDS PAGE and immunoblotting with antibodies against apoB, apoE or apoA1 (Biodesign). Alternatively, equal volumes of whole serum were subjected to immunoblotting with these antibodies.
Plasma lipoprotein isolation
Serum from mice that had been fasted for six hours was collected and equal volumes from each genotype were pooled. The lipoproteins were isolated by sequential ultracentrifugation at densities of 1.006 (VLDL), 1.019 (IDL), 1.063 (LDL) and 1.215 (HDL) as previously described (Argmann CA, 2006). Each fraction was dialyzed against a phosphate buffered saline supplemented with EDTA and sodium azide. Total cholesterol (Sigma), free cholesterol (Wako), total triglycerides (Sigma) and phospholipids (Wako) were measured enzymatically according to the manufacturer’s directions.
Atherosclerosis
During deep anesthesia, animals were perfused with saline, followed by formalin. The base of the heart with the aortic sinus was fixed, imbedded, cryosectioned, and stained with Oil-Red-O and counterstained with hematoxylin. The remainder of the aorta was dissected free of adventitial fat, stained with Sudan IV and examined microscopically for the presence or absence of atheromatous plaques. Quantitation was performed with ImageJ software.
VLDL triglyceride secretion
Mice were fasted for six hours, and then injected intravenously with either WR1339 (Sigma Aldrich) or vehicle. Plasma samples were taken prior to injection and after 90 minutes by retro-orbital bleeds, and at 180 minutes by cardiac puncture. Total triglycerides were measured (Stanbio). Equal volumes of serum taken at the 180 minute time point were pooled, and subjected to ultracentrifugation to isolate VLDL particles as above.
VLDL apoB secretion
After a 4-hour fast, the apoB secretion rates were determined as previously described (Siri et al., 2001). Mice were injected with 200 μCi [35S]-methionine (Perkin Elmer Life Sciences) and 500 mg/kg Triton WR1339 in normal saline. Plasma samples were collected after 60 minutes and subjected to SDS-PAGE and autoradiography. The apoB100 and apoB48 bands were excised and counted in scintillation fluid (Ecoscint H LS-275). The rate of apoB secretion was calculated as the amount of radiolabeled apoB present in the serum after one hour.
LDL turnover studies
LDL (d:1.025-1.060) was isolated from the plasma of C57Bl/6J male mice by sequential ultracentrifugation, labeled with [125I]-iodide monochloride (Bilheimer et al., 1972), dialyzed and filter-sterilized. 0.15 μCi of [125I]- LDL was injected via the femoral vein (n = 3 for each group), and radioactivity was measured in plasma samples collected at multiple time points thereafter. The mice had free access to food and water throughout the experiment. The radioactivity present in plasma at 30 seconds accounted for more than 98% of the injected LDL and was used to calculate the percent of injected LDL remaining in the circulation at the later time points.
Real time-PCR analysis
1 μg of RNA (RNeasy, Qiagen) was isolated from mouse liver and used as a template for cDNA synthesis (RT for PCR, Clontech)(Biddinger et al., 2005). Quantitative real time PCR was performed in the fluorescent temperature cycler (ABI) with either SYBR Green Master Mix (Roche, Mannheim, Germany, primer sequences listed in Supplemental Data Table 1) or the Taqman Universal master mix (ABI; primers purchased through Assay on Demand). Expression values were normalized to the level of input RNA.
Nuclear and Liver Extracts
Nuclear extracts were prepared using 30-60 mg of liver, per the manufacturer’s directions (NE, PER kit, Pierce). Liver extracts were prepared by homogenizing in RIPA buffer with 1% SDS. Protein concentrations were measured by Bradford assay (Biorad).
Hepatocyte Isolation
Hepatocytes were isolated from two to three month old mice using a two-step collagenase perfusion procedure, as described previously (Pertoft H, 1987). Hepatocytes were plated in collagen coated wells in DMEM containing 10% fetal bovine serum. After three hours, the medium was changed, and supplemented with 100 nM insulin and either 5 μM T090137 or vehicle. After 18 hours, cells were harvested and RNA was prepared for real time PCR analysis as above.
Statistical Analyses
Statistical significance was calculated by an unpaired Student’s t-test, and p< 0.05 was considered significant. All data are expressed as the mean ± SEM.
Supplementary Material
Acknowledgments
This work was funded in part by grants DK063696-05 (SBB), NSF Graduate Research Fellowship (JOA), DK036588 and DK073687 (MCC), DK75850 (to GS), R01 DK056626 and R01 DK048873 (DEC), 5RO1 DK053105-08 (GK), NHLBI HL 55368 and HL73030 (HNG) and DK31036 and DK45935 (CRK). This study was supported by the Lipid, Lipoprotein and Atherosclerosis Core of the Vanderbilt Mouse Metabolic Phenotyping Centers (NIH DK59637-01) (SE0160160B), and the DERC Specialized Assay Core of the Joslin Diabetes Center (DK P30DK36836).
Reference List
- Argmann CA, Houten SM, Champy MF, Auwerx J. Lipid and Bile Acid Analysis. John Wiley & Sons, Inc; 2006. Current Protocols in Molecular Biology; pp. 29B.2.1–29B.2.24. [DOI] [PubMed] [Google Scholar]
- Babaev VR, Patel MB, Semenkovich CF, Fazio S, Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in low density lipoprotein receptor-deficient mice. J Biol Chem. 2000;275:26293–26299. doi: 10.1074/jbc.M002423200. [DOI] [PubMed] [Google Scholar]
- Bartels ED, Lauritsen M, Nielsen LB. Hepatic expression of microsomal triglyceride transfer protein and in vivo secretion of triglyceride-rich lipoproteins are increased in obese diabetic mice. Diabetes. 2002;51:1233–1239. doi: 10.2337/diabetes.51.4.1233. [DOI] [PubMed] [Google Scholar]
- Baumgartl J, Baudler S, Scherner M, Babaev V, Makowski L, Suttles J, McDuffie M, Tobe K, Kadowaki T, Fazio S, Kahn CR, Hotamisligil GS, Krone W, Linton M, Bruning JC. Myeloid lineage cell-restricted insulin resistance protects apolipoprotein E-deficient mice against atherosclerosis. Cell Metab. 2006;3:247–256. doi: 10.1016/j.cmet.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddinger SB, Almind K, Miyazaki M, Kokkotou E, Ntambi JM, Kahn CR. Effects of diet and genetic background on sterol regulatory element-binding protein-1c, stearoyl-CoA desaturase 1, and the development of the metabolic syndrome. Diabetes. 2005;54:1314–1323. doi: 10.2337/diabetes.54.5.1314. [DOI] [PubMed] [Google Scholar]
- Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol. 2006;68:123–158. doi: 10.1146/annurev.physiol.68.040104.124723. [DOI] [PubMed] [Google Scholar]
- Biddinger SB, Miyazaki M, Boucher J, Ntambi JM, Kahn CR. Leptin suppresses stearoyl-CoA desaturase 1 by mechanisms independent of insulin and sterol regulatory element-binding protein-1c. Diabetes. 2006;55:2032–2041. doi: 10.2337/db05-0742. [DOI] [PubMed] [Google Scholar]
- Bilheimer DW, Eisenberg J, Levy RI. The metabolism of very low density lipoproteins. I. Preliminary in vivo and in vitro observations. Biochim Biophys Acta. 1972;26:212–221. doi: 10.1016/0005-2760(72)90034-3. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Cagen LM, Deng X, Wilcox HG, Park EA, Raghow R, Elam MB. Insulin activates the rat sterol-regulatory-element-binding protein 1c (SREBP-1c) promoter through the combinatorial actions of SREBP, LXR, Sp-1 and NF-Y cis-acting elements. Biochem J. 2005;385:207–216. doi: 10.1042/BJ20040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC, Watts GF, Barrett PH, O’Neill FH, Thompson GR. Plasma markers of cholesterol homeostasis and apolipoprotein B-100 kinetics in the metabolic syndrome. Obes Res. 2003;11:591–596. doi: 10.1038/oby.2003.83. [DOI] [PubMed] [Google Scholar]
- Chen G, Liang G, Ou J, Goldstein JL, Brown MS. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc Natl Acad Sci U S A. 2004;101:11245–11250. doi: 10.1073/pnas.0404297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Miyazaki M, Socci ND, Hagge-Greenberg A, Liedtke W, Soukas AA, Sharma R, Hudgins LC, Ntambi JM, Friedman JM. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science. 2002;297:240–243. doi: 10.1126/science.1071527. [DOI] [PubMed] [Google Scholar]
- Cusi K, Maezono K, Osman A, Pendergrass M, Patti ME, Pratipanawatr T, DeFronzo RA, Kahn CR, Mandarino LJ. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–320. doi: 10.1172/JCI7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Grooth GJ, Klerkx AH, Stroes ES, Stalenhoef AF, Kastelein JJ, Kuivenhoven JA. A review of CETP and its relation to atherosclerosis. J Lipid Res. 2004;45:1967–1974. doi: 10.1194/jlr.R400007-JLR200. [DOI] [PubMed] [Google Scholar]
- Dif N, Euthine V, Gonnet E, Laville M, Vidal H, Lefai E. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem J. 2006;400:179–188. doi: 10.1042/BJ20060499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvillard L, Pont F, Florentin E, Galland-Jos C, Gambert P, Verges B. Metabolic abnormalities of apolipoprotein B-containing lipoproteins in non-insulin-dependent diabetes: a stable isotope kinetic study. Eur J Clin Invest. 2000;30:685–694. [PubMed] [Google Scholar]
- Elam MB, Wilcox HG, Cagen LM, Deng X, Raghow R, Kumar P, Heimberg M, Russell JC. Increased hepatic VLDL secretion, lipogenesis, and SREBP-1 expression in the corpulent JCR:LA-cp rat. J Lipid Res. 2001;42:2039–2048. [PubMed] [Google Scholar]
- Fisher EA, Pan M, Chen X, Wu X, Wang H, Jamil H, Sparks JD, Williams KJ. The triple threat to nascent apolipoprotein B. Evidence for multiple, distinct degradative pathways. J Biol Chem. 2001;276:27855–27863. doi: 10.1074/jbc.M008885200. [DOI] [PubMed] [Google Scholar]
- Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care. 2005;28:1769–1778. doi: 10.2337/diacare.28.7.1769. [DOI] [PubMed] [Google Scholar]
- Ford ES, Giles WH, Mokdad AH. Increasing prevalence of the metabolic syndrome among u.s. Adults. Diabetes Care. 2004;27:2444–2449. doi: 10.2337/diacare.27.10.2444. [DOI] [PubMed] [Google Scholar]
- Garg A. Insulin resistance in the pathogenesis of dyslipidemia. Diabetes Care. 1996;19:387–389. doi: 10.2337/diacare.19.4.387. [DOI] [PubMed] [Google Scholar]
- Ginsberg HN. Diabetic dyslipidemia: basic mechanisms underlying the common hypertriglyceridemia and low HDL cholesterol levels. Diabetes. 1996;45(Suppl 3):S27–S30. doi: 10.2337/diab.45.3.s27. [DOI] [PubMed] [Google Scholar]
- Ginsberg HN, Huang LS. The insulin resistance syndrome: impact on lipoprotein metabolism and atherothrombosis. J Cardiovasc Risk. 2000;7:325–331. doi: 10.1177/204748730000700505. [DOI] [PubMed] [Google Scholar]
- Grundy SM, Brewer HB, Jr, Cleeman JI, Smith SC, Jr, Lenfant C. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Cirulation. 2004;109:433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. New England Journal of Medicine. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- Han S, Liang CP, Devries-Seimon T, Ranalletta M, Welch CL, Collins-Fletcher K, Accili D, Tabas I, Tall AR. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006;3:257–266. doi: 10.1016/j.cmet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999-2002. JAMA. 2004;291:2847–2850. doi: 10.1001/jama.291.23.2847. [DOI] [PubMed] [Google Scholar]
- Hegarty BD, Bobard A, Hainault I, Ferre P, Bossard P, Foufelle F. From The Cover: Distinct roles of insulin and liver X receptor in the induction and cleavage of sterol regulatory element binding protein-1c. Proc Natl Acad Sci U S A. 2005;102:791–796. doi: 10.1073/pnas.0405067102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard BV, Abbott WG, Beltz WF, Harper IT, Fields RM, Grundy SM, Taskinen MR. Integrated study of low density lipoprotein metabolism and very low density lipoprotein metabolism in non-insulin-dependent diabetes. Metabolism. 1987;36:870–877. doi: 10.1016/0026-0495(87)90096-5. [DOI] [PubMed] [Google Scholar]
- Isomaa B, Almgren P, Tuomi T, Forsen B, Lahti K, Nissen M, Taskinen MR, Groop L. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24:683–689. doi: 10.2337/diacare.24.4.683. [DOI] [PubMed] [Google Scholar]
- Kahn R, Buse J, Ferrannini E, Stern M. The metabolic syndrome: time for a critical appraisal: joint statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2005;28:2289–2304. doi: 10.2337/diacare.28.9.2289. [DOI] [PubMed] [Google Scholar]
- Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159–191. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- Kerouz NJ, Horsch D, Pons S, Kahn CR. Differential regulation of insulin receptor substrates-1 and -2 (IRS-1 and IRS-2) and phosphatidylinositol 3-kinase isoforms in liver and muscle of the obese diabetic (ob/ob) mouse. J Clin Invest. 1997;100:3164–3172. doi: 10.1172/JCI119872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Vicent D, Suzuma K, Yanagisawa M, King GL, Holzenberger M, Kahn CR. Knockout of insulin and IGF-1 receptors on vascular endothelial cells protects against retinal neovascularization. J Clin Invest. 2003;111:1835–1842. doi: 10.1172/JCI17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis GF, Uffelman KD, Szeto LW, Steiner G. Effects of acute hyperinsulinemia on VLDL triglyceride and VLDL apoB production in normal weight and obese individuals. Diabetes. 1993;42:833–842. doi: 10.2337/diab.42.6.833. [DOI] [PubMed] [Google Scholar]
- Lewis GF, Uffelman KD, Szeto LW, Weller B, Steiner G. Interaction between free fatty acids and insulin in the acute control of very low density lipoprotein production in humans. J Clin Invest. 1995;95:158–166. doi: 10.1172/JCI117633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis GF, Zinman B, Groenwoud Y, Vranci M, Giacca A. Hepatic glucose production is regulated both by direct hepatic and extra hepatic effects of insulin in humans. Diabetes. 1996;45:454–462. doi: 10.2337/diab.45.4.454. [DOI] [PubMed] [Google Scholar]
- Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem. 2002;277:9520–9528. doi: 10.1074/jbc.M111421200. [DOI] [PubMed] [Google Scholar]
- Liao W, Angelin B, Rudling M. Lipoprotein metabolism in the fat Zucker rat: reduced basal expression but normal regulation of heptic low density lipoprotein. Endocrinology. 1997;138:3276–3282. doi: 10.1210/endo.138.8.5337. [DOI] [PubMed] [Google Scholar]
- Lin J, Yang R, Tarr PT, Wu PH, Handschin C, Li S, Yang W, Pei L, Uldry M, Tontonoz P, Newgard CB, Spiegelman BM. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell. 2005;120:261–273. doi: 10.1016/j.cell.2004.11.043. [DOI] [PubMed] [Google Scholar]
- Lundasen T, Liao W, Angelin B, Rudling M. Leptin induces the hepatic high density lipoprotein receptor scavenger receptor B type I (SR-BI) but not cholesterol 7alpha-hydroxylase (Cyp7a1) in leptin-deficient (ob/ob) mice. J Biol Chem. 2003;278:43224–43228. doi: 10.1074/jbc.M302645200. [DOI] [PubMed] [Google Scholar]
- Maezono K, Osman A, Patti ME, Cusi K, Pendergrass M, DeFronzo RA, Mandarino L. PI 3-kinase but not map kinase signaling is insulin resistant in obesity and niddm. Diabetes. 1998;47(Supp.1):A333. [Google Scholar]
- Malmstrom R, Packard CJ, Caslake M, Bedford D, Stewart P, Yki-Jarvinen H, Shepherd J, Taskinen MR. Defective regulation of triglyceride metabolism by insulin in the liver in NIDDM. Diabetologia. 1997a;40:454–462. doi: 10.1007/s001250050700. [DOI] [PubMed] [Google Scholar]
- Malmstrom R, Packard CJ, Watson TD, Rannikko S, Caslake M, Bedford D, Stewart P, Yki-Jarvinen H, Shepherd J, Taskinen MR. Metabolic basis of hypotriglyceridemic effects of insulin in normal men. Arterioscler Thromb Vasc Biol. 1997b;17:1454–1464. doi: 10.1161/01.atv.17.7.1454. [DOI] [PubMed] [Google Scholar]
- Matsuzaka T, Shimano H, Yahagi N, Amemiya-Kudo M, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Tomita S, Sekiya M, Hasty A, Nakagawa Y, Sone H, Toyoshima H, Ishibashi S, Osuga J, Yamada N. Insulin-independent induction of sterol regulatory element-binding protein-1c expression in the livers of streptozotocin-treated mice. Diabetes. 2004;53:560–569. doi: 10.2337/diabetes.53.3.560. [DOI] [PubMed] [Google Scholar]
- Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Molecular Cell. 2000;6:87–97. [PubMed] [Google Scholar]
- Nagashima M, McLean JW, Lawn RM. Cloning and mRNA tissue distribution of rabbit cholesteryl ester transfer protein. J Lipid Res. 1988;29:1643–1649. [PubMed] [Google Scholar]
- Ono H, Shimano H, Katagiri H, Yahagi N, Sakoda H, Onishi Y, Anai M, Ogihara T, Fujishiro M, Viana AY, Fukushima Y, Abe M, Shojima N, Kikuchi M, Yamada N, Oka Y, Asano T. Hepatic Akt activation induces marked hypoglycemia, hepatomegaly, and hypertriglyceridemia with sterol regulatory element binding protein involvement. Diabetes. 2003;52:2905–2913. doi: 10.2337/diabetes.52.12.2905. [DOI] [PubMed] [Google Scholar]
- Ouguerram K, Magot T, Zair Y, Marchini JS, Charbonnel B, Laouenan H, Krempf M. Effect of atorvastatin on apolipoprotein B100 containing lipoprotein metabolism in type-2 diabetes. J Pharmacol Exp Ther. 2003;306:332–337. doi: 10.1124/jpet.103.048991. [DOI] [PubMed] [Google Scholar]
- Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- Pertoft H, Smedsrod B. Separation and Characterization of Liver Cells. Vol. 4. Academic Press; 1987. Cell Separation: Methods and Selected Applications. [Google Scholar]
- Rashid S, Barrett PH, Uffelman KD, Watanabe T, Adeli K, Lewis GF. Lipolytically modified triglyceride-enriched HDLs are rapidly cleared from the circulation. Arterioscler Thromb Vasc Biol. 2002;22:483–487. doi: 10.1161/hq0302.105374. [DOI] [PubMed] [Google Scholar]
- Reaven G. The metabolic syndrome or the insulin resistance syndrome? Different names, different concepts, and different goals. Endocrinol Metab Clin North Am. 2004;33:283–303. doi: 10.1016/j.ecl.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Reaven GM. Banting lecture: Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- Reaven GM. Banting Lecture 1988. Role of insulin resistance in human disease. Nutrition. 1997;13:65. doi: 10.1016/s0899-9007(96)00380-2. 1988 [classical article] [DOI] [PubMed] [Google Scholar]
- Reaven GM, Laws A. Insulin resistance, compensatory hyperinsulinaemia, and coronary heart disease. Diabetologia. 1994;37:948–952. doi: 10.1007/BF00400953. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CK, Liang K, Barnard RJ, Kim CH, Vaziri ND. HMG-CoA reductase, cholesterol 7 alpha-hydroxylase, LDL receptor, SR-B1, and ACAT in diet-induced syndrome X. Kidney Int. 2004;66:1503–1511. doi: 10.1111/j.1523-1755.2004.00914.x. [DOI] [PubMed] [Google Scholar]
- Saad MJA, Araki E, Miralpeix M, Rothenberg PL, White MF, Kahn CR. Regulation of insulin receptor substrate 1 in liver and muscle of animal models of insulin resistance. J Clin Invest. 1992;90:1839–1849. doi: 10.1172/JCI116060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad MJA, Folli F, Kahn JA, Kahn CR. Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J Clin Invest. 1993;92:2065–2072. doi: 10.1172/JCI116803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Linfoot P, Dare D, Aghajanian K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am J Clin Nutr. 2003;77:43–50. doi: 10.1093/ajcn/77.1.43. [DOI] [PubMed] [Google Scholar]
- Shimomura I, Bashmakov Y, Horton JD. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem. 1999a;274:30028–30032. doi: 10.1074/jbc.274.42.30028. [DOI] [PubMed] [Google Scholar]
- Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999b;96:13656–13661. doi: 10.1073/pnas.96.24.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]
- Shimomura I, Shimano H, Korn BS, Bashmakov Y, Horton JD. Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. J Biol Chem. 1998;273:35299–35306. doi: 10.1074/jbc.273.52.35299. [DOI] [PubMed] [Google Scholar]
- Siri P, Candela N, Zhang YL, Ko C, Eusufzai S, Ginsberg HN, Huang LS. Post-transcriptional stimulation of the assembly and secretion of triglyceride-rich apolipoprotein B lipoproteins in a mouse with selective deficiency of brown adipose tissue, obesity, and insulin resistance. J Biol Chem. 2001;276:46064–46072. doi: 10.1074/jbc.M108909200. [DOI] [PubMed] [Google Scholar]
- Sparks JD, Sparks CE. Insulin regulation of triacylglycerol-rich lipoprotein synthesis and secretion. Biochim Biophys Acta. 1994;1215:9–32. doi: 10.1016/0005-2760(94)90088-4. [DOI] [PubMed] [Google Scholar]
- Taniguchi CM, Kondo T, Sajan M, Luo J, Bronson R, Asano T, Farese R, Cantley LC, Kahn CR. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKClambda/zeta. Cell Metab. 2006;3:343–353. doi: 10.1016/j.cmet.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Tobin KA, Ulven SM, Schuster GU, Steineger HH, Andresen SM, Gustafsson JA, Nebb HI. Liver X receptors as insulin-mediating factors in fatty acid and cholesterol biosynthesis. J Biol Chem. 2002;277:10691–10697. doi: 10.1074/jbc.M109771200. [DOI] [PubMed] [Google Scholar]
- Wolfrum C, Stoffel M. Coactivation of Foxa2 through Pgc-1beta promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metab. 2006;3:99–110. doi: 10.1016/j.cmet.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- Zammit VA. Insulin stimulation of hepatic triacylglycerol secretion in the insulin-replete state: implications for the etiology of peripheral insulin resistance. Ann N Y Acad Sci. 2002;967:52–65. doi: 10.1111/j.1749-6632.2002.tb04263.x. [DOI] [PubMed] [Google Scholar]
- Zimmet P, Magliano D, Matsuzawa Y, Alberti G, Shaw J. The metabolic syndrome: a global public health problem and a new definition. J Atheroscler Thromb. 2005;12:295–300. doi: 10.5551/jat.12.295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.