

Abstract
Objective
MUC18 or CD146, a transmembrane glycoprotein, is mainly expressed by endothelial cells and smooth muscle cells where it serves as a cell-cell adhesion molecule. We have found MUC18 up-regulation in airway epithelial cells of patients with asthma and chronic obstructive pulmonary disease (COPD). However, the function of MUC18 in airway epithelial cells remains unclear. In the present study, we tested the hypothesis that MUC18 exerts a pro-inflammatory function during stimulation with a viral mimic polyI:C or human rhinovirus infection.
Methods
Normal human primary airway epithelial cells were transduced with lentivirus encoding MUC18 cDNA to over-express MUC18 or with GFP (control), and treated with polyI:C or HRV for detection of pro-inflammatory cytokine IL-8 and anti-viral gene IFN-β. Additionally, we performed cell culture of human lung epithelial cell line NCIH292 cells to determine the mechanisms of MUC18 function.
Results
We found that MUC18 over-expression promoted IL-8 production, while it inhibited IFN-β expression following polyI:C stimulation or HRV infection. Increased phosphorylation of MUC18 serines was observed in MUC18 over-expressing cells. Reduction of MUC18 serine phosphorylation by inhibiting ERK activity was associated with less production of IL-8 following polyI:C stimulation.
Conclusions
Our results for the first time demonstrate MUC18's pro-inflammatory and anti-viral function in human airway epithelial cells.
Keywords: Immune response, Airway epithelial cells, MUC18, Rhinovirus, IL-8, Interferon, Anti-viral, Inflammation
Introduction
Excessive inflammation is a common feature during exacerbations of various lung diseases, including asthma, chronic obstructive pulmonary disease (COPD) and cystic fibrosis. One of the major triggers in lung disease exacerbations is viral infection, in particular human rhinovirus (HRV) infection [1–6]. So far, the mechanisms underlying excessive lung inflammation of lung disease exacerbations remain unclear.
MUC18, also known as CD146 or melanoma cell adhesion molecule (MCAM), is a 113 kD transmembrane glycoprotein of the immunoglobulin superfamily [7,8]. MUC18 is composed of an extracellular domain, a single transmembrane domain, and a short (63 amino acids) cytoplasmic tail. In a normal setting, MUC18 is abundantly expressed on endothelial and smooth muscle cells, but is minimally seen or undetectable in epithelial cells and leukocytes (e.g., macrophages and neutrophils) [9]. MUC18 overexpression was initially identified in human malignant melanoma cells and was thought to promote tumor metastasis [10–12]. However, MUC18 regulation and function in inflammatory lung diseases such as asthma or COPD are poorly understood. Our recent publications [13,14] demonstrated MUC18 up-regulation in airway epithelial cells of asthmatics and COPD patients. Importantly, MUC18 is critical to bacteria-induced lung inflammation, which is in part through activation of NF-κB signaling [13,14]. However, whether MUC18 promotes inflammatory responses to Toll-like receptor (TLR) ligands mimicking viral infection or viruses has not been investigated. Moreover, the molecular mechanisms by which MUC18 promotes the inflammatory response are unclear.
In the current study, we hypothesized that MUC18 is pro-inflammatory in response to a viral mimic polyI:C as well as HRV infection. To test this hypothesis, we used the MUC18 over-expression approach in primary human airway epithelial cell cultures to determine if MUC18 exerts pro-inflammatory but anti-viral functions in response to polyI:C as well as live HRV. Furthermore, we examined the role of MUC18 serine phosphorylation and activation of ERK, a major component of the mitogen-activated protein kinases (MAPK) signaling pathway, in MUC18-mediated effects.
Methods
MUC18 over-expression in normal human primary airway epithelial cells
Lentivirus-mediated MUC18 over-expression was performed in normal human primary airway epithelial cells using our previously published method [15]. Human MUC18 cDNA (Open Biosystems; Thermo Scientific, Huntsville, AL) was cloned into a lentiviral vector pLentilox (pLL) 3.7 by removing the GFP gene, generating the pLL3.7-MUC18 vector. A pLL3.7 vector expressing GFP was used as a control. pLL3.7-GFP or pLL3.7-MUC18 was co-transfected with psPAX2 vector and envelope protein vector p-CMV-VSV-G into 293FT packaging cells using the Effectene Transfection Reagent (Qiagen, Valencia, CA). Forty-eight hours after transfection, cell supernatants containing virus were harvested and then used for infecting airway epithelial cells.
Normal human tracheobronchial epithelial cells were obtained from the tracheas and bronchi of de-identified organ donors whose lungs were not suitable for transplantation as previously described [15]. Collection and the use of these cells were approved by the Institutional Review Board (IRB) at National Jewish Health. Airway epithelial cells at passage 1 were cultured and expanded in collagen-coated 60 mm tissue culture dishes containing bronchial epithelial cell growth medium with supplements (Lonza, Walkersville, MD) at 37°C, 5% CO2, until 90% confluence. The expanded epithelial cells at passage 2 were seeded into 6-well culture plates at 1×105 cells/well and transduced with lentiviruses containing pLL3.7-GFP (GFP) or pLL3.7-MUC18 (MUC18 over-expression) by centrifugation at 920 g for 1 hour at 25°C. Seventy-two hours after transduction, cells were passed into 12-well culture plates at 1×105 cells/well. After 48 hours, cells were stimulated with polyI:C (10 μg/mL) [16] or HRV-1A (104 TCID50/mL) [17]. Additionally, cells were treated with a selective MEK/ERK inhibitor PD98059 at the same time as polyI:C. Cells were harvested for Western blot and quantitative RT-PCR at indicated time points after polyI:C or HRV-1A.
Western blot analysis
Cells were lyzed in Western lysis buffer with protease inhibitors (Fisher Scientific, Waltham, MA). The same amount of protein lysate (e.g., 30 μg) was electrophoresed on an 8% SDS-PAGE gel, transferred onto a nitrocellulose membrane, blocked with 2.5% nonfat milk, and incubated with antibodies against MUC18 (Abcam, San Francisco, CA), p-ERK1/2 and ERK1/2 (Santa Cruz Biotech Inc., Santa Cruz, CA), and GAPDH (Santa Cruz Biotech Inc., Santa Cruz, CA) overnight at 4°C. After washing, membranes were incubated with the appropriate HRP-linked secondary antibodies and Pierce ECL Western blotting substrate (Fisher Scientific, Waltham, MA).
MUC18 immunoprecipitation (IP) and Western blot of phosphorylated serines
NCI-H292 cells or normal human primary tracheobronchial epithelial cells were treated with polyI:C for 10, 15 and 30 minutes in the absence or presence of PD98059, an ERK inhibitor. Cells were then washed with ice-cold PBS once and lyzed in 500 μl ice-cold lysis buffer consisting of 50 mM Tris-HCl (pH 8.0), 120 mM NaCl, 1% Nonidet P-40, 4 mM EDTA, 50 mM NaF, 1 mM Na3VO4, and the protease inhibitor cocktail (100-fold dilution). After the cells were sonicated and centrifuged, the supernatant was transferred to a fresh tube containing 400 μl of lysis buffer and pre-cleared with protein A-agarose beads (Santa Cruz Biotechnology) for 1 hour. 0.5 μg of the rabbit anti-human MUC18 antibody was added to each tube and incubated for 2 hours in a rotator at 4°C. Immunoprecipitates were collected with protein A-agarose and dissolved in 1xLDS sample buffer for the subsequent Western blot experiments of phosphorylated serines by using an antibody against phosphorylated serine (Sigma). To ensure an equal amount of protein for each cell culture condition, an aliquot of protein samples for each condition was examined for GAPDH Western blot.
ELISA
IL-8 protein levels in cell culture supernatant were determined using the human IL-8 enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN).
Quantitative real-time RT-PCR
TaqMan gene expression assays for human IFN-β, and HRV were obtained from or custom-made by Applied Biosystems (Life Technologies, Foster City, CA). The housekeeping gene GAPDH was evaluated as an internal positive control. The comparative cycle of threshold (ΔΔCt) method was used to demonstrate the relative levels of target genes.
Statistical analysis
Data are presented as means ± SEMs. One-way ANOVA was used for multiple comparisons and a Tukey post hoc test was applied, where appropriate. The Student t test was used where only 2 groups were compared. A p value of less than 0.05 was considered significant.
Results
MUC18 over-expression (OE) in normal human primary airway epithelial cells increases pro-inflammatory cytokine production upon viral mimic polyI:C stimulation
As shown in Figure 1, MUC18 OE cells, as compared to control cells, markedly increased MUC18 protein expression at the baseline. After 4 and 24 hours of polyI:C stimulation, epithelial cells increased production of IL-8 protein in the absence or presence of MUC18 OE (Figure 2A). Notably, as compared to cells without MUC18 OE, those with MUC18 OE further amplified IL-8 production following polyI:C treatment at 24 hours (Figure 2A and 2B).
Figure 1.

Western blot showing MUC18 protein over-expression (OE) in cultured normal human primary tracheobronchial epithelial (NHTE) cells. NHTE cells were transduced with MUC18 cDNA or green fluorescence protein (GFP) gene (control) encoded in replication-deficient lentivirus. Cells were harvested after 72 hours of lentiviral infection to confirm MUC18 protein over-expression.
Figure 2.
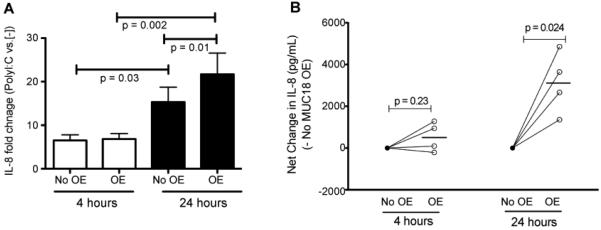
Effect of MUC18 over-expression (OE) on IL-8 protein levels in cultured normal human primary tracheobronchial epithelial (NHTE) cells. Cells were transduced with MUC18 cDNA or green fluorescence protein (GFP) gene (control) encoded in replication-deficient lentivirus. After 72 hours of transduction, cells were stimulated with polyI:C (10 μg/ml) for 4 and 24 hours. (A) IL-8 fold changes in polyI:C-stimulated NHTE cells with or without MUC18 over-expression (MUC18 OE). MUC18 OE cells, as compared to non-OE cells, show greater IL-8 induction (fold change over cells without polyI:C) following polyI:C treatment at 24, but not 4 hours. Data are expressed as means ± SEM. N=4 human donors. (B) Net changes of IL-8 after polyI:C stimulation in NHTE cells with or without MUC18 OE. At 24 hours, MUC18 OE significantly increases IL-8 levels in supernatants of polyI:C-treated cells. This trend exists at 4 hours, but is not statistically significant. N=4 human donors. The horizontal bars indicate means.
MUC18 over-expression (OE) in normal human primary airway epithelial cells decreases anti-viral gene expression upon viral mimic polyI:C stimulation
Whether MUC18 regulates the anti-viral pathway such as type I interferon (i.e., IFN-β) expression has not been addressed. As expected, polyI:C stimulation (versus cell culture medium) in both control (no MUC18 OE) and MUC18 OE cells increased IFN-β expression at both 4 hours (up to 4,000-fold, p<0.05) and 24 hours (up to 38-fold, p<0.05). Importantly, as shown in Figure 3A and 3B, MUC18 OE cells demonstrated less induction of IFN-β expression following polyI:C treatment than the control cells at 4 hours, but not at 24 hours, suggesting an early inhibitory function of MUC18 in anti-viral gene expression.
Figure 3.
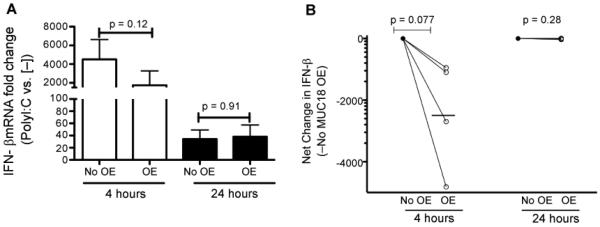
Effect of MUC18 over-expression (OE) on IL-8 protein levels in cultured normal human primary tracheobronchial epithelial (NHTE) cells. Cells were transduced with MUC18 cDNA or green fluorescence protein (GFP) gene (control) encoded in replication-deficient lentivirus. After 72 hours of transduction, cells were stimulated with polyI:C (10 μg/ml) for 4 and 24 hours. (A) IFN-β mRNA fold changes in polyI:Cstimulated NHTE cells with or without MUC18 over-expression (MUC18 OE). MUC18 OE cells, as compared to non-OE cells, show less IFN-β mRNA induction (fold change over cells without polyI:C) following polyI:C treatment at 4, but not 24 hours. Data are expressed as means ± SEM. N=4 human donors. (B) When the IFN-β mNRA data are expressed as net changes (MUC18 over-expression (OE) minus no MUC18 OE), MUC18 OE also decreases the induction of IFN-β mNRA expression 4 hours after polyI:C stimulation in NHTE cells. N=4 human donors. The horizontal bars indicate means.
Mechanisms of MUC18 function
How MUC18 functions in human airway epithelial cells remains unclear, as MUC18-related signaling pathways in inflammatory responses to TLR agonists such as polyI:C or infectious agents have not yet been examined. It has been postulated that serines (e.g., Ser606 and Ser614) of human MUC18 cytoplasmic tail may be phosphorylated, and responsible for MUC18 function [18,19]. As the first step towards understanding MUC18 signaling, we determined if there is evidence for phosphorylation of MUC18 serines. We performed a time-course (10, 15 and 30 minutes) study in lung epithelial cell line NCI-H292 cells stimulated with polyI:C. As shown in Figure 4A, at 10 minutes of polyI:C (versus cell medium alone [−]), there was no increase of MUC18 serine phosphorylation. However, at both 15 and 30 minutes following polyI:C, MUC18 serine phosphorylation was increased. As serine phosphorylation could result from ERK activation [20,21], we then determined the temporal relationship between MUC18 phosphorylation and ERK1/2 activation. Interestingly, ERK1/2 activation started at 10 minutes (Figure 4B), earlier than MUC18 phosphorylation. This suggests that ERK1/2 activation precedes MUC18 serine phosphorylation. To confirm the dependency of MUC18 serine phosphorylation on polyI:C-induced ERK1/2 activation, a selective MEK/ERK inhibitor PD98059 was added to NCI-H292 cells 30 minutes before polyI:C treatment. Indeed, PD98059 decreased MUC18 serine phosphorylation at 15 and 30 minutes after polyI:C treatment (Figure 4A). Notably, PD98059 significantly reduced IL-8 production (Figure 4C and 4D). However, PD98059 did not alter IFN-β expression in NCI-H292 cells that increased IFN-β mRNA expression following polyI:C stimulation (Figure 4E).
Figure 4.

MUC18 serine phosphorylation in polyI:C-stimulated human lung epithelial cell line NCI-H292. (A) At the indicated time points, cells were immunoprecipitated with a rabbit anti-human MUC18 antibody, and then immunoblotted with an antibody against phosphorylated serines. GAPDH immunoblot was performed using 30 μg total proteins under different conditions to verify equal loading. Densitometry was performed to measure the intensity of MUC18 p-serine and GAPDH signals. The ratio of MUC18 P-serine versus GAPDH was used to indicate the level of phosphorylated MUC18. (B) Cell lysates were immunoblotted with antibodies against phosphorylated ERK1/2 (p-ERK) and total ERK1/2. The ratio of p-ERK1/2 versus total ERK1/2 intensity as evaluated by densitometry was used to indicate the level of phosphorylated ERK1/2. (C–D) ERK inhibition by PD98059 significantly reduced IL-8 production in NCI-H292 cells at 8 hours and 24 hours after polyI:C stimulation. However, PD98059 did not inhibit IFN-β mRNA expression at 24 hours (E). N=12 replicates. *, p<0.05; #, p>0.05.
To validate MUC18 serine phosphorylation in normal human primary airway epithelial cells, cells over-expressing MUC18 or expressing GFP (control) were treated with polyI:C in the presence or absence of PD98059 for 15 min. First, cells without MUC18 OE did not show any signals of MUC18 serine phosphorylation under any conditions (data not shown). In contrast, MUC18 OE cells at the baseline showed a weak signal of MUC18 serine phosphorylation, which was amplified by polyI:C treatment (Figure 5).
Figure 5.

MUC18 serine phosphorylation in polyI:C-stimulated normal human primary tracheobronchial epithelial (NHTE) cells that over-express MUC18. Cells were treated with or without polyI:C (10 μg/ml) for 15 minutes, followed by MUC18 immunoprecipitation and immunoblotting of phosphorylated serines.
MUC18 function in normal human primary airway epithelial cells infected with live human rhinovirus
Having shown MUC18's pro-inflammatory and anti-viral function upon viral mimic polyI:C stimulation, we sought to determine if such function exists during HRV infection. MUC18 OE and control normal human primary airway epithelial cells were infected with HRV-1A, a minor group HRV involved in asthma exacerbations [22,23]. First, IL-8 production trended (p = 0.057) to be higher in MUC18 OE cells than control cells following HRV-1A infection at 4 hours (Figure 6A). Second, IFN-β mRNA induction by HRV-1A at 4 hours was significantly less in MUC18 OE cells than that in control cells (Figure 6B). However, at 24 hours post HRV-1A infection, IL-8 production and IFN-β mRNA expression were similar (p>0.10) between MUC18 OE and control cells (data not shown).
Figure 6.
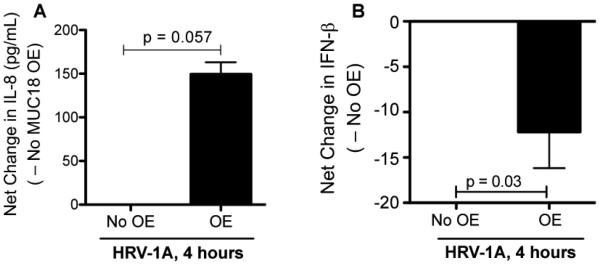
Effect of MUC18 over-expression (OE) on human rhinovirus (HRV)-induced IL-8 production in cultured normal human primary tracheobronchial epithelial (NHTE) cells. Cells were transduced with MUC18 cDNA or green fluorescence protein (GFP) gene (control) encoded in replication-deficient lentivirus. After 72 hours of transduction, cells in 12-well plated were infected under submerged condition with HRV-1A (104 TCID50/well) for 4 and 24 hours. MUC18 OE cells, as compared to non-OE cells, show a greater induction of IL-8 protein (A) following HRV-1A infection at 4 hours. Data are expressed as means ± SEM. (B) – Net changes of IFN-β mNRA expression after HRV-1A in MUC18 OE cells versus (minus) cells without MUC18 OE. At 4 hours of HRV-1A infection, MUC18 OE significantly decreases the induction of IFN-β mNRA expression. N=4 human donors.
Discussion
The current study has demonstrated the novel function of MUC18 in airway epithelial immune responses to polyI:C (a mimic of viral infection) and live HRV infection. MUC18 appears to exert a “paradoxical” role in human airway epithelial cell responses to polyI:C stimulation and HRV-1A infection. While MUC18 promotes the proinflammatory response (e.g., IL-8 production), it suppresses the anti-viral (e.g., IFN-β) gene expression.
MUC18 is classically considered to be involved in cell-cell adhesion under a normal setting. This function is particularly important in maintaining the integrity or tight junction of endothelial cells [9]. However, under a pathologic condition such as malignant melanoma or other cancers, MUC18 is significantly increased, contributing to tumor metastasis [10–12]. MUC18 expression and function in human airways have not been well studied. A previous study suggests up-regulation of MUC18 expression in brushed bronchial epithelial cells from COPD patients [24], but MUC18 function in the lung remains unclear. We have found MUC18 up-regulation in airway epithelial cells as well as in lung macrophages from human asthmatics and COPD patients [13,14]. Importantly, we demonstrated a pro-inflammatory function of MUC18 in lung macrophages in the context of bacterial (e.g., mycoplasma) infection [14].
In the current study, we have extended our previous research on MUC18 in the following areas. First, we examined the function of MUC18 in a different cell type, human primary airway epithelial cells, which is at the interface of lung and environment such as pathogens and pollutants, and is critical to the lung defense function. Second, we shifted our previous attention from bacterial infection or its mimic TLR2 agonist to respiratory rhinovirus infection and its mimic polyI:C. Third, we attempted to reveal the role of MUC18 in regulating both pro-inflammatory and anti-viral responses. By using the MUC18 over-expression approach, we have extended our previous study in lung macrophages by showing that MUC18 also promotes pro-inflammatory cytokine IL-8 production in airway epithelial cells following polyI:C or HRV treatment. This finding is important, as viral infection is a significant factor contributing to exacerbations of lung diseases, including asthma, COPD and cystic fibrosis. One of the common features in acute exacerbations of lung diseases is excessive lung inflammation such as neutrophils and IL-8, one of the most potent chemoattractants and activators for neutrophils. The fact that MUC18 enhances IL-8 production in airway epithelial cells suggests that up-regulated MUC18 expression in diseased lungs may predispose the host to exaggerated inflammatory response during viral infection. Thus, blocking the effect of MUC18 by using a neutralizing antibody may offer a novel approach to preventing or attenuating virus-mediated lung disease exacerbations.
One of the novel and exciting findings in this study is that MUC18 exerts an inhibitory role in anti-viral IFN-β gene expression. This new piece of data further indicates that MUC18 has multiple roles in lung defense. At one hand, MUC18 promotes the inflammation. On the other hand, it suppresses the anti-viral response, which may delay the viral clearance and prolong the disease process. Accompanied by the reduction of IFN-β expression is the increase (about 2 fold, data not shown) of HRV load, but such increase did not reach the level of statistical significance. The opposing effects of MUC18 on IL-8 and IFN-β expression suggest the complexity of MUC18-mediated transcriptional or translational cascade. Previous studies have demonstrated the inconsistent trend of change of pro-inflammatory response and the anti-viral interferon response following HRV or respiratory syncytial virus (RSV) infection [25,26]. Such inconsistent trend of IL-8 and IFN-β expression may be in part explained by the well-documented observation that pro-inflammatory and anti-viral responses are regulated by two distinct sets of transcription factors, NF-κB and interferon regulatory factors [IRFs], respectively [3,27,28].
How MUC18 signals during the inflammatory or infectious process has not been revealed. In the current study, we decided to examine the upstream events that MUC18 may function in the context of viral mimic polyI:C. We showed that in both lung epithelial cell line and primary airway epithelial cells, levels of phosphorylated serines were increased following polyI:C treatment, particularly in the presence of MUC18 overexpression. Our data suggest that MUC18 phosphorylation may contribute to its functions. Future studies are warranted to determine how phosphorylation of MUC18 serines regulates the downstream signaling cascades associated with MUC18 function.
One novel aspect of our study is related to the contribution of ERK activation to MUC18 serine phosphorylation. PolyI:C-mediated ERK1/2 activation has been shown by us and other investigators [29,30]. We found that ERK1/2 inhibition reduced the level of MUC18 serine phosphorylation, indicating a partial dependence of MUC18 activation on ERK activation. Whether other members (e.g., p38) of the MAPK family or other kinases phosphorylate MUC18 during viral infection or its mimic treatment remains to be explored.
There are several limitations to our current study. First, we focused on polyI:C as a surrogate of broad spectrum RNA viral infection, and only chose HRV for the viral infection model. We will consider using other strains of viruses in our future studies to further define the role of MUC18 in various virus-induced pro-inflammatory and anti-viral responses. Second, how MUC18 differentially regulates IL-8 and IFN-β expression is unclear. We plan to determine if phosphorylated MUC18 interacts with other proteins especially with adaptor proteins (e.g., TRIF) involved in pro-inflammatory cytokine and anti-viral gene expression. Third, although we demonstrated MUC18's function in airway epithelial cells infected with live HRV (i.e., HRV-1A), we have not examined MUC18 serine phosphorylation following live HRV-1A infection in the presence or absence of an ERK inhibitor. Such a mechanistic study will be performed in our future experiments to further strengthen our observation that MUC18 regulates pro-inflammatory and anti-viral responses. Lastly, a transgenic mouse model over-expressing human MUC18 in airway epithelium would be needed to dissect the in vivo function of MUC18 in airway epithelial responses to HRV or polyI:C.
In summary, our human primary airway epithelial cell culture studies have suggested a unique pro-inflammatory and anti-viral function for MUC18, a molecule that deserves future studies to broaden our knowledge in its implication in various infectious diseases or pathogen-related disease exacerbations. Therapies aimed at blocking MUC18 up-regulation or MUC18 signaling could potentially prevent or diminish excessive inflammation while enhancing host defense functions during viral infection and associated disease exacerbations.
Acknowledgments
Funding sources NIH R01AI106287 and R01HL088264.
Footnotes
Conflict of Interest The authors declare no conflict of interest.
References
- 1.Johnston SL, Pattemore PK, Sanderson G, Smith S, Campbell MJ, et al. The relationship between upper respiratory infections and hospital admissions for asthma: a time-trend analysis. Am J Respir Crit Care Med. 1996;154:654–660. doi: 10.1164/ajrccm.154.3.8810601. [DOI] [PubMed] [Google Scholar]
- 2.Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ. 1993;307:982–986. doi: 10.1136/bmj.307.6910.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gavala ML, Bertics PJ, Gern JE. Rhinoviruses, allergic inflammation, and asthma. Immunol Rev. 2011;242:69–90. doi: 10.1111/j.1600-065X.2011.01031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khetsuriani N, Kazerouni NN, Erdman DD, Lu X, Redd SC, et al. Prevalence of viral respiratory tract infections in children with asthma. J Allergy Clin Immunol. 2007;119:314–321. doi: 10.1016/j.jaci.2006.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly JT, Busse WW. Host immune responses to rhinovirus: mechanisms in asthma. J Allergy Clin Immunol. 2008;122:671–682. doi: 10.1016/j.jaci.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dulek DE, Peebles RS., Jr Viruses and asthma. Biochim Biophys Acta. 2011;1810:1080–1090. doi: 10.1016/j.bbagen.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jean D, Gershenwald JE, Huang S, Luca M, Hudson MJ, et al. Loss of AP-2 results in up-regulation of MCAM/MUC18 and an increase in tumor growth and metastasis of human melanoma cells. J Biol Chem. 1998;273:16501–16508. doi: 10.1074/jbc.273.26.16501. [DOI] [PubMed] [Google Scholar]
- 8.Lehmann JM, Riethmüller G, Johnson JP. MUC18, a marker of tumor progression in human melanoma, shows sequence similarity to the neural cell adhesion molecules of the immunoglobulin superfamily. Proc Natl Acad Sci U S A. 1989;86:9891–9895. doi: 10.1073/pnas.86.24.9891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shih IM, Nesbit M, Herlyn M, Kurman RJ. A new Mel-CAM (CD146)-specific monoclonal antibody, MN-4, on paraffin-embedded tissue. Mod Pathol. 1998;11:1098–1106. [PubMed] [Google Scholar]
- 10.Xie S, Luca M, Huang S, Gutman M, Reich R, et al. Expression of MCAM/MUC18 by human melanoma cells leads to increased tumor growth and metastasis. Cancer Res. 1997;57:2295–2303. [PubMed] [Google Scholar]
- 11.Sers C, Kirsch K, Rothbächer U, Riethmüller G, Johnson JP. Genomic organization of the melanoma-associated glycoprotein MUC18: implications for the evolution of the immunoglobulin domains. Proc Natl Acad Sci U S A. 1993;90:8514–8518. doi: 10.1073/pnas.90.18.8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson JP, Bar-Eli M, Jansen B, Markhof E. Melanoma progression-associated glycoprotein MUC18/MCAM mediates homotypic cell adhesion through interaction with a heterophilic ligand. Int J Cancer. 1997;73:769–774. doi: 10.1002/(sici)1097-0215(19971127)73:5<769::aid-ijc26>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 13.Simon GC, Martin RJ, Smith S, Thaikoottathil J, Bowler RP, et al. Up-regulation of MUC18 in airway epithelial cells by IL-13: implications in bacterial adherence. Am J Respir Cell Mol Biol. 2011;44:606–613. doi: 10.1165/rcmb.2010-0384OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Q, Case SR, Minor MN, Jiang D, Martin RJ, et al. A novel function of MUC18: amplification of lung inflammation during bacterial infection. Am J Pathol. 2013;182:819–827. doi: 10.1016/j.ajpath.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Q, Jiang D, Smith S, Thaikoottathil J, Martin RJ, et al. IL-13 dampens human airway epithelial innate immunity through induction of IL-1 receptor-associated kinase M. J Allergy Clin Immunol. 2012;129:825–833. doi: 10.1016/j.jaci.2011.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar A, Zhang J, Yu FS. Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunology. 2006;117:11–21. doi: 10.1111/j.1365-2567.2005.02258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu Q, van Dyk LF, Jiang D, Dakhama A, Li L, et al. Interleukin-1 receptor-associated kinase M (IRAK-M) promotes human rhinovirus infection in lung epithelial cells via the autophagic pathway. Virology. 2013;446:199–206. doi: 10.1016/j.virol.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 19.Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams KA, Zhang M, Xiang S, Hu C, Wu JY, et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J Biol Chem. 2013;288:33156–33170. doi: 10.1074/jbc.M113.472506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zakrzewska M, Haugsten EM, Nadratowska-Wesolowska B, Oppelt A, Hausott B, et al. ERK-mediated phosphorylation of fibroblast growth factor receptor 1 on Ser777 inhibits signaling. Sci Signal. 2013;6:ra11. doi: 10.1126/scisignal.2003087. [DOI] [PubMed] [Google Scholar]
- 22.Becker TM, Durrani SR, Bochkov YA, Devries MK, Rajamanickam V, et al. Effect of exogenous interferons on rhinovirus replication and airway inflammatory responses. Ann Allergy Asthma Immunol. 2013;111:397–401. doi: 10.1016/j.anai.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rasmussen AL, Racaniello VR. Selection of rhinovirus 1A variants adapted for growth in mouse lung epithelial cells. Virology. 2011;420:82–88. doi: 10.1016/j.virol.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulz C, Petrig V, Wolf K, Krätzel K, Köhler M, et al. Upregulation of MCAM in primary bronchial epithelial cells from patients with COPD. Eur Respir J. 2003;22:450–456. doi: 10.1183/09031936.03.00102303. [DOI] [PubMed] [Google Scholar]
- 25.Bartlett NW, Slater L, Glanville N, Haas JJ, Caramori G, et al. Defining critical roles for NF-kappaB p65 and type I interferon in innate immunity to rhinovirus. EMBO Mol Med. 2012;4:1244–1260. doi: 10.1002/emmm.201201650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hansdottir S, Monick MM, Lovan N, Powers L, Gerke A, et al. Vitamin D decreases respiratory syncytial virus induction of NF-kappaB-linked chemokines and cytokines in airway epithelium while maintaining the antiviral state. J Immunol. 2010;184:965–974. doi: 10.4049/jimmunol.0902840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vercammen E, Staal J, Beyaert R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin Microbiol Rev. 2008;21:13–25. doi: 10.1128/CMR.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadik CD, Bachmann M, Pfeilschifter J, Mühl H. Activation of interferon regulatory factor-3 via toll-like receptor 3 and immunomodulatory functions detected in A549 lung epithelial cells exposed to misplaced U1-snRNA. Nucleic Acids Res. 2009;37:5041–5056. doi: 10.1093/nar/gkp525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klettner A, Koinzer S, Meyer T, Roider J. Toll-like receptor 3 activation in retinal pigment epithelium cells - Mitogen-activated protein kinase pathways of cell death and vascular endothelial growth factor secretion. Acta Ophthalmol. 2013;91:e211–218. doi: 10.1111/aos.12031. [DOI] [PubMed] [Google Scholar]
- 30.Biswas I, Garg I, Singh B, Khan GA. A key role of toll-like receptor 3 in tissue factor activation through extracellular signal regulated kinase 1/2 pathway in a murine hypoxia model. Blood Cells Mol Dis. 2012;49:92–101. doi: 10.1016/j.bcmd.2012.05.001. [DOI] [PubMed] [Google Scholar]