

Abstract
Numerous parallelisms exist between development and cancer. In this article, I review some of the founding ideas linking development and cancer, and highlight clinical conditions exhibiting features of both developmental derangement and cancer predisposition, including cohesinopathies, rasopathies, phakomatoses, Proteus syndrome and other overgrowth disorders, recessive chromosome breakage syndromes, and dominant hereditary cancer syndromes. I suggest that these disorders encompass a continuous spectrum spanning clinical genetics and clinical oncology, and derive some general implications that might be useful in the future for the treatment of these diseases.
INTRODUCTION
As our knowledge of the mechanisms and features of epigenetic regulation is advancing, the similarities between development and cancer are becoming even more apparent, providing further support to the original ideas and founding concepts that over the past 40–50 years have linked these two apparently distinct processes.
It is in fact difficult to pinpoint the first ideas and concepts relating development to cancer, but certainly one of the earliest and clearest formulations is that of Dr. Beatrice Mintz who defined cancer as a “developmental disturbance or an error of development” [Mintz, 1978]. Dr. Mintz first hypothesized that differentiation occurs clonally; specifically, she argued that “the diversification of cell types in a multicellular organism is presumably due to differential functioning of specific genetic loci in the various cells of an individual” and that “the many cells that comprise each specialized type are mitotically descended from some much smaller number of precursor cells in which the tissue-specific loci first became active, with subsequent retention of that activity – or of that capacity for activity – in the mitotic progeny of each of the precursor cells” [Mintz, 1971]. She concluded that there should be at least two differentiating precursor cells (due to the likely lethality of a single-cell system) in any given adult tissue, and went on to estimate the number of such cells in various tissue types through the analysis of “allophenic” mice (otherwise defined as chimeric mice, a less precise term that eventually stuck), i.e. morphologically normal mice derived from aggregated eggs of different genotype [Mintz, 1971]. The logical consequence of this line of reasoning was that these precursor or stem cells, involved in the growth and differentiation of tissues, are the perfect candidate target for transformation: “If this were in fact the case, it would account not only for the proliferative aspects of malignancy, but also for the common presence in tumors of a variety of cell types, the whole comprising a kind of aberrant or abortive differentiation of that tissue from its stem-cell population. Moreover, it would account for shifts toward a more benign or more malignant state. In this sense, neoplasia is indeed a derangement of differentiation.” [Mintz, 1978]
We can then see how normal development and cancer are, in general terms, two opposing processes: during development, an ordered unfolding of epigenetic instructions allows an undifferentiated cell, the fertilized egg, to generate all the differentiated tissues of the organism; to the contrary, in cancer, due to a subversion or malfunctioning of epigenetic instructions, there is a reduction in differentiation with the acquisition of undifferentiated features, in a process that is often progressive during tumorigenesis, from benign, well differentiated tumors to more malignant, less differentiated lesions, owing to the emergence and selection of more aggressive and undifferentiated clonal variants (Fig. 1). Thus, even when taking into consideration the obviously opposing features of normalcy and cancer, it is not surprising that similarities exist between normal tissue stem cells and cancer stem/progenitor cells, that in fact share comparable epigenetic signatures, including DNA methylation profiles and chromatin modifications [Baylin, 2009; Widschwendter et al., 2007]. In fact, those similarities extend beyond the normal stem cell/cancer stem cell dualism. Signaling pathways used in normal morphogenesis - tissue invaginations, ingressions, egressions, angiogenesis, patterning and, at the cellular level, proliferation, differentiation, apoptosis, polarity - are coopted and deranged in tumorigenesis, affecting cancer initiation, progression, invasion and metastasis. Mutation(s) affecting abnormal prenatal patterning, hence a malformation syndrome, through effects on tissue homeostasis (“gatekeeper” genes), genomic stability (“caretaker genes”), gene expression modulation (epigenetic regulators and genetic modifiers), gene-environment interactions, local factors (microenvironment, stroma and inflammation) or systemic factors (endocrine and growth factor milieu), will have an impact in the postnatal context on the specific phenotypic pattern of the emerging cancer, influence its molecular profile and even constrain its clinical evolution [Ponder, 2001].
Figure 1.

Schematic depicting the opposing processes of development and cancer in which epigenetic instructions guide differentiation or, when altered in cancer, promote loss of differentiation.
This comparable but contrasting biological and molecular landscape necessarily influences the respective disease manifestations. Thus, within the extremes of diseases that are strictly developmental or strictly malignant, there exists a continuum of conditions with varying prevalence of developmental and tumor features that span the fields of clinical genetics and clinical oncology (Fig. 2). Here I review these conditions and highlight common characteristics within each subgroup.
Figure 2.

Schematic showing a continuous clinical spectrum of diseases characterized by varying degrees of developmental (dev) or cancer (ca) manifestations.
A CONTINUOUS CLINICAL SPECTRUM LINKING DEVELOPMENTAL DISEASE AND CANCER
Cohesinopathies and defects of epigenetic regulation
Cohesinopathies represent examples of conditions in which developmental defects dominate the clinical framework over cancer predisposition (Fig. 2). The cohesin complex is a ring structure encircling the DNA that mediates cohesion of sister chromatids from the time of their replication until their separation at the metaphase to anaphase transition. Its core subunits are SMC1, SMC3, RAD21 and STAG1/2/3, whereas cohesin accessory factors that regulate the function of the core components include NIPBL (a.k.a. SCC2, involved in loading onto DNA, along with SCC4), PDS5A/B (involved in cohesion maintenance along with WAPL) and ESCO2 (an acetyltransferase that acetylates SMC3 and PDS5-WAPL, and affects cohesion establishment and maintenance) [Xiong and Gerton, 2010]. Cornelia de Lange syndrome is caused by mutations in NIPBL, SMC1A and SMC3, and is characterized by striking craniofacial features (synophrys, arched eyebrows, long eyelashes, upturned nose, microcephaly), intellectual disability and autistic tendencies, short stature, upper limb defects (from absence of forearms to oligodactyly), gastroesophageal reflux and other abnormalities. Roberts/SC Phocomelia Syndrome is caused by mutation of both alleles of ESCO2, and is characterized by prenatal growth retardation, limb defects (bilateral symmetric tetraphocomelia or hypomelia, oligodactyly and syndactyly), and craniofacial abnormalities (microcephaly, ear malformation, cleft lip and palate, micrognathia, hypertelorism, downslanting palpebral fissures) [Bose and Gerton, 2010; Mannini et al., 2010].
A low predisposition to cancer is reported for cohesinopathies and involves esophageal adenocarcinoma (Cornelia de Lange), and melanoma, rhabdomyosarcoma and oculomotor nerve angioma (Roberts) [Kline et al., 2007; Mannini et al., 2010]. Somatic mutations or altered expression levels of cohesin core and accessory components have been reported in several human cancer types [Mannini et al., 2010]. Thus, it is possible that disease manifestations in cohesinopathies are caused by genomic instability (vide infra), not unlike constitutional aneuploidy syndromes [Ganmore et al., 2009]; specifically, defective cohesin activity could cause chromosome missegregation and aneuploidy [Mannini et al., 2010]. However, an alternative, not mutually exclusive hypothesis, that perhaps better explains the developmental features of cohesinopathies, posits that cohesins play a role in the control of gene expression by affecting long-range chromatin organization [Bose and Gerton, 2010]. According to this model, cohesinopathies are associated with abnormal epigenetic regulation, and therefore are related to other developmental diseases caused by defective chromatin modification/remodeling and exhibiting variable risk of cancer, such as Coffin-Siris syndrome (mutations in genes encoding components of the SWI/SNF remodeling complex), Kabuki syndrome (caused by mutations in KMT2D and KDM6A, encoding a histone lysine methyltransferase and demethylase, respectively) and Rubinstein-Taybisyndrome (caused by mutations in CBP and EP300, encoding histone acetyltransferases).
Rasopathies
In comparison to cohesinopathies, cancer predisposition becomes a more relevant issue in rasopathies, developmental disorders resulting from germline mutations in genes of the RAS-RAF-MEK-ERK signaling pathway. Costello syndrome, Cardiofaciocutaneous (CFC) syndrome, Noonan Syndrome and LEOPARD syndrome are clinically overlapping dominant disorders characterized by dysmorphic facial features, cardiovascular defects, short stature and other manifestations. In general, they all appear to be caused by gain-of-function mutations that activate the RAS-RAF-MEK-ERK pathway, by targeting HRAS (Costello), KRAS (CFC, Noonan), BRAF (CFC, Noonan, LEOPARD), MAP2K1 (MEK1), MAP2K2 (MEK2) (CFC), NRAS, SOS1 (Noonan), PTPN11 and RAF1 (Noonan, LEOPARD) [Schubbert et al., 2007; Tartaglia and Gelb, 2010; Tidyman and Rauen, 2009]. Importantly, strong activation of this critical signaling pathway is associated with lethality, therefore all the gain-of-function mutations in the germline of these patients are functionally and biochemically less powerful compared to the corresponding somatic mutations detected in sporadic cancer [Schubbert et al., 2007]. These causative mutations do impart predisposition to tumors and in fact all these syndromes show a mildly increased risk of cancer, with cumulative incidence by age 20 years ranging from 4% (Noonan) to 15% (Costello) [Kratz et al., 2011]. Tumors include rhabdomyosarcoma, neuroblastoma, ganglioneuroblastoma, bladder carcinoma and juvenile myelomonocytic leukaemia (the latter usually associated with Noonan syndrome) [Kratz et al., 2011; Schubbert et al., 2007; Tartaglia and Gelb 2010; Tidyman and Rauen 2009].
Phakomatoses
Next in the clinical spectrum are the phakomatoses, diseases with a presentation equally characterized by developmental defects and cancer predisposition (Table I). The name derives from phakos (φακός), i.e. “spot”, “lens” in Greek, as used for the first time by the Dutch ophthalmologist Jan van der Hoeve in 1920, to describe characteristic retinal lesions in tuberous sclerosis patients.
Table I.
Phakomatoses
| Syndrome | Clinical appearance | Gene |
|---|---|---|
| Neurofibromatosis 1a | Café-au-lait spots, Lisch nodules, axillary freckling, dermal and plexiform neurofibromas, tibial dysplasia, predisposition to malignant peripheral nerve sheath tumors, gliomas, astrocytomas, pheochromocytomas and juvenile chronic myelogenous leukemia | NF1 |
| Neurofibromatosis 2 | Neurofibromas, bilateral vestibular Schwannomas (previously, acoustic neurinomas) and hearing loss, cranial meningiomas, spinal astrocytomas and ependymomas, juvenile cataract | NF2 |
| Tuberous Sclerosis Complex | Cortical tubers and seizures, subependymal nodules and giant cell astrocytomas, renal angiomyolipomas, cysts and carcinomas, cardiac rhabdomyomas, pulmonary lymphangioleiomyomatosis, retinal phakomas, intellectual disability and autistic behavior, angiofibromas, hypomelanotic spots | TSC1, TSC2 |
| Von-Hippel-Lindau Syndrome | Retinal angiomas, central nervous system hemangioblastomas, renal cysts and tumors, pancreatic cysts and tumors, endolymphatic sac tumors, pheochromocytomas | VHL |
| Gorlin Syndrome or Basal Cell Nevus Syndrome | Multiple basal cell carcinomas, jaw cysts, medulloblastoma, macrocephaly, bifid ribs and other skeletal anomalies, palmar and plantar pits, ocular hypertelorism, enlarged jaw, calcification of falxcerebri | PTCH |
| Cowden Diseaseb | Trichilemmomas, papillomatous papules, acral keratosis; breast fibroadenomas and adenocarcinoma, macrocephaly, multinodular goiter, thyroid adenomas and follicular carcinoma, hamartomatous gastrointestinal polyps, multiple uterine leiomyomas, bicornate uterus and endometrial carcinoma | PTEN |
| Bannayan-Riley-Ruvalcaba Syndromeb | Macrocephaly, lipomatosis, hemangiomatosis, and speckled penis; cancer risk and spectrum probably similar to Cowden Disease | PTEN |
| Peutz Jeghers Syndrome | Hamartomatous gastrointestinal polyps, mucocutaneous pigmentation, increased risk of gastrointestinal, breast, ovarian (sex cord tumor), cervical, lung and pancreatic cancer | STK11 (LKB1)c |
| Juvenile Polyposis Syndrome | Multiple gastrointestinal juvenile polyps, gastrointestinal cancer | SMAD4, BMPR1A |
| Adenomatous Polyposis | Colonic adenomas (polyps) and carcinomas, extracolonic tumors (upper gastrointestinal tract, desmoid, osteoma, thyroid, brain), congenital hypertrophy of the retinal pigment epithelium, epidermoid cysts, supernumerary teeth | APC |
| BAP1 Cancer Susceptibility Syndrome | Atypical melanocytic tumors, atypical Spitz tumors, uveal and cutaneous melanomas, malignant mesothelioma, renal cell carcinoma | BAP1 |
Neurofibromatosis 1 can also be classified as a rasopathy
Cowden Disease and Bannayan-Riley-Ruvalcaba Syndrome can be defined as PTEN Tumor Hamartoma Syndromes
Alternative gene name
The phakomatoses are classical tumor suppressor gene (TSG) syndromes that follow the elegant and illuminating two-hit paradigm originally proposed by Dr. Alfred Knudson to explain paradigmatically the genetics of retinoblastoma [Knudson, 1971]. As such, they are inherited in an autosomal dominant fashion because the first hit is a germline mutation of the relevant TSG, but the disease manifests recessively at the cellular/tissue level only after a somatic mutation/deletion/silencing event inactivates the second copy of the TSG.
The causative TSGs of phakomatoses are critical regulators of cellular proliferation, differentiation and apoptosis, that form a tight signaling network (Fig. 3) and effectively act as “gate-keepers” for the relevant target tissue [Kinzler and Vogelstein, 1997]. Thus, when the second hit takes place, as a result of altered coordination of proliferation, differentiation and apoptosis, the normal tissue homeostasis is subverted and characteristic benign focal lesions appear, generally defined as phakomas, which are either benign tumors (i.e. adenomas) or hamartomas (Table I). However, the phakomas can undergo additional mutations and evolve into malignant lesions, generally carcinomas, as epithelial tissues typically require more than two mutations for malignant transformation [Knudson, 2001]. In other words, the developmental manifestations of phakomatoses are due to the two-hit inactivation of the relevant TSG, whereas the malignant manifestations are caused by subsequent mutations in other TSGs and oncogenes. In keeping with their critical role in regulating tissue homeostasis, homozygous inactivation of the phakomatoses’ TSGs, as assessed in mouse models, causes embryonic lethality. Based on these considerations, the recently identified BAP1 Cancer Susceptibility Syndrome that predisposes to uveal and cutaneous melanomas, mesotheliomas and renal cancer, and is caused by mutations of the BAP1(BRCA1-associated protein-1) gene [Carbone et al., 2013; Farley et al., 2013; Testa et al., 2011; Testa et al., 2013; Wiesner et al., 2011], encoding a component of the polycomb group repressive deubiquitinase complex, should be considered a bona fide phakomatosis: i) its phakomas are atypical melanocytic proliferations in which both alleles of BAP1 have been inactivated; ii) these focal lesions can evolve into cutaneous melanoma; iii) functional consequences of BAP1 inactivation at the cellular level include deregulated proliferation; iv) homozygous inactivation of BAP1 in the mouse germline leads to embryonic lethality [Carbone et al., 2013; Dey et al., 2012; Testa et al., 2013].
Figure 3.

Network of oncoproteins (red) and tumor suppressor proteins (green); proteins encoded by phakomatoses’ genes are shown in dark green. Broken and continuous lines indicate indirect and direct biochemical action, respectively. GF and GFR, growth factor and growth factor receptor respectively.
Proteus syndrome, SOLAMEN syndrome and other disorders of the PI3K-AKT-PTEN pathway
Proteus syndrome is similar to the classical phakomatoses Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome caused by PTEN mutations [Eng 2003], but exhibits distinct clinical and molecular features. Proteus syndrome is characterized by segmental overgrowth that is progressive, disproportionate and distorting; hyperplasia of multiple tissues and organs derived from all three germ layers; linear epidermal nevi and nearly pathognomonic cerebriform connective tissue nevi; “lipomatous” overgrowth and lipoatrophy; and susceptibility to numerous tumors, in particular ovarian cystadenomas, meningiomas and parotid monomorphic adenoma [Biesecker and Sapp, 1993]. In this disorder, the developmental overgrowth is often accompanied by tumors over half the body. Unlike the inherited phakomatoses associated with TSG mutations, Proteus Syndrome is not inherited and is caused by a postzygotic mosaic activating mutation of the AKT1 protooncogene; the Glu17Lys mutation is present in lesions from more than 90% of Proteus Syndrome patients and is also frequently detected in cancer [Lindhurst et al., 2011; Opitz and Jorde, 2011]. The oncogenic kinase AKT1 acts downstream of phosphatidylinositol 3′-kinase (PI3K) and controls critical cellular processes, such proliferation, survival, and intermediary metabolism, in particular glucose metabolism and insulin signaling [Bellacosa et al., 2005]. A critical step in the activation of AKT family members (AKT1, AKT2 and AKT3) is translocation to the membrane through binding of their pleckstrin homology (PH) domain to the PI3K-generated 3′-phosphorylated phosphoinositide lipids PIP3 and PIP2, a process that is directly antagonized by the lipid phosphatase activity of PTEN [Bellacosa et al., 2005]. The AKT1 Glu17Lys mutation is predicted to increase the positive charges of the PH domain and therefore the affinity for 3′-phosphorylated phospholipids [Bellacosa et al., 1998], thus leading to constitutive activation of the kinase [Lindhurst et al., 2011]. Clearly, this oncogenic mutation is tolerated only in a mosaic state, hence the segmental manifestations. Similarly to classical phakomatoses, subsequent somatic mutational events need to occur, in addition to the AKT1 mutation, for cancer to arise.
SOLAMEN Syndrome (Segmental Overgrowth, Lipomatosis, Arteriovenous Malformation and Epidermal Nevus) has also been defined as Proteus-like syndrome [Eng 2003] or Type 2 Segmental Cowden Disease [Happle 2007], but its pathogenesis is clearly different. In fact, it arises in a background of Cowden Disease (germline PTEN mutation) through the occurrence of a mosaic, second hit of PTEN, hence yielding segmental manifestations resembling Proteus syndrome [Caux et al., 2007; Happle 2007]. Other conditions caused by activating mutations of the PI3K-AKT axis variously mimic the disorders above (Fig. 4). Germline or mosaic Glu17Lys mutation of AKT2 leads to asymmetrical overgrowth and hypoglycemia [Hussain et al., 2011]. Activating germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA (both encoding catalytic subunits of PI3K) cause two overgrowth syndromes, Megalencephaly-capillary malformation (MCAP) and Megalencephaly-polymicrogyria-polydactyly-hydrocephalus (MPPH) syndromes, associated with a mild tumor predisposition (Wilms tumor, leukemia, medulloblastoma and meningioma) [Riviere et al., 2012]. Mosaic Glu17Lys mutation in AKT3 is associated with hemimegalencephaly, a condition resembling tuberous sclerosis complex [Poduri et al., 2012]. Finally, a syndrome of fibroadipose overgrowth is caused by mosaic activating mutations in PIK3CA [Lindhurst et al., 2012]. This pathway is activated in another overgrowth syndrome, Beckwith-Wiedemann syndrome (macrosomia, macroglossia, abdominal wall defects, visceromegaly, ear creases and/or pits, renal malformations, neonatal hypoglycemia, increased risk of Wilmstumor, hepatoblastoma and other embryonal tumors), through biallelic expression, due to loss of imprinting, of IGF2, encoding a growth factor upstream of PI3K-AKT and RAS-RAF-MEK-ERK signaling [Choufani et al., 2013; Pollak 2008].
Figure 4.
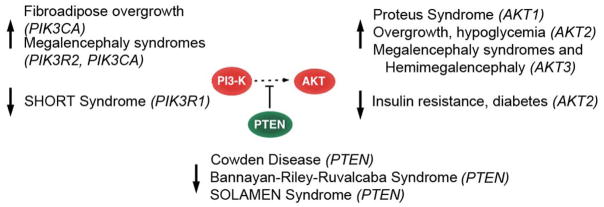
Disorders of the PI3K-AKT-PTEN pathway. Up- and down-arrows indicate activating (gain-of-function) and inhibiting (loss-of-function) mutations, respectively.
Interestingly, the PI3K-AKT axis is also targeted by loss-of-function mutations, leading to conditions that are the phenotypic opposite of overgrowth syndromes (Fig. 4). SHORT syndrome is caused by inactivating mutation of the PIK3R1 gene, encoding a catalytic subunit of PI3K, and is characterized by short stature, hyperextensibility of joints, ocular depression, Rieger anomaly, teething delay, lipodystrophy and insulin resistance or diabetes [Chudasama et al., 2013; Dyment et al., 2013; Thauvin-Robinet et al., 2013]. Lipoatrophy, insulin resistance and diabetes were also described in a family segregating an inactivating mutation of AKT2 [George et al., 2004].
Recessive chromosome breakage syndromes associated with cancer predisposition
Further right on the clinical spectrum (Fig. 2) are the classically defined chromosome breakage syndromes inherited as recessive disorders (Table II). These syndromes have in recent years expanded beyond the classical four: Ataxia Teleangectasia, Fanconi Anemia, Bloom syndrome and Xeroderma Pigmentosum. Cancer predisposition dominates the clinical framework, but notable dysmorphic features and birth defects are present (Table II). In fact, there is a “spectrum within the spectrum”, because the extent of the developmental defects varies within this disease group; for instance, the recently identified MUTYH-associated polyposis, an autosomal recessive disease caused by defective base excision repair (Table II), lacks developmental features. On the other hand, some related recessive syndromes, such as Cockayne syndrome, Trichothiodystrophy and Seckelsyndrome, do not display any cancer predisposition and are not discussed here.
Table II.
Chromosome Breakage Syndromes Inherited as Recessive Disorders and Associated with Cancer Predisposition
| Syndrome | Clinical Appearance | Chromosome Effects | Gene |
|---|---|---|---|
| Ataxia-Telangiectasia | Progressive cerebellar ataxia; oculo-cutaneous telangiectases, immune deficiency; predisposition to cancers, especially leukemias and lymphomas; elevated cancer incidence in heterozygotes | Chromatid breaks; formation of dicentric and acentric chromosomes, radiosensitivity | ATM |
| Xeroderma Pigmentosum | Sensitivity to sunlight, leading to “parchment skin” with pigmentary disturbances, multiple skin cancers (basal cell and squamous cell carcinoma, melanoma), leukemias, sarcomas; mental deterioration, ocular abnormalities | Defective nucleotide excision repair, elevated number of crossovers between sister chromatids | XPA-XPG, XPV |
| Bloom Syndrome | Short stature, disfiguring facial rashes made worse by sunlight; immunological problems; predisposition to leukemias and lymphomas, Wilms tumor, medulloblastoma and other solid tumors | Chromosome breaks; elevated number of crossovers between sister chromatids | BLM |
| Fanconi Anemia | Anemia and pancytopenia; short stature; radial aplasia and other skeletal defects; hyper- or hypopigmentation and cafe-au-lait spots; renal and genitourinary malformations; cardiac, gastrointestinal, and central nervous system abnormalities, predisposition to cancer, especially leukemia but also solid tumors | Chromatid breaks, gaps; exchange of segments between chromosomes; hypersensitivity to cross-linking agents | FANCA, FANCB, FANCC, BRCA2 a (FANCD1)b, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL |
| AT-like disorder | Ataxia, immune deficiency, neurological problems, predisposition | Chromosome breaks, radiosensitivity | MRE11A |
| Nijmegen Breakage Syndrome | Microcephaly, distinct facial appearance, short stature, immunodeficiency, radiation sensitivity, predisposition to lymphoma, glioma, medulloblastoma, rhabdomyosarcoma | Chromosome breaks | NBS1 |
| Mosaic Variegated Aneuploidy | Microcephaly, short stature, eye abnormalities, CNS abnormalities, seizures, intellectual disability, predisposition to Wilms tumor, nephroblastoma, rhabdomyosarcoma, leukemia | Anueploidy | BUB1B |
| Werner Syndrome | Premature aging, short stature, cancer predisposition (especially sarcomas) | Chromosomal deletions and translocations | WRN |
| Rothmund-Thomson Syndrome | Skin atrophy, telangiectasia, hyper- and hypopigmentation, skeletal abnormalities, elevated risk of cancer (especially osteosarcomas and skin cancer) | Chromosomal instability | RECQL4 |
| LIG4 Syndrome | Unusual facies, microcephaly, developmental delay, pancytopenia, immune deficiency, predisposition to leukemia | Defects in DNA strand break repair, radiosensitivity, | LIG4 |
| 46BR Syndrome | Immune deficiency, sun sensitivity, stunt growth, predisposition to lymphoma | Chromosomal instability | LIG1 |
| MUTYH-Associated Polyposis | Colonic adenomas and cancer, duodenal and gastric adenomas and cancer, elevated risk of ovarian, bladder, breast, endometrial and thyroid cancer | Defects in excision repair of adenine opposite 8-oxoguanine, resulting in elevated C:G to A:T transversions | MUTYH |
Biallelic mutations in BRCA2 cause Fanconi Anemia (recessive), whereas monoallelic mutations are associated with BRCA2 breast/ovarian cancer syndrome (dominant).
Alternative gene name
Genes involved in recessive chromosome breakage syndromes encode proteins involved in DNA repair or DNA damage response and therefore their role in preventing developmental manifestations and cancer is related to their function in the maintenance of genome stability. Not only exogenous but also endogenous genotoxic damage is a significant problem; e.g. it is estimated that approximately 50 endogenous double strand DNA breaks are produced per cell per cell cycle and need to be repaired efficiently, because even a single double strand DNA break can cause cell killing [Vilenchik and Knudson, 2003]. It is possible that the rapidly proliferating tissues of the developing organism may be particularly sensitive to the lack of DNA damage response and repair; alternatively, accumulation of DNA damage in progenitor/stem cells may cause their apoptosis with consequent clinical manifestations.
Dominant hereditary cancer syndromes
The last group of conditions along the clinical spectrum (Fig. 2) is represented by the dominant hereditary cancer syndromes. Unlike the previous group, these syndromes are dominantly inherited but are recessive at the cellular level, i.e. they follow the Knudson’s two-hit paradigm. Some of these conditions, such as the BRCA1-BRCA2 breast/ovarian cancer syndrome and the Hereditary Non-Polyposis Colorectal Cancer or Lynch syndrome are caused by mutations in genome stability genes, involved in DNA double strand break or mismatch repair, respectively. Since these TSGs act as “caretakers” of the genome [Kinzler and Vogelstein 1997], rather than “gatekeepers” like the phakomatoses’ genes, the largely predominant clinical feature is cancer predisposition, while major developmental defects are absent. Still, some minor morphogenetic defects might be present. For instance, 85% of the BRCA2 carriers (i.e. BRCA2+/− women) exhibit papillomatosis and invaginations of the ovarian surface epithelium with inclusion cysts [Salazar et al., 1996]. Similarly, increased hyalinization of the intralobular stroma and alteration in the branching pattern of breast lobules I have been detected in BRCA1+/− women [Russo et al., 2001]. These single-hit effects might be related to altered gene expression detected in morphologically normal epithelial cells from carriers of BRCA1 or BRCA2 mutations [Bellacosa et al., 2010]. On the other hand, mild developmental signs reminiscent of those in neurofibromatosis 1 (café-au-lait spots) are apparent in children with constitutional mismatch repair-deficiency syndrome, in which biallelic inheritance of mismatch repair gene mutations leads to childhood cancers (leukemias/lymphomas, brain cancer, in particular glioblastoma) [Wimmer and Etzler, 2008]. Finally, the recently identified germline mutations of the developmental gene HOXB13 in hereditary prostate cancer families [Ewing et al., 2012], suggest that cancer predisposition may stem from altered prostate morphogenesis. In fact, targeted disruption of HoxB13 in the murine germline is associated with overgrowth of tail bud structures (spinal cord and tail vertebrae) and lobe-specific abnormalities of the prostate gland without evidence of preneoplastic lesions [Economides and Capecchi, 2003; Economides et al., 2003].
DISCUSSION
The clinical vignettes presented above indicate that there exists a large number of clinical conditions exhibiting varying levels of developmental defects and cancer predisposition, thus spanning the now-closer-than-ever fields of clinical genetics and clinical oncology. While the organization of these diseases along this continuum may help derive, as I have tried to do, general considerations on their molecular and clinicopathological features, it would be also beneficial to develop new therapeutic approaches dictated by the overlapping molecular and clinical features.
Thus, given the molecular and clinical overlaps described above, it appears that a possible strategy in the future would be to utilize cancer drugs for developmental conditions. The mTOR inhibitor rapamycin that blocks the signaling pathway downstream of AKT-PTEN shows great promise for the treatment of Cowden syndrome, Tuberous Sclerosis Complex and perhaps Proteus syndrome [Marsh et al., 2011; Marsh et al., 2008; Truchuelo et al., 2012]; in the future, PI3K and/or AKT inhibitors might be useful for the treatment of overgrowth syndromes. Along these lines, BRAF and/or MEK and MAPK inhibitors may be very effective in the treatment of rasopathies. A related approach may involve correcting the underlying metabolic defect(s). This approach has proven effective in experimental systems; for instance, catecholamine administration rescues Gata3-mutant murine embryos beyond their nominal time of death at embryonic day (E)11 up to E16.5 [Lim et al., 2000].
Vice versa, developmental approaches may become very effective for cancer treatment. In pioneering experiments in the mouse, malignant cells were reprogrammed to normalcy when injected into a blastocyst, thus providing proof-of-principle of the success of this approach [Mintz, 1978]. As our understanding of the epigenetic mechanisms governing normal development and reprogramming increases, it should be possible to attempt the application of differentiation therapy to several tumor types perhaps through the use of small molecules or gene therapy approaches. Such an approach may receive a success reminiscent of the differentiation effects observed with all-trans retinoic acid in patients with acute promyelocytic leukemia [Huang et al., 1988].
Acknowledgments
Grant sponsor: National Cancer Institute CA078412 to AB, CA06927 to Fox Chase Cancer Center, a grant with the Pennsylvania Department of Health, and an appropriation from the Commonwealth of Pennsylvania.
This article is dedicated to Prof. Giovanni Neri, my former Professor of Genetics at the Catholic University Medical School in Rome, who influenced my academic development in so many different ways, through both professional mentorship and personal advice, and in particular by giving equal emphasis to two parallel lines of research in the Department of Human Genetics, later Medical Genetics, that he leads: developmental diseases and cancer.
Many concepts and considerations presented in this review originated from fruitful and rewarding conversations with Dr. Alfred Knudson (including on the concept of Fig. 3), Dr. Beatrice Mintz, and the speakers of the Symposium on “Order and ‘Disorder’ in Development and Cancer”, held in their honor at the Fox Chase Cancer Center on November 1–2, 2012.
I would like to thank past and present members of my research group for thoughtful comments and open discussions over the years; Drs. A. G. Knudson, A.T. Meadows, F. Roegiers, J. R. Testa and R. Tricarico for critical reading of the manuscript; and R. Sonlin for secretarial assistance. The author apologizes to colleagues for having failed to cite their work due to space constraints.
References
- Baylin S. Stem Book (Internet) Stem cells, cancer, and epigenetics. Cambridge MA: Harvard Stem Cell Institute; 2009. [PubMed] [Google Scholar]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Godwin AK, Peri S, Devarajan K, Caretti E, Vanderveer L, Bove B, Slater C, Zhou Y, Daly M, Howard S, Campbell KS, Nicolas E, Yeung AT, Clapper ML, Crowell JA, Lynch HT, Ross E, Kopelovich L, Knudson AG. Altered gene expression in morphologically normal epithelial cells from heterozygous carriers of BRCA1 or BRCA2 mutations. Cancer Prev Res. 2010;3:48–61. doi: 10.1158/1940-6207.CAPR-09-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- Biesecker LG, Sapp JC. Proteus Syndrome. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. GeneReviews. Seattle WA: University of Washington, Seattle; 1993. [Google Scholar]
- Bose T, Gerton JL. Cohesinopathies, gene expression, and chromatin organization. The Journal of cell biology. 2010;189:201–210. doi: 10.1083/jcb.200912129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nature reviews Cancer. 2013;13:153–159. doi: 10.1038/nrc3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caux F, Plauchu H, Chibon F, Faivre L, Fain O, Vabres P, Bonnet F, Selma ZB, Laroche L, Gerard M, Longy M. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur J Hum Genet. 2007;15:767–773. doi: 10.1038/sj.ejhg.5201823. [DOI] [PubMed] [Google Scholar]
- Choufani S, Shuman C, Weksberg R. Molecular findings in Beckwith-Wiedemann syndrome. American journal of medical genetics Part C, Seminars in medical genetics. 2013;163:131–140. doi: 10.1002/ajmg.c.31363. [DOI] [PubMed] [Google Scholar]
- Chudasama KK, Winnay J, Johansson S, Claudi T, Konig R, Haldorsen I, Johansson B, Woo JR, Aarskog D, Sagen JV, Kahn CR, Molven A, Njolstad PR. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet. 2013;93:150–157. doi: 10.1016/j.ajhg.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, Kirkpatrick DS, Pham VC, Lill JR, Bakalarski CE, Wu J, Phu L, Katavolos P, LaFave LM, Abdel-Wahab O, Modrusan Z, Seshagiri S, Dong K, Lin Z, Balazs M, Suriben R, Newton K, Hymowitz S, Garcia-Manero G, Martin F, Levine RL, Dixit VM. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012;337:1541–1546. doi: 10.1126/science.1221711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyment DA, Smith AC, Alcantara D, Schwartzentruber JA, Basel-Vanagaite L, Curry CJ, Temple IK, Reardon W, Mansour S, Haq MR, Gilbert R, Lehmann OJ, Vanstone MR, Beaulieu CL, Majewski J, Bulman DE, O’Driscoll M, Boycott KM, Innes AM. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. 2013;93:158–166. doi: 10.1016/j.ajhg.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economides KD, Capecchi MR. Hoxb13 is required for normal differentiation and secretory function of the ventral prostate. Development. 2003;130:2061–2069. doi: 10.1242/dev.00432. [DOI] [PubMed] [Google Scholar]
- Economides KD, Zeltser L, Capecchi MR. Hoxb13 mutations cause overgrowth of caudal spinal cord and tail vertebrae. Dev Biol. 2003;256:317–330. doi: 10.1016/s0012-1606(02)00137-9. [DOI] [PubMed] [Google Scholar]
- Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- Ewing CM, Ray AM, Lange EM, Zuhlke KA, Robbins CM, Tembe WD, Wiley KE, Isaacs SD, Johng D, Wang Y, Bizon C, Yan G, Gielzak M, Partin AW, Shanmugam V, Izatt T, Sinari S, Craig DW, Zheng SL, Walsh PC, Montie JE, Xu J, Carpten JD, Isaacs WB, Cooney KA. Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med. 2012;366:141–149. doi: 10.1056/NEJMoa1110000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley MN, Schmidt LS, Mester JL, Pena-Llopis S, Pavia-Jimenez A, Christie A, Vocke CD, Ricketts CJ, Peterson J, Middelton L, Kinch L, Grishin N, Merino MJ, Metwalli AR, Xing C, Xie XJ, Dahia PL, Eng C, Linehan WM, Brugarolas J. Germline BAP1 mutation predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res. 2013 doi: 10.1158/1541-7786.MCR-13-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganmore I, Smooha G, Izraeli S. Constitutional aneuploidy and cancer predisposition. Hum Mol Genet. 2009;18:R84–93. doi: 10.1093/hmg/ddp084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, Dunger DB, Barford D, Umpleby AM, Wareham NJ, Davies HA, Schafer AJ, Stoffel M, O’Rahilly S, Barroso I. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happle R. Type 2 segmental Cowden disease vs. Proteus syndrome. Br J Dermatol. 2007;156:1089–1090. doi: 10.1111/j.1365-2133.2007.07818.x. [DOI] [PubMed] [Google Scholar]
- Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, Gu LJ, Wang ZY. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72:567–572. [PubMed] [Google Scholar]
- Hussain K, Challis B, Rocha N, Payne F, Minic M, Thompson A, Daly A, Scott C, Harris J, Smillie BJ, Savage DB, Ramaswami U, De Lonlay P, O’Rahilly S, Barroso I, Semple RK. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334:474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386:761–763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- Kline AD, Grados M, Sponseller P, Levy HP, Blagowidow N, Schoedel C, Rampolla J, Clemens DK, Krantz I, Kimball A, Pichard C, Tuchman D. Natural history of aging in Cornelia de Lange syndrome. American journal of medical genetics Part C, Seminars in medical genetics. 2007;145C:248–260. doi: 10.1002/ajmg.c.30137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. American journal of medical genetics Part C, Seminars in medical genetics. 2011;157:83–89. doi: 10.1002/ajmg.c.30300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KC, Lakshmanan G, Crawford SE, Gu Y, Grosveld F, Engel JD. Gata3 loss leads to embryonic lethality due to noradrenaline deficiency of the sympathetic nervous system. Nat Genet. 2000;25:209–212. doi: 10.1038/76080. [DOI] [PubMed] [Google Scholar]
- Lindhurst MJ, Parker VE, Payne F, Sapp JC, Rudge S, Harris J, Witkowski AM, Zhang Q, Groeneveld MP, Scott CE, Daly A, Huson SM, Tosi LL, Cunningham ML, Darling TN, Geer J, Gucev Z, Sutton VR, Tziotzios C, Dixon AK, Helliwell T, O’Rahilly S, Savage DB, Wakelam MJ, Barroso I, Biesecker LG, Semple RK. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44:928–933. doi: 10.1038/ng.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L, Blumhorst C, Brockmann K, Calder P, Cherman N, Deardorff MA, Everman DB, Golas G, Greenstein RM, Kato BM, Keppler-Noreuil KM, Kuznetsov SA, Miyamoto RT, Newman K, Ng D, O’Brien K, Rothenberg S, Schwartzentruber DJ, Singhal V, Tirabosco R, Upton J, Wientroub S, Zackai EH, Hoag K, Whitewood-Neal T, Robey PG, Schwartzberg PL, Darling TN, Tosi LL, Mullikin JC, Biesecker LG. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannini L, Menga S, Musio A. The expanding universe of cohesin functions: a new genome stability caretaker involved in human disease and cancer. Hum Mutat. 2010;31:623–630. doi: 10.1002/humu.21252. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Trahair TN, Kirk EP. Mutant AKT1 in Proteus syndrome. N Engl J Med. 2011;365:2141–2142. doi: 10.1056/NEJMc1111367. author reply 2142. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Trahair TN, Martin JL, Chee WY, Walker J, Kirk EP, Baxter RC, Marshall GM. Rapamycin treatment for a child with germline PTEN mutation. Nat Clin Pract Oncol. 2008;5:357–361. doi: 10.1038/ncponc1112. [DOI] [PubMed] [Google Scholar]
- Mintz B. Clonal basis of mammalian differentiation. Symp Soc Exp Biol. 1971;25:345–370. [PubMed] [Google Scholar]
- Mintz B. Gene expression in neoplasia and differentiation. Harvey Lect. 1978;71:193–246. [PubMed] [Google Scholar]
- Opitz JM, Jorde LB. Hamartoma syndromes, exome sequencing, and a protean puzzle. N Engl J Med. 2011;365:661–663. doi: 10.1056/NEJMe1107384. [DOI] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, Barry BJ, Bourgeois BF, Riviello JJ, Barkovich AJ, Black PM, Ligon KL, Walsh CA. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nature reviews Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- Ponder BA. Cancer genetics. Nature. 2001;411:336–341. doi: 10.1038/35077207. [DOI] [PubMed] [Google Scholar]
- Riviere JB, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, Worthylake T, Sullivan CT, Ward TR, Butler HE, Kramer NA, Albrecht B, Armour CM, Armstrong L, Caluseriu O, Cytrynbaum C, Drolet BA, Innes AM, Lauzon JL, Lin AE, Mancini GM, Meschino WS, Reggin JD, Saggar AK, Lerman-Sagie T, Uyanik G, Weksberg R, Zirn B, Beaulieu CL, Majewski J, Bulman DE, O’Driscoll M, Shendure J, Graham JM, Jr, Boycott KM, Dobyns WB. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo J, Lynch HT, Russo IH. Mammary gland architecture as a determining factor in the susceptibility of the human breast to cancer. Breast J. 2001;7:278–291. doi: 10.1046/j.1524-4741.2001.21033.x. [DOI] [PubMed] [Google Scholar]
- Salazar H, Godwin AK, Daly MB, Laub PB, Hogan WM, Rosenblum N, Boente MP, Lynch HT, Hamilton TC. Microscopic benign and invasive malignant neoplasms and a cancer-prone phenotype in prophylactic oophorectomies. J Natl Cancer Inst. 1996;88:1810–1820. doi: 10.1093/jnci/88.24.1810. [DOI] [PubMed] [Google Scholar]
- Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Gelb BD. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann N Y Acad Sci. 2010;1214:99–121. doi: 10.1111/j.1749-6632.2010.05790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, Cox NJ, Dogan AU, Pass HI, Trusa S, Hesdorffer M, Nasu M, Powers A, Rivera Z, Comertpay S, Tanji M, Gaudino G, Yang H, Carbone M. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43:1022–1025. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa JR, Malkin D, Schiffman JD. Connecting molecular pathways to hereditary cancer risk syndromes. Am Soc Clin Oncol Educ Book. 2013;2013:81–90. doi: 10.1200/EdBook_AM.2013.33.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J, Le Merrer M, Le Luyer B, Heron D, Mathieu-Dramard M, Bitoun P, Petit JM, Odent S, Amiel J, Picot D, Carmignac V, Thevenon J, Callier P, Laville M, Reznik Y, Fagour C, Nunes ML, Capeau J, Lascols O, Huet F, Faivre L, Vigouroux C, Riviere JB. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet. 2013;93:141–149. doi: 10.1016/j.ajhg.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truchuelo T, Diaz-Ley B, Rios L, Alcantara J, Jaen P. Facial angiofibromas treated with topical rapamycin: an excellent choice with fast response. Dermatol Online J. 2012;18:15. [PubMed] [Google Scholar]
- Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci USA. 2003;100:12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, Windpassinger C, Wackernagel W, Loy S, Wolf I, Viale A, Lash AE, Pirun M, Socci ND, Rutten A, Palmedo G, Abramson D, Offit K, Ott A, Becker JC, Cerroni L, Kutzner H, Bastian BC, Speicher MR. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43:1018–1021. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet. 2008;124:105–122. doi: 10.1007/s00439-008-0542-4. [DOI] [PubMed] [Google Scholar]
- Xiong B, Gerton JL. Regulators of the cohesin network. Annu Rev Biochem. 2010;79:131–153. doi: 10.1146/annurev-biochem-061708-092640. [DOI] [PubMed] [Google Scholar]