

Summary
Background
Constitutive activation of kit contributes to pathogenesis of acute myeloid leukemia (AML) and targeting Kit may be of therapeutic benefit. APcK110, a novel inhibitor of Kit, has potent proapoptotic and antiproliferative activity in AML cell lines and primary AML samples. Here we extend our studies to the activity of APcK110 in a xenograft mouse model.
Methods
After sub-lethal whole body radiation, OCI/AML3 cells were injected intravenously in NOD-SCID mice. Ten days later, either APcK110 or phosphate buffered saline (PBS) was injected intraperitoneally every other day. Kaplan-Meier estimates were used to calculate survival.
Results
We show that 1) all mice injected with OCI/AML3 cells developed a clinical and histological picture consistent with myelomonocytic AML; and 2) survival of APcK110-treated mice was significantly longer compared with mice injected with PBS (p=.02).
Conclusions
APcK110 is a novel kit kinase inhibitor with anti-AML activity in vitro and in vivo. Further evaluation in toxicology and clinical studies is warranted.
Keywords: APcK110, Acute myeloid leukemia, Kit inhibitor
Introduction
Acute myeloid leukemias (AML) are characterized by dysregulated hematopoiesis leading to expansion of hematopoietic progenitor cells in marrow, blood and other organs. This process is driven by acquisition of mutations inside hematopoietic progenitor cells that impair differentiation and stimulate proliferation thus providing a select group of cells a survival advantage over its undisturbed counterparts [5, 11, 17]. Although initiating events remain obscure and it can be assumed that several molecular hits are involved, a crucial role has been ascribed to abnormally activated protein tyrosine kinases [6].
This may not surprise as protein kinases function in key positions of almost all aspects of cell biology and therefore require tight control and regulation. Kit [stem cell factor (SCF) receptor] is a class III transmembrane receptor tyrosine kinase [9, 15]. Binding of its ligand SCF to kit results in receptor dimeriziation, tyrosine phosphorylation and activation of downstream signaling pathways including the PI3K/AKT, Ras/MAP kinase, and JAK/STAT systems [14]. Kit expression is identified on most blasts of patients with AML (63% to 90%) [8, 16]. Whereas controlled activation of kit and its downstream partners plays a major role in maintaining normal hematopoiesis, constitutive activation of the receptor has been implicated in AML pathogenesis. Gain-of-function mutations of KIT are one mechanism resulting in unchecked signaling. Although these mutations are rare in unselected patients with AML they are found in 17% to 46.1% of patients with core binding factor (CBF) leukemias with translocations t(8;21), inv(16), or translocation t (16;16) and where they have been associated with worse outcome [2, 3, 12, 13]. Early clinical studies using tyrosine kinase inhibitors imatinib and dasatinib in combination with chemotherapy have been reported, and there is growing interest in the development of stronger and more specific kit inhibitors in AML therapy [1, 7]. We have recently demonstrated that APcK110, a novel inhibitor of kit, inhibited proliferation of the SCF-responsive human AML cell line OCI/AML3 more potently than either imatinib or dasatinib and also inhibited proliferation of primary AML blasts in a clonogenic assay, but having little effect on normal colony-forming cells [4]. Although OCI/AML3 cell lines do not express mutations of KIT, we chose this cell line because in our view it represents most closely processes that are relevant in AML pathobiology. We here extend our investigations of APcK110 to an AML xenograft mouse model to further characterize the activity of this compound.
Materials and methods
Cell line
AML cell line OCI/AML3 was provided by M.D. Minden (Ontario Cancer Institute, Toronto, ON, Canada). OCI/AML3 cells proliferate in the presence of culture medium and FCS without exogenous growth factors and do not harbor mutations of KIT[16]. Cells were maintained in RPMI 1640 (Life Technologies) supplemented with 10% FCS (Flow Laboratories), grown in plastic tissue culture flasks (Falcon Plastics; Becton Dickinson), and split twice weekly.
Xenograft animal studies
All animal work was done in accordance with a protocol approved by the Institutional Animal Care and Use Committee. Eight-weeks-old female NOD-SCID mice (strain NOD.CB17-PRKDS SCID\J) were obtained from the Jackson Laboratory (Bar Harbor, ME) and maintained in our institutional animal facility. After exposure to 30 cGy sub-lethal whole-body radiation, 1×105 OCI/AML3 cells were injected intravenously. Ten days later, the mice were injected intraperitoneally every other day either with 500 nM ApCK110 [diluted in phosphate buffered saline (PBS)] or PBS at the same volume. The mice were sacrificed in accordance with the experimental protocol or when they became moribund or unable to obtain food or water or if they lost >20% of their body weight. Necropsy was performed on all animals. Bone marrow, blood, and organs (liver spleen, lungs, heart, kidneys, and long and spinal bones) of all mice were preserved in formalin for histological analysis.
Histology, cyto- and immune-histochemistry
Bone and the various organ specimens were processed for histological analysis according to standard procedure [10]. Bone marrow cells were obtained from the tibia and both glass slides and cytospins were prepared. Glass slides of blood and marrow smears and with cytospun mouse blood or bone marrow cells were stored at 4°C. For analysis, the slides were incubated with normal mouse serum (Sigma, St. Louis, MO) for 1 h in a humid environment and washed 3 times for 5 min in PBS (Gibco). Then, 20 μl of either rabbit anti-human CD45 or phospho-Kit antibodies (R&D Systems Minneapoplis, MN) or their isotypes were added and the glass slides were covered with a plastic coverslip and the slides were incubated in the dark at room temperature for 1 h. After incubation, the slides were washed 3 times in PBS, dried and mounted with Vectashield mounting media for fluorescence (Vector Laboratories, Burlingame, CA). All slides were scanned, analyzed and photographed using a fluorescence microscope (Olympus, Center Valley, PA).
Statistical analysis
Kaplan-Meier estimates were used to calculate survival. Survival was defined as from the day of injection of either APcK110 or PBS until death.
Results
We first demonstrate that following intravenous infusion of OCI/AML3 cells all 20 immunodeficient mice developed a picture consistent with AML. Figure 1a shows high-power views of representative samples from blood and marrow. Wright stained marrow and blood highlight aggregates of heterogeneous large blast cells with basophilic cytoplasm, prominent nucleoli and nuclear indentations. Characterizing the blasts further, immunostains for myeloperoxidase (MPO) were negative, but positive for butyrate and anti-human CD45. Figure 1b shows low-power views of Wright and anti-human CD45 stained marrow, lung, and liver. The marrow sample was hypercellular with sheets of blasts throughout the cut. Sections of lung and liver showed diffuse infiltration of the tissues with immature blast cells. The mice were then randomized to be injected intraperitoneally with either APcK110 or PBS. Necropsy was performed on all animals for the criteria outlined above. Although sections of marrow samples after treatment with APcK110 (Wright stains and stains for anti-human CD45) still revealed evidence of residual AML at the time of death (Fig. 1c), we found that those mice which were injected with the kit inhibitor APcK110 lived significantly longer than those treated with PBS (P=.02) (Fig. 2).
Fig. 1.
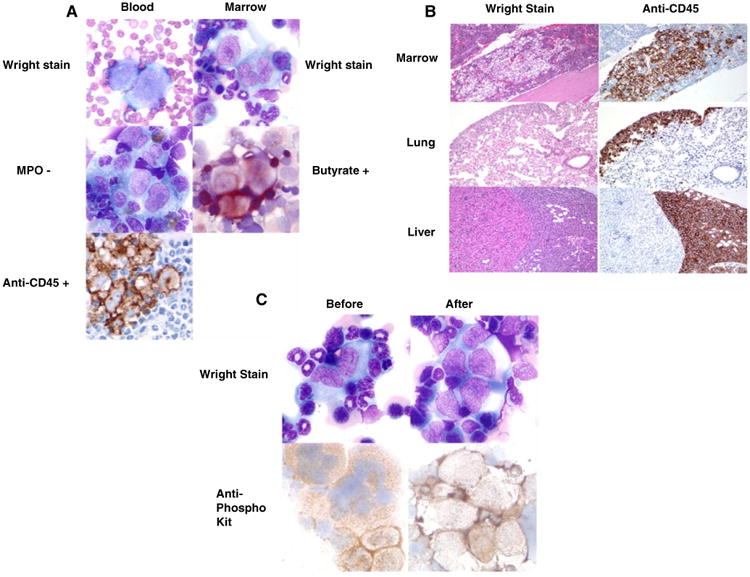
a) Marrow and blood stains demonstrating infiltration with myeloperoxidase-negative, butyrate- and anti-human CD45-positive blasts. b) Wright stain and anti-human CD45 stains in marrow, lung, and liver samples showing diffuse tissue involvement by AML. c) Marrow stains before and after intraperitoneal injection with APcK110
Fig. 2. Effect of APcK110 on survival of OCI/AML3-injected Nod-SCID mice.
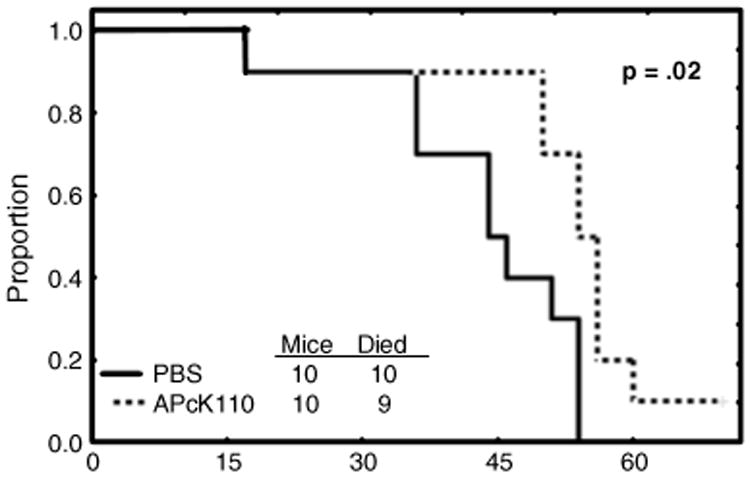
Intense daily monitoring did not detect any obvious drug-related toxicities. The mice lost weight, became moribund, and the necropsy showed extensive infiltrate in all organs. The findings were similar in treated and untreated animals.
Discussion
In past experiments we detected CD117 (kit) by flow cytometry in 15% of OCI/AML3 cells (unpublished data) and could also not identify mutations of KIT although in previous in vitro studies inhibition of proliferation of OCI/AML3 cells following exposure to APcK110 was similar to what we observed in a KIT mutation-positive mastocytosis cell line (HMC1.2) [4]. This finding could be explained by the fact that OCI/AML3 cells, that produce SCF, are SCF-responsive in that growth is stimulated autologously by SCF in an autocrine manner. By the same token, proliferation can be effectively inhibited by blocking the effect of SCF through its receptor kit even in the absence of detectable KIT mutations. This finding is of relevance for two reasons: 1) most blasts of patients with AML do not carry KIT mutations, but express kit at high percentages; and 2) effective inhibition of kit and abrogation of downstream signaling pathways can negatively impact AML proliferation. In in vitro studies of APcK110 we could indeed demonstrate inhibition of AML blast colony-forming cell proliferation in all but one sample which was characterized by absence of cell surface CD117 (0.3% by flow cytometry) [4]. The extent of inhibition of kit-positive AML blasts was not significantly different from what was observed in three samples of blasts, which carried D816V KIT mutations. Furthermore, phosphorylation of kit, Stat3, Stat5, and Akt in OCI/AML3 cells was effectively inhibited and caspase-dependent apopotosis induced by APcK110. Even though APcK110 was screened for its activity against kit, kinase profiling revealed a small number of other kinases which were also inhibited and included TGFBR2, MEK2, p38γ, RET, MEK4 and others. An off target effect can therefore not be excluded to explain at least partly the activity of the compound. In either case, the results appear to vindicate our choice of OCI/AML3 cells even in the absence of mutations of KIT. The cell lines represent closely of what is going on in AML. Our data that inhibition of Kit nevertheless prolonged the life of the animals suggest that inhibition of Kit in the vast majority of AML cases might be adventitious.
Clinical experience with attempts to inhibit kit has been mixed. In a study of 40 patients with AML or high-risk myelodysplastic syndrome (median age 73 years) who were either not eligible for myelosuppressive therapy and/or had relapsed disease, imatinib was combined with low-dose cytarabine [7]. Of 38 evaluable patients, a “blast response” was observed in 6 (16%) with an early mortality rate of 18.9%. The combination was not worse than myelosup-pressive therapy in a historic comparison, but was not superior to low-dose cytarabine alone either. In another study of 21 AML patients in first relapse with a median age of 47 years, standard dose cytarabine plus daunorubicin was combined with escalating doses of imatinib [1]. Eleven of 19 evaluable patients achieved either CR or CRp (58%), which in a couple of patients have been durable. Interestingly, whereas kit expression did not correlate with CR, patients with absent nuclear phospho-stat3 expression had a trend for a higher CR rate (82%) compared to patients with nuclear phospho-stat3 expression (33%; p=.18). Although small, the studies highlight the need for more accurate selection of patient populations based on molecular features. In addition, both studies used imatinib, which in our experience was a significantly weaker inhibitor of kit than APcK110 [4].
In the current study, we have extended our investigations of APcK110 to a xenograft mouse model where we also demonstrate improved survival of mice injected with the human AML cell line OCI/AML3. Further development of APcK110 in toxicology and clinical studies is therefore warranted.
References
- 1.Advani A, Copelan EA, Sobecks R, et al. A phase 1 trial of imatinib mesylate with daunorubicin and cytarabine for patients with c-kit positive relapsed AML. Blood. 2008;112:351. [Google Scholar]
- 2.Cairoli R, Beghini A, Grillo G, et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood. 2006;107:3463–3468. doi: 10.1182/blood-2005-09-3640. [DOI] [PubMed] [Google Scholar]
- 3.Du J, Schenk RF, Corbacioglu A, et al. Detection of RAS, KIT, and FLT3 gene mutation in t(8;21)-positive acute myeloid leukemia (AML): evaluation of the clinical relevance. Blood. 2006;108:652a. abstract. [Google Scholar]
- 4.Faderl S, Pal A, Bornmann W, et al. Kit inhibitor APcK110 induces apoptosis and inhibits proliferation of acute myeloid leukemia. Cancer Res. 2009;69:3910–3917. doi: 10.1158/0008-5472.CAN-08-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fröhling S, Scholl C, Gilliland DG, Levine RI. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23:6285–6294. doi: 10.1200/JCO.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 6.Gilliland DG, Speck NA. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. 2002;2:502–513. doi: 10.1038/nrc840. [DOI] [PubMed] [Google Scholar]
- 7.Heidel F, Cortes J, Rücker FG, et al. Results of a multicenter phase II trial for older patients with c-kit-positive acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (HR-MDS) using low-dose ara-C and imatinib. Cancer. 2007;109:907–914. doi: 10.1002/cncr.22471. [DOI] [PubMed] [Google Scholar]
- 8.Kanakura Y, Ikeda H, Kitayama H, et al. Expression, function and activation of the proto-oncogene c-kit product in human leukemia cells. Leuk Lymphoma. 1993;10:35–41. doi: 10.3109/10428199309147354. [DOI] [PubMed] [Google Scholar]
- 9.Kitamura Y, Hirota S. Kit as a human oncogenic tyrosine kinase- the stem cell factor receptor. Cell Mol Life Sci. 2004;61:2924–2931. doi: 10.1007/s00018-004-4273-y. [DOI] [PubMed] [Google Scholar]
- 10.Konoplev S, Huang X, Drabkin HA, et al. Cytoplasmic localization of nucleophosmin in bone marrow blasts of acute myeloid leukemia patients is not completely concordant with NPM1 mutation and is not predictive of prognosis. Cancer. 2009;115:4737–4744. doi: 10.1002/cncr.24543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mrózek K, Marcucci G, Paschka P, et al. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109:431–448. doi: 10.1182/blood-2006-06-001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nanri T, Matsuno N, Kawakita T, et al. Mutations in the receptor tyrosine kinase pathway are associated with clinical outcome in patients with acute myeloblastic leukemia harboring t (8;21)(q22;q22) Leukemia. 2005;19:1361–1366. doi: 10.1038/sj.leu.2403803. [DOI] [PubMed] [Google Scholar]
- 13.Paschka P, Marcucci G, Ruppert AS, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol. 2006;107:3904–3911. doi: 10.1200/JCO.2006.06.9500. [DOI] [PubMed] [Google Scholar]
- 14.Rönnstrand L. Signal transduction via the stem cell factor receptor/c-kit. Cell Mol Life Sci. 2004;61:2535–2548. doi: 10.1007/s00018-004-4189-6. [DOI] [PubMed] [Google Scholar]
- 15.Roskoski R., Jr Structure and regulation of Kit protein-tyrosine kinase—the stem cell factor receptor. Biochem Biophys Res Commun. 2005;338:1307–1315. doi: 10.1016/j.bbrc.2005.09.150. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz S, Heinecke A, Zimmermann M, et al. Expression of the c-kit receptor (CD117) is a feature of almost all subtypes of de novo acute myeloblastic leukemia (AML), including cytogenetically good-risk AML, and lacks prognostic significance. Leuk Lymphoma. 1999;34:85–94. doi: 10.3109/10428199909083383. [DOI] [PubMed] [Google Scholar]
- 17.Wouters BJ, Löwenberg B, Delwel R. A decade of genome-wide gene expression profiling in acute myeloid leukemia: flashback and prospects. Blood. 2009;113:291–298. doi: 10.1182/blood-2008-04-153239. [DOI] [PMC free article] [PubMed] [Google Scholar]