

Abstract
CGRP and specific CGRP receptors are found in the heart where they produce positive-inotropic and anti-apoptotic effects, key adaptations to exercise and cardiovascular disease. PI3K/Akt and MAPK signaling imbalances are associated with cardiomyocyte pathologies; however, the effects of CGRP on these pathways are unclear. Therefore, we hypothesized that CGRP modulates inotropic and apoptotic adaptations of cardiomyocytes by regulating PI3K/Akt and MAPK/ERK signaling balances. We treated cardiomyocytes with combinations of CGRP, PI3K/Akt and MAPK signaling agonists and antagonists. We evaluated expression of the mRNA and proteins levels of survival signaling molecules related to the PI3K/Akt and MAPK and measured apoptosis by caspase 3/7 activity. CGRP1-37 decreased Akt, NFκB, SOD-3 and increased ERK1/2 and p38 MAPK expressions, which was antagonized by CGRP8-37. Akt-negative construct transfection, Ad.Akt(K179M), inhibited the CGRP1-37-induced increment in MAPK expressions. A PI3K-antagonist treatment with LY294002 or CGRP1-37/Ad.Akt(K179M) co-treatment alleviated the CGRP-increased caspase activity and -decrements in SOD-3.
These findings demonstrate a CGRP negative effect on the PI3K/Akt signaling pathway and CGRP receptor-induced crosstalk between PI3K/Akt and MAPK in normal cardiomyocytes. Future studies to differentiate CGRP effects on intracellular signal transduction mechanisms in pathological conditions will elucidate the significance of CGRP in, and provide novel therapeutic targets for, heart failure.
Keywords: CGRP, PI3K/Akt signaling, MAPK/ERK signaling, Cardiac myocytes, Rat
INTRODUCTION
Activity of CGRP and specific CGRP receptors in the heart produce positive-inotropic [1,2] and anti-apoptotic [3,4] effects, which are key adaptations to exercise and cardiovascular disease. CGRP is a 37-aminoacid, regulatory peptide derived by alternative splicing of the calcitonin gene located on chromosome 11 and one of a family of multifunctional peptides that includes amylin and adrenomedullin (AM) [5]. Amylin is also a 37-aminoacid peptide, named for its deposition of amyloid and role in glycemic control, released from the pancreas with insulin. Amylin inhibits gastric motility and appetite, thereby regulating blood glucose [6–8]. AM is a 52-aminoacid peptide, named for the pheochromocytoma cell in which it was originally discovered, is highly expressed in cardiac and vascular tissues and, like CGRP, is a potent vasodilator [9]. CGRP is also synthesized in and released from sensory neurons, a mediator of pain signaling and plays a central role in sensitizing the trigeminal ganglion to Ca2+ in migraine headache [10,11]. AM has both positive- and a negative-inotropic effects in cardiac myocytes [12], decreases papillary muscle contractile force (Bell et al 2010) and increases cell resistance to oxidative stress and production of NO [13]; whereas, CGRP increases cardiomyocyte contractile force [1] and is released by K+ induction of Ca2+ currents [14] as well as by NO [15] and the pro-inflammatory cytokine TNF-α [16]. These calcitonin regulatory peptides appear to regulate Ca2+ fluxes, activate adenylate cyclase and, therefore, increase cellular cAMP activity [17,18] but by actions on different receptor motifs.
The two forms of CGRP are α-CGRP and β-CGRP are different by three aminoacids; however, β-CGRP is expressed from a separate gene that does not produce calcitonin [5,19–21]. Activity of CGRP depends on the calcitonin receptor-like receptor (CL), associated with G proteins, and three distinct receptor activity modifying proteins (RAMP1, RAMP2 and RAMP3). These RAMPs are determinants of membrane localization and binding specificity of CL receptors. A CL-RAMP1 complex constitutes the CGRP-1 receptor, activated by α-CGRP and CL-RAMP2 and CL-RAMP3 complexes are receptors for AM [22]. Although the nonfunctional CGRP8-37 molecule antagonizes the CGRP-1 receptor, CGRP also binds to the CGRP-2 receptor that is not affected by CGRP8-37 [23].
Abnormal plasma levels of AM and CGRP are reported in pre-eclampsia and other cardiovascular diseases associated with endothelial dysfunction [24]. Moreover, both AM and CGRP appear to mediate positive-inotropy in cardiac myocytes [1,12]. These findings suggest that CGRP receptors could provide novel, specific targets for preventing and treating cardiovascular disease. Moreover, AM is reported to exert its effects by MAPK/ERK [25] and CGRP by PI3K/Akt intracellular signaling pathways, shared by other regulators of positive-inotropy [1,2]. There is also substantial crosstalk between these pathways in experimental models [4]. The early signs of cardiovascular disease include hypertension with increased contractile force and Ca2+ fluxes, leading to cardiac remodeling, fueled by oxidative stress with apoptosis [26]. However, the linkages between CGRP receptors and intracellular signal transduction pathways for positive-inotropy and anti-apoptosis remain unclear [27]. The present study was, therefore, designed to determine the relationships between specific CGRP-1 receptors and PI3K/Akt and MAPK/ERK pathways for signaling positive-inotropic and anti-apoptotic effects in cardiomyocytes.
MATERIALS AND METHODS
Conformity statement
All the procedures used in this study conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH) publication No. 85-23, revised 1996. The animal protocol has been independently approved by Howard University institutional animal care and usage committee.
Animal preparation
Male Sprague-Dawley rats, 200–250 g body weight, were purchased from Harlan Laboratories (Madison, WI). The animals were allowed to recover and become familiar with their new environment upon arrival to the animal house of the Howard University College of Medicine, for 1 week. The animals were housed in secure, clean and environmentally-controlled room temperature (70°F–74°F) with a 6:00 h to 18:00 h light cycle and were fed food and water ad libitum.
Tissue samples and treatment conditions
Cardiac tissue was obtained from adult male Sprague Dawley rats. Hearts were removed from the Sprague Dawley anesthetized rats (halothane) and perfused with either 10 μM CGRP1-37, 10 μM CGRP8-37, 1 μM LY249002; or adenovirus strain with a modified construct: Ad:Akt(K179M) or Ad:myrAkt alone or in combination in a perfusion buffer (11.9 mM NaCl, 46.9 mM KCl, 9.4 mM MgSO4, 12.2 mM KH2PO4, 1 mM Ascorbic acid, 250 mM NaHCO3, 115.4 mM Glucose, and 1 mM CaCl2) for 45 min. The Ad:Akt(K179M) and Ad:MyrAkt both are adenoviral construct that expresses a kinase-inactive, dominant negative Akt mutant. The CGRP and LY249002 concentrations used were similar to previously reported effects of these molecules in the rodent cardiovascular system [1,28–31].
qRT -PCR
Total mRNA (from perfused heart tissue) was isolated using the Aurum Total RNA Fatty and Fibrous tissue Kit (Biorad; Hercules, CA) according to the manufacturer’s manual. 1 μg of total mRNA was then used for reverse transcription and amplification using the SuperScript-III One-step RT-PCR kit (Life Technologies; Grand Island, NY) following the manufacturer’s protocol. PCR was performed using Akt, SOD-3, NFκB, ERK1, and p38 MAPK primers. Rat beta-actin forward 5′-TCGTGCGTGACATTAAGGAG-3′ and reverse 5′-GTCAGGCAGCTCGTAGCTCT-3′; endogenous rat sod3 forward 5′-GACCTGGAG ATCTGG ATGGA-3′ and reverse 5′-GTGGTTGGAGGTGTTCTGCT-3′; AKT-1 forward 5′-CTGGGTTACCCCGGTGTGT-3′ and reverse 5′-GCACATCCGAGAAACAAAA-3′; ERK1 forward 5′-GAGCCCAGGGGAACTGCT-3′ and reverse 5′-CTGGAAGCGGGCTGTCTC-3′; P38/MAPK14 forward 5′-AGGAGAGGCCCACGTTCTAC-3′ and reverse 5′-TCAGGCTCTTCCATTCGTCT-3′. β-actin was employed as an internal control. The Biorad iQ5 cycler was used for the qRT-PCR.
Caspase 3/7 activity assay
Caspase 3/7 activity has been measured according to the manufacturer instructions, Promega (WI). Accordingly, protein extracts from treated homogenized cardiac tissue were incubated for 30 minutes in 96-well plates. Caspase-3/7 activity reagent (Promega, WI) was added to samples in 1:1 dilutions. This reagent causes lysis of the cell and cleavage of the DEVD-aminoluciferin substrate, which is freed and degraded by luciferase enzyme. Thus, a luminescent signal is emitted corresponding to caspase-3/7 activity. The samples were analyzed using Victor V3 multiplate plate reader (Perkin Elmer) at the excitation wavelength of 485 nm.
Western blotting assay
Total protein was isolated from rat hearts and exposed to RIPA lysis buffer which was composed of: EGTA(1 mM),) EDTA (2mM), DTT (2 mM), benzamidine (10 mM), b-glycerophosphate (20 mM), Na3VO4 (0.2 mM), NaF (20 mM), NaVO3 (0.5 mM), 0.6% deoxycholate, 0.1% Triton X-100, and 1 tablet/10 mL of complete protease inhibitors. The lysates were incubated on ice for 15 min and centrifuged for 20 min at a speed of 14,000 rpm. Protein concentrations were recorded from the samples, separated by SDS-PAGE and transferred onto nitrocellulose membranes where NFκB, ERK1/2, phospho-ERK1/2, p38 MAPK, GAPDH (as control) antibody probes were used to display protein expression. The above mentioned probes along with the secondary anti-rabbit monoclonal antibody were employed in this protocol (Cell signaling). Bands were visualized by chemiluminescence. Membranes from three separate experiments were scanned and the densities of the bands were evaluated using the NIH “Image J” software package.
Statistical methods
Statistical analyses were performed using Prism 6.0 (Graphpad) software and verified using Microsoft Excel, which gave the same results. Paired Student’s t-tests were used to compare the pre- and post-treatment data for the same animal group. The heteroscedastic two-sample unpaired Student’s t-test, assuming unequal variances, was used to compare treatment effects between two different animal groups. Using the null hypothesis, P ≤ 0.05 was significant.
RESULTS
Effects of CGRP on the survival and the proliferative pathways gene expression
We initially evaluated the direct effects of CGRP on Akt gene expression in hearts perfused with CGRP1-37 alone and in combination with the PI3K/Akt activator (IGF-1) or the PI3K inhibitor LY294002. We also incorporated an adenoviral construct containing coding for kinase-inactive dominant negative Akt mutant in cardiac tissue using Ad.MyrAkt or Ad.Akt(K179M). As shown in (Figure 1A), the CGRP1-37 treatment decreased Akt mRNA expression (−1.48 ± 0.36 fold, P<0.05). Inhibition of PI3K or transfection with Ad.MyrAkt also decreased Akt mRNA expression in the presence of CGRP1-37 (−1.78 ± 0.67 fold and −1.20 ± 0.84 fold, respectively, P<0.05). The IGF-1 treatment increased Akt mRNA expression (1.41 ± 1.08 fold, P=0.03), even in the presence of CGRP1-37, thereby counteracting the effects of CGRP1-37. As expected from an acute effect, changes in gene expression are small but significant. We also evaluated the effects CGRP1-37 on NFκB mRNA expression, downstream of Akt. (Figure 1B) shows that the CGRP1-37 treatment decreased NFκB mRNA expression (−3.36 ± 0.81 fold, P<0.05). This CGRP1-37-induced decrement in NFκB mRNA expression was blocked by LY294002 and by Ad.Akt(K179M) treatments (1.70 ± 0.66 fold, P<0.05 as compared to CGRP1-37 alone).
Figure 1. Effects of CGRP on the survival and the proliferative pathways gene expression.

A. The CGRP1-37 + IGF-1 treatment increased Akt mRNA expression. All other treatments decreased Akt mRNA expression. B. The CGRP1-37 + LY294002 and the CGRP1-37 + Ad.Akt(K179M) treatments both increased SOD-3 mRNA expression, C. All the treatments decreased p38 MAPK mRNA expression. D. The CGRP1-37 treatment decreased NFκB mRNA expression CGRP1-37. NFκB expression was increased by the CGRP1-37 + LY294002, the CGRP1-37 + IGF-1 and the CGRP1-37 + Ad.Akt(K179M) treatments. E. The CGRP1-37 + Ad.Akt(K179M) treatment decrease ERK1 mRNA expression. (N=6, * P<0.05).
The effects of CGRP1-37 on SOD-3 mRNA expression are shown in (Figure 1C). The CGRP1-37 treatment decreased SOD-3 mRNA expression (−2.03 ± 0.68 fold, P<0.05). To evaluate the associations of CGRP1-37, Akt and SOD activities, we treated hearts with CGRP1-37 in combination with either Ad.Akt(K179M), or LY294002. These co-treatments decreased the CGRP1-37-induced decrement in SOD-3 mRNA expression (−0.675± 0.99 fold, P=0.04 and −1.16 ± 0.78 fold, P=0.02 compared to CGRP1-37 alone, respectively). The IGF-1 treatment also decreased the CGRP1-37- induced decrement in SOD-3 mRNA expression.
Parallel MAPK signaling molecules such as ERK1 and p38 MAPK are shown to respond to stress stimuli associated with apoptosis, growth factors, interleukins, and interferons. Therefore, we evaluated the effects of CGRP on mRNA expression of these MAPKs. As shown in (Figure 1D), the CGRP1-37 treatment increased ERK1 mRNA expression (1.54 ± 0.80 fold, P<0.05). This CGRP1-37-induced increment in ERK1 mRNA expression was effectively antagonized by co-treatment using the dominant-negative Ad.Akt(K179M) (−2.88 ± 1.00 fold, P=0.007). The CGRP1-37 and IGF-1 co-treatment failed to further modulate the CGRP1-37-induced increment in ERK1 mRNA expression. The CGRP1-37 treatment decreased p38 MAPK mRNA expression as depicted in (Figure 1E) (−2.14 ± 0.32 fold, P<0.05). The CGRP1-37 co-treatments using LY204002 and Ad.Akt(K179M) failed to further modulate the CGRP1-37-induced decrement in p38 MAPK mRNA expression. The IGF-1 treatment decreased the CGRP1-37-induced decrement in p38 MAPK mRNA expression (−0.90 ± 0.38 fold, P=0.02).
Effects of CGRP on survival and proliferative pathways activities
Recently, we have shown that CGRP1-37 treatment similar to that used in this study decreased the expression of Akt protein [1]. This CGRP1-37 induced decrement in Akt expression was effectively antagonized by the CGRP8-37 treatment. IGF-1 cotreatment also decreased the CGRP1-37-induced decrement in Akt protein expression. In this study, the CGRP1-37 treatment increased ERK1 protein activity (56.02 ± 14.15%, P=0.03 compared to control). This CGRP1-37-induced increment in ERK1 activity was antagonized by the CGRP8-37 or by the Ad.Akt(K179M) co-treatments (Figure 2). IGF-1 co-treatment did not further affect the CGRP1-37-induced increment in ERK1 activity (77.88 ± 6.09%, P=0.02 compared to control). There was no significant effect of CGRP1-37 on ERK2 protein activity.
Figure 2. Effects of CGRP on the proliferative pathways activities, ERK1 and ERK2.

Upper: sample western blots for ERK1 (left) and ERK2 (right) total and phosphorylated form with the corresponding loading control GAPDH. Lower: Bar graphs depicted the effects of CGRP1-37 in the presence or absence of antagonists (CGRP8-37) or Akt activator- (IGF-1) or down-regulator (Ad.Akt(K179M)). (N= 6, * p<0.05).
Figure 3A shows that the CGRP1-37 treatment increased p38 MAPK protein expression (75.72 ± 1.62%). The CGRP1-37-induced increment in p38 MAPK expression was effectively antagonized by the CGRP8-37 or Ad.Akt(K179M) co-treatments, but not by the LY294002 co-treatment. The co-treatment with IGF-1 reduced but did not alleviate the CGRP1-37 effect on p38 MAPK protein expression (46.48 ± 3.86%; P=0.05 compared to control; which is −56.42 ± 1.16%, P=0.05 compared to CGRP1-37 alone). (Figure 3B) shows that the CGRP1-37 treatment did not change NFκB protein expression significantly (−8.02 ± 0.34%, P>0.1). The LY294002 or the Ad.Akt(K179M) co-treatments decreased NFκB protein expression (−14.77 ± 0.36% and −19.67 ± 0.46%, P=0.05). The IGF-1 co-treatment increased NFκB protein expression marginally (9.95 ± 0.60%, P= 0.10).
Figure 3. Effects of CGRP on the survival pathways activity, p38 MAPK and NFkB.

A. The CGRP1-37, the CGRP1-37 + LY294002 and the CGRP1-37 + IGF-1 treatments increased p38 MAPK protein expression. B. CGRP1-37 decreased NFκB protein expression and the CGRP1-37 + IGF-1 treatment increased NFκB protein expression (N=6, * P<0.05).
Effects of CGRP1-37 on cellular apoptosis
Figure 4 demonstrates that the CGRP1-37 treatment and the CGRP1-37/IGF-1 co-treatment had no direct effects on caspase 3/7 activity. In order to verify the functionality or the responsiveness of the caspase 3/7 in our preparation, we inhibited the PI3K/Akt pathway with LY294002 treatment which increased caspase 3/7 activity (27.0 ± 11.2%, P=0.02) and with CGRP1-37/Ad.Akt(K179M) co-treatment which also increased caspase 3/7 activity (19.2 ± 1.7%, P=0.02).
Figure 4. Effects of CGRP1-37 on cellular apoptosis.
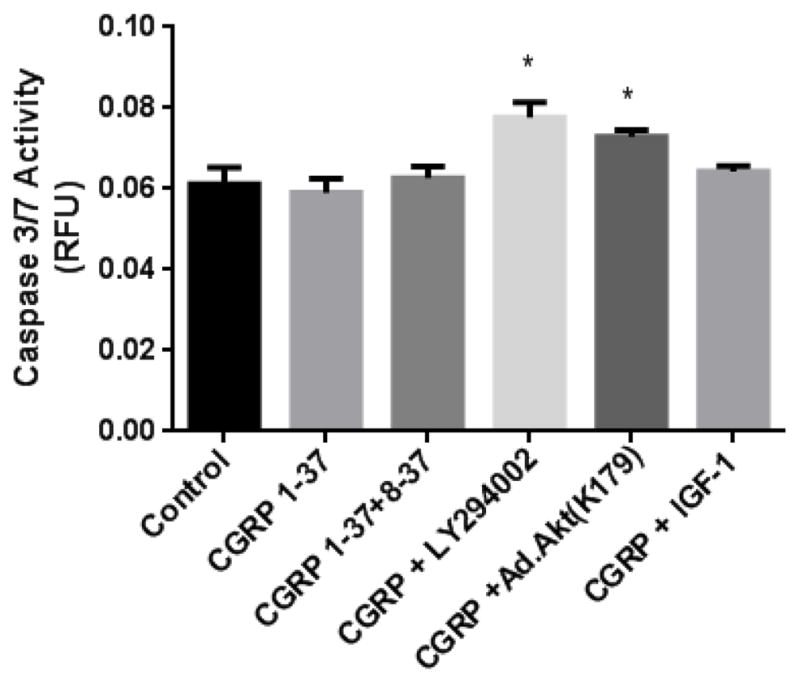
Caspase 3/7 activity from protein extracts that were treated alone with CGRP1-37 or in combination with CGRP8-37, LY294002, Ad.Akt (K179M) and IGF-1. Caspase activity is expressed as relative fluorescence units (RFU). The CGRP1-37 treatment did not directly affect caspase 3/7 activity. The CGRP1-37 + LY294002 and CGRP1-37 + Ad.Akt(K179M) treatments increased caspase 3/7 activity (P<0.05). The IGF-1 + CGRP1-37 treatment had no effect on caspase 3/7 activity (N=3; * P<0.05).
DISCUSSION
The main finding of this study is that physiologically-active CGRP1-37 treatments shifted the intracellular signaling balance in normal cardiomyocytes. These effects of CGRP1-37 were, largely, antagonized by pretreatments with the physiologically-inactive specific CGRP-1 receptor blocker CGRP8-37 which decreased specific activities of PI3K/Akt cell survival signal transduction molecules and increased specific activities of MAPK/ERK, oxidative stress and apoptosis transduction molecules. The effects of CGRP1-37 not antagonized by CGRP8-37 suggest that some of the effects of CGRP were mediated by the CGRP-2 receptor.
In this study we have shown that the CGRP1-37 has a detrimental effect on the survival signaling pathway related to PI3K/Akt in the heart. To that effect we have demonstrated that CGRP1-37 induces a reduction in Akt gene expression that corroborates with a lower Akt protein activation level. This effect seems to be a direct effect of CGRP1-37 as transfection with the dominant negative Akt or inhibition of its direct upstream effector, PI3K, induced the same level of decrement in Akt gene expression similar to what we have recently found with its protein activity level [1]. Interestingly, IGF-1 offsets the CGRP1-37 effect, which may indicates that Akt is sufficient and necessary signaling switch for the CGRP effects. It has been recently shown that nerve growth factor (NGF) improves neurite outgrowth [32,33] mainly through PI3K/Akt activation of cGMP in CGRP-containing DRG neurons [34,35]. Furthermore, NGF is reported to induces expression of CGRP in DRG [36]. Thus, in accordance with our present data, it seems likely that the CGRP is part of a regulatory mechanism that monitors the NGF activation of the PI3K/Akt signaling pathway. No comparable studies have yet been performed on cardiac myocytes which makes the present report novel and significant. Akt signaling is central to many cellular survival mechanisms and decreased Akt expression or activation could, therefore, be a key factor in a number of pathophysiological events and sequelae [37]. Accordingly, the present study demonstrates that the CGRP1-37 treatment also decreased mRNA expressions of the anti-oxidant enzyme SOD-3, as well as the anti-apoptotic Akt-downstream effector, NFκB. We realize that changes in mRNA levels are limited, but this is expected from a short-term acute effect. These anti-survival effects were produced by CGRP-induced down-regulation of Akt because they were prevented by either PI3K inhibition or dominant-negative Akt. The PI3K/Akt signaling agonist IGF-1 also counteracted the CGRP-induced decrements in NFκB and SOD3 mRNA expression, thereby corroborating the central role of Akt. This finding is also consistent with previously reported CGRP effects on cell survival and cardiac inotropic function [1]. Nonetheless, this finding contradicts a previous report that CGRP alleviated SOD activity in a model of hyperoxia-induced lung injury [38]. However, our findings agree with those of others demonstrating that exogenous CGRP decreased NFκB and induced apoptosis in thymocytes [39]. These disparate findings suggest tissue-specificity in the downstream apoptotic and/or oxidative effects of CGRP. Thus, the finding that the CGRP1-37 treatment decreased both Akt and NFκB mRNA expression, the latter downstream of Akt mRNA expression, suggests that such NFκB mRNA expression is indicative of the capacity for CGRP to employ the entire PI3K/Akt cell survival signaling pathway that includes an anti-apoptotic effect. This interpretation is bolstered by the finding that LY294002 and Ad.Akt(K179M) decreased basal NFKB mRNA expression.
In cardiac myocytes, we previously demonstrated cross reactivity, also called crosstalk, between the PI3K/Akt and MAPK signaling pathways [4]. The MAPK pathway, particularly ERK1 and p38 MAPK, has been implicated in signaling cellular proliferation such as that which occurs in the development of cardiac hypertrophy, [28,29,40,41]. Thus, it was important to probe such interactions in the context of the CGRP1-37 deactivation of the Akt activity. We found that the CGRP1-37 treatment increased ERK1 mRNA and protein expression in an Akt-dependent manner. This was evidenced by the finding that CGRP1-37-induced increase in ERK1 mRNA expression was inhibited by dominant-negative Akt co-transfection. These findings may suggest that CGRP modulates ERK1 partly via Akt signaling. Similar findings are reported in hepatocytes and PC12 cells, suggesting PI3K positive crosstalk with ERK1/2 [42,43]. On the other hand, the CGRP1-37 treatment noticeably reduced p38 MAPK mRNA expression, independently of PI3K/Akt, but enhanced p38 MAPK protein expression in an Akt-dependent manner. This peculiar interaction suggests an auto-regulatory translational mechanism involving Akt, whereby a CGRP induced reduction in Akt activation may have relieved an Akt-driven inhibition of p38 MAPK protein synthesis, perhaps by an epigenetic mechanism. This was evidenced by the findings that PI3K inhibition (which decreases Akt activation) mimicked the CGRP1-37 treatment effect on p38 MAPK and that the CGRP1-37 and IGF-1 co-treatment significantly dampened the CGRP1-37-enhanced p38 MAPK protein expression. The fact that the Ad.Akt(K179M) co-transfection blocked this CGRP1-37-induced p38 MAPK effect suggests that Akt activation rather than the Akt protein expression level is relevant here. An epigenetic hypothesis for exogenous CGRP signaling is also consistent with the finding that although the CGRP1-37 treatment decreased NFκB mRNA expression, it had no effect on NFκB protein expression. Therefore, it is suggestive that IGF-1 has the capacity to counteract the CGRP-induced decrement in p38 MAPK mRNA expression.
All these CGRP1-37 induced effects on PI3K/Akt, MAPK and NFκB were blocked by the calcitonin receptor-like receptor (CALCRL) antagonist CGRP8-37 thereby indicating that CGRP1-37 was acting via its membrane receptor on the cardiomyocytes. These results imply that CGRP1-37 weakens the anti-apoptotic and strengthens the proliferative signaling pathways, notably in an Akt-dependent manner.
The cellular biomarkers for apoptosis, caspase 3/7 activity were apparently not modulated directly by exogenous CGRP. However, inhibition of Akt by either the LY294002 or the Ad:Akt(K179M) treatment increased the caspase 3/7 activity, irrespective of CGRP1-37, indicating responsiveness of the caspases to changes in Akt expression. Thus, it seems that enhancement of signaling in a pathway parallel to PI3K/Akt, the MAPK/ERK pathway, may have counter-balanced the decrement in anti-apoptotic signaling via the PI3K/Akt pathway. A compensatory activation of ERK1 induced by down-regulation of PI3K/Akt signaling is reported in transgenic mice [44]. To the extent that, as we describe herein, MAPK/ERK signaling enhancement can be Akt-dependent, Akt appears to be playing an auto-regulatory role in maintaining cell survival in the presence of CGRP1-37.
In summary, this is the first study to demonstrate the effects of CGRP on the PI3K/Akt and the MAPK pathways for cell survival, apoptosis and stress. As depicted in the diagram in (Figure 5), on one hand CGRP induces down-regulation of the PI3K/Akt/SOD pathway which may lead to elevated oxidative stress. On the other hand, this CGRP effect does not affect NFκB nor caspase 3/7 activity, which could be due to the observed enhancement of the anti-apoptotic MAPK (ERK1/2 and p38) pathways [45]. Furthermore, in our setting ERK1/2 activation seems to be Akt dependent, whereas p38 is mostly Akt-independent. P38-MAPK is known to respond to environmental stress such as the oxidative ones induced by CGRP [45]. Thus, the activation levels of the both MAPKs versus the level of oxidative stress may dictate the overall cellular response to CGRP. These effects of CGRP treatments demonstrate that the PI3K/Akt cell survival and MAPK cell anti-apoptotic (ERK) and stress (p38) signaling pathways are not exclusive, exhibiting substantial interdependence, connectivity and crosstalk. These findings together with those of previous studies from our laboratory, showing CGRP1-37-induced positive-inotropic effects correlated with changes in Ca2+ fluxes in cardiomyocyte, sarcomere and whole heart preparations; suggest that CGRP receptors could be useful targets for preventing and treating cardiovascular disease. Future studies to differentiate the effects of CGRP on the intracellular signal transduction mechanisms in pathological conditions, such as cardiac hypertrophy will, no doubt, help elucidate the significance of CGRP dysregulation in, and provide novel therapeutic targets for, heart failure.
Figure 5. Diagram showing the inhibitory effect of CGRP on the PI3K/Akt pathway protein expression via the CGRP-1 receptor.

P38-MAPK and ERK1/2-MAPK are activated by CGRP1-37 in an Akt-independent and dependent manner, respectively. The activation levels of the latter versus the level of oxidative stress may dictate the overall cellular response to CGRP1-37. Filled arrows designate an inhibitory action, while unfilled arrows denote an activation process.
Acknowledgments
This work was supported in part by grants 1 R15 AA019816-01A1, GM08016-38 NIGMS/NIH, and 2G12 RR003048 RCMI, Division of Research Infrastructure to GEH. The authors would like to thank Dr. Joanne Allard for making the western blot Kodak Imager available.
References
- 1.Al-Rubaiee M, Gangula PR, Millis RM, Walker RK, Umoh NA, Cousins VM, et al. Inotropic and lusitropic effects of calcitonin gene-related peptide in the heart. Am J Physiol Heart Circ Physiol. 2013;304:H1525–1537. doi: 10.1152/ajpheart.00874.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katori T, Hoover DB, Ardell JL, Helm RH, Belardi DF, Tocchetti CG, et al. Calcitonin gene-related peptide in vivo positive inotropy is attributable to regional sympatho-stimulation and is blunted in congestive heart failure. Circ Res. 2005;96:234–43. doi: 10.1161/01.RES.0000152969.42117.ca. [DOI] [PubMed] [Google Scholar]
- 3.Huang MH, Nguyen V, Wu Y, Rastogi S, Lui CY, Birnbaum Y, et al. Reducing ischaemia/reperfusion injury through delta-opioid-regulated intrinsic cardiac adrenergic cells: adrenopeptidergic cosignalling. Cardiovasc Res. 2009;84:452–60. doi: 10.1093/cvr/cvp233. [DOI] [PubMed] [Google Scholar]
- 4.Zhao A, Alvin Z, Laurence G, Li C, Haddad GE. Cross-talk between MAPKs and PI-3K pathways alters the functional density of I(K) channels in hypertrophied hearts. Ethn Dis. 2010;20:S1-219-24. [PMC free article] [PubMed] [Google Scholar]
- 5.Höppener JW, Steenbergh PH, Zandberg J, Adema GJ, Geurts van Kessel AH, Lips CJ, et al. A third human CALC (pseudo)gene on chromosome 11. FEBS Lett. 1988;233:57–63. doi: 10.1016/0014-5793(88)81355-3. [DOI] [PubMed] [Google Scholar]
- 6.Mayer AP, Durward A, Turner C, Skellett S, Dalton N, Tibby SM, et al. Amylin is associated with delayed gastric emptying in critically ill children. Intensive Care Med. 2002;28:336–340. doi: 10.1007/s00134-002-1224-7. [DOI] [PubMed] [Google Scholar]
- 7.Mulder H, Ekelund M, Ekblad E, Sundler F. Islet amyloid polypeptide in the gut and pancreas: localization, ontogeny and gut motility effects. Peptides. 1997;18:771–783. doi: 10.1016/s0196-9781(97)00008-9. [DOI] [PubMed] [Google Scholar]
- 8.cherbaum WA. The role of amylin in the physiology of glycemic control. Exp Clin Endocrinol Diabetes. 1998;106:97–102. doi: 10.1055/s-0029-1211958. [DOI] [PubMed] [Google Scholar]
- 9.Asghar MS, Hansen AE, Kapijimpanga T, van der Geest RJ, van der Koning P, Larsson HB, et al. Dilation by CGRP of middle meningeal artery and reversal by sumatriptan in normal volunteers. Neurology. 2010;75:1520–1526. doi: 10.1212/WNL.0b013e3181f9626a. [DOI] [PubMed] [Google Scholar]
- 10.Ceruti S, Villa G, Fumagalli M, Colombo L, Magni G, Zanardelli M, et al. Calcitonin gene-related peptide-mediated enhancement of purinergic neuron/glia communication by the algogenic factor bradykinin in mouse trigeminal ganglia from wild-type and R192Q Cav2.1 Knock-in mice: implications for basic mechanisms of migraine pain. J Neurosci. 2011;31:3638–3649. doi: 10.1523/JNEUROSCI.6440-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durham PL, Masterson CG. Two mechanisms involved in trigeminal CGRP release: implications for migraine treatment. Headache. 2013;53:67–80. doi: 10.1111/j.1526-4610.2012.02262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mittra S, Bourreau JP. Gs and Gi coupling of adrenomedullin in adult rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2006;290:H1842–1847. doi: 10.1152/ajpheart.00388.2005. [DOI] [PubMed] [Google Scholar]
- 13.Pires AL, Pinho M, Sena CM, Seica R, Leite-Moreira AF. Intermedin elicits a negative inotropic effect in rat papillary muscles mediated by endothelial-derived nitric oxide. Am J Physiol Heart Circ Physiol. 2012;302:1131–1137. doi: 10.1152/ajpheart.00877.2011. [DOI] [PubMed] [Google Scholar]
- 14.Amrutkar DV, Ploug KB, Olesen J, Jansen-Olesen I. Role for voltage gated calcium channels in calcitonin gene-related peptide release in the rat trigeminovascular system. Neuroscience. 2011;172:510–517. doi: 10.1016/j.neuroscience.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 15.Zhang YM, Peng J, Hu CP, Jiang QT, Jiang GL, Li YJ. Clonidine induces calcitonin gene-related peptide expression via nitric oxide pathway in endothelial cells. Peptides. 2009;30:1746–1752. doi: 10.1016/j.peptides.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Oprée A, Kress M. Involvement of the proinflammatory cytokines tumor necrosis factor-alpha, IL-1 beta, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene-related peptide release from rat skin. J Neurosci. 2000;20:6289–6293. doi: 10.1523/JNEUROSCI.20-16-06289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishikawa T, Okamura N, Saito A, Masaki T, Goto K. Positive inotropic effect of calcitonin gene-related peptide mediated by cyclic AMP in guinea pig heart. Circ Res. 1988;63:726–734. doi: 10.1161/01.res.63.4.726. [DOI] [PubMed] [Google Scholar]
- 18.Pan CS, Jin SJ, Cao CQ, Zhao J, Zhang J, Wang X, et al. The myocardial response to adrenomedullin involves increased cAMP generation as well as augmented Akt phosphorylation. Peptides. 2007;28:900–909. doi: 10.1016/j.peptides.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 19.Meens MJ, Mattheij NJ, van Loenen PB, Spijkers LJ, Lemkens P, Nelissen J, et al. G-protein βγ subunits in vasorelaxing and anti-endothelinergic effects of calcitonin gene-related peptide. Br J Pharmacol. 2012;166:297–308. doi: 10.1111/j.1476-5381.2011.01774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogoshi M, Inoue K, Naruse K, Takei Y. Evolutionary history of the calcitonin gene-related peptide family in vertebrates revealed by comparative genomic analyses. Peptides. 2006;27:3154–3164. doi: 10.1016/j.peptides.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 21.Watkins HA, Rathbone DL, Barwell J, Hay DL, Poyner DR. Structure-activity relationships for α-calcitonin gene-related peptide. Br J Pharmacol. 2013;170:1308–1322. doi: 10.1111/bph.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson SD, Aitken JF, Bailey RJ, Poyner DR, Hay DL. Novel peptide antagonists of adrenomedullin and calcitonin gene-related peptide receptors: identification, pharmacological characterization, and interactions with position 74 in receptor activity-modifying protein 1/3. J Pharmacol Exp Ther. 2009;331:513–521. doi: 10.1124/jpet.109.156448. [DOI] [PubMed] [Google Scholar]
- 23.Woolley MJ, Conner AC. Comparing the molecular pharmacology of CGRP and adrenomedullin. Curr Protein Pept Sci. 2013;14:358–374. doi: 10.2174/13892037113149990053. [DOI] [PubMed] [Google Scholar]
- 24.Fei X, Hongxiang Z, Qi C, Daozhen C. Maternal plasma levels of endothelial dysfunction mediators including AM, CGRP, sICAM-1 and tHcy in pre-eclampsia. Adv Clin Exp Med. 2012;21:573–579. [PubMed] [Google Scholar]
- 25.Kato K, Yin H, Agata J, Yoshida H, Chao L, Chao J. Adrenomedullin gene delivery attenuates myocardial infarction and apoptosis after ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2003;285:H1506–1514. doi: 10.1152/ajpheart.00270.2003. [DOI] [PubMed] [Google Scholar]
- 26.Roe ND, Ren J. Oxidative activation of Ca(2+)/calmodulin-activated kinase II mediates ER stress-induced cardiac dysfunction and apoptosis. Am J Physiol Heart Circ Physiol. 2013;304:H828–839. doi: 10.1152/ajpheart.00752.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma YX, Guo Z, Sun T. CGRP inhibits norepinephrine induced apoptosis with restoration of Bcl-2/Bax in cultured cardiomyocytes of rat. Neurosci Lett. 2013;549:130–134. doi: 10.1016/j.neulet.2013.05.028. [DOI] [PubMed] [Google Scholar]
- 28.Alvin Z, Laurence GG, Coleman BR, Zhao A, Hajj-Moussa M, Haddad GE. Regulation of L-type inward calcium channel activity by captopril and angiotensin II via the phosphatidyl inositol 3-kinase pathway in cardiomyocytes from volume-overload hypertrophied rat hearts. Can J Physiol Pharmacol. 2011;89:206–215. doi: 10.1139/Y11-011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvin ZV, Laurence GG, Coleman BR, Zhao A, Hajj-Moussa M, Haddad GE. Regulation of the instantaneous inward rectifier and the delayed outward rectifier potassium channels by Captopril and Angiotensin II via the Phosphoinositide-3 kinase pathway in volume-overload-induced hypertrophied cardiac myocytes. Med Sci Monit. 2011;17:165–172. doi: 10.12659/MSM.881843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gangula PR, Lanlua P, Bukoski RD, Wimalawansa SJ, Yallampalli C. Mesenteric arterial relaxation to calcitonin gene-related peptide is increased during pregnancy and by sex steroid hormones. Biol Reprod. 2004;71:1739–1745. doi: 10.1095/biolreprod.104.031369. [DOI] [PubMed] [Google Scholar]
- 31.Gangula PR, Lanlua P, Wimalawansa S, Supowit S, DiPette D, Yallampalli C. Regulation of calcitonin gene-related peptide expression in dorsal root ganglia of rats by female sex steroid hormones. Biol Reprod. 2000;62:1033–1039. doi: 10.1095/biolreprod62.4.1033. [DOI] [PubMed] [Google Scholar]
- 32.Hashikawa-Hobara N, Hashikawa N, Yutani C, Zamami Y, Jin X, Takatori S, et al. The Akt-nitric oxide-cGMP pathway contributes to nerve growth factor-mediated neurite outgrowth in apolipoprotein E knockout mice. J Pharmacol Exp Ther. 2011;338:694–700. doi: 10.1124/jpet.111.181487. [DOI] [PubMed] [Google Scholar]
- 33.Hashikawa-Hobara N, Hashikawa N, Zamami Y, Takatori S, Kawasaki H. The mechanism of calcitonin gene-related peptide-containing nerve innervation. J Pharmacol Sci. 2012;119:117–121. doi: 10.1254/jphs.12r02cp. [DOI] [PubMed] [Google Scholar]
- 34.Cunha TM, Roman-Campos D, Lotufo CM, Duarte HL, Souza GR, Verri WA, Jr, et al. Morphine peripheral analgesia depends on activation of the PI3Kgamma/AKT/nNOS/NO/KATP signaling pathway. Proc Natl Acad Sci U S A. 2010;107:4442–4447. doi: 10.1073/pnas.0914733107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han G, Ma H, Chintala R, Miyake K, Fulton DJ, Barman SA, et al. Nongenomic, endothelium-independent effects of estrogen on human coronary smooth muscle are mediated by type I (neuronal) NOS and PI3-kinase-Akt signaling. Am J Physiol Heart Circ Physiol. 2007;293:314–321. doi: 10.1152/ajpheart.01342.2006. [DOI] [PubMed] [Google Scholar]
- 36.Lindsay RM, Harmar AJ. Nerve growth factor regulates expression of neuropeptide genes in adult sensory neurons. Nature. 1989;337:362–364. doi: 10.1038/337362a0. [DOI] [PubMed] [Google Scholar]
- 37.Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011;23:1515–1527. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Dang H, Yang L, Wang S, Fang F, Xu F. Calcitonin gene-related peptide ameliorates hyperoxia-induced lung injury in neonatal rats. Tohoku J Exp Med. 2012;227:129–138. doi: 10.1620/tjem.227.129. [DOI] [PubMed] [Google Scholar]
- 39.Millet I, Phillips RJ, Sherwin RS, Ghosh S, Voll RE, Flavell RA, et al. Inhibition of NF-kappaB activity and enhancement of apoptosis by the neuropeptide calcitonin gene-related peptide. J Biol Chem. 2000;275:15114–15121. doi: 10.1074/jbc.275.20.15114. [DOI] [PubMed] [Google Scholar]
- 40.Millis RM, Alvin ZV, Zhao A, Haddad GE. Effects of IGF-1 on I(K) and I(K1) Channels via PI3K/Akt Signaling in Neonatal Cardiac Myocytes. Int J Cell Biol. 2012;2012:712153. doi: 10.1155/2012/712153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teos LY, Zhao A, Alvin Z, Laurence GG, Li C, Haddad GE. Basal and IGF-I-dependent regulation of potassium channels by MAP kinases and PI3-kinase during eccentric cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2008;295:H1834–1845. doi: 10.1152/ajpheart.321.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boaglio AC, Zucchetti AE, Toledo FD, Barosso IR, Sánchez Pozzi EJ, Crocenzi FA, et al. ERK1/2 and p38 MAPKs are complementarily involved in estradiol 17β-D-glucuronide-induced cholestasis: crosstalk with cPKC and PI3K. PLoS One. 2012;7:e49255. doi: 10.1371/journal.pone.0049255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yung LY, Tso PH, Wu EH, Yu JC, Ip NY, Wong YH. Nerve growth factor-induced stimulation of p38 mitogen-activated protein kinase in PC12 cells is partially mediated via G(i/o) proteins. Cell Signal. 2008;20:1538–1544. doi: 10.1016/j.cellsig.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 44.Ham YM, Mahoney SJ. Compensation of the AKT signaling by ERK signaling in transgenic mice hearts overexpressing TRIM72. Exp Cell Res. 2013;319:1451–1462. doi: 10.1016/j.yexcr.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 45.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]