

Abstract
Porphyrin macrocycles have been the subject of intense study in the last century because they are widely distributed in nature, usually as metal complexes of either iron or magnesium. As such, they serve as the prosthetic groups in a wide variety of primary metabolites, such as hemoglobins, myoglobins, cytochromes, catalases, peroxidases, chlorophylls, and bacteriochlorophylls; these compounds have multiple applications in materials science, biology and medicine. This article describes current methodology for preparation of simple, symmetrical model porphyrins, as well as more complex protocols for preparation of unsymmetrically substituted porphyrin macrocycles similar to those found in nature. The basic chemical reactivity of porphyrins and metalloporphyrin is also described, including electrophilic and nucleophilic reactions, oxidations, reductions, and metal-mediated cross-coupling reactions. Using the synthetic approaches and reactivity profiles presented, eventually almost any substituted porphyrin system can be prepared for applications in a variety of areas, including in catalysis, electron transport, model biological systems and therapeutics.
Keywords: Aromaticity, porphyrin, pyrrole, reactivity, substitutions, synthesis
1. INTRODUCTION
In this article we review the widely-used methodology which has been utilized to synthesize tetrapyrrole macrocycles in the porphyrin series from monopyrrole precursors; we also review selected porphyrin reactivity profiles.
1.1. Monographs
The porphyrin area has been honored, over time, with a significant number of Nobel prizes for its heroes, and is thus a research field that is rich in its history. Between 1934 and 1940 Hans Fischer and the Munich porphyrin group published three volumes that became a roadmap for synthesis of porphyrin compounds [1-3]; in particular, these three volumes of Die Chemie des Pyrrols contained a compendium of laboratory methods. The tradition of laboratory methods within porphyrin monographs was continued in 1964 when Falk’s book “Porphyrins and Metalloporphyrins” was published [4]; this contained a fairly detailed laboratory methods section but also attempted, for the first time, to apply modern principles of chemistry and electronic theory to the porphyrin field. This was followed in 1974 by a new version of “Porphyrins and Metalloporphyrins” (edited by one of us), which also contained a laboratory methods section, and then by Dolphin’s more traditional series called “The Porphyrins” (1978-80). An authoritative and highly detailed description of the synthetic art of porphyrin chemistry and of reactivity profiles in the “octa-alkyl” and “tetra-aryl” porphyrin series can be found in “The Porphyrin Handbook”, which consists of two ten-volume sets containing no less than 122 chapters [5]. Since then, the follow-on series (30 volumes to date) by the same editors entitled “The Handbook of Porphyrin Science” has featured no less than six complete volumes with the word “synthesis” in their titles [6-11]. By and large, however, these more recent volumes [6-11] have dealt with syntheses of porphyrins with specific arrays of substituent for use in commercial applications, rather than on the development of new ways to synthesize the porphyrin core. A very thorough book dealing with syntheses and properties of porphyrin isomers, as well as contracted and expanded systems (outside the purview of this review) has also appeared [12].
1.2. Nomenclature
The Fischer system for nomenclature of porphyrins is shown in structure 1. Along with this simple numbering system, Fischer also created a host of trivial names for porphyrins [e.g. protoporphyrin (the “first” porphyrin), deuteroporphyrin (the “second” porphyrin), etioporphyrin] and for chlorophyll derivatives (chlorin-e6, rhoding7, etc.). Fischer also created a “type” isomer system of nomenclature (see later for the etioporphyrin examples) which was used to define the nature of the substituent array in certain porphyrins. For example, a porphyrin made from tetramerization of a monopyrrole containing one methyl and one ethyl group can have four isomers - types I-IV. Enumeration of isomers of substituted porphyrins, and the construction of combinatorial libraries, has been authoritatively reviewed by Taniguchi and Lindsey [13]. The Fischer system for porphyrin nomenclature (structure 1) provides a link back to the rich history of porphyrin chemistry. As biosynthetic studies of vitamin B12 and porphyrins began to assume a dominant position in organic and biological chemistry, and as unique radiochemical and stable isotope labeling became the norm in these studies, it became necessary to identify every carbon atom in the porphyrin ring uniquely, but the Fischer system (1) leaves several carbon atoms unassigned; to be sure, these unassigned carbons never carry a substituent in a porphyrin, but they assumed importance as the biological origin of every carbon in porphyrins and vitamin B12 was investigated. The IUPAC subsequently adopted a system of nomenclature (structure 2); this is the officially adopted nomenclature system, and is used in this article (Fig. 1).
Fig. (1).

Porphyrin nomenclature.
2. PORPHYRIN SYNTHESES
A full description of all currently available routes to porphyrins is outside the scope of this brief article. Such a treatment can be found elsewhere [14]. Instead, with increasing complexity, three of the most typical synthetic approaches will be described.
The choice of which synthetic route to use to synthesize any particular porphyrin depends upon the symmetry features of the product itself. Syntheses of porphyrins by simple polymerization of monopyrroles will be discussed first; typical examples of porphyrins which can be made from this approach are octaethylporphyrin 3 and 5,10,15,20-tetraphenylporphyrin 4 (Fig. 1). Next we shall progress to the so-called MacDonald route using two dipyrromethanes (the [2+2] approach) and one of its variants (the [3+1] method); porphyrins 5 and 6 are typical examples that can be approached synthetically in this way (Fig. 1). Finally, we shall finish the synthetic part of the article with porphyrin synthesis through cyclization of an open-chain tetrapyrrole (the so-called a,c-biladienes); this approach yields porphyrins such as 7, and is truly general because it possesses no symmetry restrictions.
A multitude of preparative routes to monopyrroles exist; reviews of this literature have been published [1, 15-17]. Methods for elaboration of monopyrroles into dipyrromethane, tripyrrane, and a,c-biladiene compounds will be discussed in the sections related to the porphyrins in which they are used as essential intermediates.
Porphyrin syntheses from monopyrroles are best described with reference to two classical porphyrins - octaethylporphyrin (3; OEP) and tetraphenylporphyrin (4; TPP). Simply based on the symmetry properties of these two porphyrins, the most economical approach must involve some kind of monopyrrole tetramerization, either using monopyrroles bearing a 2-substituent which can provide the four interpyrrolic carbons in the product, or using a 2,5-di-unsubstituted monopyrrole with a separate reagent which will furnish the one-carbon bridging atoms. Attempting to synthesize OEP 3 by laborious multi-step construction of an open-chain tetrapyrrolic intermediate would be wasteful in time and resources.
2.1. Porphyrins via Monopyrrole Tetramerization
Everyone in the porphyrin field agrees that the easiest porphyrin to synthesize is TPP, 4. One simply needs to react pyrrole 8 with benzaldehyde under acidic conditions. The route was first reported by Rothemund [18, 19], who preferred to carry out the reaction in sealed glass tubes at high temperature. Roald Hoffmann has related to one of us that explosion of such a sealed glass tube converted him into a theoretician, and subsequently, a 1981 Nobel Laureate in Chemistry. A modification by Adler, Longo and their colleagues (involving use of refluxing propionic acid instead of sealed tube chemistry) [20], reproducibly affords a 20-25% yield of TPP from these simple shelf chemicals). This trivial procedure involves addition of equimolar amounts of pyrrole 8 and benzaldehyde to refluxing propionic acid (Scheme 1); after heating for about one half hour, the mixture is allowed to cool and the TPP is filtered off. The reaction also works with a limited number of substituted benzaldehydes that are able to survive refluxing propionic acid. The synthesis was finally optimized by Lindsey’s group [21, 22], which showed that excellent yields of a wide variety of tetraarylporphyrins can be obtained using a high dilution two step reaction between arylaldehydes and pyrrole, in presence of a Lewis acid catalyst (usually BF3-etherate). In the second step of the reaction, the initial product (a colorless porphyrinogen, 9) is oxidized to porphyrin 4 using a quinone such as 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ). Material isolated from the Rothemund and Adler/Longo approaches is highly crystalline but is nonetheless contaminated [23] with about 5% or less of 5,10,15,20-tetraphenylchlorin 10. Brief treatment [24] of the product with DDQ (which is the final step in the more recent Lindsey procedure) accomplishes transformation of 10 into 4; the separation of these two components by chromatography is much more demanding than is transformation of 10 into 4.
Scheme 1.

A majority of naturally occurring porphyrins (i.e. those not derived by degradation of chlorophylls or bacteriochlorophylls) do not contain meso-substituents, and there are certainly none that possess four such substituents. This has led many to question the utility of TPP as a “natural” mimic. Thus, over the years, OEP has been favored as a model for natural porphyrins more than has TPP. Synthetic approaches to porphyrins such OEP are a more complex because of the increased difficulty of preparing the 3,4-disubstituted pyrrole starting materials; pyrrole 8 can, of course simply be purchased and only needs to be distilled.
In the synthesis of TPP, the interpyrrolic carbons are provided by the aldehyde group of the corresponding arylaldehyde. In the case of octaalkylporphyrins (such as 3), the future meso- (i.e. interpyrrolic) carbons can either be added separately by being not attached to the pyrrole (as for TPP) or can be already present in the form of a 2-substituent on the pyrrole. It is important to note that monopyrrole tetramerizations can only produce a structurally unique product if the 3- and 4-substituents in the monopyrrole are identical (Scheme 2).
Scheme 2.

There are two major routes to OEP 3. Firstly, tetramerization of 2,5-di-unsubstituted pyrroles in the presence of reagents which can provide the four meso methine carbons of the product can be used. Thus, cyclization of 3,4-diethylpyrrole 11 with formaldehyde affords 55-75% yields of OEP (3) (Scheme 2) [25]. Secondly, but in an approach which was chronologically developed first, tetramerization of pyrroles bearing 2-CH2R substituents gives acceptable yields of porphyrin; the “R” group on the “benzylic” methylene must be a good leaving group because the methylene carbon will eventually be the source of the interpyrrolic carbons of the product after a nucleophilic attack upon it. Once the condensation reaction has taken place, an oxidation step is necessary in order to afford good yields of symmetrical porphyrin from an intermediate porphyrinogen. The required CH2R groups can be attached to pyrrolic intermediates by used of the Mannich reaction of pyrrole 11 with formaldehyde and dimethylamine [or with commercially available (N,N-dimethylmethylene)ammonium iodide (Eschenmoser’s reagent [26, 27])] to give the 2-(N,N-dimethylaminomethyl)pyrrole 12; heating of 12 in acetic acid affords about a 50% yield of 3 (Scheme 2) [28, 29]. Alternatively, pyrrole 13 can be cyclotetramerized to give a similar yield of 3 by heating in acetic acid containing potassium ferricyanide [30, 31]. The Barton-Zard pyrrole synthesis [32] has afforded a very efficient access to monopyrroles of the type 14, which can be reduced (to the carbinol 15) and then tetramerized under acidic conditions to give 3 in high yield [33, 34].
Surprise is often expressed when relatively good yields of porphyrins are obtained from monopyrrole polymerizations - would not infinite linear polymers be preferred? If one constructs molecular models of the open-chain tetrapyrrole seco-porphyrinogen, one can see that once one has achieved an open-chain tetrapyrrole polymer (16) the head of the polymer is virtually biting the tail (Scheme 3).
Scheme 3.

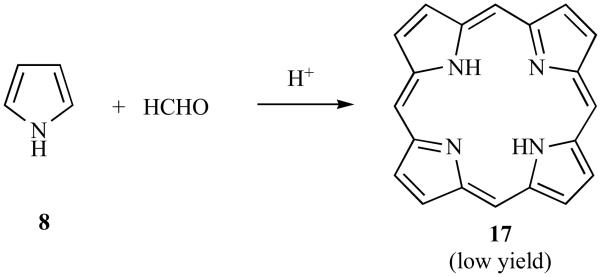
This is particularly true if the monopyrrole precursor has fairly large 3- and 4-substituents because these enforce a helical geometry upon the elongating molecule. For this reason too, attempts to prepare the unsubstituted porphyrin 17 from pyrrole and formaldehyde usually gives a very low yield, probably due to a preference for infinite elongation due to the small 3- and 4-substituents in 8 (Scheme 4) and due to solubility problems of the product 17. However, several groups have synthesized substituted porphyrins and can be desubstituted and this simplifies the problems associated with the low solubility in almost all solvents, of 17. An example is the synthesis of the soluble tetra-tert-butylporphyrin 18 from the Mannich pyrrole 19, followed by removal of the four tert-butyl groups using aqueous sulfuric acid to give 17 in 64% yield (Scheme 5) [35].
Scheme 4.
Scheme 5.

Monopyrrole tetramerization works best for pyrroles with identical 3- and 4-substituents. If the 3- and 4-substituents on the monopyrrole reactant are not identical, mixtures will usually result due to acid-catalyzed pyrrole ring scrambling reactions [13, 36]. As an example, and as mentioned earlier in this article, acid-catalyzed tetramerization of pyrrole 20 will result in a mixture of the four so-called etioporphyrin type isomers (21-24) (Scheme 6). With appropriate care to avoid acidic conditions, etioporphyrin I 21 can be obtained by tetramerization of a monopyrrole; pyrrole 25 has a leaving group which is labile even under neutral conditions [37, 38], and when reacted in methanol containing potassium ferricyanide gives a good yield of pure etioporphyrin I 21 (Scheme 7).
Scheme 6.

Scheme 7.

2.2. The MacDonald [2+2] Porphyrin Synthesis
The development of the so-called MacDonald synthesis was a game-changer for porphyrin synthesis [39]. At about the same time, Woodward and coworkers reported their landmark synthesis of chlorophyll-a that also employed a similar [2+2] approach in the fabrication of the initial porphyrin macrocycle [40].
Dipyrromethanes are the key intermediates in the MacDonald approach.
2.2.1. Syntheses of Dipyrromethanes
Dipyrromethanes can be synthesized using fairly straightforward methods. These important intermediates for porphyrin syntheses are categorized as either symmetrical or unsymmetrical about the interpyrrolic 5-carbon. Symmetrically substituted dipyrromethanes such as 26 can be prepared in one step by self-condensation of bromomethylpyrroles such as 27 in hot methanol [41], or by heating 2-acetoxymethylpyrroles such as 28 in methanol/hydrochloric acid [42]. The pyrrole 28 is obtained from the corresponding methylpyrrole 29 by oxidation of the 2-methyl with lead tetra-acetate. Catalytic hydrogenation cleaves the terminal benzyl esters to afford the corresponding 1,9-dicarboxylic acid 30 which can be formylated using the Vilsmeier reagent (dimethylformamide with either phosphoryl chloride or benzoyl chloride) to give 31 (Scheme 8).
Scheme 8.

These two formyl groups provide the bridging carbons in the MacDonald [2+2] porphyrin macrocyclization (see later). Unsymmetrically substituted dipyrromethanes such as 32, are obtained by condensation of 2-acetoxymethylpyrroles, e.g. 33, with 2-unsubstituted pyrroles 34 in acetic acid and a catalytic amount of p-toluenesulfonic acid (Scheme 9) [43]. Montmorillonite K-10 clay has also been shown to be a very useful acid catalyst in dipyrromethane syntheses [44, 45].
Scheme 9.

Condensation of two unsymmetrical dipyrromethanes 35 and 36 with appropriate bridging carbons will result in two porphyrin products (37 and 38) because the dipyrromethanes can react in either of two orientations related by a 180° rotation of one of the dipyrromethanes (Scheme 10).
Scheme 10.

These symmetry complications in a porphyrin synthesis involving an A-B and a C-D dipyrromethane can be avoided if the A-B or C-D dipyrromethane unit (only one of them!) is symmetrical about its interpyrrolic (5-) carbon atom. Thus, MacDonald and coworkers generically showed [39] that a symmetrical 1,9-diformyldipyrromethane such as 31, can be reacted with a 1,9-di-unsubstituted dipyrromethane 39 (obtained from 32), or the corresponding 1,9-dicarboxylic acid 40, in the presence of an hydriodic acid catalyst to give pure porphyrin, such as 41, in very high yields (Scheme 11). The method can be modified such that a monoformyldipyrromethane such as 42 is self-condensed to give centrosymmetrical porphyrins (e.g. 43) (Scheme 12). The initially formed intermediate is a porphodimethene salt 44; conversion to porphyrin requires an oxidation step that is usually provided by adventitious aerial oxygen; chemical oxidants such as o-chloranil or DDQ can also be used. Since MacDonald’s report [39], others have shown that p-toluenesulfonic acid is a much better choice as the catalyst [46, 47].
Scheme 11.

Scheme 12.

A modification of the MacDonald [2+2] approach that avoids the final oxidation step has been published [48, 49]. Treatment of the 1,9-di-iododipyrromethane 45 with the known 1,9-diformyldipyrromethane 31 in acetic acid and acetic anhydride containing trifluoromethylsulfonic acid gave a 76% yield of the diacetylporphyrin 46. Porphyrin 46 was then further transformed into hematoporphyrin-IX dimethyl ester 47 (by double reduction) and protoporphyrin-IX dimethyl ester 48 (by double dehydration of 47) (Scheme 13).
Scheme 13.

The [2+2] route using dipyrromethanes is probably the most widely used pathway to synthetic porphyrins.
2.3. From Tripyrrolic Intermediates: The [3+1] Route
The so-called [3+1] route to porphyrins employs the fundamental chemistry developed in the MacDonald approach. However, instead of condensing two dipyrromethanes (hence [2+2]), a tripyrrane 49 is reacted with a monopyrrole 50 bearing the two “bridging” carbon atoms. This macrocyclization of a 2,5-difunctionalized monopyrrole unit with an open-chain tripyrrole to produce a porphyrin 51 has a number of useful applications (Scheme 14).
Scheme 14.

It is often the case that the purpose of a particular porphyrin synthesis is to attach or conjugate the porphyrin to another species via unique functionality on the porphyrin. Thus, access to a uniquely functionalized point on the porphyrin will enable the application. Using the [3+1] protocol permits efficient synthesis of a porphyrin in which the required functional handle group is situated on the monopyrrole component to be inserted at the final stage. This kind of chemistry has been facilitated by Sessler’s recent advance in tripyrrane syntheses [50]. In its simplest form Sessler’s route only yields symmetrically substituted tripyrranes, but the ease with which tripyrranes can be made using this method far outweighs the symmetry limitation. Thus, reaction of two equivalents of pyrrole 52 with pyrrole 53 affords the dibenzyl tripyrrane-1,14-dicarboxylate 54 in excellent yield. Catalytic debenzylation then affords the required tripyrrane-1,14-dicarboxylic acid 55 (Scheme 15).
Scheme 15.

The [3+1] approach has symmetry limitations because the terminal rings of the tripyrrane are usually identical. Removal of this limitation will require an advance in tripyrrane synthesis.
The first example of the [3+1] approach to porphyrins was reported by Boudif and Momenteau. A 2,5-diformylpyrrole and a tripyrrane were used [51]. Though the approach clearly has general applicability, Boudif and Momenteau targeted only bis-acrylic or bis-propionic porphyrins, e.g. 56, from tripyrrane 57 and diformylpyrrole 58 (Scheme 16). Lash’s group expanded the generality of the approach in their syntheses of porphyrins such as 59 (Fig. 2) [52]. Sessler and his group also showed the applicability of the approach and also reported syntheses of porphyrins such as 60 (Fig. 2) [53].
Scheme 16.

Fig. (2).

Lash’s 3+1 products.
Our own group developed a [3+1] procedure which does not need acid catalysis, and which guarantees the integrity of the tripyrrane reactant against pyrrole ring-scrambling reactions [54]; treatment of tripyrrane 61, with the 2,5-bis(N,N-dimethylaminomethyl)pyrrole 62 in methanol containing ferricyanide gave the porphyrin 63, in good yield (Scheme 17).
Scheme 17.

2.4. From a,c-Biladiene Salts
The synthesis of a porphyrin (e.g. 7) with a completely unsymmetrical array of substituents must progress through open-chain tetrapyrrolic intermediates. Though a number of open-chain tetrapyrroles have been investigated and utilized with success [14] over the years, the most used open-chain precursors of choice have been shown to be 1,19-dimethyl-a,c-biladiene salts such as 64.
Symmetrical 1,19-dimethyl-a,c-biladienes, e.g. 64, can be synthesized by condensation of dipyrromethane-1,9-dicarboxylic acids 30 with two equivalents of a 2-formyl-5-methylpyrrole 65. The method of choice in earlier studies for cyclization of the a,c-biladiene to give porphyrin, always as its copper(II) complex 66, has involved use of copper(II) chloride or acetate in hot dimethylformamide. Demetalation with H2SO4/ TFA then affords, for example, the metal free porphyrin 67 [55, 56] (Scheme 18). Other cyclization protocols have been studied [57-65] including other metal salts or even electrochemical oxidation. The mechanism of this cyclization, which involves loss of (at least) one of the terminal methyl groups of the a,c-biladiene is not immediately obvious, and a full review of the evidence and conclusions has been published [56]. A breakthrough in the mechanistic investigation of a,c-biladiene cyclizations was achieved when it was shown [56, 60, 61] that electrochemical oxidation of, for example, a,c-biladiene dihydrobromide 68 proceeds to give metal free porphyrin 69 via the cyclic dihydroporphyrin intermediate 70 (Scheme 19) which then eliminates the quaternary methyl group (presumably by an SN2 attack that involves the newly aromatized porphyrin as the leaving group) after macrocyclization.
Scheme 18.

Scheme 19.

The electrochemical cyclization [60, 61] has the advantage that it gives the metal free porphyrin (e.g. 69) as the product, thereby eliminating the need to treat the copper(II) complex [from the copper(II) chloride route] with concentrated sulfuric and trifluoroacetic acids. Also, the reaction progress can be monitored using 1H-NMR spectroscopy in the absence of the paramagentic copper(II) ion. But scaling-up of electrochemical reactions to preparative quantities is not easy. However, it has been found that use of chromium(III) for the oxidative cyclization results in the production of the metal-free porphyrin product [66]. Many researchers had assumed that the metal salts used in these cyclization provided template around which the a,c-biladiene, or some subsequent intermediate on the pathway to porphyrin, could assemble. Recent results however show [65] that the primary function of the metal ion is oxidation, and not templation, and this is also confirmed by the success of the electrochemical approach that uses no transition metal salts at all. Several mechanistic studies [57-59], and experiments involving oxidation with different metal ions have been published, and these have been reviewed and analyzed [56].
Symmetry limitations associated with the earlier a,c-biladiene syntheses were removed by development of routes to unsymmetrically substituted a,c-biladienes [14, 67, 68]. There now exists two complementary methods, which involve “clockwise” or “counterclockwise” elongation of the developing a,c-biladiene (Scheme 20). The counterclockwise route was developed first [67] and involves catalytic hydrogenation of a benzyl tert-butyl dipyrromethane-1,9-dicarboxylate 32 to give a monocarboxylic acid 71 which reacts under mild conditions with a 2-formyl-5-methylpyrrole 65 to afford a high yield of a so-called tripyrrene salt 72. Treatment with TFA (which serves to cleave the tert-butyl ester) and a second, different 2-formylpyrrole 73, gives a high yield of 1,19-dimethyl-a,c-biladiene salt 74 with a completely unsymmetrical array of substituents; cyclization using, for example, the copper(II) method, followed by demetalation with concentrated sulfuric acid in trifluoroacetic acid gives the porphyrin 75. Any other method for cyclization, e.g. the chromium(III) method [54, 66], or electrochemical methods [60, 61] which give directly the metal free porphyrin can be used.
Scheme 20.

The second, so-called clockwise approach (Scheme 20), involves [68] treatment of the dipyrromethane mixed ester 32 with TFA to give the dipyrromethane 76; treatment with formylpyrrole 73 affords the tripyrrene salt 77. Catalytic hydrogenation cannot be used for cleavage of the benzyl ester because this would also saturate the tripyrrene double bond. Thus, sulfuric acid/HBr is used, and this is followed by addition of formylpyrrole 65 to give the a,c-biladiene salt 74, identical with that obtained using the other tripyrrene route. a,c-Biladiene cyclization proceeds as earlier to give porphyrin. There is an advantage to having two different routes to the same a,c-biladiene and porphyrin, particularly when very valuable monopyrroles are being used (e.g. carbon-13 or carbon-14 labeled) and good synthetic strategy requires that this precious pyrrole be added in the last possible step.
1,19-Dimethyl-a,c-biladienes are highly effective intermediates for porphyrin synthesis; good yields of porphyrin are obtained and there are no obvious symmetry or substituent difficulties. The use of a,c-biladienes bearing 1- and 19-substituents other than methyls [62-65] has been investigated, and these reactions yield the predictable meso-substituted porphyrins, along with a host of macrocycles in which either the 1- or the 19-substituent in the original a,c-biladiene has migrated around the macrocycle. These results are also discussed in detail elsewhere [56, 62-65].
3. PORPHYRIN FUNCTIONALIZATION
The porphyrin macrocycle contains 22 conjugated π-electrons, but only 18 of these are required for its conjugated aromatic network. As a consequence, porphyrins can undergo additions and substitution reactions without loss of their aromaticity, as well as cross-couplings, oxidations and reductions, to produce a variety of functionalized macrocycles and linear tetrapyrroles, that continue to find multiple applications in biology, medicine and materials science.
The most electronically reactive positions at the porphyrin periphery are the meso-positions, and generally the preferential sites for electrophilic aromatic substitutions, additions and radical reactions. On the other hand, the β-pyrrolic positions are the most sterically accessible and can also undergo the same type of reactions. Substitution, cross-couplings and radical reactions have been used to produce a variety of functionalized porphyrins, whereas additions to β-β’ peripheral double bonds lead to the formation of chlorins, bacteriochlorins, or isobacteriochlorins, and to the meso positions produce phlorins, porphodimethenes, and expanded tetrapyrrolic macrocycles. The inner pyrrolenine nitrogen atoms of porphyrins can be protonated to give the corresponding mono- or di-cations, and the NH groups can be deprotonated to produce di-anions. Porphyrins can also be readily metalated with a wide variety of metal ions and all the naturally occurring porphyrins are metal complexes. The metal ions have an important inductive effect on the π-electron system and strongly influence the chemical reactivity, photophysical properties and biological functions of porphyrin macrocycles. Closed-shell configuration metals incapable of dπ-pπ back-bonding, such as Zn(II) and Cd(II), induce the highest negative charge onto the porphyrin periphery, conferring them the lowest one-electron oxidation potentials. On the other hand, metals capable of π-back-bonding decrease the electron density on the macrocycle, for example Cu(II) and Ni(II) with d6-d9 configurations, and in particular metals with d1-d5 configurations, such as Sn(IV) and Fe(III), strongly reduce the electron density at the porphyrin periphery.
3.1. Halogenation and Pd(0)-Catalyzed Coupling Reactions
Electron-rich porphyrins normally undergo electrophilic reactions at unsubstituted peripheral positions, when the inner pyrrolenine nitrogens are “protected” by metalalation. Fluorination reagents include N-fluoropyridinium triflates [69], cesium fluoroxysulfate [70], and fluorides of cobalt or silver [71]. N-Chlorosuccinimide (NCS) [72-77] or chlorine [64, 65] are normally used for chlorination, although HCl/H2O2 [78] and PhSeCl3 [79] have also been reported. Similarly, N-bromosuccinimide (NBS) [63, 64, 67, 80-84], bromine [64, 73, 85, 86], HBr/H2O2 [66, 71], copper(II) bromide [85] and PhSeBr3 [69] have been used for bromination of porphyrin macrocycles. Polybromination can lead to mixtures of regioisomers when unsubstituted β- and meso-positions are available at the macrocycle periphery. Nevertheless, in the case of meso-tetraarylporphyrins, such as TPP 4, tetra-bromination can be achieved regioselectively using a slight excess of NBS in refluxing CHCl3, leading to the corresponding 2,3,12,13-tetrabromoporphyrins, such as 78 in about 80% yield (Scheme 21) [88].
Scheme 21.

This regioselectivity is only observed in metal-free porphyrins, where bond fixation occurs after the first substitution, therefore promoting bromination on antipodal pyrrolic units. β-Octabromination can also be obtained using an excess NBS in refluxing o-dichlorobenzene or bromine in chloroform/carbon tetrachloride; in this case the best yields are obtained with the metal complexes of porphyrins (usually Cu, which can be demetalated under acid conditions) due to their higher stability under the reaction conditions, to produce e.g. 79 [89]. Mono- and di-iodination have been accomplished with bis(trifluoroacetoxy)-iodobenzene-iodine in chloroform/pyridine [90, 91]. Using meso-5,15-diphenylporphyrin 80, the first iodination occurs selectively at one of the free meso-positions while the second substitution occurs at one of the β positions leading to a mixture of diiodo regioisomers. However, the meso-dibrominated porphyrin 81 can be obtained in high yield (96%) and regioselectively, by reacting 80 with NBS in chloroform and pyridine at 0 °C for one hour (Scheme 22) [92]. Two different halogens can also be inserted into porphyrin 80 by performing consecutive bromination and iodination reactions (in this order), giving porphyrin 82 in 66% yield [80]. Regioselective di-iodination was also accomplished when bulky meso-aryl groups are present, such as in 5,15-di(3’,4’,5’-trimethoxyphenyl)porphyrin 83, the meso-diiodo porphyrin 84 is obtained regioselectively (Scheme 23).
Scheme 22.

Scheme 23.

Side reactions of brominations and iodinations have been reported, via the formation of porphyrin π-cation radicals, which can react in multiple ways. For example, OEP 3 reacted with NBS in the presence of 2,2’-azobis(2-methylpropionitrile) (AIBN) to produce trans-(2-bromovinyl)porphyrin 85 (Scheme 24) by way of a (1-bromoethyl) intermediate, which can be trapped as the ether derivative in the presence of alcohols [66, 93].
Scheme 24.

The nucleophilic substitution of halogen substituents has also been reported, by for example cyanide [76, 94, 95], nitrite [96], and thiolate ions [88]. The main use of bromo- and iodo-porphyrins is as starting materials for cross-coupling reactions, via palladium(0)-catalyzed Suzuki, Sonogashira, Heck and Stille type reactions to produce various aryl, ethynyl and alkenyl-functionalized porphyrin systems; dimeric and oligomeric porphyrin systems have also been prepared using these methodologies [97-108]. For example, dodecaarylporphyrins 86 were prepared in 14-65% yields using a Suzuki coupling reaction of metal-free 79 with arylboronic acids (Scheme 25) [109-111].
Scheme 25.

This methodology has been used to produce a variety of functionalized porphyrins, for example carborane-containing porphyrin 87 in 78% from bromoporphyrin 78 (Scheme 26) [112] and carboxylate-functionalized 88 from 80 (Scheme 27) [70, 81].
Scheme 26.

Scheme 27.

Metalloporphyrins are often used in palladium(0)-catalyzed coupling reactions to avoid macrocycle metalation by palladium and/or copper, sometimes used as co-catalyst. Allenylboronate has also been used to produce allenyl and alkynyl-functionalized porphyrins from the corresponding bromoporphyrins [113]. Both meso- and β-bromoporphyrins have been functionalized by palladium(0)-catalyzed couplings with amines, amides, alcohols, sulfides, carbazates, hydrazones and phosphines [114-123]. Examples are the synthesis of porphyrins 89 [88] and 90 [102] from 81 or its zinc(II) complex (Scheme 27).
Brominated porphyrins have also been coupled with iodo-alkyl fluorides in the presence of tris(dibenzylideneacetone)dipalladium(0), triphenylarsine and copper(0) in DMSO to produce meso- and β-perfluoroalkylated porphyrins in 40-85% yields [124], and with potassium organotrifluoroborates in the presence of Pd(dppf)Cl2 and Cs2CO3 to produce a variety of functionalized porphyrins [125].
Suzuki-type couplings have also been performed on borylated porphyrins, which can be prepared from palladium-catalyzed reactions of bromoporphyrins with pinacol borane or bispinacol borane. Borylated porphyrins undergo a variety of palladium-catalyzed coupling reactions with aryl halides [126-132]. One example is the borylation of the zinc(II) complex of meso-diphenylporphyrin 80, followed by palladium(0)-catalyzed coupling to N-Boc protected 4-iodo-phenylalanine to give porphyrin 91 (Scheme 28) [112].
Scheme 28.

Sonogashira coupling reactions are used for the preparation of a variety of ethynyl-substituted porphyrins, usually by reaction of a meso- or β-halogenated porphyrin with acetylene compounds [117, 133-138]. Using this methodology, the zinc(II) complex of di-iodoporphyrin 84 was converted into ethynylporphyrins 92 in 25-72% yields (Scheme 29) [81], and nickel(II)-porphyrin 81 gave 93 in 66% yield upon coupling with ferrocenylethyne (Scheme 30) [119].
Scheme 29.

Scheme 30.

Different functionalizations can be introduced onto a porphyrin by performing successive halogenation-coupling-halogenation-coupling reactions or by taking advantage of the different reactivity of two halogens (e.g. Br and I) that are first introduced into the macrocycle, followed by two coupling reactions. For example, porphyrins 94 were prepared from zinc(II) porphyrin 82 by consecutive Sonogashira and Stille coupling reactions (Scheme 31) [81]. An interesting cross-annulative coupling reaction has also reported between meso-diarylporphyrin 95 and various acetylenes in the presence of Pd2(dba)3, tris(o-tolyl)phosphine and dicyclohexylmethylamine in refluxing toluene, to give porphyrins 96 in 78-87% yields (Scheme 32) [139].
Scheme 31.

Scheme 32.

Heck and Stille coupling reactions are used to prepare alkenyl and aryl-substituted porphyrins [140-144]. Stille-type reactions were first used in porphyrin functionalization for the preparation of protoporphyrin IX dimethyl ester 98 from dibromo-deuteroporphyrin IX dimethyl ester 97 in 85% yield (Scheme 33) [87].
Scheme 33.

Mono-substituted alkenes under Heck conditions tend to produce the corresponding trans-alkenylporphyrins, particularly for large, sterically demanding substituents. For example, bromoporphyrins 99 gave the corresponding trans-alkenylporphyrins 100 in >65% yields (the cis-isomer was also observed when R = CN) under Heck conditions [123] and zinc(II)-porphyrin 101 was produced in 86% yield under Stille conditions (Scheme 34) [125]. Mono-di- and tetra-benzoporphyrins have been prepared via vicinal Heck couplings of di-, tetra-, and octa-bromoporphyrins followed by in situ 6π-electrocyclization and aromatization [145-147].
Scheme 34.

For example, dibenzoporphyrins 103 were prepared from tetrabromoporphyrins 102 in a single step, in 48-60% yields (Scheme 35) [130]. Using a Stille cross-coupling reaction, porphyrin 104 was prepared in 60% yield from porphyrin 78 (Scheme 36) [129].
Scheme 35.

Scheme 36.

Silylmethyl-substituted porphyrins, such as 105, are prepared by palladium(0)-catalyzed Kumada cross-coupling reactions of both meso- and β-bromoporphyrins with silylmethyl Grignard reagents [148]. For example, 105 was obtained from porphyrin 81 in 82% yield (Scheme 37). The silylmethylporphyrins can be converted into a variety of functional groups, including CHO, CH2OH, and CH2F.
Scheme 37.

3.2. Nitration and Reactions of Nitroporphyrins
The nitration of porphyrins can be achieved with nitric acid [149-153], nitrate salts (usually of copper, zinc or silver) [154], or with N2O4 [155-158] or nitrite ion [159-161] via the corresponding porphyrin π-cation radicals. The peripheral nitro substituents of porphyrins can be reduced to the corresponding amino derivatives using SnCl2/HCl [162], or with NaBH4 and 10% Pd/C in methanol [163], and β-aminoporphyrins have been shown to undergo acylation, diazotization and various reactions with carbonyl compounds [164-167].
Meso- and β-nitroporphyrins undergo ipso substitution of the nitro group to produce a variety of functionalized porphyrin macrocycles [106,168-172]. In addition, β-nitroporphyrins also undergo nucleophilic Michael-type additions at the adjacent β-position [92, 105, 116, 158, 173-178] to produce functionalized porphyrins and chlorins. For example, β-nitroporphyrin 106 reacted with alkylmagnesium bromides or organolithium reagents to give alkylated porphyrins 107 [156] and with sodium methoxide or benzaldoxime to produce the corresponding 2-methoxy- or 2-hydroxy-porphyrin, respectively, via chlorin intermediates (Scheme 38). Several functionalized macrocycles have been obtained using this methodology, including 2-and 2,3-substituted porphyrins and chlorins. For example, trans-chlorins 109 were produced by reaction with active methylene compounds in the presence of base, via a fused cyclopropane chlorin [163]. β-Fused pyrroloporphyrins, such as 108, were also obtained from metallo-2-nitro-5,10,15,20-tetraphenylporphyrin by reaction with α-isocyanoacetic esters in the presence of a base [179, 180]. On the other hand, 5-nitro-octaethylporphyrin 110 reacted by ipso-substitution of the nitro group in the presence of soft nucleophiles to produce porphyrins 111, whereas hard nucleophiles, such as methylmagnesium bromide and benzyl oxide gave the corresponding 15-substituted porphyrins (Scheme 39) [157], due to the higher stability of the intermediate, conferred by the 5-nitro group.
Scheme 38.

Scheme 39.

A β-nitro substituent on a porphyrin macrocycle, such as 106, directs electrophilic substitutions to the antipodal pyrrole ring, and this strategy has been used to regioselectively prepare 2,3-dibromo-meso-tetrarylporphyrins after reductive denitration [181].
3.3. Formylation and Acylation Reactions
The Vilsmeier formylation of porphyrins using DMF/POCl3 followed by treatment with aqueous base introduces a formyl group at the meso- or the β-position of porphyrin macrocycles [182-185]. The use of a sterically hindered Vilsmeier reagent, such as that prepared from N,N-diisobutylformamide/POCl3 favors substitution at the β-positions [186] whereas N,N-(dimethylamino)acrolein (3-DMA)/POCl3 produces 2-formylvinylporphyrins in good yields [187, 188].
Other regioselective formylation methods use trimethyl orthoformate in TFA [189, 190], and via oxidation of 1,3-dithianylporphyrins [191, 192] or (2-pyridyldimethylsilyl)methylporphyrins [193].
Formyl groups at the periphery of porphyrins can undergo a variety of reactions, including reductions, oxidations and nucleophilic additions, leading to a variety of functionalized macrocycles [194-206]. meso-Hydroxymethylporphyrins undergo dimerization under acidic conditions, producing dimers which upon dehydrogenation in acetic acid are transformed into trans-ethylene porphyrin dimers [207-210]. 1,2-Ethylene porphyrin dimers (cis- and trans-isomers), such as 113, are obtained directly by reductive coupling of metalloformylporphyrins (e.g. 112) using low-valent titanium complexes via a McMurry coupling reaction (Scheme 40) [89, 139, 140, 211-214]. Formyl-porphyrins also undergo Grignard and Wittig reactions [215-221] to produce a variety of cis- and trans-(2-substituted-vinyl)porphyrins, which can be further functionalized (see also section below). The electrophilic acylation of metalloporphyrins can be achieved with acetic anhydride in the presence of Lewis acid catalysts, such as SnCl4, and occurs preferentially at the β-positions due to steric factors [222, 223]. The acetyl substituents of porphyrins can be reduced to vinyl groups with NaBH4 followed by dehydration using p-toluenesulfonic acid in 1,2-dichlorobenzene or alternatively by use of benzoyl chloride/DMF [224]. Alternatively, the enantioselective reduction of mono- and diacetyldeuteroporphyrin IX dimethyl ester can be accomplished with borane dimethyl sulfide in the presence of methyloxazaborolidine as the homochiral catalyst [225-227]. Acetylporphyrins also react under Vilsmeier conditions to produce 1-chloro-2-(formyl)vinyl derivatives, which can be converted to ethynyl-functionalized porphyrins by treatment with base [228, 229]. Under Wittig conditions zinc(II) acetylporphyrins react to produce isopropenylporphyrins [230, 231].
Scheme 40.

3.4. Reactions with Nucleophiles
Porphyrin macrocycles react with strong nucleophiles, such as Grignard and organolithium reagents preferentially at the meso-positions, with formation of meso-functionalized porphyrins as well as phlorins and porphodimethenes, or at the β-positions with formation of chlorins. The best known nucleophilic addition reactions occur on the π-cation radicals of metalloporphyrins, which can be obtained by chemical or electrochemical oxidation. The Mg(II) and Zn(II) complexes of porphyrins are usually employed since these display the lowest oxidation potentials and I2 (usually in the presence of AgClO4 or AgPF6), tris(p-bromophenyl)ammoniumyl hexachloroantimonate (TBAH) or N-chorobenzotriazole (CBT) are the oxidizing agents of choice. Although relatively stable in methanol, the π-cation radicals of metalloporphyrins react with a variety of soft nucleophiles to produce the corresponding meso- and β-substituted metalloporphyrins. For example, meso-nitroporphyrins are obtained by treatment of metalloporphyrins with thallium(III) nitrate or cerium(IV) ammonium nitrate [232]. Pyridinium porphyrin salts are produced from the reaction of π-cation radicals with various pyridines [233-237]. Other nucleophiles (CN−, SCN−, Cl−, CH3CO2−, imidazole, PPh3) were also shown to react with the π-cation radicals of porphyrins [202, 238]; dimeric and higher oligomeric porphyrin systems have been obtained from bidentate nucleophiles, such as bipyridine [239-241]. Chemical and electro-chemical oxidative couplings of Zn(II) 5,15-diarylporphyrins lead to the formation of meso-meso and meso-β linked porphyrin arrays, via nucleophilic attack of the π-cation radicals by neutral porphyrins [161, 242, 243]. Treatment of Zn(II) 5,15-diarylporphyrins with AgPF6 and I2 in the presence of pyridine led to regioselective meso-iodination [244]. The more electrophilic π-dications of porphyrins are formed under stronger oxidation conditions and react with, for example, methanol, preferentially at the meso-positions, leading to isoporphyrin derivatives [245, 246].
Reactions of organometallic reagents with both free-base and metalloporphyrins at unsubstituted meso-positions leads to the formation of intermediate phlorins and porphodimethenes, which are oxidized to the corresponding meso-substituted porphyrins [247, 248]. For example OEP 3 and several of its metal complexes produced the corresponding meso-functionalized porphyrins 115 in 40-90% yields (Scheme 41). Di-, tri-, and tetra-meso-alkylated porphyrins can also be obtained via this methodology in overall yields of 20-40%, by successive one equivalent additions of alkyllithium at low temperature, followed by hydrolysis and oxidation with DDQ [249]. Alkyl- and aryl-lithium agents showed meso-regioselectivity when 5,15-disubstituted porphyrins, such as 80 were used, producing the corresponding porphyrins 116 in 56-94% yields (Scheme 42) [250-252].
Scheme 41.

Scheme 42.

meso-Tetrasubstituted porphyrins react with alkyllithiums to produce porphodimethenes, phlorins or chlorins [253]. Regioselective 15-meso-alkylation of zinc(II) 5-formyloctaethylporphyrin 112 was accomplished by addition of methyl or ethyl magnesium bromide to give porphyrins 114, via a phlorin intermediate (Scheme 40) [254]; under the same conditions the free-base and Ni(II) derivatives react preferentially at the meso-formyl group with formation of the 1-hydroxylalkyl derivatives. Nucleophilic addition of hydroxide ion to Au(III) 5,10,15,20-tetraphenylporphyrin also occurs at a meso-position with formation of the corresponding hydroxyphlorin, whereas the Cu(II), Pd(II), Cd(II) and Mn(III) complexes of TPP were unreactive under the same conditions [255]. Reactions of organometallic reagents with iron(III) porphyrins occur preferentially at the metal center with formation of σ-bonded species [256, 257]; migration of the σ-bonded groups (alkyl, vinyl, aryl) from the metal ion to the nitrogen atom is induced by oxidation or by acid treatment [258-264].
3.5. Oxidations and Reductions
Porphyrins and metalloporphyrins (usually zinc complexes) can be oxidized to oxophlorins and oxochlorins, or to ring-opened products such as formyl- and benzoyl-biliverdins. 5-Oxophlorins (such as 117) are formed by hydrolysis of meso-trifluoroacetoxyporphyrins, obtained from reaction of Zn(II) and Mg(II) β-substituted porphyrins with thallium(III) trifluoroacetate (TTFA) (Scheme 43) [265, 266].
Scheme 43.

Hydrogen peroxide in concentrated sulfuric acid [267, 268] and benzoyl peroxide can also be used for meso-oxidation [269, 270]. Reactions at the periphery of 5-oxophlorins occur preferentially at the electron-rich position-15 [271]. Neutral π-radicals of 5-oxophlorins are easily formed under mild conditions, and were shown to be stabilized by bulky 15-substituents [272, 273]. Extensive meso-oxidation can lead to formation of di-, tri-, and tetraoxoporphyrins (the so-called xanthoporphyrinogens). Osmium tetroxide reacts selectively at the β-β’ double bonds of porphyrins to produce the corresponding cis-dihydroxychlorins; for example 118 is obtained from reaction of OEP 3 (Scheme 43) [274-277]. The β-substituted cis-dihydroxychlorins undergo pinacol-pinacolone rearrangement in perchloric acid or fuming sulfuric acid to give β-oxochlorins (for example 119 from 118). The regioselectively of the osmylation reaction and the migratory aptitudes of different peripheral substituents in the pinacol-pinacolone rearrangement have been investigated [278-282]. Metal-free oxochlorins such as 119 react further with osmium tetroxide regioselectively at the opposite pyrrole ring, whereas metal derivatives react preferentially at the adjacent β-β’ double bond producing the corresponding vic-dihydroxy- and oxo-bacteriochlorins and isobacteriochlorins [283-287]. meso-Tetraarylporphyrins such as TPP react with benzoyl peroxide producing β-benzoyloxyporphyrins, which are converted into β-hydroxyporphyrins such as 120 by metalation, basic hydrolysis and demetalation (Scheme 44). Compound 120 can also be obtained from 2-nitro-meso-tetraphenylporphyrin by nucleophilic displacement of the nitro group with the sodium salt of E-benzaldoxime [156, 157]. Oxidation of 120 to the 2,3-dioneporphyrin 121 can be accomplished under a variety of conditions, including CrO3/acetic acid [288], photooxidation [157], and SeO2/dioxane [157]; dioneporphyrin 121 reacts with aromatic o-diamines and with aromatic aldehydes in the presence of NH4OAc/AcOH in CHCl3, producing a variety of pyrazino- and imidazole-porphyrin monomers and oligomeric systems [151, 289-292].
Scheme 44.

Open-chain oxygenated tetrapyrroles are obtained under several conditions, including O2 in the presence of ascorbic acid or hydrazine (the so-called coupled oxidation of iron porphyrins) [293, 294], using thallium(III) or cerium(IV) trifluoroacetate salts [232, 295], N2O4 or sodium nitrate and trifluoroacetic acid [296, 297], hydrogen peroxide [298], or meta-chloroperoxybenzoic acid (mCPBA) in pyridine [299], by reactions of metalloporphyrin π-cation radicals with nucleophiles [236], or from photo-oxidation by singlet oxygen, in the presence of light and molecular oxygen to give bilitrienones [300-308]. Porphyrins can also be reduced to dihydro- (chlorins, phlorins, porphodimethenes), tetrahydro- (isobacteriochlorins, bacteriochlorins, porphomethenes), and hexahydro-porphyrins (porphyrinogens). Reduction to cis-chlorins is normally accomplished with diimide (obtained from p-toluenesulfonylhydrazine) and potassium carbonate [309-312]. On the other hand, trans-chlorins can be obtained from the reduction of Fe(III) β-substituted porphyrins with sodium metal in isopentyl alcohol [313, 314]. Porphyrins can also be photo-reduced by light in the presence of amines, ascorbic acid, and other compounds [315-320]. Regioselective photoreductions to cis-chlorins are achieved with ascorbic acid in the presence of diazabicyclo[2.2.2]octane (DABCO) as catalyst. Tetrahydroporphyrins are obtained by further reduction of chlorins under the conditions above. Metalloporphyrins often give isobacteriochlorins, whereas metal-free porphyrins preferentially give bacteriochlorins. Reduction to porphyrinogen can be accomplished by catalytic hydrogenation and this methodology has been used to prepare unsymmetrical macrocycles from symmetric porphyrins [321]. Metalloporphyrins can be reduced to π-anion radicals and π-dianions with, for example, sodium in THF or with sodium anthracenide [322-326]. Protonation and alkylation of porphyrin π-anions occurs preferentially at the meso-positions with formation of phlorins and porphodimethenes, which can rearrange to chlorins and be oxidized to porphyrins.
ACKNOWLEDGEMENTS
We acknowledge continuous research support from the National Institutes of Health, current grant number R01 CA132861.
LIST OF ABBREVIATIONS
- TPP
meso-tetraphenylporphyrin
- OEP
octaethylporphyrin
- DDQ
2,3-dichloro-5,6-dicyanobenzoquinone
- TFA
trifluoroacetic acid
- TTFA
thallium(III) trifluoroacetate
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- dppf
bis(diphenylphosphino)ferrocene
- dba
dibenzylideneacetone
- DPEphos
(oxydi-2,1-phenylene)bis (diphenylphosphine)
- DMSO
dimethylsulfoxide
- DMF
dimethylformamide
- DME
dimethoxyethane
- AIBN
2,2’-azobis(2-methylpropionitrile)
- Boc
tert-butoxycarbonyl
- THF
tetrahydrofuran
- BHT
butylated hydroxytoluene
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- TBAH
tris(p-bromophenyl)ammoniumyl hexachloroantimonate
- CBT
N-chorobenzotriazole
- DABCO
diazabicyclo[2.2.2]octane
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
DISCLOSURE
This article is an update of a previous article published in 2000 in Current Organic Chemistry VOLUME: 4 ISSUE: 2 DOI: 10.2174/1385272003376346.
REFERENCES
- [1].Fischer H, Orth H. Die Chemie des Pyrrols. Vol. 1. Akademische Verlagsgesellschaft; Leipzig: 1934. [Google Scholar]
- [2].Fischer H, Orth H. Die Chemie des Pyrrols. Vol. 2. Akademische Verlagsgesell-schaft; Leipzig: 1937. part 1. [Google Scholar]
- [3].Fischer H, Stern A. Die Chemie des Pyrrols. Vol. 2. Akademische Verlagsgesellschaft; Leipzig: 1940. part 2. [Google Scholar]
- [4].Smith KM, editor. Porphyrins and Metalloporphyrins. Elsevier; Amsterdam: 1975. [Google Scholar]
- [5].Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. 1-10. Academic Press; Boston: 1999. and Vols. 11-20, 2003. [Google Scholar]
- [6].Kadish KM, Smith KM, Guilard R, editors. Synthesis and Coordination Chemistry (Part 1) Vol. 2. World Scientific Press; Singapore: 2010. Handbook of Porphyrin Science. [Google Scholar]
- [7].Kadish KM, Smith KM, Guilard R, editors. Synthetic Methodology. Vol. 3. World Scientific Press; Singapore: 2010. Handbook of Porphyrin Science. [Google Scholar]
- [8].Kadish KM, Smith KM, Guilard R, editors. Synthesis and Structural Studies. Vol. 13. World Scientific Press: Singapore; 2011. Handbook of Porphyrin Science. [Google Scholar]
- [9].Kadish KM, Smith KM, Guilard R, editors. Synthetic Developments (Part 1) Vol. 16. World Scientific Press; Singapore: 2012. Handbook of Porphyrin Science. [Google Scholar]
- [10].Kadish KM, Smith KM, Guilard R, editors. Synthetic Developments (Part 2) Vol. 17. World Scientific Press; Singapore: 2012. >Handbook of Porphyrin Science. [Google Scholar]
- [11].Kadish KM, Smith KM, Guilard R, editors. Synthesis. Vol. 23. World Scientific Press; Singapore: 2012. Handbook of Porphyrin Science. [Google Scholar]
- [12].Sessler JL, Weghorn SJ. Expanded, contracted and isomeric porphyrins. Pergamon Press; Oxford: 1997. [Google Scholar]
- [13].Taniguchi M, Lindsey JS. Enumeration of isomers of substituted tetrapyrrole macrocycles: from classical problems in biology to modern combinatorial libraries. In: Kadish KM, Smith KM, Guilard R, editors. Handbook of Porphyrin Science. Vol. 23. World Scientific Press; Singapore: 2012. pp. 3–80. [Google Scholar]
- [14].Smith KM. Strategies for the synthesis of octaalkylporphyrin systems. In: Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Vol. 1. Academic Press; Boston: 1999. pp. 1–43. [Google Scholar]
- [15].Jackson AH, Smith KM. Total synthesis of pyrrole pigments. In: ApSimon JW, editor. Total Synthesis of Natural Products. Vol. 1. Wiley; New York: 1973. pp. 143–278. and 1984; Vol. 6, pp 237-280. [Google Scholar]
- [16].Baltazzi E, Krimen LI. Recent advances in chemistry of pyrrole. Chem. Rev. 1963;63:511–556. [Google Scholar]
- [17].Gossauer A. Die Chemie der Pyrrole. Springer Verlag; Berlin: 1974. [Google Scholar]
- [18].Rothemund P. Formation of porphyrins from pyrrole and aldehydes. J. Am.Chem. Soc. 1935;57:2010–2011. [Google Scholar]
- [19].Rothemund P, Menotti AR. The synthesis of α,β,γ,δ-tetraphenylporphine. J. Am. Chem. Soc. 1941;63:267–270. doi: 10.1021/ja01185a047. [DOI] [PubMed] [Google Scholar]
- [20].Adler AD, Longo FR, Finarelli JD, Goldmacher J, Assour J, Korsakoff L. A simplified synthesis for meso-tetraphenylporphine. J. Org. Chem. 1967;32:476–477. [Google Scholar]
- [21].Lindsey JS, Schreiman IC, Hsu HC, Kearney PC, Marguerettaz AM. Rothemund and Adler-Longo reactions revisited: synthesis of tetra-phenylporphyrins under equilibrium conditions. J. Org. Chem. 1987;52:827–836. [Google Scholar]
- [22].Lindsey JS. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 1. Academic Press; Boston: 1999. pp. 45–118. [Google Scholar]
- [23].Badger GM, Jones RA, Laslett RL. Porphyrins. VII. The synthesis of porphyrins by the Rothemund reaction. Aust. J. Chem. 1964;17:1028–1035. [Google Scholar]
- [24].Barnett GH, Hudson MF, Smith KM. Concerning meso-tetraphenylporphyrin purification. J. Chem. Soc. Perkin Trans., 1. 1975:1401–1403. [PubMed] [Google Scholar]
- [25].Sessler JL, Mozaffari A, Johnson MR. 3,4-Diethylpyrrole and 2,3,7,8,12,13,17,18-octaethylporphyrin. Org. Synth. 1991;70:68–77. [Google Scholar]
- [26].Ono M, Lattmann R, Imomota K, Lehmann C, Früh T, Eschenmoser A. Monopyrrole precursors for the synthesis of uroporphyrinogenoctanitrile. Croat. Chem. Acta. 1985;58:627–638. [Google Scholar]
- [27].Schreiber J, Maag H, Hashimoto N, Eschenmoser A. Dimethyl(methylene)ammonium iodide. Angew. Chem. Int. Ed. Engl. 1971;10:330–331. [Google Scholar]
- [28].Whitlock HW, Hanauer R. Octaethylporphyrin. J. Org. Chem. 1968;33:2169–2171. [Google Scholar]
- [29].Eisner U, Lichtarowicz A, Linstead RP. Chlorophylls and related compounds. Part VI. The synthesis of octaethylchlorin. J. Chem. Soc. 1957:733–739. [Google Scholar]
- [30].Inhoffen HH, Fuhrhop J-H, Voigt H, Brockmann H., Jr. Formylierung der meso-Kohlenstoffeatome von alkylsubstituierten porphyrinen. Liebigs Ann. Chem. 1966;695:133–145. [Google Scholar]
- [31].Siedel W, Winkler F. Oxydation von pyrrolderivaten mit bleitetraacetat. Liebigs Ann. Chem. 1943;554:162–201. [Google Scholar]
- [32].a Barton DHR, Zard SZ. A new synthesis of pyrroles from nitroalkenes. J. Chem. Soc. Chem. Commun. 1985:1098–1099. [Google Scholar]; b Barton DHR, Kervagoret J, Zard SZ. A useful synthesis of pyrroles from nitroolefins. Tetrahedron. 1990;46:7587–7598. [Google Scholar]
- [33].Ono N, Kawamura H, Bougauchi M, Maruyama K. Porphyrin synthesis from nitrocompounds. Tetrahedron. 1990;46:7483–7496. [Google Scholar]
- [34].Aoyagi K, Haga T, Toi H, Aoyama Y, Mizutani T, Ogoshi H. Electron deficient porphyrins. III. Facile syntheses of perfluoroalkylporphyrins including water soluble porphyrin. Bull. Chem. Soc. Jpn. 1997;70:937–943. [Google Scholar]
- [35].Neya S, Quan J, Hoshino T, Hata M, Funasaki N. Convenient synthesis of porphine from b-tetra(tert-butyl)porphyrin. Tetrahedron Lett. 2004;45:8629–8630. [Google Scholar]
- [36].Mauzerall D. The thermodynamic stability of porphyrinogens. J. Am. Chem. Soc. 1960;82:2601–2603. [Google Scholar]
- [37].Nguyen LT, Smith KM. Syntheses of Type-I porphyrins via monopyrrole tetramerization. Tetrahedron Lett. 1996;37:7177–7180. [Google Scholar]
- [38].Nguyen LT, Senge MO, Smith KM. One-pot synthesis of regiochemically pure porphyrins from two different pyrroles. Tetrahedron Lett. 1994;35:7581–7584. [Google Scholar]
- [39].Arsenault GP, Bullock E, MacDonald SF. Pyrromethanes and porphyrins therefrom. J. Am. Chem. Soc. 1960;82:4384–4389. [Google Scholar]
- [40].a Woodeard RB. Totalsynthese des Chlorophylls. Angew. Chem. 1960;72:651–662. [Google Scholar]; b Woodward RB, Ayer WA, Beaton JM, Bickelhaupt F, Bonnett R, Buchschacher P, Closs GL, Dutler H, Hannah J, Hauck FP, Ito S, Langemann A, Le Goff E, Leimgruber W, Lwowski W, Sauer J, Valenta Z, Volz H. The total synthesis of chlorophyll a. Tetrahedron. 1990;46:7599–7659. [Google Scholar]
- [41].Fischer H, Orth H. Die Chemie des Pyrrols. Vol. 1. Akademische Verlagsgesell-schaft; Leipzig: 1934. p. 333. [Google Scholar]
- [42].Mironov AF, Ovsepyan TR, Evstigneeva RP, Preobrazenskii NA. Synthetic studies in the dipyrrylmethane series. Synthesis of 4,4'-dimethyl- 3,3'-bis(β-carbomethoxyethyl)dipyrrylmethane. Zh. Obshch. Khim. 1965;35:324–328. [Google Scholar]
- [43].Cavaleiro JAS, Gonsalves A.M.d’A.R., Kenner GW, Smith KM. Synthesis of pyrromethanes and a tripyrrane. J. Chem. Soc. Perkin Trans. 1. 1973:2471–2478. [Google Scholar]
- [44].Jackson AH, Pandey RK, Rao KRN, Roberts E. Reactions on solid supports, Part II: a convenient method for synthesis of pyrromethanes using a Montmorillonite clay as catalyst. Tetrahedron Lett. 1985;26:793–796. [Google Scholar]
- [45].Freeman BA, Smith KM. Alternative syntheses of pyrromethanes and porphyrins using acid-modified Montmorillonite K-10 Clay. Synth. Commun. 1999;29:1843–1855. [Google Scholar]
- [46].Cavaleiro JAS, Gonsalves A.M.d’A.R., Kenner GW, Smith KM. Total synthesis of deuteriated derivatives of drotoporphyrin-IX for NMR studies of haemoproteins. J. Chem. Soc. Perkin Trans. 1. 1974:1771–1781. [PubMed] [Google Scholar]
- [47].Cavaleiro JAS, Kenner GW, Smith KM. Biosynthesis of protoporphyrin-IX from coproporphyrinogen-III. J. Chem. Soc. Perkin Trans. 1974;1:1188–1194. doi: 10.1039/p19740001188. [DOI] [PubMed] [Google Scholar]
- [48].Martin P, Mueller M, Flubacher D, Boudier A, Blaser H-U, Spielvogel D. Total synthesis of hematoporphyrin and protoporphyrin: A conceptually new approach. Org. Pro. Res. Dev. 2010;14:799–804. doi: 10.2533/chimia.2013.204. [DOI] [PubMed] [Google Scholar]
- [49].Martin P, Mueller M, Flubacher D, Boudier A, Blaser H-U, Spielvogel D. Total synthesis of hematoporphyrin and protoporphyrin: A conceptually new approach. Chimia. 2013;67:204–206. doi: 10.2533/chimia.2013.204. [DOI] [PubMed] [Google Scholar]
- [50].a Sessler JL, Lynch V, Johnson MR. Synthesis and crystal structure of a novel tripyrrane-containing porphyrinogen-like macrocycle. J. Org. Chem. 1987;52:4394–4397. [Google Scholar]; b Sessler JL, Cyr MJ, Lynch V. Synthetic and structural studies of sapphyrin, a 22-p-electron pentapyrrolic "expanded porphyrin". J. Am. Chem. Soc. 1990;112:2810–2813. [Google Scholar]
- [51].a Boudif A, Momenteau M. Synthesis of a porphyrin-2,3-diacrylic acid using a new ‘3 + 1’ type procedure. J. Chem. Soc. Chem. Commun. 1994:2069–2070. [Google Scholar]; b Boudif A, Momenteau M. A new convergent method for porphyrin synthesis based on a ‘3 + 1’ condensation. J. Chem. Soc. Perkin Trans. 1. 1996:1235–1242. [Google Scholar]
- [52].a Lash TD, Novak BH. Tetraphenanthro[9,10-b:9,10-g:9,10-l:9,10-q]-porphyrin, a new highly conjugated porphyrin system. Angew. Chem. Int. Ed. Engl. 1995;34:683–685. [Google Scholar]; b Lash TD, Novak BH. New highly conjugated porphyrin chromophores: Synthesis of mono- and diphenanthroporphyrins. Tetrahedron Lett. 1995;36:4381–4284. [Google Scholar]; c Lin Y, Lash TD. Porphyrin synthesis by the “3 + 1” methodology: A superior approach for the preparation of porphyrins with fused 9,10-phenanthroline subunits. Tetrahedron Lett. 1995;36:9441–9444. [Google Scholar]; d Lash TD. Porphyrin synthesis by the “3+1” approach: new applications for an old methodology. Chem. Eur. J. 1996;2:1197–1200. [Google Scholar]; e Lash TD. Porphyrins with exocyclic rings. Part 9. Synthesis of porphyrins by the ‘3+1’ approach. J. Porphyrins Phthalocyanines. 1997;1:29–144. [Google Scholar]
- [53].Sessler JL, Genge JW, Urbach A, Sanson P. A ‘3+1' approach to monofunctionalized alkyl porphyrins. Synlett. 1996;2:187–188. [Google Scholar]
- [54].Nguyen LT, Senge MO, Smith KM. Simple methodology for syntheses of porphyrins possessing multiple peripheral substituents with an element of symmetry. J. Org. Chem. 1996;61:998–1003. [Google Scholar]
- [55].Johnson AW, Kay IT. The formation of porphyrins by the cyclisation of bilenes. J. Chem. Soc. C. 1961:2418–2423. [Google Scholar]
- [56].Smith KM. Cyclizations of a,c-biladiene salts to give porphyrins and their derivatives. In: Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Vol. 1. Academic Press; Boston: 1999. pp. 119–148. [Google Scholar]
- [57].Smith KM, Kehres LA. Syntheses of methyl devinylporphyrins related to protoporphyrin-IX. Initial studies on the mechanism of the copper(II) catalyzed cyclizations of 1',8'-dimethyl-a,c-biladienes. J. Chem. Soc. Perkin Trans. 1. 1983:2329–2335. [Google Scholar]
- [58].Smith KM, Minnetian OM. Novel porphyrins from copper(II) mediated cyclizations of 1',8'-dimethyl-a,c-biladiene salts: Mechanism of the cyclization reaction. J. Org. Chem. 1985;50:2073–2080. [Google Scholar]
- [59].Smith KM, Minnetian OM. Cyclizations of 1',8'-dimethyl-a,c-biladiene salts to give porphyrins: A study with various oxidizing agents. J. Chem. Soc. Perkin Trans. 1. 1986:277–280. [Google Scholar]
- [60].Jeyakumar D, Snow KM, Smith KM. Electrosynthesis of porphyrins from a,c-biladienes. J. Am. Chem. Soc. 1988;110:8562–8564. [Google Scholar]
- [61].Swanson KL, Snow KM, Jeyakumar D, Smith KM. Tetrapyrrole products from electrochemical cyclization of 1',8'-disubstituted-a,c-biladiene salts. Tetrahedron. 1991;47:685–696. [Google Scholar]
- [62].Liddell PA, Gerzevske KR, Lin JJ, Olmstead MM, Smith KM. Novel macrocycles from metal-catalyzed oxidative cyclizations of a,c-biladiene salts. J. Org. Chem. 1993;58:6681–6691. doi: 10.1021/jo9619138. [DOI] [PubMed] [Google Scholar]
- [63].Gerzevske KR, Lin JJ, Smith KM. Kinetic vs. thermodynamic product distributions of macrocyclic tetrapyrrole cyclization products from 1,19- disubstituted a,c-biladiene salts. Heterocycles. 1994;37:207–212. [Google Scholar]
- [64].Lin JJ, Gerzevske KR, Liddell PA, Senge MO, Olmstead MM, Khoury RG, Weeth BE, Tsao SA, Smith KM. Metal-catalyzed oxidative cyclizations of a,c-biladiene salts bearing 1- and/or 19-arylmethyl substituents: Macrocyclic products and their chemistry. J. Org. Chem. 1997;62:4266–4276. doi: 10.1021/jo9619138. [DOI] [PubMed] [Google Scholar]
- [65].Lin JJ, Holmes RT, Smith KM. Oxidative cyclization of a,c-biladienes: The role of the metal ion. J. Porphyrins Phthalocyanines. 1998;2:363–368. [Google Scholar]
- [66].Boschi T, Paolesse R, Tagliatesta P. Transition-metal-catalyzed cyclization of [a,c] biladiene salts as an efficient route to the synthesis of alkyl porphyrins. Inorg. Chim. Acta. 1990;168:83–87. [Google Scholar]
- [67].Almeida JAPB, Kenner GW, Rimmer J, Smith KM. A general stepwise synthesis of unsymmetrically substituted porphyrins. Tetrahedron. 1976;32:1793–1799. [Google Scholar]
- [68].Smith KM, Craig GW. Porphyrin synthesis through tripyrrins: An alternate approach. J. Org. Chem. 1983;48:4302–4306. [Google Scholar]
- [69].Naruta Y, Tani F, Maruyama K. Meso-perfluorination of porphyrins with N-fluoropyridinium triflate. Tetrahedron Lett. 1992;33:1069–1072. [Google Scholar]
- [70].Andrews LE, Bonnett R, Kozyrev AN, Appelman EH. meso-Reactivity of porphyrins and related compounds. Part 10. Direct fluorination of octaethylporphyrin with caesium fluoroxysulphate. J. Chem. Soc. Perkin Trans. 1. 1988:1735–1738. [Google Scholar]
- [71].Tsuchiya S, Seno M. Novel synthetic method of phenol from benzene catalysed by perfluorinated hemin. Chem. Lett. 1989:263–266. [Google Scholar]
- [72].Wijesekera T, Matsumono A, Dolphin D, Lexa D. Perchlorinated and highly chlorinated meso-tetraphenylporphyrins. Angew. Chem. Int. Ed. Engl. 1990;29:1028–1030. [Google Scholar]
- [73].Gonsalves A.M.d’A.R., Johnstone RAW, Pereira MM, Shaw J, Sobral JFN. Metal-assisted reactions. Part 22. Synthesis of perhalogenated prophyrins and their use as oxidation catalysts. Tetrahedron Lett. 1991;32:1355–1358. [Google Scholar]
- [74].Chorghade MS, Dolphin D, Dupré D, Hill DR, Lee EC, Wijesekera TP. Improved protocols for the synthesis and halogenation of sterically hindered metalloporphyrins. Synthesis. 1996:1320–1324. [Google Scholar]
- [75].Wijesekera T, Dupré D, Cader MR, Dolphin D. Porphyrins bearing halogens at the meso-phenyl and beta-pyrrolic positions: Synthesis and spectral properties. Bull. Soc. Chim. Fr. 1996;133:765–765. [Google Scholar]
- [76].Vicente MGH, Smith KM. Functionalizations of the alkyl substituents in octa-alkylporphyrins. Tetrahedron. 1991:6887–6894. [Google Scholar]
- [77].Hoffmann P, Robert A, Meunier B. Preparation and catalytic activities of the manganese and iron derivatives of Br8TMP and Cl12TMP, two robust porphyrin ligands obtained by halogenation of tetramesitylporphyrin. Bull. Soc. Chim. Fr. 1992;129:85–97. [Google Scholar]
- [78].Bonnett R, Gale IAD, Stephenson GF. The meso-reactivity of porphyrins and related compounds. Part II. Halogenation. J. Chem. Soc. C. 1966:1600–1604. [Google Scholar]
- [79].Ali H, van Lier JE. Phenylselenyl halides: efficient reagents for the selective halogenation and nitration of porphyrins. Tetrahedron Lett. 1991:5015–5018. [Google Scholar]
- [80].Hoffmann P, Labat G, Robert A, Meunier B. Highly selective bromination of tetramesitylporphyrin: an easy access to robust metalloporphyrins, M-Br8TMP and M-Br8TMPS. Examples of application in catalytic oxygenation and oxidation reactions. Tetrahedron Lett. 1990;31:1991–1994. [Google Scholar]
- [81].DiMagno SG, Lin VS-Y, Therien MJ. Facile elaboration of porphyrins via metal-mediated cross-coupling. J. Org. Chem. 1993;58:5983–5993. [Google Scholar]
- [82].Bonnett R, Campion-Smith IH, Kozyrev AN, Mironov AF. J. Chem. Res. (S) 1990:138–14. [Google Scholar]; Bonnett R, Campion-Smith IH, Kozyrev AN, Mironov AF. J. Chem. Res. (M) 1990:1015–1026. [Google Scholar]
- [83].Tagliatesta P, Li J, Autret M, Van Caemelbecke E, Villard A, D’Souza F, Kadish KM. Electrochemistry and spectral characterization of oxidized and reduced (TPPBrx)FeCl where TPPBrx is the dianion of β-pyrrole brominated tetraphenylporphyrin and x varies from 0 to 8. Inorg. Chem. 1996;35:5570–5576. doi: 10.1021/ic960148c. [DOI] [PubMed] [Google Scholar]
- [84].Shea KM, Jaquinod L, Khoury RG, Smith KM. Dodecasubstituted chlorins (dihydroporphyrins) Chem. Commun. 1998:759–760. [Google Scholar]
- [85].Bhyrappa P, Krishnan V. Octabromotetraphenylporphyrin and its metal derivatives: electronic structure and electrochemical properties. Inorg. Chem. 1991;30:239–245. [Google Scholar]
- [86].Hariprasad G, Dahal S, Maiya BG. meso-Substituted octabromoporphyrins: synthesis, spectroscopy, electrochemistry and electronic structure. J. Chem. Soc. Dalton Trans. 1996:3429–3436. [Google Scholar]
- [87].Minnetian OM, Morris IK, Snow KM, Smith KM. New syntheses and reactions of some halogenated porphyrins. J. Org. Chem. 1989;54:5567–5574. [Google Scholar]
- [88].Crossley MJ, Burn PL, Chew SS, Cuttance FB. Newsom. I.A. Regiospecific introduction of four substituents to porphyrin systems at antipodal pyrrolenic positions. J. Chem. Soc., Chem. Commun. 1991:1564–1566. [Google Scholar]
- [89].Spyroulias GA, Despotopoulos AP, Raptopoulous CP, Terzis A, Montauzon D, Poilblanc R, Coutsolelos AG. Comparative study of structure-properties relationship for novel beta-halogenated lanthanide porphyrins and their nickel and free bases precursors, as a function of number and nature of halogens atoms. Inorg. Chem. 2002;41:2648–2659. doi: 10.1021/ic000738h. [DOI] [PubMed] [Google Scholar]
- [90].Boyle RW, Johnson CK, Dolphin D. Iodination and Heck alkynylation of 5,15-diphenylporphyrin. A convenient entry to asymmetrically meso-substituted porphyrins. J. Chem. Soc. Chem. Commun. 1995:527–528. [Google Scholar]
- [91].Shanmugathasan S, Johnson CK, Edwards C, Matthews EK, Dolphin D, Boyle RW. Regioselective halogenation and palladium-catalysed couplings on 5,15-diphenylporphyrin. J. Porphyrins Phthalocyanines. 2000;4:228–232. [Google Scholar]
- [92].Monteiro CJP, Pereira MM, Vicente MGH, Arnaut LG. Photophysical properties of unsymmetric meso-substituted porphyrins synthesized via the Suzuki coupling reaction. Tetrahedron. 2012;68:8783–8788. [Google Scholar]
- [93].Vicente MGH, Smith KM. Functionalization of alkyl substituents in octaalkylporphyrins. Synlett. 1990:579–581. [Google Scholar]
- [94].Callot HJ. Bromination of meso-tetraphenylporphine. Structure and reactivity of products. Tetrahedron Lett. 1973;50:4987–4990. [Google Scholar]
- [95].Callot HJ. Bomination of m-tetraphenylporphine. Preparation of alkyl derivatives and polycyanoporphines. Bull. Soc. Chim. Fr. 1974:1492–1496. [Google Scholar]
- [96].Arnold DP, Bott RC, Eldridge H, Elms FM, Smith G, Zojaji M. Functionalization of 5,15-diphenylporphyrin: preparation and x-ray crystal structures of meso nitro, bromo, and trimethylsilylethynyl derivatives. Aust. J. Chem. 1997;50:495–503. [Google Scholar]
- [97].Gauler R, Risch N. Synthesis of carbon-carbon linked porphyrinoligomers by utilization of Heck-type coupling of natural tetrapyrroles. Tetrahedron Lett. 1997;38:223–224. [Google Scholar]
- [98].Shultz DA, Gwaltney KP, Lee H. A modified procedure for Sonogashira couplings: synthesis and characterization of a bisporphyrin, 1,1-bis[zinc(II) 5‘-ethynyl-10‘,15‘, 20‘-trimesitylporphyrinyl]methylenecyclohexane. J. Org. Chem. 1998;63:4034–4038. [Google Scholar]
- [99].Vicente MGH, Jaquinod L, Smith KM. Oligomeric porphyrin arrays. Chem. Commun. 1999:1771–1782. [Google Scholar]
- [100].Setsune J-I. Palladium chemistry in recent porphyrin research. J. Porphyrins Phthalocyanines. 2004;8:93–102. [Google Scholar]
- [101].Fazekas M, Pintea M, Senge MO, Zawadzka M. Synthetic strategies and porphyrin building blocks for unsymmetrical multichromophores. Tetrahedron Lett. 2008;49:2236–2239. [Google Scholar]
- [102].Esdaile LJ, Senge MO, Arnold DP. New palladium catalysed reactions of bromoporphyrins: synthesis and crystal structures of nickel(II) complexes of primary 5-aminoporphyrin, 5,5'-bis(porphyrinyl) secondary amine, and 5-hydroxyporphyrin. Chem. Commun. 2006:4192–4194. doi: 10.1039/b608365j. [DOI] [PubMed] [Google Scholar]
- [103].Gao G-Y, Ruppel JV, Fields KB, Xu X, Chen Y, Zhang XP. Synthesis of diporphyrins via palladium-catalyzed C-O bond formation: effective access to chiral diporphyrins. J. Org. Chem. 2008;73:4855–4858. doi: 10.1021/jo800443n. [DOI] [PubMed] [Google Scholar]
- [104].Bringmann G, Gotz DCG, Gulder TAM, Gehrke TH, Bruhn T, Kupfer T, Radacki K, Braunschweig H, Heckmann A, Lambert C. Axially chiral β,β'-bisporphyrins: synthesis and configurational stability tuned by the central metals. J. Am. Chem. Soc. 2008;130:17812–17825. doi: 10.1021/ja8055886. [DOI] [PubMed] [Google Scholar]
- [105].Aratani N, Osuka A. Synthesis of meso-meso linked hybrid porphyrin arrays by Pd-catalyzed cross-coupling reaction. Org. Lett. 2001;3:4213–4216. doi: 10.1021/ol0168770. [DOI] [PubMed] [Google Scholar]
- [106].Peng X, Nakamura Y, Aratani N, Kim D, Osuka A. 1,4-Phenylene-bridged meso-meso linked diporphyrin array. Tetrahedron Lett. 2004;45:4981–4984. [Google Scholar]
- [107].Frampton MJ, Akdas H, Cowley AR, Rogers JE, Slagle JE, Fleitz PA, Drobizhev M, Rebane A, Anderson HL. Synthesis, crystal structure and nonlinear optical behavior of β-unsubstituted meso-meso E-vinylene-linked porphyrin dimers. Org. Lett. 2005;7:5365–5368. doi: 10.1021/ol0520525. [DOI] [PubMed] [Google Scholar]
- [108].Tokuji S, Yurino T, Aratani N, Shinokubo H, Osuka A. Palladium-catalyzed dimerization of meso-bromoporphyrins: highly regioselective meso-β coupling via unprecedented remote C-H bond cleavage. Chem. Eur. J. 2009;15:12208–12211. doi: 10.1002/chem.200902143. [DOI] [PubMed] [Google Scholar]
- [109].Chan KS, Zhou X, Au MT, Tam CY. Synthesis of beta-aryl substituted porphyrins by palladium catalyzed Suzuki cross-coupling reactions. Tetrahedron. 1995;51:3129–3136. [Google Scholar]
- [110].Zhou X, Tse MK, Wan TSM, Chan KS. Synthesis of βmono-, tetra-, and octasubstituted sterically bulky porphyrins via Suzuki cross coupling. J. Org. Chem. 1996;61:3590–3593. doi: 10.1021/jo952205+. [DOI] [PubMed] [Google Scholar]
- [111].Muzzi CM, Medforth CJ, Voss L, Cancilla M, Lebrilla C, Ma J-G, Shelnutt JA, Smith KM. Novel dodecaarylporphyrin: synthesis and dynamic properties. Tetrahedron Lett. 1999;40:6159–6162. [Google Scholar]
- [112].Hao EH, Fronczek FR, Vicente MGH. Carborane functionalized pyrroles and porphyrins via the Suzuki cross-coupling reaction. Chem. Commun. 2006:4900–4902. doi: 10.1039/b611571c. [DOI] [PubMed] [Google Scholar]
- [113].Locos OB, Dahms K, Senge MO. Allenylporphyrins: a new motif on the porphyrin periphery. Tetrahedron Lett. 2009;50:2566–2569. [Google Scholar]
- [114].Chen Y, Zhang XP. Facile and efficient synthesis of meso-arylamino- and alkylamino-substituted porphyrins via palladium-catalyzed amination. J. Org. Chem. doi: 10.1021/jo034063m. [DOI] [PubMed] [Google Scholar]
- [115].Gao G-Y, Colvin AJ, Chen Y, Zhang XP. Versatile synthesis of meso-aryloxy- and alkoxy-substituted porphyrins via palladium-catalyzed C-O cross-coupling reactions. Org. Lett. 2003;5:3261–3264. doi: 10.1021/ol035081t. [DOI] [PubMed] [Google Scholar]
- [116].Takanami T, Hayashi M, Hino F, Suda K. Palladium-catalyzed meso-amination and amidation of porphyrins: marked acceleration with the Ni(II) central metal ion. Tetrahedron Lett. 2003;44:7353–7357. [Google Scholar]
- [117].Esdaile LJ, McMurtrie JC, Turner P, Arnold DP. Modified porphyrinoids from carbazates and hydrazones and the first crystal structure of a diiminoporphodimethene. Tetrahedron Lett. 2005;46:6931–6935. [Google Scholar]
- [118].Gao G-Y, Chen Y, Zhang XP. General synthesis of meso-amidoporphyrins via palladium-catalyzed amidation. Org. Lett. 2004;6:1837–1840. doi: 10.1021/ol049440b. [DOI] [PubMed] [Google Scholar]
- [119].Gao G-Y, Colvin AJ, Chen Y, Zhang XP. Versatile synthesis of meso-aryloxy- and alkoxy-substituted porphyrins via palladium-catalyzed C-O cross-coupling reactions. Org. Lett. 2003;5:3261–3264. doi: 10.1021/ol035081t. [DOI] [PubMed] [Google Scholar]
- [120].Chen Y, Gao GY, Zhang XP. Palladium-mediated synthesis of novel meso-chiral porphyrins: cobalt-catalyzed cyclopropanation. Tetrahedron Lett. 2005;46:4965–4969. [Google Scholar]
- [121].Gao G-Y, Colvin AJ, Chen Y, Zhang XP. Synthesis of meso-arylsulfanyl- and alkylsulfanyl-substituted porphyrins via palladium-mediated C-S bond formation. J. Org. Chem. 2004;69:8886–8892. doi: 10.1021/jo048552d. [DOI] [PubMed] [Google Scholar]
- [122].Atefi F, McMurtrie JC, Turner P, Duriska M, Arnold DP. meso-Porphyrinylphosphine oxides: mono- and bidentate ligands for supramolecular chemistry and the crystal structures of monomeric {[10,20-diphenylporphyrinatonickel(II)-5,15-diyl]-bis-[P(O)Ph2]} and polymeric self-coordinated {[10,20-diphenylporphyrinatozinc(II)-5,15-diyl]-bis-[P(O)Ph2]} Inorg. Chem. 2006;45:6479–6489. doi: 10.1021/ic060372u. [DOI] [PubMed] [Google Scholar]
- [123].Matano Y, Matsumoto K, Nakao Y, Uno H, Sakaki S, Imahori H. Regioselective β-metalation of meso-phosphanylporphyrins. Structure and optical properties of porphyrin dimers linked by peripherally fused phosphametallacycles. J. Am. Chem. Soc. 2008;130:4588–4589. doi: 10.1021/ja710542e. [DOI] [PubMed] [Google Scholar]
- [124].Liu C, Chen QY. General and efficient synthesis of meso- and β-perfluoroalkylated porphyrins via Pd-catalyzed cross-coupling reaction. Synlett. 2005:1306–1310. [Google Scholar]
- [125].Horn S, Cundell B, Senge MO. Exploration of the reaction of potassium organotrifluoroborates with porphyrins. Tetrahedron Lett. 2009;50:2562–2565. [Google Scholar]
- [126].Hyslop AG, Kellett MA, Iovine PM, Therien MJ. Suzuki porphyrins: New synthons for the fabrication of porphyrin-containing su- pramolecular assemblies. J. Am. Chem. Soc. 1998120:12676–12677. [Google Scholar]
- [127].Iovine PM, Kellett MA, Redmore NP, Therien MJ. Syntheses and 1H NMR spectroscopy of rigid, cofacially aligned, porphyrin-bridge-quinone systems in which the interplanar separations between the porphyrin, aromatic bridge, and quinone are less than the sum of their respective van der Waals radii. J. Am. Chem. Soc. 2000;122:8717–8727. [Google Scholar]
- [128].Chng LL, Chang CJ, Nocera DG. meso-Tetraaryl cofacial bisporphyrins delivered by Suzuki cross-coupling. J. Org. Chem. 2003;68:4075–4078. doi: 10.1021/jo026610u. [DOI] [PubMed] [Google Scholar]
- [129].Chang LL, Chang CJ, Nocera DG. Catalytic O-O activation chemistry mediated by iron hangman porphyrins with a wide range of proton-donating abilities. Org. Lett. 2003;5:2421–2424. doi: 10.1021/ol034581j. [DOI] [PubMed] [Google Scholar]
- [130].Chang CJ, Chang LL, Nocera DG. Proton-coupled O-O activation on a redox platform bearing a hydrogen-bonding scaffold. J. Am. Chem. Soc. 2003;125:1866–1876. doi: 10.1021/ja028548o. [DOI] [PubMed] [Google Scholar]
- [131].Zhang T-G, Zhao Y, Asselberghs I, Persoons A, Clays K, Therien MJ. Design, synthesis, linear, and nonlinear optical properties of conjugated (porphinato)zinc(II)-based donor-acceptor chromophores featuring nitrothiophenyl and nitrooligothiophenyl electron-accepting moieties. J. Am. Chem. Soc. 2005;127:9710–9720. doi: 10.1021/ja0402553. [DOI] [PubMed] [Google Scholar]
- [132].Sazanovich IV, Balakumar A, Muthukumaran K, Hindin E, Kirmaier C, Diers JR, Lindsey JS, Bocian DF, Holten D. Excited-state energy-transfer dynamics of self-assembled imine-linked porphyrin dyads. Inorg. Chem. 2003;42:6616–6628. doi: 10.1021/ic034558u. [DOI] [PubMed] [Google Scholar]
- [133].Ali H, van Lier JE. Synthesis of β-substituted porphyrins using palladium catalysed reactions. Tetrahedron. 1994;50:11933–11944. [Google Scholar]
- [134].Lee C-W, Lu H-P, Lan C-M, Huang Y-L, Liang Y-R, Yen W-N, Liu Y-C, Lin Y-S, Diau EW-G, Yeh C-Y. Novel zinc porphyrin sensitizers for dye-sensitized solar cells: synthesis and spectral, electrochemical, and photovoltaic properties. Chem. Eur. J. 2009;15:1403–1412. doi: 10.1002/chem.200801572. [DOI] [PubMed] [Google Scholar]
- [135].Poon KW, Liu W, Chan P-K, Yang Q, Chan T-WD, Mak TCW, Ng DKP. Tetrapyrrole derivatives substituted with ferrocenylethynyl moieties. Synthesis and electrochemical studies. J. Org. Chem. 2001;66:1553–1559. doi: 10.1021/jo0004308. [DOI] [PubMed] [Google Scholar]
- [136].Huang X, Zhu C, Zhang S, Li W, Guo Y, Zhan X, Liu Y, Bo Z. Porphyrin-dithienothiophene π-conjugated copolymers: synthesis and their applications in field-effect transistors and solar cells. Macromolecules. 2008;41:6895–6902. [Google Scholar]
- [137].Jiblaoui A, Baudequin C, Chaleix V, Ducourthial G, Louradour F, Ramondenc Y, Sol V, Leroy-Lhez S. An easy one-pot desilylation/copper-free Sonogashira cross-coupling reaction assisted by tetrabutylammonium fluoride (TBAF): synthesis of highly π-conjugated porphyrins. Tetrahedron. 2013;69:5098–5103. [Google Scholar]
- [138].Yu Z, Ptaszek M. Multifunctional bacteriochlorins from selective palladium-coupling reactions. Org. Lett. 2012;14:3708–3711. doi: 10.1021/ol3015545. [DOI] [PubMed] [Google Scholar]
- [139].Sahoo AK, Mori S, Shinokubo H, Osuka A. Facile peripheral functionalization of porphyrins by Pd-catalyzed [3+2] annulation with alkynes. Angew. Chem. Int. Ed. 2006;45:7972–7975. doi: 10.1002/anie.200603580. [DOI] [PubMed] [Google Scholar]
- [140].Ishizuka T, Yamasaki H, Osuka A, Furuta H. Syntheses of aryl- and arylethynyl-substituted N-confused porphyrins. Tetrahedron. 2007;63:5137–5147. [Google Scholar]
- [141].Locos OB, Arnold DP. The Heck reaction for porphyrin functionalisation: synthesis of meso-alkenyl monoporphyrins and palladium-catalysed formation of unprecedented meso-β ethene-linked diporphyrins. Org. Biomol. Chem. 2006;4:902–916. doi: 10.1039/b516989e. [DOI] [PubMed] [Google Scholar]
- [142].Odobel F, Suzenet F, Blart E, Quintard J-P. An efficient synthetic approach to highly conjugated porphyrin-based assemblies containing a bipyridine moiety. Org. Lett. 2000;2:131–133. doi: 10.1021/ol990356j. [DOI] [PubMed] [Google Scholar]
- [143].Bhyrappa P, Velkannan V. β-Tetrakis(2-thienyl)-meso-tetraphenylporphyrins: Synthesis, structural and electrochemical redox properties. Inorg. Chim. Acta. 2012;387:64–73. [Google Scholar]
- [144].Smith MJ, Clegg W, Nguyen KA, Rogers JE, Pachter R, Fleitz PS, Anderson HL. Synthesis and crystal structure of a push-pull quinoidal porphyrin: a nanoporous framework assembled from cyclic trimer aggregates. Chem. Commun. 2005:2433–2435. doi: 10.1039/b502776d. [DOI] [PubMed] [Google Scholar]
- [145].Deshpande R, Jiang L, Schmidt G, Rakovan J, Wang X, Wheeler K, Wang H. A concise approach to the synthesis of opp-dibenzoporphyrins through the Heck reaction. Org. Lett. 2009;11:4251–4253. doi: 10.1021/ol901615f. [DOI] [PubMed] [Google Scholar]
- [146].Jiang L, Engle JT, Sirk L, Hartley CS, Ziegler CJ, Wang H. Triphenylene-fused porphyrins. Org. Lett. 2011;13:3020–3023. doi: 10.1021/ol200853g. [DOI] [PubMed] [Google Scholar]
- [147].Jiang L, Zaenglein RA, Engle JT, Mittal C, Hartley CS, Ziegler CJ, Wang H. Water-soluble ionic benzoporphyrins. Chem. Commun. 2012:6927–6929. doi: 10.1039/c2cc31057k. [DOI] [PubMed] [Google Scholar]
- [148].Sugita N, Hayashi S, Hino F, Takanami T. Palladium-catalyzed Kumada Coupling Reaction of bromoporphyrins with silylmethyl Grignard reagents: Preparation of silylmethyl-substituted porphyrins as a multipurpose synthon for fabrication of porphyrin systems. J. Org. Chem. 2012;77:10488–10497. doi: 10.1021/jo302122f. [DOI] [PubMed] [Google Scholar]
- [149].Dahal S, Krishnan V. Charge transfer excited states of zinc(II) derivatives of β-substituted dinitrotetraphenylporphyrin. Photochem. Photobiol. A: Chem. 1995;89:105–112. [Google Scholar]
- [150].Bartoli JF, Battioni P, De Foor WR, Mansuy D. Synthesis and remarkable properties of iron β-polynitroporphyrins as catalysts for monooxygenation reactions. J. Chem. Soc. Chem. Commun. 1994:23–24. [Google Scholar]
- [151].Johnson AW, Oldfield D. The nitration and hydroxylation of ætioporphyrim I. J. Chem. Soc. 1965:4303–4312. [Google Scholar]
- [152].Drach JE, Longo FR. Electrophilic substitution on porphin. I. Nitration. J. Org. Chem. 1974;39:3282–3284. doi: 10.1021/jo00936a027. [DOI] [PubMed] [Google Scholar]
- [153].Ozette K, Battioni P, Leduc P, Bartoli J-F, Mansuy D. A new manganese-β-heptanitro-porphyrin with extreme redox potentials: spectral, electrochemical and catalytic properties. Inorg. Chim. Acta. 1998;272:4–6. [Google Scholar]
- [154].Watanabe E, Nishimura S, Ogoshi H, Yoshida Z. Orientation of electrophilic meso-substitution in metallooctaethylporphyrins. Tetrahedron. 1975;31:1385–1390. [Google Scholar]
- [155].Fanning JC, Mandel FS, Gray TL. Datta-Gupta, N. The reaction of metalloporphyrins with nitrogen dioxide. Tetrahedron. 1979;35:1251–1255. [Google Scholar]
- [156].Gong L-C, Dolphin D. Nitrooctaethylporphyrins: synthesis, optical and redox properties. Can. J. Chem. 1985;63:401–405. [Google Scholar]
- [157].Catalano MM, Crossley MJ, Harding MM, King LG. Control of reactivity at the porphyrin periphery by metal ion co-ordination: a general method for specific nitration at the β-pyrrolic position of 5,10,15,20-tetraarylporphyrins. J. Chem. Soc. Chem. Commun. 1984:1535–1536. [Google Scholar]
- [158].Shea KM, Jaquinod L, Smith KM. Dihydroporphyrin synthesis: new methodology. J. Org. Chem. 1998;63:7013–7021. doi: 10.1021/jo980965p. [DOI] [PubMed] [Google Scholar]
- [159].Baldwin JE, Crossley MJ, DeBernardis JF. Efficient peripheral functionalization of porphyrins. Tetrahedron. 1982;38:685–693. [Google Scholar]
- [160].Barnett GH, Smith KM. Reactions of some metalloporphyrin and metallochlorin π-cation radicals with nitrite. J. Chem. Soc. Chem. Commun. 1974:772–773. [Google Scholar]
- [161].Osuka A. Shimidzu, H. meso,meso-Linked porphyrin arrays. Angew. Chem. Int. Ed. Engl. 1997;36:135–137. [Google Scholar]
- [162].Crossley MJ, Govenlock LJ, Prashar JK. Synthesis of porphyrin-2,3,12,13- and -2,3,7,8-tetraones: building blocks for the synthesis of extended porphyrin arrays. J. Chem. Soc. Chem. Commun. 1995:2379–2380. [Google Scholar]
- [163].Baldwin JE, DeBernardis JF. Efficient peripheral functionalization of capped porphyrins. J. Org. Chem. 1977;42:3986–3987. [Google Scholar]
- [164].Hombrecher HK, Gherdan VM, Ohm S, Cavaleiro JAS, Neves MGPMS, Condesso MF. Synthesis and electrochemical investigation of β-alkyloxy substituted meso-tetraphenylporphyrins. Tetrahedron. 1993;49:8569–8578. [Google Scholar]
- [165].Crossley MJ, Hambley TW, Mackay LG, Try AC, Walton RJ. Porphyrin analogues of Tröger's base: large chiral cavities with a bimetallic binding site. Chem. Soc. Chem. Commun. 1995:1077–1079. [Google Scholar]
- [166].Crossley MJ, King GK, Newsom IA, Sheehan CS. Investigation of a ‘reverse’ approach to extended porphyrin systems. Synthesis of a 2,3- diaminoporphyrin and its reactions with α-diones. J. Chem. Soc. Perkin Trans. 1. 1996:2675–2684. [Google Scholar]
- [167].Johnson CK, Dolphin D. Syntheses of chlorins possessing fused nitrogen-containing rings. Tetrahedron Lett. 1998;39:4619–4622. [Google Scholar]
- [168].Crossley MJ, King LG, Pyke SM. A new and highly efficient synthesis of hydroxyporphyrins. Tetrahedron. 1987;43:4569–4577. [Google Scholar]
- [169].Crossley MJ, Burn PL, Langford SJ, Pyke SM, Stark AG. A new method for the synthesis of porphyrin-α-diones that is applicable to the synthesis of trans-annular extended porphyrin systems. J. Chem. Soc. Chem. Commun. 1991:1567–1568. [Google Scholar]
- [170].Crossley MJ, Gosper JJ, Wilson MG. A general reductive denitration method for regiospecific deuteriation of the porphyrin nucleus: synthesis of [20-2.1]mesoporphyrin IX dimethyl ester. J. Chem. Soc. Chem. Commun. 1985:1798–1799. [Google Scholar]
- [171].Beavington R, Rees PA, Burn PL. A study on the oxidation of 2-hydroxyporphyrins to porphyrin-α-diones. J. Chem. Soc. Perkin Trans. 1. 1998:2847–2852. [Google Scholar]
- [172].Crossley MJ, King LG, Pyke SM, Tansey CW. Reaction of 5-nitrooctaethylporphyrins with nudeophiles. J.Porphyrins Phthalocyanines. 2002;6:685–694. [Google Scholar]
- [173].Crossley MJ, King LG. A new method for regiospecific deuteration and reduction of 5,10,15,20-tetraphenylporphyrins: nucleophilic reaction of borohydride ion with 2-nitro-5,10,15,20-tetraphenylporphyrins. J. Org. Chem. 1993;58:4370–4375. [Google Scholar]
- [174].Crossley MJ, Harding MM, Tansey CW. A convenient synthesis of 2-Alkyl-5,10,15,20-tetraphenylporphyrins: reaction of metallo-2-nitro-5,10,15,20-tetraphenylporphyrins with Grignard and organolithium reagents. J. Org. Chem. 1994;59:4433–4437. [Google Scholar]
- [175].Crossley MJ, King LG. Reaction of metallo-2-nitro-5,10,15,20-tetraphenylporphyrins with oxyanions. Temperature-dependent competition between nucleophilic addition and single-electron transfer processes. J. Chem. Soc. Perkin Trans. 1. 1996:1251–1260. [Google Scholar]
- [176].Crossley MJ, King LG, Simpson JL. Solvent-dependent ambident nucleophilicity of phenoxide ion towards nitroporphyrins: synthesis of 2-hydroxyaryl- and 2-aryloxy-5,10,15,20-tetraphenylporphyrins by displacement of a nitro group. J. Chem. Soc. Perkin Trans. 1. 1997:3087–3096. [Google Scholar]
- [177].Jaquinod L, Gros CP, Khoury RG, Smith KM. A convenient synthesis of functionalized tetraphenylchlorins. Chem. Commun. 1996:2581–2582. [Google Scholar]
- [178].Luguya R, Fronczek FR, Smith KM, Vicente MGH. Synthesis of novel carboranychlorins with dual application in boron neutron capture therapy (BNCT) and photodynamic therapy (PDT) J. Appl. Rad. Isotopes. 2004;61:1117–1123. doi: 10.1016/j.apradiso.2004.05.068. [DOI] [PubMed] [Google Scholar]
- [179].Jaquinod L, Gros CP, Olmstead MM, Antolovich M, Smith KM. First syntheses of fused pyrroloporphyrins. Chem. Commun. 1996:1475–1476. [Google Scholar]
- [180].Gros CP, Jaquinod L, Khoury RG, Olmstead MM, Smith KM. Approaches to β-fused porphyrinoporphyrins: pyrrolo- and dipyrromethanoporphyrins. J. Porphyrins Phthalocyanines. 1997;1:201–212. [Google Scholar]
- [181].Jaquinod L, Khoury RG, Shea KM, Smith KM. Regioselective syntheses and structural characterizations of 2,3-dibromo- and 2,3,7,8,12,13-hexabromo-5,10,15,20-tetraphenylporphyrins. Tetrahedron. 1999;55:13151–13158. [Google Scholar]
- [182].Smith KM, Bisset GMF, Case JJ, Tabba HD. Polyformylation of copper(II) porphyrins. Tetrahedron Lett. 1980;21:3747–3750. [Google Scholar]
- [183].Smith KM, Bisset GMF, Tabba HD. Polyformylation of copper(II) complexes of octa-alkyl porphyrins. J. Chem. Soc. Perkin Trans. 1. 1982:581–585. [Google Scholar]
- [184].Momenteau M, Loock B, Bisagni E. Five-coordinate iron(II) porphyrins derived from meso-α,β,γ,δ tetraphenylporphin: synthesis, characterization, and coordinating properties. Can. J. Chem. 1979;57:1804–1813. [Google Scholar]
- [185].Buchler JW, Dreher C, Herget G. Metallkomplexe mit tetrapyrrolliganden, XLVIII. Vilsmeier-formylierung von metallporphyrinen mit CoII, NiII, PdII, PtII, CuII, ZnII, CoIII, CrIII, MnIII, FeIII, AlIII, SiIV und PtIV in abhängigkeit vom zentralmetall. Liebigs Ann. Chem. 1988:43–54. [Google Scholar]
- [186].Smith KM, Langry KC. Electrophilic substitution reactions of derivatives of deuteroporphyrin-IX: deuteriation and Vilsmeier formylation. J. Chem. Soc. Perkin Trans. 1. 1983:439–444. [Google Scholar]
- [187].Vicente MGH, Rezzano IN, Smith KM. Efficient new syntheses of benzochlorins, benzoisobacteriochlorins and benzobacteriochlorins. Tetrahedron Lett. 1990;31:1365–1368. [Google Scholar]
- [188].Vicente MGH, Smith KM. Vilsmeier reactions of porphyrins and chlorins with 3-(dimethylamino)acrolein. New syntheses of benzochlorins, benzoisobacteriochlorins and dibenzobacteriochlorins and reductive coupling of porphyrins and chlorins using low-valent titanium complexes. J. Org. Chem. 1991;56:4407–4418. [Google Scholar]
- [189].Montforts F-P, Scheurich G, Meier A, Haake G, Hoper F. An improved method for the preparation of formyldeuteroporphyrins. Synthesis of biologically relevant porphyrins. Tetrahedron Lett. 1991;32:3477–3480. [Google Scholar]
- [190].Haake G, Meier A, Montforts F-P, Scheurich G, Zimmermann G. Synthese von chlorinen aus dem roten blutfarbstoff hämin. Liebigs Ann. Chem. 1992:325–336. [Google Scholar]
- [191].Senge MO, Hatscher SS, Wiehe A, Dahms K, Kelling A. The dithianyl group as a synthon in porphyrin chemistry: condensation reactions and preparation of formylporphyrins under basic conditions. J. Am. Chem. Soc. 2004;126:13634–13635. doi: 10.1021/ja045223u. [DOI] [PubMed] [Google Scholar]
- [192].Dahms K, Senge MO, Bakri Bakar M. Exploration of meso-substituted formylporphyrins and their Grignard and Wittig reactions. Eur. J. Org. Chem. 2007:3833–3848. [Google Scholar]
- [193].Takanami T, Wakita A, Sawaizumi A, Iso K, Onodera H, Suda K. One-pot synthesis of meso-formylporphyrins by SNAr reaction of 5,15-disubstituted porphyrins with (2-pyridyldimethylsilyl)methyllithium. Org. Lett. 2008;10:685–687. doi: 10.1021/ol703107t. [DOI] [PubMed] [Google Scholar]
- [194].Clezy PS, Lim CL, Shannon JS. Chemistry of pyrrolic compounds. XXVII. Some aspects of the mass spectra and chemistry of meso-substituted porphyrins. Aust. J. Chem. 1974;27:1103–1120. [Google Scholar]
- [195].Arnold DP, Johnson AW, Mahendran M. Some reactions of meso- formyloctaethylporhyrin. J. Chem. Soc. Perkin Trans. 1. 1978:366–370. [Google Scholar]
- [196].Runge S, Senge MO. Reaction of β-formylporphyrins with organometallicreagents. A facile method for the preparation of porphyrins with exocyclic double bonds. Tetrahedron. 1999;55:10375–10390. [Google Scholar]
- [197].Hoper F, Montforts F-P. A simple synthesis of functionalized rac-chlorins via allylboration of porphyrin aldehydes. Liebigs Ann. Chem. 1995:1033–1038. [Google Scholar]
- [198].Smith KM, Bisset GMF, Bushell MJ. meso-Methylporphyrins and chlorins. Bioorg. Chem. 1980;9:1–26. [Google Scholar]
- [199].Smith NW, Smith KM. Preparation of bacteriopetroporphyrins by partial synthesis from the Chlorobium chlorophylls. Energy Fuels. 1990;4:675–688. [Google Scholar]
- [200].Smith KM, Bisset GMF. General synthesis of hydrocarbon-soluble porphyrins. J. Org. Chem. 1979;44:2077–2081. [Google Scholar]
- [201].Smith KM, Bisset GMF, Bushell MJ. Recent studies on the Chloro- bium chlorophylls (bacteriochlorophylls-c) Int. J. Biochem. 1980;12:695–700. doi: 10.1016/0020-711x(80)90146-9. [DOI] [PubMed] [Google Scholar]
- [202].Smith KM, Bisset GMF. Meso (methine) functionalization of octaalkylporphyrins. J. Chem. Soc. Perkin Trans. 1. 1981:2625–2630. [Google Scholar]
- [203].Bonfantini EE, Officer DL. The synthesis of butadiene-bridged porphyrindimers and styryl porphyrins using a porphyrin-derived Wittig reagent. Tetrahedron Lett. 1993;34:8531–8534. [Google Scholar]
- [204].Burrell AK, Officer DL. Functionalizing porphyrins via Wittig reactions: a building block approach. Synlett. 1998:1297–1307. [Google Scholar]
- [205].Yeh C-Y, Miller SE, Carpenter SD, Nocera DG. Structurally homologous beta- and meso-amidinium porphyrins. Inorg. Chem. 2001;40:3643–3646. doi: 10.1021/ic001387+. [DOI] [PubMed] [Google Scholar]
- [206].Ol'shevskaya VA, Zaitsev AV, Luzgina VN, Kondratieva TT, Ivanov OG, Kononova EG, Petrovskii PV, Mironov AF, Kalinin VN, Hofmann J, Shtil AA. Novel boronated derivatives of 5,10,15,20-tetraphenylporphyrin: Synthesis and toxicity for drug-resistant tumor cells. Biorg. Med. Chem. 2006;14:109–120. doi: 10.1016/j.bmc.2005.07.067. [DOI] [PubMed] [Google Scholar]
- [207].Ponomarev GV, Borovkov VV, Sugiura K-I, Sakata Y, Shul’ga AM. Synthesis and properties of cis-1,2-bis(octaethylporphyrinyl)ethylene. Tetrahedron Lett. 1993;34:2153–2156. [Google Scholar]
- [208].Shul’ga AM, Ponomarev GV. Porphyrins. 23. Synthesis and properties of ethane-bis(porphyrins) Khim. Geterotsikl. Soedin. 1988:339–344. [Google Scholar]
- [209].Higuchi H, Shimizu K, Ojima J, Sugiura K-I, Sakata Y. Synthesis and properties of etheno-bridged porphyrin trimers. Tetrahedron Lett. 1995;36:5359–5362. [Google Scholar]
- [210].Yashunsky DV, Ponomarev GV, Arnold DP. Synthesis of meso-monosubstituted ethane- and trans-ethylenebis(porphyrins) Tetrahedron Lett. 1995;36:8485–8488. [Google Scholar]
- [211].Zhilina ZI, Ishkov YV, Voloshanovskii IS, Andronati ASA. Reductive dimerization of the cuprous complex of 2-formyl-5,10,15,20-tetraphenylporphyrin. Dokl. Akad. Nauk USSR. 1988;303:377–380. [Google Scholar]
- [212].Senge MO, Gerzevske KR, Vicente MGH, Forsyth TP, Smith KM. Models for the photosynthetic reaction center - synthesis and structure of 1,2-cis- and -trans-ethene and skewed 1,1-carbinol bridged porphyrin dimers. Angew. Chem. Int. Ed. Engl. 1993;32:750–753. [Google Scholar]
- [213].Jaquinod L, Nurco DJ, Medforth CJ, Pandey RK, Forsyth TP, Olmstead MM, Smith KM. Synthesis and characterization of bis(chlorin)s from the McMurry reaction of formylchlorins. Angew. Chem. Int. Ed. Engl. 1996;35:1013–1016. [Google Scholar]
- [214].Tokuji S, Awane H, Yorimitsu H, Osuka A. Direct arylation of meso-formyl porphyrin. Chem. Eur. J. 2013;19:64–68. doi: 10.1002/chem.201203742. [DOI] [PubMed] [Google Scholar]
- [215].Callot HJ, Castro B, Selve C. Porphyrines synthetiques porteuses dechaines laterales peptidiques. I. Atropoisomérie d'amides cis(mésotétraphénylporphyrinyl)-3 propénoïques. Tetrahedron Lett. 1978;32:2877–2880. [Google Scholar]
- [216].Momenteau M, Loock B, Rougee M. Five-coordinate iron(II) porphyrins derived from meso-α,β,γ,δ tetraphenylporphin: synthesis, characterization, and coordinating properties. Can. J. Chem. 1979;57:1804–1813. [Google Scholar]
- [217].Dolphin D, Sivasothy R. The preparation of porphyrin S-411 (dehydroco-prophyrin) and harderoporphyrin from protoporphyrin-IX. Can. J. Chem. 1981;59:779–85. [Google Scholar]
- [218].Arnold DP, Nitschinsk LJ. Porphyrin dimers linked by conjugated butadiynes. Tetrahedron. 1992;48:8781–8792. [Google Scholar]
- [219].Gosper JJ, Ali M. A conformationally constrained conjugated porphyrindimer. J. Chem. Soc. Chem. Commun. 1994:1707–1708. [Google Scholar]
- [220].Kahl SB, Schaeck JJ, Koo M-S. Improved methods for the synthesis of porphyrin alcohols and aldehydes from protoporphyrin IX dimethyl ester and their further modification. J. Org. Chem. 1997;62:1875–1880. [Google Scholar]
- [221].Screen TEO, Blake IM, Rees LH, Clegg W, Borwick SJ, Anderson HL. Making conjugated connections to porphyrins: a comparison of alkyne, alkene, imine and azo links. J. Chem. Soc., Perkin Trans. 1. 2002:320–329. [Google Scholar]
- [222].Omote M, Ando A, Koyama M, Takagi T, Kumadaki I. Synthesis of fluorinated analogs of hematoporphyrin II. Heterocycles. 1994;39:381–384. [Google Scholar]
- [223].Jeandon C, Bauder C, Callot HJ. Selective acetylation of porphyrins possessing a free pyrrolic position as a tool for petroporphyrin analysis. A reinvestigation. Energy Fuels. 1990;4:665–667. [Google Scholar]
- [224].Shiau F-Y, Whyte BJ, Castelfranco PA, Smith KM. Partial syntheses of the isomerically pure magnesium(II) protoporphyrin IX mono-methyl esters and their identification. J. Chem. Soc. Perkin Trans. 1. 1991:1781–1785. [Google Scholar]
- [225].Kusch D, Montforts F-P. Enantioselective synthesis of hematoporphyrin stereoisomers. Tetrahedron: Asymm. 1995;6:867–870. [Google Scholar]
- [226].Kusch D, Tollner E, Lincke A, Montforts F-P. A simple, chirogenic, enantioselective synthesis of chlorins and isobacteriochlorins. Angew. Chem. Int. Ed. Engl. 1995;34:784–787. [Google Scholar]
- [227].Kusch D, Montforts F-P. Synthesis of enantiomerically pure hydroxuyeth- ylporphyrins and their transformation into optically active chlorin derivatives. Part 4. Liebigs Ann. Chem. 1996:83–86. [Google Scholar]
- [228].Jiang X, Smith KM. 3,8-Di-ethynyl-3,8-devinylprotoporphyrin IX and its hemin. J. Chem. Soc. Chem. Commun. 1993:1054–1056. [Google Scholar]
- [229].Jiang X, Smith KM. Syntheses of some β-substituted alkyne porphyrins related to protoporphyrin-IX. J. Chem. Soc. Perkin Trans. 1996;1:1601–1606. [Google Scholar]
- [230].Bauder C, Ocampo R, Callot HJ. Total synthesis of cyclopentenoporphyrins of sedimentary origin: deoxophylloerythroetioporphyrin, chlorophyll c fossils and related compounds. Tetrahedron. 1992;48:5135–5150. [Google Scholar]
- [231].Malinen PK, Tauber AY, Hynninen PH, Montforts F-P. An unexpected Wittig reaction of dimethyl 3,3'-(3-formyl-2,7,12,18-tetramethyl-21H, 23H-porphyrin-13,17-diyl)-dipropionate. Tetrahedron Lett. 1997;38:3381–3382. [Google Scholar]
- [232].Evans B, Smith KM, Cavaleiro JAS. Bile pigment studies. Part 4. Some novel reactions of metalloporphyrins with thallium(III) and cerium(IV) salts. Ring cleavage of meso-tetraphenylporphyrin. J. Chem. Soc. Perkin Trans. 1. 1978:768–773. [Google Scholar]
- [233].Barnett GH, Evans B, Smith KM, Besecke S, Fuhrhop J-H. Synthesis of meso-pyridinium porphyrin salts. Tetrahedron Lett. 1976:4009–4012. [Google Scholar]
- [234].Fuhrhop J-H, Wanja U, Bunzel M. Metalloporphyrins in polymeric matrices, micelles and vesicles, V. 5-(1'-Methyl-4,4'-bipyridinium-1-yl)octaethylporphyrin dichloride, a meso-viologenporphyrinate. Liebigs Ann. Chem. 1984:426–434. [Google Scholar]
- [235].Fuhrhop J-H, Kruger W, Meding U. Synthesis and reactivity of meso-(3-carbamoylpyridinio)-porphyrins. Liebigs Ann. Chem. 1981:1367–1377. [Google Scholar]
- [236].Shine HJ, Padilla AG, Wu S-M. Ion radicals. 45. Reactions of zinc tetraphenylporphyrin cation radical perchlorate with nucleophiles. J. Org. Chem. 1979;44:4069–4074. [Google Scholar]
- [237].El Kahef L, Giraudeau A. P-Substitution de la méso-tétraphénylporphyrine de zinc par voie électrochimique. Can. J. Chem. 1991;69:1161–1165. [Google Scholar]
- [238].Smith KM, Barnett GH, Evans B, Martynenko Z. Novel meso-substitution reactions of metalloporphyrins. J. Am. Chem. Soc. 1979;101:5953–5961. [Google Scholar]
- [239].Ruhlman L, Giraudeau A. One-pot electrochemical generation of a porphyrin dimer with a bis(diphenylphosphonium)acetylene bridge. Chem. Commun. 1996:2007–2008. [Google Scholar]
- [240].Giraudeau A, Ruhlman L, El Kahef L, Gross M. Electrosynthesis and characterization of symmetrical and unsymmetrical linear porphyrin dimers and their precursor monomers. J. Am. Chem. Soc. 1996;118:2969–2979. [Google Scholar]
- [241].Ruhlman L, Lobstein S, Gross M, Giraudeau A. An electrosynthetic path toward pentaporphyrins. J. Org. Chem. 1999;64:1352–1355. [Google Scholar]
- [242].Ogawa T, Nishimoto Y, Yoshida N, Ono N, Osuka A. Completely regioselective synthesis of directly linked meso, meso and meso, β porphyrin dimers by one-pot electrochemical oxidation of metalloporphyrins. Angew. Chem. Int. Ed. 1999;38:176–179. [Google Scholar]
- [243].Yoshida N, Shimidzu H, Osuka A. Meso-meso linked diporphyrins from 5,10,15-trisubstituted porphyrins. Chem. Lett. 1998:55–56. [Google Scholar]
- [244].Nakano A, Shimidzu H, Osuka A. Facile regioselective meso-iodination of porphyrins. Tetrahedron Lett. 1998;39:9489–9492. [Google Scholar]
- [245].Dolphin D, Felton RH, Borg DC, Fajer J. Isoporphyrins. J. Am. Chem. Soc. 1970;92:743–745. doi: 10.1021/ja00714a038. [DOI] [PubMed] [Google Scholar]
- [246].Lee WA, Bruice TC. Transfer of oxygen from percarboxylic acids and alkyl hydroperoxides to (meso-tetraphenylporphinato)cobalt(III) chloride. Inorg, Chem. 1986;25:131–135. [Google Scholar]
- [247].Senge MO. Stirring the porphyrin alphabet soup - functionalization reactions for porphyrins. Chem. Commun. 2011;47:1943–1960. doi: 10.1039/c0cc03984e. [DOI] [PubMed] [Google Scholar]
- [248].Senge MO. Nucleophilic substitution as a tool for the synthesis of unsymmetrical porphyrins. Acc. Chem. Res. 2005;38:733–743. doi: 10.1021/ar0500012. [DOI] [PubMed] [Google Scholar]
- [249].Senge MO, Feng X. Synthesis of directly meso-meso linked bisporphyrins using organolithium reagents. Tetrahedron Lett. 1999;40:4165–4168. [Google Scholar]
- [250].Senge MO, Feng X. Regioselective reaction of 5,15-disubstituted porphyrins with organolithium reagents - synthetic access to 5,10,15-trisubstituted porphyrins and directly meso-meso-linked bisporphyrins. J. Chem. Soc., Perkin Trans. 1. 2000:3615–3621. [Google Scholar]
- [251].Feng X, Senge MO. An efficient synthesis of highly functionalized asymmetric porphyrins with organolithium reagents. J. Chem. Soc., Perkin Trans. 1. 2001:1030–1038. [Google Scholar]
- [252].Feng X, Bischoff I, Senge MO. Mechanistic studies on the nucleophilic reaction of porphyrins with organolithium reagents. J. Org. Chem. 2001;66:8693–8700. doi: 10.1021/jo010559x. [DOI] [PubMed] [Google Scholar]
- [253].Krattinger B, Callot HJ. New routes from porphyrins to stable phlorins. Meso-alkylation and reduction of meso-tetraphenyl- and octaalkylporphyrins. Tetrahedron Lett. 1996;37:7699–7702. [Google Scholar]
- [254].Jiang X, Nurco DJ, Smith KM. Direct meso-alkylation of meso-formylporphyrins using Grignard reagents. Chem. Commun. 1996:1759–1760. [Google Scholar]
- [255].Segawa H, Azumi R, Shimidzu T. Direct hydroxylation at the meso position of gold(III) tetraphenylporphyrin by nucleophilic addition: novel hydroxyphlorin derivatives. J. Am. Chem. Soc. 1992;114:7564–7565. [Google Scholar]
- [256].Ogoshi H, Sugimoto H, Yoshida Z-I, Kobayashi H, Sakai H, Maeda Y. Syntheses and magnetic properties of aryliron(III) complexes of octaethylporphyrins. J. Organomet. Chem. 1982;234:185–195. [Google Scholar]
- [257].Cocolios P, Lagrange G, Guilard R. Synthese et caracterisation par RMN 1H d'alkyl(aryl)ferriporphyrines a liaison σ metal carbone. J. Organomet. Chem. 1983;253:65–79. [Google Scholar]
- [258].Callot HJ, Schaeffer E. Model for the in vivo transformation of cyto- chrome P-450 into “green pigments”. Tetrahedron Lett. 1980;21:1335–1338. [Google Scholar]
- [259].Dolphin D, Halko DJ, Johnson E. Reversible cobalt-nitrogen alkyl and acyl group migration in cobalt porphyrins. Inorg. Chem. 1981;20:4348–4351. [Google Scholar]
- [260].Lange M, Mansuy D. N-Substituted porphyrin formation from carbene iron-porphyrin complexes: a possible pathway for cytochrome P450 heme destruction. Tetrahedron Lett. 1981;22:2561–2564. [Google Scholar]
- [261].Ortiz de Montellano PR, Kunze KL, Augusto O. Hemoprotein destruction. Iron-nitrogen shift of a phenyl group in a porphyrin complex. J. Am. Chem. Soc. 1982;104:3545–3546. [Google Scholar]
- [262].Callot HJ, Metz F. J. Acid-catalysed intramolecular cobalt to nitrogen aryl migration and intramolecular reverse reaction in cobalt porphyrins. Synthesis of N-phenylporphyrins. Chem. Soc. Chem. Commun. 1982:947–948. [Google Scholar]
- [263].Setsune J-I, Dolphin D. Organometallic aspects of cytochrome-p-450 metabolism. Can. J. Chem. 1987;65:459–467. [Google Scholar]
- [264].Kadish KM, Caemelbecke EV, D’Souza F, Medforth CJ, Smith KM, Tabard A, Guilard R. Generation of a stable 8-bonded Fe(IV) porphyrin. Formation and reactivity of [(OETPP)FeIV(C6H5)n+ where n = 1,2 or 3 and OETPP is the dianion of 2,3,7,8,12,13,17,18-octaethyl-5,10,15,20-tetraphenylporphyrin. Organometallics. 1993;12:2411–2413. [Google Scholar]
- [265].McCombie SW, Smith KM. Oxophlorin (oxyporphyrin) synthesis. Tetrahedron Lett. 1972;24:2463–2464. [Google Scholar]
- [266].Barnett GH, Hudson MF, McCombie SW, Smith KM. Synthesis of oxophlorins (oxyporphyrins) from zinc and magnesium porphyrins. J. Chem. Soc. Perkin Trans. 1. 1973:691–696. [Google Scholar]
- [267].Stolzenberg AM, Glazer PA, Foxman BM. Structure, reactivity, and electrochemistry of free-base β-oxoporphyrins and metallo-β-oxoporphyrins. Inorg. Chem. 1986;25:983–991. [Google Scholar]
- [268].Chang CK, Wu W. On the hydrogen peroxide/sulfuric acid oxidation of mesoporphyrin. Synthesis of mesoporphyrindiones. J. Org. Chem. 1986;51:2134–2138. [Google Scholar]
- [269].Bonnett R, Cornell P, McDonagh AF. The meso-reactivity of porphyrins and related compounds. Part VII. Benzoyloxylation of phenylpyrroles and of octaethylporphyrin. J. Chem. Soc. Perkin Trans. 1976;1:794–800. [PubMed] [Google Scholar]
- [270].Jackson AH, Rao KRN, Wilkins M. Synthetic and biosynthetic studies of porphyrins. Part 11. The synthesis of meso oxygenated protoporphyrins. J. Chem. Soc. Perkin Trans. 1. 1987:307–312. [Google Scholar]
- [271].Fuhrhop J-H, Besecke S, Subramanian J. A stable octaethyloxophlorin radical. J. Chem. Soc. Chem. Commun. 1973:1–2. [Google Scholar]
- [272].Fuhrhop J-H, Besecke S, Subramanian J, Mengersen C, Riesner D. Reactions of oxophlorins and their π radicals. J. Am. Chem. Soc. 1975;97:7141–7152. [Google Scholar]
- [273].Khoury RG, Jaquinod L, Shachter AM, Nelson NY, Smith KM. Stabilization of neutral oxophlorin π-radicals by bulky meso-alkyl groups. Chem. Commun. 1997:215–216. [Google Scholar]
- [274].Bonnett R, Nizhnik AN, Berenbaum MC. Second generation tumour photosensitisers: the synthesis of octa-alkyl chlorins and bacteriochlorins with graded amphiphilic character. J. Chem. Soc. Chem. Commun. 1989:1822–1823. [Google Scholar]
- [275].Adams KR, Berenbaum MC, Bonnett R, Nizhnik AN, Salgado A, Vallés MA. Second generation tumour photosensitisers: the synthesis and biological activity of octaalkyl chlorins and bacteriochlorins with graded amphiphilic character. J. Chem. Soc. Perkin Trans. 1. 1992:1465–1470. [Google Scholar]
- [276].Bruckner C, Dolphin D. 2,3-vic-Dihydroxy-meso-tetraphenylchlorins from the osmium tetroxide oxidation of meso-tetraphenylporphyrin. Tetrahedron Lett. 1995;36:3295–3298. [Google Scholar]
- [277].Chang CK, Sotiriou C. C-Hydroxy- and C-methylchlorins. A convenient route to heme d and bonellin model compounds. J. Org. Chem. 1985;50:4989–4991. [Google Scholar]
- [278].Chang CK, Sotiriou C. Migratory aptitudes in pinacol rearrangement of vic-dihydroxychlorins. J. Heterocycl. Chem. 1985;22:1739–1741. [Google Scholar]
- [279].Nakamura K, Osamura Y. MO study of the possibility of a concerted mechanism in the pinacol rearrangement. J. Phys. Org. Chem. 1990;3:737–745. [Google Scholar]
- [280].Kozyrev AN, Pandey RK, Medforth CJ, Zheng G, Dougherty TJ, Smith KM. Syntheses of stable bacteriochlorophyll-a derivatives as potential photosensitizers for photodynamic therapy. Tetrahedron Lett. 1996;37:747–751. [Google Scholar]
- [281].Pandey RK, Isaac M, MacDonald I, Medforth CJ, Senge MO, Dougherty TJ, Smith KM. Pinacol-pinacolone rearrangements in vic-dihydroxychlorins and bacteriochlorins: effect of substituents at the peripheral positions. J. Org. Chem. 1997;62:1463–1472. [Google Scholar]
- [282].Chang CK, Sotiriou C. A novel method of functionalizing the ethyl chain of octaethylporphyrin. J. Org. Chem. 1987;52:926–929. [Google Scholar]
- [283].Chang CK, Sotiriou C, Wu W. Differentiation of bacteriochlorin and isobacteriochlorin formation by metallation. High yield synthesis of porphyrindiones via OsO4 oxidation. J. Chem. Soc. Chem. Commun. 1986:1213–1215. [Google Scholar]
- [284].Bruckner C, Dolphin D. β,β'-Dihydroxylation of meso-tetraphenylchlorins and metallochlorins. Tetrahedron Lett. 1995;36:9425–9428. [Google Scholar]
- [285].Pandey RK, Shiau F-Y, Isaac M, Ramaprasad S, Dougherty TJ, Smith KM. Substituent effects in tetrapyrrole subunit reactivity and pinacolpinacolone rearrangements: vic-dihydroxychlorins and vic-dihydroxybacterio-chlorins. Tetrahedron Lett. 1992;33:7815–7818. [Google Scholar]
- [286].Pandey RK, Shiau F-Y, Sumlin AB, Dougherty TJ, Smith KM. Syntheses of new bacteriochlorins and their antitumor activity. Bioorg. Med. Chem. Lett. 1994;4:1263–1267. [Google Scholar]
- [287].Pandey RK, Constantine S, Tsuchida T, Zheng G, Medforth CJ, Aoudia M, Kozyrev AN, Rodgers MAJ, Kato H, Smith KM, Dougherty TJ. Synthesis, photophysical properties. in vivo photosensitizing efficacy, and human serum albumin (HSA) binding properties of some novel bacteriochlorins. J. Med. Chem. 1997;40:2770–2779. doi: 10.1021/jm9702894. [DOI] [PubMed] [Google Scholar]
- [288].Beavington R, Rees PA, Burn PL. A study on the oxidation of 2-hydroxyporphyrins to porphyrin-α-diones . J. Chem. Soc. Perkin Trans. 1. 1998:2847–2852. [Google Scholar]
- [289].Crossley MJ, Prashar JK. Thiophene-appended porphyrin systems. Tetrahedron Lett. 1997;38:6751–6754. [Google Scholar]
- [290].Gulyas PT, Langford SJ, Lokan NR, Ranasinghe MG, Paddon-Row MN. Convenient synthetic route to rigid donor-{bridge}-acceptor systems involving porphyrin and phenanthroline annulation of norbornylogous bridges via 2,3-norbornanediones. J. Org. Chem. 1997;62:3038–3039. doi: 10.1021/jo970279a. [DOI] [PubMed] [Google Scholar]
- [291].Sessler JL, Brown CT, O’Connor D, Springs SL, Wang R, Sathiosatham M, Hirose T. A rigid chlorin-naphthalene diimide conjugate. A possible new noncovalent electron transfer model system. J. Org. Chem. 1998;63:7370–7374. doi: 10.1021/jo9810112. [DOI] [PubMed] [Google Scholar]
- [292].Crossley MJ, McDonald JA. Fused porphyrin-imidazole systems: new building blocks for synthesis of porphyrin arrays. J. Chem. Soc. Perkin Trans. 1. 1999:2429–2431. [Google Scholar]
- [293].Bonnett R, McDonagh AF. The meso-reactivity of porphyrins and related compounds. Part VI. Oxidative cleavage of the haem system. The four isomeric biliverdins of the IX series. J. Chem. Soc., Perkin Trans. 1. 1973:881–888. doi: 10.1039/p19730000881. [DOI] [PubMed] [Google Scholar]
- [294].Crusats J, Suzuki A, Mizutani , Ogoshi H. Regioselective porphyrin bridge cleavage controlled by electronic effects. Coupled oxidation of 3-demethyl-3-(trifluoromethyl)mesohemin IX and identification of its four biliverdin derivatives. J. Org. Chem. 1998;63:602–607. doi: 10.1021/jo9714728. [DOI] [PubMed] [Google Scholar]
- [295].Wasser PKW, Fuhrhop J-H. The photooxygenation of metalloporphyrins and metallochlorins. Ann. N.Y. Acad. Sci. 1973;206:533–547. doi: 10.1111/j.1749-6632.1973.tb43235.x. [DOI] [PubMed] [Google Scholar]
- [296].Catalano MM, Crossley MJ, Harding MM, King LG. Control of reactivity at the porphyrin periphery by metal ion co-ordination: a general method for specific nitration at the β-pyrrolic position of 5,10,15,20-tetraarylporphyrins. J. Chem. Soc., Chem. Commun. 1984:1535–1536. [Google Scholar]
- [297].Ongayi O, Vicente MGH, Ghosh B, Fronczek FR, Smith KM. Bilitrienones from the chemical oxidation of dodecasubstituted porphyrins. Tetrahedron. 2010;66:63–67. doi: 10.1016/j.tet.2009.10.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Bonnett R, Dimsdale MJ. The meso-reactivity of porphyrins and related compounds. Part V. The meso-oxidation of metalloporphyrins. J. Chem. Soc., Perkin Trans. 1. 1972:2540–2548. doi: 10.1039/p19720002540. [DOI] [PubMed] [Google Scholar]
- [299].Ongayi O, Vicente MGH, Ou Z, Kadish KM, Kumar MR, Fronczek FR, Smith KM. Synthesis and electrochemistry of undecasubstituted metallo-benzoylbiliverdins. Inorg. Chem. 2006;45:1463–1470. doi: 10.1021/ic050841c. [DOI] [PubMed] [Google Scholar]
- [300].Fuhrhop J-H, Wasser PKW, Subramanian J, Schrader U. Formylbiliverdine und ihre metallkomplexe. Liebigs Ann. Chem. 1974:1450–1466. [Google Scholar]
- [301].Cox GS, Whitten DG. Mechanisms for the photooxidation of protoporphyrin IX in solution. J. Am. Chem. Soc. 1982;104:516–521. [Google Scholar]
- [302].Brault D, Aveline B, Delgado O, Vever-Bizet C. Chlorin-type photosensitizers derived from vinyl porphyrins. Proc. SPIE. 1994;2325:40–47. [Google Scholar]
- [303].Iakovides P, Smith KM. Synthesis of oxygen analogues of the sulfchlorins. Tetrahedron Lett. 1990;31:3853–3856. [Google Scholar]
- [304].Iakovides P, Smith KM. Syntheses of oxygen analogues of sulfhemes-A and -C. Tetrahedron. 1996;52:1123–1148. [Google Scholar]
- [305].Smith KM, Brown SB, Troxler RF, Lai J-J. Mechanism of photo-oxygenation of meso-tetraphenylporphyrin metal complexes. Tetrahedron Lett. 1980;21:2763–2766. [Google Scholar]
- [306].Smith KM, Brown SB, Troxler RF, Lai J-J. Photo-oxygenation of meso-tetraphenylporphyrin metal complexes. Photochem. Photobiol. 1982;36:147–152. doi: 10.1111/j.1751-1097.1982.tb04356.x. [DOI] [PubMed] [Google Scholar]
- [307].Matsuura T, Inoue K, Ranade AC, Saito I. Photooxygenation of magnesium meso-tetraphenylporphyrin. Photochem. Photobiol. 1980;31:23–26. [Google Scholar]
- [308].Cavaleiro JAS, Neves MGPMS, Hewlins MJE, Jackson AH. The photo-oxidation of meso-tetraphenylporphyrins. J. Chem. Soc., Perkin Trans. 1. 1990:1937–11943. [Google Scholar]
- [309].Whitlock HW, Jr., Hanauer R, Oester MY, Bower BK. Diimide reduction of porphyrins. J. Am. Chem. Soc. 1969;91:7485–7489. [Google Scholar]
- [310].Ulman A, Gallucci J, Fisher D, Ibers JA. Facile syntheses of tetraalkylchlorin and tetraalkylporphyrin complexes and comparison of the structures of the tetramethylchlorin and tetramethylporphyrin complexes of nickel(II) J. Am. Chem. Soc. 1980;102:6852–6854. [Google Scholar]
- [311].Vasudevan J, Stibrany RT, Bumby J, Knapp S, Potenza JA, Emge TJ, Schugar HJ. J. An edge-over-edge Zn(II) bacteriochlorin dimer having an unshifted Qy band. The importance of π-Overlap. J. Am. Chem. Soc. 1996;118:11676–11677. [Google Scholar]
- [312].Stolzenberg AM, Simerly SW, Steffey BD, Haymond GH. An edge-over-edge Zn(II) bacteriochlorin dimer having an unshifted Qy band. The importance of π-overlap. J. Am. Chem. Soc. 1997;119:11843–11854. [Google Scholar]
- [313].Smith KM, Lai J-J. Syntheses of heme-d models. J. Am. Chem. Soc. 1984;106:5746–5748. [Google Scholar]
- [314].Burns DH, Lai J-J, Smith KM. Syntheses of chlorins from unsymmetrically substituted iron(III) porphyrins. J. Chem. Soc. Perkin Trans. 1. 1988:3119–3131. [Google Scholar]
- [315].Whitten DG, Yau JC, Carroll FA. Photochemistry and oxidation-reduction reactions of tin porphyrins. J. Am. Chem. Soc. 1971;93:2291–2296. [Google Scholar]
- [316].Fuhrhop J-H, Lumbantobing T. The redox properties of tin(IV) and germanium(IV) octaethylporphinato-dihydroxides as compared to other metallooctaethylporphins. Tetrahedron Lett. 1970;32:2815–2818. [Google Scholar]
- [317].Harel Y, Manassen J. Photoreduction of tetraphenylporphyrins by amines in the visible. Photochemical syntheses of reduced tetraphenylporphyrins and the mechanism of photoreduction. J. Am. Chem. Soc. 1978;100:6228–6234. [Google Scholar]
- [318].Wolf H, Scheer H. Photochemische hydrierung von phäoporphyrinen: 7,8-cis-phäophorbide. Liebigs Ann. Chem. 1973:1710–1740. [Google Scholar]
- [319].Smith KM, Simpson DJ. Site-specific reduction of unsymmetrically substituted porphyrins to give isomerically pure chlorins. J. Chem. Soc. Chem. Commun. 1987:613–614. [Google Scholar]
- [320].Iakovides P, Simpson DJ, Smith KM. Regioselective photoreduction of zinc(II) porphyrins to give chlorins. Photochem. Photobiol. 1991;54:335–343. doi: 10.1111/j.1751-1097.1991.tb02025.x. [DOI] [PubMed] [Google Scholar]
- [321].Kämpfen U, Eschenmoser A. Porphyrin synthesis by ring transplantation. Tetrahedron Lett. 1985;26:5899–5902. [Google Scholar]
- [322].Closs GL, Closs LE. Negative ions of porphin metal complexes. J. Am. Chem. Soc. 1963;85:818–819. [Google Scholar]
- [323].Buchler JW, Schneehage HH. Metallkomplexe mit tetrapyrrol-liganden. V. Zum einfluβ des zentralmetalls auf die hydrierung des octaäthylporphin-systems. Tetrahedron Lett. 1972:3803–3806. [Google Scholar]
- [324].Buchler JW, Puppe L. Metallkomplexe mit Tetrapyrrol-Liganden, II1) Metallchelate des α.γ-Dimethyl-α.γ-dihydro-octaäthylporphins durch reduzierende Methylierung von Octaäthylporphinato-zink. Liebigs Ann. Chem. 1970;740:142–163. [Google Scholar]
- [325].Dwyer PN, Buchler JW, Scheidt WR. Crystal structure and molecular stereochemistry ofα,γ-dimethyl-α,γ-dihydrooctaethylporphinatonickel(II) J. Am. Chem. Soc. 1974;96:2789–2795. [Google Scholar]
- [326].Shulga AM, Sinyakov GN, Filatov IV, Gurinovich GP, Dzilinski K. Study of electronic structure of Zn-porphyrin and Zn-chlorin π-anions J. Mol. Struct. 1995;348:65–68. [Google Scholar]