

Abstract
Background
Angiotensin II (ANG II) stimulates fetal heart growth, though little is known regarding changes in cardiomyocyte endowment or the molecular pathways mediating the response. We measured cardiomyocyte proliferation and morphology in ANG II treated fetal sheep and assessed transcriptional pathway responses in ANG II and losartan (an ANG II type 1 receptor antagonist) treated fetuses.
Methods
In twin gestation pregnant sheep, one fetus received ANG II (50 μg/kg/min iv) or losartan (20 mg/kg/d iv) for 7 days; non-instrumented twins served as controls.
Results
ANG II produced increases in heart mass, cardiomyocyte area (left ventricle (LV) and right ventricle mononucleated and LV binucleated cells) and the percentage of Ki-67-positive mononucleated cells in the LV (all p< 0.05). ANG II and losartan produced generally opposing changes in gene expression, affecting an estimated 55% of the represented transcriptome. The most prominent significantly effected biological pathways included those involved in cytoskeletal remodeling and cell cycle activity.
Conclusion
ANG II produces an increase in fetal cardiac mass via cardiomyocyte hypertrophy and likely hyperplasia, involving transcriptional responses in cytoskeletal remodeling and cell cycle pathways.
INTRODUCTION
Remodeling responses to biomechanical and neurohumoral stimuli have been extensively documented in the fetal, neonatal and adult heart (1, 2). Each of these developmental stages poses a different functional environment for the heart, resulting in distinct morphologic, genomic and proteomic responses. For example, only the fetal heart is composed of a substantial number of mononucleated rather than multinucleated cardiomyocytes (3). Underlying developmental differences accompanied by respective diversity in protein expression, likely result in differential cardiac adaptive responses to growth promoting stimuli.
Angiotensin II (ANG II) exerts multiple direct and indirect effects on the heart. ANG II stimulates type 1 (AT1) and type 2 (AT2) receptors on cardiomyocytes and vascular smooth muscle, resulting in altered cardiac structure and function from direct myocardial effects as well as from increased afterload. Direct effects of ANG II on cardiomyocytes have been described for fetal, neonatal and adult tissues (reviewed in Schlüter and Wenzel (4)). The majority of these studies have been performed in isolated cardiomyocytes, which may or may not reflect what occurs in vivo. We previously demonstrated that elevated ANG II levels result in an increase in fetal sheep left ventricular mass (5). However, whether this in vivo effect was related to cardiomyocyte hyperplasia, hypertrophy, or both, as occurs with increased afterload produced by fetal pulmonary artery banding or chronic plasma infusion, is not known (3, 5). Based upon these previous studies, we hypothesized chronic infusion of ANG II increases fetal cardiac mass through cardiomyocyte hyperplasia and hypertrophy. We additionally sought to investigate potential mechanisms regulating cardiomyocyte fate in fetuses exposed to ANG II by determining the levels of terminal proteins of the mitogen activated protein kinase (MAPK) and phosphatidylinositol 3-kinase/protein kinase B (AKT) signaling pathways.We further undertook a transcriptome-wide discovery approach to provide information on the i biological pathways regulating myocardial responses in fetuses treated with ANG II and losartan, an AT1 receptor antagonist. The effects of reduced systolic load on cardiomyocyte morphometry were not examined in the losartan treated animals as this strategy has previously been studied using enalaprilat (6).
METHODS
Animals and surgical preparation
All procedures were performed within the regulations of the Animal Welfare Act and the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of Iowa Animal Care and Use Committee. Time-bred pregnant ewes of mixed Dorset-Suffolk breed were obtained from a local supplier and acclimated to the laboratory over several days.
Pregnant ewes at 125-126 days gestation (term 145 days) with twin fetal pregnancies were used for the study (n = 15 ewes). Anesthesia was induced with 12 mg/kg of thiopental sodium (Pentothal Sodium, Abbott Laboratories) and maintained with a mixture of isoflurane (1-3%), oxygen (30%) and nitrous oxide. Under sterile conditions, the uterus was opened over the fetal hind limbs. Indwelling catheters (PE-90, ID = 0.86 mm, OD = 1.27 mm, Intramedic, Franklin Lakes, NJ) were placed into the right fetal femoral artery and vein and a catheter for measurement of amniotic pressure was secured to the fetal skin. Control twin fetuses were not instrumented to avoid a second uterine incision and inadvertent manipulation of the first fetus that would increase the risk for loss of the surgical preparation. All incisions were closed in separate layers and catheters were exteriorized through a subcutaneous tunnel and placed in a cloth pouch on the ewe's flank. Ampicillin sodium (Wyeth Laboratories, Philadelphia) was administered to the ewe prior to surgery (2 g), intra-amniotically at the completion of surgery and daily for three days. Pregnant ewes were returned to individual pens and allowed free access to food and water. Butorphanol (0.1 mg/kg i.v., Torbugesic; Fort Dodge Animal Health, Fort Dodge, IA) was given for 24 h postoperatively for analgesia. Animals were allowed 24 h after surgical preparation before physiologic measurements were begun.
Experimental protocols
To address the separate aims of the study, two sets of experiments were performed. To examine heart and cardiomyocyte growth in response to ANG II, the catheterized fetus received a continuous intravenous infusion of ANG II (50 ug /kg/min, 0.5 ml/hr) using a battery operated Pegasus VARIO micro-piston pump (Instech Lab Inc., Plymouth Meeting, PA). Physiological measurements were obtained prior to initiating the infusion, then daily for 5 days. Ewes were confined to stanchions during the recording periods though afforded free access to food and water. Pressures were recorded with Transpac pressure transducers (Abbott, Abbott Park, IL) on a calibrated computerized system (ADInstruments, Colorado Springs, CO). Fetal arterial pressures were referenced to amniotic fluid pressure. Arterial pressure tracings were used to determine fetal heart rate. Fetal arterial blood samples were taken daily for blood gases and pH (Gem Premier 3000, Instrumentation Laboratory, Bedford MA).
Tissue collection, cardiac dissociation and cardiomyocyte analysis
At the completion of the infusion, ewes were anesthetized, the fetuses exteriorized, administered heparin (5000 U i.v.) and saturated potassium chloride (10 ml i.v.) to arrest their hearts in diastole. Fetuses and excised hearts were weighed. Fetal hearts were then enzymatically dissociated on a Langendorff apparatus, and the cardiomyocytes fixed for morphometric analysis with the long-axis (length) and maximal cross-sectional diameter (width) dimensions of cardiomyocytes measured as previously described (3). Fixed myocytes were prepared in a wet mount with methylene blue, and selected for measurement according a random, non-repeating and unbiased method employing a counting frame. Myocytes were photographed at 40x on a light microscope (Zeiss Axiophot, Bellevue, WA), and photomicrographs analyzed using calibrated Optimas software (Optimas, Seattle, WA) and Image J software (National Institutes of Health, Bethesda, MD). At least 50 cells of each type (mononucleated or binucleated) were measured per ventricle per fetus. Separately, cells were stained with hematoxylin and eosin and at least 300 myocytes from each ventricle of each fetus were counted to determine the number of nuclei per cardiomyocyte.
Cell cycle activity
The anti-Ki-67 antibody MIB-1 (DAKO, Carpinteria, CA) was used to immunohistologically detect cell cycle activity in dissociated cardiomyocytes as described (3). No fewer than 500 mononucleated myocytes were counted per ventricle per fetus for cell cycle activity analysis. Ki-67 positive myocytes are expressed as a percent of total mononucleated cardiomyocytes.
Morphometry data analysis
Values are presented as means ± SEM. Statistical comparisons were performed by Student's unpaired, two-tailed t test or ANOVA with Tukey's post hoc test if the F statistic was found to be significant. A value of P < 0.05 was considered significant.
The second series of studies investigated mechanisms regulating adaptive changes in the fetal heart in response to loading and unloading of the heart with ANG II and the AT1 receptor antagonist, losartan, respectively. Catheterized fetuses received either an infusion of ANG II, as described above, or losartan (20 mg/kg est. fetal weight, iv) on a daily basis for 5 days with the twin serving as a control. Physiological measurements were obtained daily for 5 days, following which fetuses were euthanized as described above. Fetuses were weighed and their hearts were removed. A portion of the left ventricular free wall, one cm below the AV groove, 0.5 cm in width and extending to within 0.5 cm of the septum was quickly excised, weighed, frozen in liquid nitrogen and stored at −80°C. This tissue was used for RNA isolation and microarray analysis as described below. The remainder of the heart was dissected into anatomical components weighed and immediately frozen in liquid nitrogen. Ewes were euthanized by intravenous administration of pentobarbital sodium (Euthasol Solution, Virbac Corp., Fort Worth TX).
Quantitative Immunoblot
Immunoblots were performed to quantify protein expression (7). Primary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA) specific to total ERK1/2 (sc-93), phosphorylated ERK1/2 (sc-7383), total JNK1/2 (sc-1648), phosphorylated JNK1/2 (sc-6254) and Cell Signaling Technology (Beverly, MA) specific to Akt (9272), phosphorylated (Ser-473) Akt (9271), p38 (9212) and phospho-p38 (9211).
RNA Isolation and Microarray Assay
Frozen tissues were shipped to MOgene, Inc. (St. Louis, MO) on dry ice. High quality RNA was isolated (RNA Integrity Number of 7.5-10 and 28S/18S ratio of 1.6-2.1) and confirmed with an Agilent Bioanalyzer at MOgene, Inc. Five hundred ng of total RNA was amplified using Agilent QuickAmp Labeling Kit (no dye) and purified using Zymo Research RNA Clean and Concentrator spin columns. Three μg of amplified RNA was Cy-labeled using the Kreatech ULS Labeling Kit, fragmented according to Agilent specifications and hybridized to the Agilent sheep gene expression microarray (Agilent Technologies, Santa Clara CA) for 17 hours at 65°C and 10 rpm. Slides were scanned on an Agilent C scanner (20 bit) at 5 μm resolution. Sixteen microarray hybridizations were performed, each with samples from an ANG II, losartan infused fetus (n = 4 for each), or a paired twin control (n = 8), using a mixed design on 2 slides with 8 microarrays per slide. Microarray data were submitted to the NCBI Gene Expression Omnibus (GEO), and can be accessed at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE45463 (project accession no. GSE45463).
Microarray Quality Control
Hybridization scans were free of visual artifacts. Spike-in probe sets exhibited strong linear relationships between concentration and signal intensity for all 16 microarrays (each microarray R2 > 0.99).
Microarray Gene Annotation
At the time of analysis, public annotations of gene identities for the sheep transcriptome were largely incomplete. Therefore, identities of sheep microarray oligonucleotide sequences lacking public annotation were assigned, where possible, based on inter-species homology (8) using blastn (9) on the NCBI ‘nr’ database accessed December 1-4, 2011. The accession of each top scoring hit was then cross-referenced for a match at Entrez Gene (10), and if none present then at Entrez Nucleotide (11).
Microarray Analysis
Microarray analysis was performed using the open source R (R Development Core Team, University of Auckland, Auckland NZ) and Bioconductor statistical environments (12). Pre-processing employed background correction using a saddle-point approximation to maximum likelihood (13) and quantile normalization using the “limma” package (14). ComBat, an empirical Bayes framework robust with small sample sizes, was used to minimize batch effects (15). Data filtering, partitioning around medoids, principal component analysis, and hierarchical clustering were performed in R. Significant differences among group means was assessed using analysis of variance (ANOVA). The proportion of the transcriptome experiencing differential expression was estimated by the method of Storey (16). To account for multiple comparisons, the positive false discovery rate was calculated using the “qvalue” package (16).
Pathway Analysis
Enrichment of significantly affected genes among pathways was investigated using the MetaCore database within the GeneGo pathway analysis software (Thomas Reuters, Carlsbad, CA). MetaCore does not offer pathway analysis for sheep specific pathways, therefore human homologs were used for the analysis. MetaCore pathways with a probabilistic over-representation of statistically significant genes were determined using Fisher's Exact Test, controlling for a false discovery rate of 0.05.
RESULTS
Thirty fetuses from 15 pregnant ewes were studied, with 10 fetuses receiving ANG II, 5 fetuses receiving losartan, and 15 fetuses serving as twin-matched controls. In the first group of studies, infusion of ANG II produced an increase in fetal mean arterial pressure from 44 ± 2 mmHg on day 0 to 58 ± 3 mmHg on day 6 (Table 1, p<0.05). No significant changes in fetal heart rate or arterial blood gas values were identified. For the second group of studies, which provided the opportunity to determine ventricular mass and isolation of protein and RNA, ANG II produced a similar increase in fetal mean arterial pressure from 42 ± 3 mmHg on day 0 to 60 ± 2 mmHg on day 6 (Table 1, p<0.05). Administration of losartan resulted in a decrease in fetal mean blood pressure from 43 ± 3 on day 0 to 28 ± 3 mmHg on day 6 (Table 1, p<0.05). No statistical differences in fetal heart rate were observed over this period of time. Arterial blood gas values, including pH, PCO2 and PO2 did not change over time in the ANG II infused fetuses, whereas PO2 decreased in the losartan-treated fetuses with PCO2 and pH remaining unchanged (Table 1).
Table 1.
Somatic, hemodynamic and arterial blood values in fetuses infused with angiotensin II or losartan.
| Angiotensin II (n=5) † | Control (n = 10) | Losartan (n=5) | Angiotensin II (n=5) | |
|---|---|---|---|---|
| Sex | 2m, 3f | 6m, 4f | 2m, 3f | 3m, 2f |
| Age, day of gestation | 131 ± 1 | 131 ± 1 | 131 ± 1 | 131 ± 1 |
| MABP, mmHg | ||||
| d0 | 44 ±2 | – | 43 ± 3 | 42 ± 3 |
| d6 | 58 ±3 | – | 28 ± 3† | 60 ± 2† |
| Heart rate, bpm | ||||
| d0 | 166 ±9 | – | 159 ± 6 | 158 ± 8 |
| d6 | 155 ±6 | – | 154 ± 10 | 161 ± 10 |
| pH | ||||
| d0 | 7.38 ±0.11 | – | 7.36 ± 0.1 | 7.35 ± 0.00 |
| d6 | 7.39 ±0.01 | – | 7.34 ± 0.2 | 7.35 ± 0.00 |
| PO2, torr | ||||
| d0 | 17 ±1 | – | 20 ± 1 | 19 ± 2 |
| d6 | 18 ± 2 | – | 17 ± 1* | 20 ± 2 |
| PCO2, torr | ||||
| d0 | 55 ± 3 | – | 52 ± 1 | 56 ± 2 |
| d6 | 54 ± 2 | – | 56 ± 3 | 56 ± 2 |
| Fetal weight (FW), kg | 3.74 ± 0.15 | 3.80 ± 0.12 | 3.54 ± 0.12 | 3.36 ± 0.01 |
| Heart, g | – | 16.66 ± 1.03 | 14.00 ± 0.25 | 18.27 ± 0.90 |
| Heart, g/kg FW | – | 4.38 ± 0.21 | 3.96 ± 0.07 | 5.44 ± 0.27* |
| LV, g/kg FW | – | 1.74 ± 0.06 | 1.69 ± 0.05 | 2.16 ± 0.11* |
| RV, g/kg FW | – | 1.96 ± 0.15 | 1.60 ± 0.09 | 2.35 ± 0.20* |
MABP, mean arterial blood pressure; d, day; n, sample size for each group; m, male; f, female. Heart weight represents sum of left ventricle (LV) and right ventricle (RV) free wall + septum weights.
Initial group of angiotensin II infused fetuses utilized for isolated cardiomyocyte morphology.
p< 0.05 compared to control and losartan values.
p < 0.05 compared to d0 values.
Fetal weight was similar among all groups of animals. The effects of altering arterial pressure by ANG II and losartan on the fetal heart mass are depicted in Table 1. Because one group of ANG II infused fetuses (n=5) and their matched controls were utilized for enzymatic cardiomyocyte dissociation, weights of individual heart components were not obtained. For animals in the second group of studies, LV, RV, septum, and total heart weight (LV + RV + septum), expressed per kg fetal body weight, were significantly greater in ANG II fetuses compared to control or losartan animals. Cardiac weights following losartan were not statistically different from controls.
ANG II fetuses displayed significant increases in the area of LV mono- and bi-nucleated cardiomyocytes and RV mononucleated cardiomyocytes (Figure 1) resulting from increases in cell width but not length (Figure 1). The percent binucleation (used as an index of terminal differentiation) of cardiomyocytes was not different between groups (Figure 2). Ki-67 staining, a marker of cell cycle activity, was significantly greater in the LV of ANG II infused animals compared to controls, while no difference was identified in the RV (Figure 2).
Figure 1.

Effect of angiotensin II infusion on fetal ventricular cardiomyocyte (a) cell area, (b) cell length, and (c) cell width in left ventricle mononucleated cells (white bars); left ventricle binucleated cells (grey bars); right ventricle mononucleated cells (diagonal stripe bars); and right ventricle binucleated cells (black bars). Values expressed as means ± SE. *p <0.05 compared to control of similar cell type.
Figure 2.
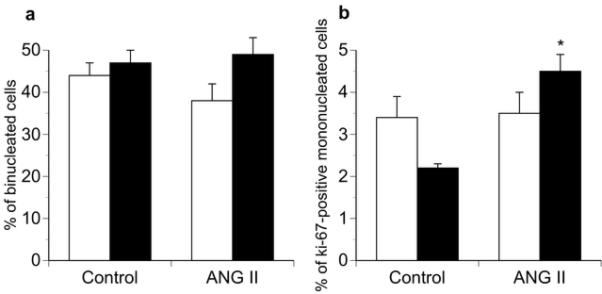
Effect of angiotensin II (ANG II) infusion on a) fetal cardiomyocyte binucleation (maturation) and b) Ki-67 (+) immunostaining of mononucleated cells. White bars, right ventricle; black bars, left ventricle. Values expressed as means ± SE (n = 5 for each group). *p <0.05 compared to control in similar ventricle.
MAP Kinase and AKT Signaling Pathways
ANG II and losartan had no effect on LV steady-state protein levels on total or the phosphorylated, active forms of ERK1/2, JNK, p38 or Akt1 compared to controls (Figure 3).
Figure 3.

Effect of angiotensin II (ANG II) and losartan infusion on steady-state protein levels of total (panels a-d) and activated (panels e-h) mitogen-activated protein kinases (ERK ½, JNK ½, p38) and AKT 1 in fetal left ventricular myocardium. Control (twin of ANG II infused, white bar); ANG II infused diagonal stripe bar); Control (twin of losartan infused, black bar); losartan infused (vertical stripe bars). Values expressed as means ± SE (n = 5 for each group).
Impact of renin-angiotensin modulation on the cardiac transcriptome
Cardiac gene expression was assessed for four losartan- and four ANG II-infused fetuses and their control twins using the Agilent G4813A ovine microarray. The global impact of ANG II and losartan on gene expression segregated into separate groups (Figure 4A), suggesting that ANG II and losartan had distinct effects on gene expression. To further characterize the origins of this clustering pattern, we assessed principal expression components among those microarray features most impacted by ANG II and/or losartan. Interestingly, this demonstrated grouping of the ANG II versus losartan hearts onto opposite sides of the principal component space (Figure 4B), with the control hearts located in the intervening space. By contrast, there was no such apparent grouping in control analyses in which the same procedures were performed on (i) those genes whose expression was most likely to not be impacted by losartan and ANG II and (ii) the dataset after randomization of group identities. Thus, among genes most affected by ANG II and/or losartan, the effects of these two interventions on gene expression tended to be in opposite directions, whereas the control hearts from the two groups tended to be similar to each other with intermediary expression.
Figure 4.

Impact of renin-ANG II modulation on the cardiac transcriptome. (A) Clustering analysis on all 15,208 microarray features. The effects of ANG II (red triangle) or losartan (blue circle) were isolated by subtracting control expression from expression in its corresponding intervention twin fetus. Components 1 and 2 explained 77% of the point variability. (B) Principal component analysis of genes whose expression was most likely affected by ANG II (red circle) versus its twin (grey), and/or losartan (blue circle) versus its control twin (aqua). Genes most likely affected were identified by p-value < 0.0015 by paired t-test for either intervention, which led to selection of 89 microarray features. Red lines indicate ANG II twinships; blue lines indicate losartan twinships. Axes represent principal components 1, 2, and 3 (PC1, PC2, and PC3 respectively), which accounted for 67% of the overall variance. (C) Hierarchical clustering of expression values of probes meeting stringent statistical significance defined by ANOVA based q-value < 0.01. A total of 319 genes met this criteria and were clustered in both dimensions using McQuitty's method and the maximum distance function in R. A – ANG II, L – losartan, AC – ANG II control, LC – losartan control; numbers refer to twinships (e.g., L3 and LC3 were twins).
ANOVA was used to identify the swath genes impacted by renin-angiotensin modulation by grouping samples as ANG II, losartan, and control. Combining of control fetuses from both experimental interventions was appropriate because all 8 control fetus expression features clustered together (Figure 4B) and received identical treatment. Although the ANOVA-based analysis estimated that 55% of the represented transcripts differed among the three groups, only ¼ of these (14% of represented transcriptome) could be confidently identified at a positive false discovery rate (pFDR) of < 0.05. Hierarchical clustering was used to visualize the expression structure of the most significantly affected genes (pFDR<0.01, 319 spots). Among these, genes upregulated by losartan tended to be downregulated by ANG II, and vice-versa (Figure 4C).
Of the 15,208 probe sequences on the sheep microarray, 11,087 were well annotated by inter-species homology, meeting the following criteria: (i) stringent homology demonstrating E-value < 0.001, (ii) blastn accession mapped to a single entry, (iii) identification of both gene symbol and organism. Of these, 8,638 were non-redundant. These annotations were based on closest homology to transcripts from Bos taurus (N=5,953), Ovis aries (N=717), Sus scrofa (N=525), Pan troglodytes (N=254), Canis lupus familiaris (N=242), and other species. Homology to mammalian species accounted for 99.8% of the annotations. There were 1,520 well annotated transcripts exhibiting differential expression among groups (pFDR < 0.05, Supplemental Table 1, online). Of these, 675 were upregulated by ANG II and downregulated by losartan, 723 exhibited the opposing pattern, 69 were upregulated by both, and 53 were downregulated by both. The transcripts with the strongest statistical changes in expression in these four categories are detailed in Supplemental Tables 2-5 (online).
Biological pathways with expression impacted by renin-ANG modulation
Of the 643 pathways in the MetaCore database, 243 demonstrated an over-representation of the genes significantly affected by ANG II and losartan by Fisher's Exact Test, controlling for FDR of 0.05. The top 25 pathways are shown in Table 2 and all statistically significant pathways are provided in Supplemental Table 6 (online).
Table 2.
Top 25 scoring pathways altered by renin-angiotensin system modulation.
| Pathway | qValue |
|---|---|
| Cell cycle_Role of APC in cell cycle regulation | 2.97E-09 |
| Development_TGF-beta-dependent induction of EMT via RhoA, PI3K and ILK. | 7.50E-09 |
| Cell cycle_The metaphase checkpoint | 7.50E-09 |
| Development_Regulation of epithelial-to-mesenchymal transition (EMT) | 7.50E-09 |
| Immune response_IL-6 signaling pathway | 7.12E-08 |
| Cytoskeleton remodeling_Cytoskeleton remodeling | 8.46E-08 |
| Immune response_CD137 signaling in immune cell | 2.33E-07 |
| Cell cycle_Initiation of mitosis | 3.13E-07 |
| Cytoskeleton remodeling_TGF, WNT and cytoskeletal remodeling | 3.70E-07 |
| Cell cycle_Chromosome condensation in prometaphase | 5.40E-06 |
| Development_TGF-beta-dependent induction of EMT via MAPK | 1.69E-05 |
| Immune response_Oncostatin M signaling via MAPK in mouse cells | 1.91E-05 |
| Signal transduction_AKT signaling | 3.08E-05 |
| Immune response_Oncostatin M signaling via MAPK in human cells | 3.11E-05 |
| Immune response_ETV3 affect on CSF1-promoted macrophage differentiation | 3.11E-05 |
| Development_Thrombopoietin-regulated cell processes | 4.46E-05 |
| Cell adhesion_Chemokines and adhesion | 4.76E-05 |
| Cell cycle_Spindle assembly and chromosome separation | 5.29E-05 |
| Development_PIP3 signaling in cardiac myocytes | 6.47E-05 |
| Apoptosis and survival_BAD phosphorylation | 9.78E-05 |
| Development_Growth hormone signaling via PI3K/AKT and MAPK cascades | 9.78E-05 |
| Apoptosis and survival_HTR1A signaling | 0.000119062 |
| Immune response_Signaling pathway mediated by IL-6 and IL-1 | 0.000121599 |
| Development_VEGF signaling via VEGFR2 - generic cascades | 0.000167581 |
| Development_IGF-1 receptor signaling | 0.000167581 |
DISCUSSION
The fetal heart is in a continuous state of remodeling, undergoing cardiomyocyte proliferation, enlargement and differentiation. During the last third of gestation, these coordinated processes result in a marked increase in cardiac mass. We previously demonstrated that increased systolic load from ANG II infusion increases cardiac mass in fetal sheep, though specific effects on the cardiomyocyte were not investigated (5). In the current study, we demonstrate that increased fetal cardiac mass following ANG II mediated increased cardiac load results primarily from hypertrophy of both mononucleated and binucleated cardiomyocytes. There also appears to be a contribution from cellular proliferation, as evidenced increased staining of the cell cycle activity marker, Ki-67. Because the ratio of mononucleated to binucleated cells did not significantly differ between groups despite an increase in left ventricle cardiomyocyte cell cycle activity, ANG II appears to have little effect on the proportion of cells undergoing terminal differentiation. Although evidence for MAPK involvement was not found, microarray analysis from both ANG II and losartanexposed hearts identified several alternative pathways that may be involved in the fetal hearts response to altered load.
Normal physiologic fetal cardiac growth during the latter third of gestation primarily occurs through cellular proliferation and enlargement of myocytes as they undergo terminal differentiation (binucleation) with limited contribution from cellular hypertrophy (17). By contrast, pathologic cardiac adaptions to altered fetal cardiac load likely include both alterations in proliferation and growth of mononucleated and binucleated cardiomyocytes. Pulmonary artery banding in fetal sheep results in increased cardiac mass, RV but not LV cardiomyocyte enlargement and increased RV cardiomyocyte binucleation, suggestive of hyperplastic and hypertrophic growth (18). Increased fetal cardiac preload and afterload load induced by daily intravascular plasma infusion increased in cardiac mass associated with cardiomyocyte hypertrophy, increased cell cycle activity and increased percentage of binucleated cardiomyocytes (3). In the present study, we found ANG II was associated with increased cardiomyocyte area, and presumably volume. Additionally, increased Ki-67 staining in the LV suggests upregulation of cell cycle activity and thus proliferation. Evidence for more rapid progression of cardiomyocyte terminal differentiation was not seen. To study the effects of reduced systolic load on fetal cardiomyocyte development, O'Tierney et al. utilized an eight-day infusion of enalaprilat, an angiotensin converting enzyme inhibitor (6). Enalaprilat decreased arterial pressure by over 20 mmHg and significantly decreased heart weight-to-body weight ratio, suggesting attenuation of fetal heart growth. Cardiomyocyte size was not different between groups, though there was a significant decrease in LV and RV cardiomyocyte cell cycle activity, suggesting slowing of normal hyperplastic growth. Taken together, these studies emphasize the importance of cardiac load on cardiac growth.
Despite growing understanding of the pathologic changes that occur in the fetal heart with alterations in load, the molecular mechanisms regulating these changes remain poorly defined. Montgomery and colleagues observed that RV loading from pulmonary artery banding altered expression of connexin isoforms involved in gap junction formation (19). In both aortic and pulmonary artery banded fetuses, levels of phosphorylated p38 were significantly increased compared to controls, whereas no differences were identified in the levels of phosphorylated (activated) ERK or JNK (7). The role of p38 in the fetal heart has not been clearly defined, though evidence suggests it promotes exit from the cell cycle, inducing myocyte differentiation and hypertrophy (20, 21). In the present study, we found that neither loading the heart with ANG II or unloading the heart with losartan resulted in significant changes in the levels of total or activated ERK, JNK, p38 or Akt. The absence of change in the expression of activated terminal kinases does not rule out their involvement in the observed responses , as we examined protein expression at single time point, well after the adaptive process has been initiated. The lack of change in the levels of activated MAPKs, though which ANG II is thought to directly signal cardiac hypertrophy, may also suggest that the effects of ANG II on the fetal heart result more from increased load of the heart rather than a direct effect mediated through cardiomyocyte ANG II receptors.
We utilized gene expression arrays performed on RNA isolated from a single area of the LV in ANG II- and losartan-infused fetal sheep and twin controls to explore molecular changes associated with manipulation of the fetal RAS. These studies show that losartan and ANG II infusion have generally opposing effects on the fetal cardiac transcriptome. The importance of the renin-ANG II axis on gene expression in the fetal heart is illustrated by the estimation that expression of over 50% of the transcriptome was quantitatively altered by ANG II and/or losartan (Supplementary Table 1, online). Interestingly, blockade of the renin-ANG system in WKY rats for 4 weeks with candesartan, resulting in a significant decrease in systolic blood pressure (135 vs. 104 mmHg), produced no significant changes in cardiac gene expression on a 9000 gene array (22). In contrast, we found AT1 receptor blockade, which produced a significant decrease in fetal blood pressure, resulted in a large number of changes in gene expression. Our finding is consistent with the apparent dependence of load in regulating fetal cardiomyocyte proliferation and hypertrophy (6).
To further explore mechanisms contributing to load-related remodeling of the fetal heart, we utilized results from the gene expression arrays to perform biological pathway analysis. This approach allows for identification of potential biochemical and molecular processes in the fetal heart affected by manipulation of the renin-ANG II system. Our model holds a particular uniqueness in that studies in adult animals suggest pathologic cardiac hypertrophy is accompanied by activation of “fetal cardiac genes” and that a similar transcriptional program regulates hypertrophic cardiac growth and controls early cardiac development. Whether fetal cardiac hypertrophy represents overactivity of transcriptional regulators of normal development or involves separate pathways is not known. Chief among the pathways identified as highly impacted by fetal RAS modulation are those involved in cytoskeletal remodeling, transcriptional regulation and cell cycle activity (Table 2). An impact on skeletal remodeling pathways has been previously observed in the loaded or unloaded heart in several adult species, including flies and rodents (23-26). Murine cardiac transcriptome analysis following chronic ANG II infusion found the largest group of altered genes to be related to extracellular matrix, cell membrane and nuclear pathways (27). Among the 25 top scoring pathways altered by RAS modulation were also a large number involving transforming growth factor beta (TGF-B) signaling and inflammatory pathways. Identification of TGF-B pathways is not surprising given their known importance in the cardiac response to pressure overload and ANG II (28, 29). There is growing evidence that inflammation plays a role in cardiac hypertrophy, and in particular cytokines activated downstream of nuclear factor kB (30). Along these lines, NFkB and the transcription factor AP-1 are activated by ANG II and in the postnatal heart contribute to ANG II-induced hypertrophy (31). The importance of the IL-6 pathway (#5 on Table 2) in several different models of cardiac hypertrophy has also recently been highlighted (32, 33). Taken together, these findings suggest that many of the molecular adaptations that take place in pathologic hypertrophy in the adult heart are present in response to increased pressure load in the fetal heart.
The ability to discern biological pathways regulating 1) cardiomyocyte hyperplasia from those of hypertrophy and 2) cardiac responses to mechanical stress from those of humoral factors would mark tremendous progress in understanding the adaptive response of the fetal heart to increased load. As yet, however, we are unable to identify such links. Isolated and cultured neonatal and fetal myocytes have been primarily used to study the regulation of cardiomyocyte proliferation. Both chemical and mechanical factors, including pressure load, ANG II, isoproterenol, cortisol, growth hormone, and IGF-1 have been shown to stimulate proliferation utilizing overlapping and complementary pathways (2, 34-36). Signaling pathways associated with these factors involve Akt, phosphoinositol 3-kinase (PI3K), ERK, Ras, and gp130 (37, 38). Our findings of significant changes in a number of signaling pathways involving these factors (e.g., Akt signaling, PIP3 signaling in cardiac myocytes, IL-6 signaling pathways, IGF-1 receptor signaling, Table 2) supports the importance of these pathways in the adaptive changes we observed in the fetal heart.
Our study has a number of limitations. We are unable to differentiate the routes of influence by which ANG II affects cardiomyocytes. More specifically, we cannot separate the direct effects of activation or blockade of cardiac AT1 receptors from indirect effects on the heart by changes in pressure load. Our control animals, which were paired twins, were not surgically instrumented as were study animals, and thus not perfect controls. We also did not perform validation of specific gene changes by quantitative PCR. Such studies will be important as we move forward in understanding molecular regulation of physiologic and pathologic fetal heart growth. Furthermore, the microarray studies were performed on cardiac ventricular wall tissue homogenates and not isolated myocytes. The potential contribution of non-myocyte tissue to the expression data is recognized. We also did not perform any histological evaluation of the cardiac tissue to address whether an inflammatory process was present and potentially contributing to the transcriptome and signaling pathway results. Lastly, gene expression and protein levels were assessed at only a single time point in the development of increased cardiac mass. Had measurements been performed at an earlier time point, a different profile of gene expression likely would be evident.
In summary, our studies indicate that in vivo, ANG II increases fetal left ventricle mass primarily by induction of cardiomyocyte proliferation and hypertrophy. Despite a well described role for MAPKs in regulating pathologic cardiac growth, no involvement of terminal MAPK proteins was identified at the time point measured. To our knowledge, this study represents the first to examine changes in the transcriptome of the fetal heart in response to pressure load in an animal with a similar developmental pattern to the human. The use of gene expression profiling to identify potential signaling pathways altered in response to pressure loading and unloading that may help delineate molecular mechanisms of adaptive growth of the fetal heart. These findings provide a foundation for future experiments in the under-explored area of fetal heart growth.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by National Institutes of Health DK097820 and University of Iowa Fraternal Order of Eagles Diabetes Research Center pilot grant (to A.W. Norris) and NIH HL080657 (to J.L. Segar).
Footnotes
Disclosure: The authors declare no conflicts of interest
REFERENCES
- 1.Barry SP, Davidson SM, Townsend PA. Molecular regulation of cardiac hypertrophy. Int J Biochem Cell Biol. 2008;40:2023–39. doi: 10.1016/j.biocel.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 2.Thornburg K, Jonker S, O'Tierney P, et al. Regulation of the cardiomyocyte population in the developing heart. Prog Biophys Mol Biol. 2011;106:289–99. doi: 10.1016/j.pbiomolbio.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jonker SS, Faber JJ, Anderson DF, Thornburg KL, Louey S, Giraud GD. Sequential growth of fetal sheep cardiac myocytes in response to simultaneous arterial and venous hypertension. Am J Physiol Regul Integr Comp Physiol. 2007;292:R913–R9. doi: 10.1152/ajpregu.00484.2006. [DOI] [PubMed] [Google Scholar]
- 4.Schluter KD, Wenzel S. Angiotensin II: a hormone involved in and contributing to pro-hypertrophic cardiac networks and target of anti-hypertrophic cross-talks. Pharmacol Ther. 2008;119:311–25. doi: 10.1016/j.pharmthera.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 5.Segar JL, Dalshaug GB, Bedell KA, Smith OM, Scholz TD. Angiotensin II in cardiac pressure-overload hypertrophy in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2001;281:R2037–47. doi: 10.1152/ajpregu.2001.281.6.R2037. [DOI] [PubMed] [Google Scholar]
- 6.O'Tierney PF, Anderson DF, Faber JJ, Louey S, Thornburg KL, Giraud GD. Reduced systolic pressure load decreases cell-cycle activity in the fetal sheep heart. Am J Physiol Regul Integr Comp Physiol. 2010;299:R573–8. doi: 10.1152/ajpregu.00754.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson AK, Protheroe KN, Scholz TD, Segar JL. The mitogen-activated protein kinases and Akt are developmentally regulated in the chronically anemic fetal sheep heart. J Soc Gynecol Investig. 2006;13:157–65. doi: 10.1016/j.jsgi.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Goyal R, Longo LD. Gene expression in sheep carotid arteries: major changes with maturational development. Pediatr Res. 2012;72:137–46. doi: 10.1038/pr.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 10.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2005;33:D54–8. doi: 10.1093/nar/gki031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wheeler DL, Barrett T, Benson DA, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2007;35:D5–12. doi: 10.1093/nar/gkl1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silver JD, Ritchie ME, Smyth GK. Microarray background correction: maximum likelihood estimation for the normal-exponential convolution. Biostatistics. 2009;10:352–63. doi: 10.1093/biostatistics/kxn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer; New York, NY: 2005. pp. 397–420. [Google Scholar]
- 15.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 16.Storey JD. A direct approach to false discovery rates. J R Stat Soc. 2002;64:479–98. [Google Scholar]
- 17.Jonker SS, Zhang L, Louey S, Giraud GD, Thornburg KL, Faber JJ. Myocyte enlargement, differentiation, and proliferation kinetics in the fetal sheep heart. J Appl Physiol. 2007;102:1130–42. doi: 10.1152/japplphysiol.00937.2006. [DOI] [PubMed] [Google Scholar]
- 18.Barbera A, Giraud GD, Reller MD, Maylie J, Morton MJ, Thornburg KL. Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1157–64. doi: 10.1152/ajpregu.2000.279.4.R1157. [DOI] [PubMed] [Google Scholar]
- 19.Montgomery MO, Jiao Y, Phillips SJ, et al. Alterations in sheep fetal right ventricular tissue with induced hemodynamic pressure overload. Basic Res Cardiol. 1998;93:192–200. doi: 10.1007/s003950050086. [DOI] [PubMed] [Google Scholar]
- 20.Braz JC, Bueno OF, Liang Q, et al. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–86. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao P, Georgakopoulos D, Kovacs A, et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001;98:12283–8. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato N, Liang YQ, Ochiai Y, Birukawa N, Serizawa M, Jesmin S. Candesartan-induced gene expression in five organs of stroke-prone spontaneously hypertensive rats. Hypertens Res. 2008;31:1963–75. doi: 10.1291/hypres.31.1963. [DOI] [PubMed] [Google Scholar]
- 23.Brooks WW, Bing OH, Conrad CH, et al. Captopril modifies gene expression in hypertrophied and failing hearts of aged spontaneously hypertensive rats. Hypertension. 1997;30:1362–8. doi: 10.1161/01.hyp.30.6.1362. [DOI] [PubMed] [Google Scholar]
- 24.Jin H, Yang R, Awad TA, et al. Effects of early angiotensin-converting enzyme inhibition on cardiac gene expression after acute myocardial infarction. Circulation. 2001;103:736–42. doi: 10.1161/01.cir.103.5.736. [DOI] [PubMed] [Google Scholar]
- 25.Kang BY, Hu C, Ryu S, et al. Genomics of cardiac remodeling in angiotensin II-treated wild-type and LOX-1-deficient mice. Physiol Genomics. 2010;42:42–54. doi: 10.1152/physiolgenomics.00009.2010. [DOI] [PubMed] [Google Scholar]
- 26.Montana ES, Littleton JT. Expression profiling of a hypercontraction-induced myopathy in Drosophila suggests a compensatory cytoskeletal remodeling response. J Biol Chem. 2006;281:8100–9. doi: 10.1074/jbc.M512468200. [DOI] [PubMed] [Google Scholar]
- 27.Hu C, Dandapat A, Sun L, et al. Modulation of angiotensin II-mediated hypertension and cardiac remodeling by lectin-like oxidized low-density lipoprotein receptor-1 deletion. Hypertension. 2008;52:556–62. doi: 10.1161/HYPERTENSIONAHA.108.115287. [DOI] [PubMed] [Google Scholar]
- 28.Koitabashi N, Danner T, Zaiman AL, et al. Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J Clin Invest. 2011;121:2301–12. doi: 10.1172/JCI44824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenkranz S. TGF-Beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63:423–32. doi: 10.1016/j.cardiores.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 30.Smeets PJ, Teunissen BE, Planavila A, et al. Inflammatory pathways are activated during cardiomyocyte hypertrophy and attenuated by peroxisome proliferator-activated receptors PPARalpha and PPARdelta. J Biol Chem. 2008;283:29109–18. doi: 10.1074/jbc.M802143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valente AJ, Clark RA, Siddesha JM, Siebenlist U, Chandrasekar B. CIKS (Act1 or TRAF3IP2) mediates Angiotensin-II-induced Interleukin-18 expression, and Nox2-dependent cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2012;53:113–24. doi: 10.1016/j.yjmcc.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.del Vescovo CD, Cotecchia S, Diviani D. A-kinase-anchoring protein-Lbc anchors IkappaB kinase beta to support interleukin-6-mediated cardiomyocyte hypertrophy. Mol Cell Biol. 2013;33:14–27. doi: 10.1128/MCB.00887-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papay RS, Shi T, Piascik MT, Naga Prasad SV, Perez DM. alpha(1)A-adrenergic receptors regulate cardiac hypertrophy in vivo through interleukin-6 secretion. Mol Pharmacol. 2013;83:939–48. doi: 10.1124/mol.112.084483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191–227. doi: 10.1016/j.pharmthera.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Bruel A, Christoffersen TE, Nyengaard JR. Growth hormone increases the proliferation of existing cardiac myocytes and the total number of cardiac myocytes in the rat heart. Cardiovasc Res. 2007;76:400–8. doi: 10.1016/j.cardiores.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 36.Tseng YT, Yano N, Rojan A, et al. Ontogeny of phosphoinositide 3-kinase signaling in developing heart: effect of acute beta-adrenergic stimulation. Am J Physiol Heart Circ Physiol. 2005;289:H1834–42. doi: 10.1152/ajpheart.00435.2005. [DOI] [PubMed] [Google Scholar]
- 37.MacLellan WR, Schneider MD. Genetic dissection of cardiac growth control pathways. Annu Rev Physiol. 2000;62:289–319. doi: 10.1146/annurev.physiol.62.1.289. [DOI] [PubMed] [Google Scholar]
- 38.Sundgren NC, Giraud GD, Schultz JM, Lasarev MR, Stork PJ, Thornburg KL. Extracellular signal-regulated kinase and phosphoinositol-3 kinase mediate IGF-1 induced proliferation of fetal sheep cardiomyocytes. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1481–9. doi: 10.1152/ajpregu.00232.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.