

Abstract
Hepatitis C virus (HCV) is a global public health problem involving chronic infection of the liver in over 170 million people. Chronic HCV causes liver disease and is linked with liver cancer. Viral innate immune evasion strategies and human genetic determinants underlie the transition of acute HCV infection to viral persistence and the support of chronic infection. Host genetic factors, such as sequence polymorphisms in IFNL3, a gene in the host interferon system, can influence both the outcome of infection and response to antiviral therapy. Recent insights into how HCV regulates innate immune signaling within the liver reveal a complex interaction of patient genetic background with viral and host factors of innate immune triggering and control that impart the outcome of HCV infection and immunity.
HCV is highly successful at establishing a chronic infection
Hepatitis C virus (HCV) is a major public health problem that infects approximately 170 million people worldwide1. Infection occurs within hepatocytes, the chief parenchymal cell of the liver. HCV typically causes a chronic infection, and is a major cause of liver disease, liver transplantation, and liver cancer2. The first line of immune defense against HCV relies on cell-intrinsic innate immunity within hepatocytes. This innate immune response serves to recognize HCV as non-self and induces local antiviral defenses in the infected cell and liver tissue which can function to recruit and modulate the actions of immune cells of the adaptive immune response. Hepatic innate immunity is therefore essential for controlling the outcome of HCV infection and immunity. However, the majority of those infected with HCV fail to mount a productive immune response that clears infection. Underscoring this problem is that HCV has evolved multiple mechanisms to regulate and evade innate immunity, thus providing a foundation for chronic infection. The standard of care therapy for HCV infection has been treatment with pegylated IFN-α plus ribavirin. As an antiviral cytokine, IFN is an innate immune therapeutic but is very unpleasant for the patient and for those infected with the difficult to treat HCV genotypes (1 and 4) it is only curative 40-50% of the time3. While first-generation direct acting antiviral drugs are now being introduced into the clinic, they are applied in combination with IFN to enhance efficacy and reduce chances for viral resistance breakthrough4-5. Moreover, underlying HCV infection outcome are polymorphisms in IFNL3 (also referred to as IL28B), a human gene of the host system of innate antiviral defense, and these polymorphisms can influence both the outcome of infection and response to therapy6-10. Understanding how HCV regulates hepatic innate immune responses is essential for guiding the improvement of HCV therapy, developing effective vaccine applications, and designing new therapeutic strategies to control infection and suppress liver disease. In this perspective, we will describe how both virus and host control of innate immune factors contribute to HCV persistence in the liver and how these virus/host interactions impact the outcome of infection, immunity, and current IFN-based antiviral therapy.
The host response to HCV infection
HCV is a hepacivirus and member of the Flaviviridae family. The viral genome is a single copy of positive-sense RNA. After entry into hepatocytes, the virus uncoats and the viral genome is translated into a single polyprotein that is co- and post-translationally processed into structural and nonstructural proteins by a combination of host peptidases and two viral-encoded proteases. The HCV nonstructural proteins assemble as a replication complex onto modified intracellular membranes to replicate the HCV genome. This process involves the production of an antigenomic replication intermediate RNA and likely an accumulation of double stranded (ds) RNA intermediate products. The new viral genomes are packaged into viral particles by the viral structural proteins. The resulting virus is released from the hepatocyte in association with host lipoproteins, such that in the blood HCV is present as a lipoprotein-coated virus11. During the viral replication process HCV is sensed as non-self by pathogen recognition receptors (PRRs) in the host cell that identify and bind to pathogen associated molecular pattern (PAMP) motifs within viral products, leading to coordinated activation of the innate immune response and adaptive immune responses. Both the innate and adaptive arms of immunity, including cross-talk between liver-resident and infiltrating cells, such as hepatocytes, Kupffer cells, pDCs, natural killer (NK) cells, and other immune cells, contribute to the host’s ability to resolve HCV infection12-14. However, despite these immune defenses HCV infection becomes chronic in about 70-80% of those who are acutely infected2. This outcome is due to a combination of host and viral factors that regulate the intracellular innate immune response against HCV. Importantly, inactivation of this intracellular innate immune response by HCV may also result in a non-functional adaptive immune response. Indeed, PRR signaling (specifically MAVS) is required for functional innate and adaptive responses during infection with West Nile virus, a related virus of the Flaviviridae family15. While outside of the scope of this perspective, a functional adaptive immune response is also critical to resolving HCV infection (reviewed in 14,16).
Detection of HCV
A variety of PRRs sense viruses as foreign invaders within the host cell through specific PAMP recognition to activate innate immune signaling. The RIG-I-like receptors (RLRs), retinoic acid inducible gene-I (RIG-I) and melanoma differentiation antigen 5 (MDA5), are cytosolic PRRs that sense RNA viruses. Toll-like receptors (TLRs) mediate virus sensing from within endosomal compartments to signal innate defenses, while Nod-like receptors (NLRs) serve to sense cytosolic viral products or viral metabolites to drive inflammatory responses. Activation of these PRRs drives the innate antiviral and proinflammatory responses that limit virus replication and spread while also serving to recruit adaptive immune cells and enhance their effector actions at the site of infection. A variety of other nucleic acid binding proteins can also serve as putative PRRs where their engagement of viral nucleic acid results in their interaction with and regulation of specific PRR signaling pathways. Protein kinase R (PKR) is an example of these nontraditional PRRs, as its dsRNA binding activity promotes its interaction with mitochondrial antiviral signaling protein (MAVS) impart PRR signaling of innate immunity 17-18
We have shown that HCV is recognized by RIG-I within hours of infection and activates downstream signaling prior to extensive viral protein synthesis19. RIG-I signaling during HCV infection is initiated upon its binding of PAMP RNA that includes an exposed 5’ triphosphate and the 3’ non-translated region of the HCV genome RNA rich in poly U/UC ribonucleotides20-21. This multi-component PAMP motif presents a non-self RNA signature that is detected by RIG-I to activate innate immunity. While the 5’ triphosphate and poly U/UC region are at opposite ends of the viral genome, known intra-genome interactions between the 5’ and 3’ ends of the RNA22 can bring both into proximity for RIG-I binding and activation by HCV replication intermediates. HCV PAMP could be presented to RIG-I either from incoming genomes after viral uncoating or during HCV genome amplification. HCV RNA binding induces a RIG-I conformational change that promotes oligomerization and translocation from the cytosol into intracellular membranes23-25. This process requires interactions with the chaperone protein 14-3-3ε and the E3 ubiquitin ligase TRIM25, which together with RIG-I comprise a translocon that facilitates the interaction of RIG-I with the signaling adaptor protein MAVS24,26. The RIG-I/MAVS interaction promotes the formation of a MAVS signalosome that propagates activation of downstream effector molecules, including the transcription factors IRF-3 and NF-κB, and a variety of proinflammatory cytokines27 (Fig. 1).
Figure 1. Sensing of HCV can activate innate antiviral defenses through IFN induction in hepatocytes.
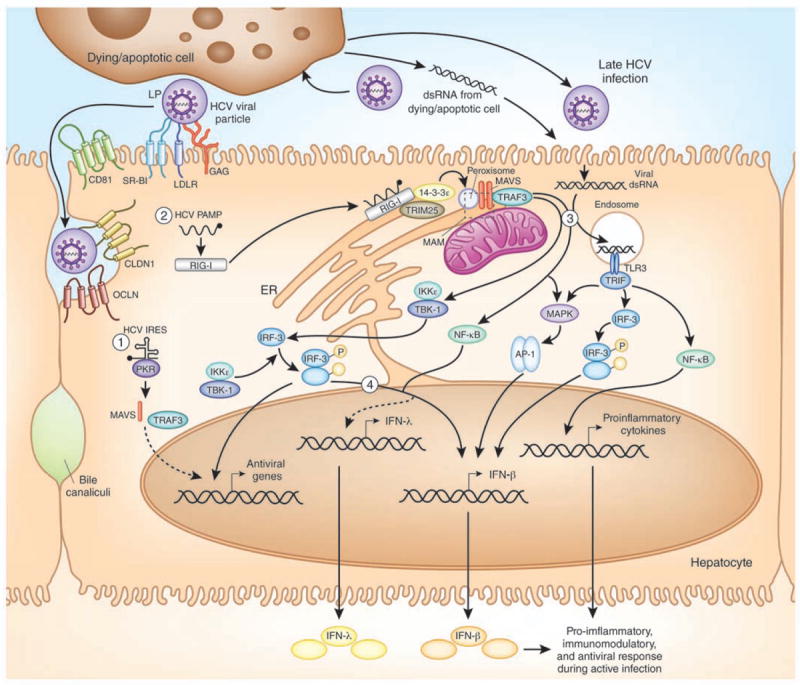
Innate sensing of HCV in the infected hepatocyte occurs through the combined actions of (1) PKR (2) RIG-I, and (3) TLR3. These proteins recognize specific features of HCV, including the dsRNA in the HCV IRES and the HCV poly U/UC PAMP early during infection; and dsRNA that accumulates following HCV infection (virus particle coated with apolipoprotein (LP) is depicted) or by uptake of HCV dsRNA from dying cells later during infection. This recognition leads to downstream signaling, as indicated by the arrows, that results in the induction of antiviral/immunomodulatory genes, IFN-β, and other proinflammatory cytokines. See the text for mechanistic details. The dashed box marks the location of the mitochondrial-associated ER membrane (MAM), a site of MAVS signaling. (4) The mechanisms leading to hepatic activation of IFN-λ during HCV infection have yet to be fully characterized.
TLR3 has also been implicated as a PRR that senses HCV, although its role in HCV detection and immunity is still not fully understood. TLR3 is an endosomal sensor of dsRNA expressed in a number of cell types within the liver, including hepatocytes and the liver resident macrophage Kupffer cells28-29. TLR3 signals are transmitted through the adaptor protein TRIF, which activates IRF-3 and NF-κB for the production of type I IFN, proinflammatory cytokines, and chemokines, as well as for apoptotic signaling30-31 (Fig. 1). Whereas synthetic dsRNA ligands of TLR3 can induce IRF-3 dependent signaling in cells expressing ectopic TLR3 within 24 hours28, HCV infection in these cells activates this signaling, including cytokine production, at later times (3-4 days) after infection28,32. The TLR3 ligand of HCV has recently been identified as HCV dsRNA replication intermediates that accumulate late during HCV replication and interact with the dsRNA binding domain of TLR332. It is unlikely that incoming viral genomes or early replication products serve as TLR3 ligands, as TLR3-based cytokine production requires several days to be induced. Instead, the HCV ligands of TLR3 are probably present either following uptake of extracellular HCV dsRNA (either from dying cells or from the extracellular milieu) by scavenger receptors on nearby uninfected cells33 or by accumulation of dsRNA viral replication intermediates exposed to TLR3 in either the endosome or autophagic vesicles34. Therefore, during HCV infection TLR3-mediated signaling could serve as a secondary innate immune detection or surveillance system for uninfected cells after initial RIG-I detection of HCV, possibly involved in setting up an antiviral state within regions of the liver or chemokine induction for recruitment of T cells to the liver during HCV infection32. Interestingly, the cytokine induction profile following TLR3 activation by HCV is distinct from that induced by the dsRNA mimic polyinosine-cytosine, suggesting a level of specificity attributed to the HCV/TLR3 interaction28,32. It is possible that the induction of the antiviral and proinflammatory responses by TLR3 and RIG-I could contribute to HCV-mediated pathogenesis, including liver inflammation and fibrosis progression during chronic infection, although the mechanisms of such immunopathogenesis are unknown. Additionally, the roles of TLR3 and RIG-I signaling in mediating HCV liver disease in vivo are not known.
Recent studies have now reclassified the well-known HCV antiviral protein kinase PKR as a genuine PRR for HCV that activates and contributes to innate immune signaling and IFN production35. PKR is a dsRNA binding protein whose kinase activity can be induced by binding to HCV dsRNA to phosphorylates the alpha subunit of eukaryotic initiation factor 2 (eIF2α) to suppress host mRNA translation, but not HCV translation because HCV uses an internal ribosome entry site (IRES) translation mechanism that is insensitive to the levels of eIF2α phosphorylation that suppress Cap-dependent translation of host mRNAs36-39. It is now known that PKR binding to HCV dsRNA also activates a kinase-independent signal transduction cascade that drives induction of specific ISGs and IFN-β production by signaling through MAVS, TRAF3, IRFs, and NF-κB, all prior to RIG-I activation17-18,35 (Fig. 1). The HCV ligand for PKR is the structured RNA at the IRES of the HCV RNA35,39. While the mechanisms governing PKR and RIG-I signaling cross-talk are still being defined, these studies reveal a role for PKR as a PRR that can cooperate with RIG-I to drive downstream gene expression that mediates antiviral defenses.
Innate immune effectors against HCV
Antiviral defenses are triggered by pathogen recognition and signaling that induces IFN and drives the expression of hundreds of ISGs encoding innate immune effectors that impart control of virus replication and spread. A recent study that directly compared the antiviral activity of nearly 400 ISGs has revealed that groups of ISGs or specific ISG “biosets” exhibit coordinate and synergistic virus-specific antiviral function40. For HCV, this bioset includes IRF family members, other signal transduction factors, and specific ISGs with potent viral regulatory activity, many with still undefined mechanisms of action (Table 1). While direct antiviral ISG action is important to control HCV replication41, full restriction of HCV infection is mediated by additional actions of innate immune signaling amplification through IRFs and other virus-responsive pathways42.
Table 1.
Anti-HCV interferon stimulated genes
| Gene Symbol | Proposed anti-HCV mechanism | Ref. |
|---|---|---|
| ADAR | editing viral RNA | 41 |
| DDIT4 | unknown | 41 |
| DDX58 (RIG-I) | activates IFN-β pathway signaling, including IRFs | 41,102 |
| DDX60 | enhances RIG-I signaling | 41 |
| EIF2AK2 (PKR) | inhibits protein translation (vie eIF2α); activates RIG-I | 35,103-105 |
| GBP1 | unknown | 106 |
| IFI44L | unknown | 41 |
| IFI6 | unknown | 106 |
| IFIT1 | sequesters viral nucleic acids; suppresses translation | 107-109 |
| IFIT3 | enhances MAVS/TBK1 signaling | 73,110 |
| IFITM1 | inhibits HCV receptor-mediated entry | 73,108,111 |
| IFITM3 | unknown, possibly inhibits intracellular trafficking | 73,112-113 |
| IRF-1 | IFN induction, direct induction of ISGs | 40,104 |
| IRF-7 | IFN induction, direct induction of ISGs | 41 |
| ISG12 | unknown | 106 |
| ISG20 | exonuclease activity | 103 |
| MAP3K14 (NIK) | unknown, possibly NF-κB pathway activation | 41 |
| MOV10 | unknown | 41 |
| MS4A4A | unknown | 41 |
| MX1 (MxA) | unknown | 106 |
| NOS2 | unknown | 73 |
| NT5C3 | unknown | 41 |
| OAS1 | activates RNaseL | 106 |
| OASL | unknown | 40,114 |
| PLSCR1 | unknown, possibly prevents membrane rearrangements | 73 |
| RNAseL | cleaves viral genome, activates RIG-I signaling | 73,115 |
| RSAD2 (viperin) | interacts with NS5A and VAP-A to block viral replication | 73,103,116-117 |
| SSBP3 | unknown | 41 |
| TRIM14 | unknown | 73 |
HCV evasion of innate antiviral immunity
Primary acute HCV infection can be cleared spontaneously when there is a high initial viremia43, suggesting that high level and rapid PRR signaling and innate immune induction following sensing of HCV PAMPs can control acute HCV infection. However, despite the effective non-self detection of HCV by RIG-I, TLR3, and PKR to trigger innate immune programs, as many as 80% of people with acute HCV infection do not effectively control the virus and develop a chronic infection2. This high frequency of chronic infection reflects the fact that HCV has evolved several mechanisms to evade and suppress innate immunity, resulting in HCV progression to chronicity (reviewed in42). The viral NS3/4A protease is a central component of the HCV innate immune evasion strategy. The multifunctional NS3/4A protease is required for HCV replication, during which it processes the HCV polyprotein at several sites to liberate the viral NS proteins44. The NS3/4A protease complex is anchored to intracellular membranes through the NS4A transmembrane domain and an amphipathic α-helix at the NS3 amino terminus that facilitates membrane association and cleavage of membrane-anchored substrates45-46. NS3/4A can block RIG-I signaling, because in addition to proteolytically processing the HCV polyprotein, NS3/4A targets and cleaves MAVS from intracellular membranes to prevent signal transduction19,47-51 (Fig. 2). As MAVS must be anchored to membranes for downstream signaling, this cleavage event prevents activation of the RIG-I pathway during acute infection, abrogates IFN induction, and supports the progression to chronic infection. However, we note that other hepatotropic viruses, including hepatitis A virus (HAV) and GB virus B (GVB-B), encode proteases that also cleave MAVS52-53. While GVB-B can mediate a chronic infection in marmosets54, HAV does not generally become chronic. Thus MAVS cleavage is probably necessary but not sufficient for viral chronicity.
Figure 2. HCV control of IFN induction and immune evasion.
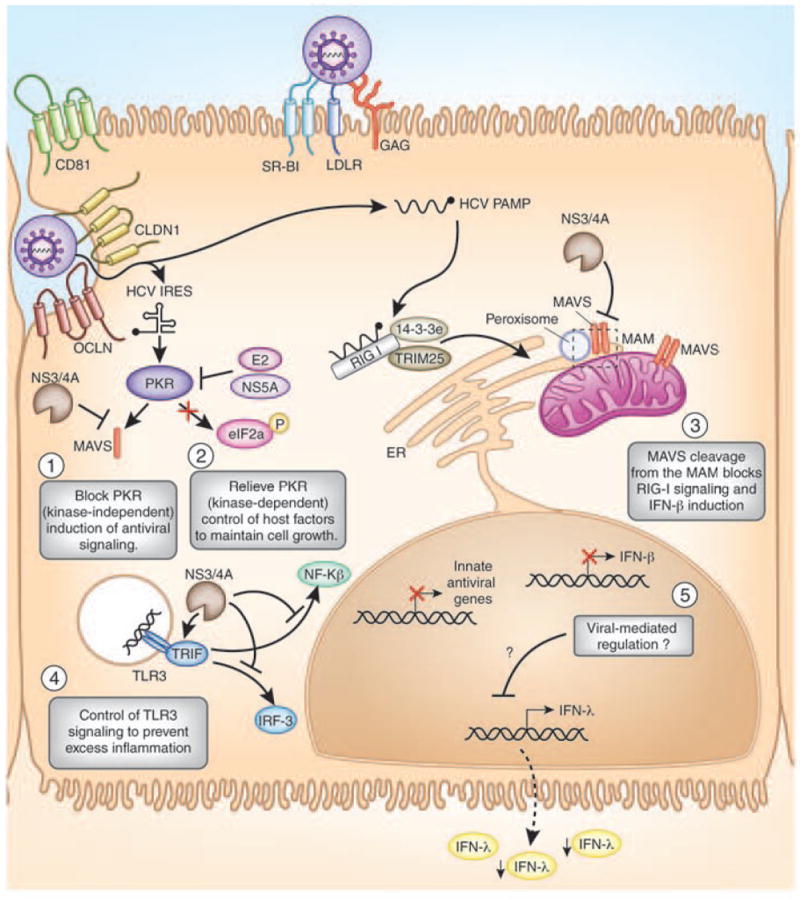
Immune evasion by HCV in the hepatocyte occurs at several points during viral infection. The proposed regulation is shown here, where the HCV NS3/4A protease cleaves the signaling adaptors MAVS (on the mitochondrial-associated ER membrane (MAM; in the region indicated by the dashed box) and TRIF to inactivate (1) PKR, (3) RIG-I, and (4) TLR3 signaling pathways to prevent induction of immunomodulatory innate antiviral genes and IFN-β allowing for HCV replication. (5) HCV infection control of IFN-λ induction is not yet defined; (2) HCV E2 and NS5A proteins inactivate PKR kinase-dependent activation of the host translation factor eIF2α to reactivate protein translation during infection.
NS3/4A cleavage of MAVS was previously thought to occur at the mitochondrial outer membrane, a primary site of MAVS localization19,51,55-56. However, we now know that the MAVS transmembrane domain also anchors the protein at diverse sites within an intracellular membrane network at peroxisomes and on mitochondrial-associated membranes (MAM), an ER membrane subdomain residing at junctions between the ER and mitochondria57-58. The NS3/4A protease complex localizes to all of these membranes during HCV infection58. However, rather than cleaving MAVS from the outer mitochondrial membrane, NS3/4A targets and cleaves the MAM-localized MAVS. This process abrogates RIG-I signaling despite a level of intact MAVS remaining on the mitochondria58. Therefore, during HCV infection, the MAM-resident MAVS likely transduces RIG-I pathway signaling. Cleavage of MAVS during HCV infection has also been shown in vivo in the liver of patients with chronic HCV infection19,59. Importantly, patients with cleaved MAVS exhibited lower levels of IFN pathway activation59. Thus, MAVS cleavage by the HCV NS3/4A protease disrupts RIG-I signaling of innate antiviral immunity and attenuates IFN production.
The HCV NS3/4A protease also proteolytically targets TRIF60, the TLR3 signaling-adaptor protein. Cell culture studies have shown that TRIF is cleaved by NS3/4A in vitro60. While specific TRIF proteolytic fragments have not been detected during HCV infection, the relative abundance of TRIF protein is decreased, likely due to destabilization and degradation following cleavage28. NS3/4A cleavage of TRIF suggests that control of TLR3 signaling must be important for successful HCV infection, perhaps by preventing excess inflammation that could impart viral suppression or by blocking chemokine induction required for vigorous cytotoxic T cell (CTL) responses known to be important for HCV clearance16. TLR3-independent RNA sensing mechanisms that signal through TRIF to impart innate immunity and inflammatory responses have also been described61, and NS3/4A targeting of TRIF may also contribute to HCV persistence by blocking their actions during infection.
HCV regulates PKR activity during viral infection; however this regulation is complex and need to be redefined in the context of PKR having seemingly opposing pro- and anti-HCV roles during infection35-37. PKR-mediated translational suppression of host mRNAs during HCV infection and IFN therapy can inhibit translation of host factors important for HCV replication and cellular growth (PKR functions as an antiviral molecule) but it also can inhibit translation of ISGs and IFN (PKR functions as a proviral molecule)36-37. Furthermore, PKR-kinase independent signaling during HCV infection to activate specific ISGs and IFN-β would be considered to be antiviral. We know that HCV has several PKR-inactivation strategies, both at the level of PKR/PRR signaling (NS5A/E2 direct actions on PKR; NS3/4A cleavage of MAVS) and at the level of PKR-regulated translational inhibition (NS5A/E262-64) that likely contribute to viral persistence and/or IFN therapy responses, but the exact mechanisms, timing, and how they would support this viral persistence are still unclear (Fig. 2). It is possible that NS5A and E2 inactivation of PKR-translational suppression could function only at specific times during infection (for example, after NS3/4A suppression of MAVS-dependent signaling) to ensure the requisite synthesis of host factors required to maintain cellular growth, while supporting an environment for persistent HCV replication.
Innate immune regulation in the HCV patient might determine the response to IFN therapy
IFN-based therapies are the standard course of therapy for HCV. IFN is an antiviral cytokine that induces the expression of hundreds of ISGs, many of which have antiviral or immunomodulatory activities that can limit virus replication and spread, as well as prime the adaptive immune response to HCV infection65. In fact, in acutely-infected patients, IFN-therapy is extremely successful at preventing chronic infection66. However, in chronically infected patients, the response to IFN-based therapies is variable, and many patients maintain high levels of HCV viremia in spite of IFN treatment. We propose that the low effectiveness of IFN therapy in those chronically infected with HCV is attributed to a combination of viral and host factors that contribute to regulation of IFN action and treatment-induced clearance (Fig. 3). The viral factor that most highly predicts IFN treatment response is the viral genotype. There are 6 major HCV genotypes defined by their sequence conservation and variation that further divides them as subtypes and viral quasispecies67. Interestingly patients infected with different HCV genotypes have differential therapy responses. Patients infected with HCV genotypes 2 and 3 exhibit the highest response rates to therapy, with 70-80% of these patients achieving a sustained virologic response (SVR) while only 45-60% of patients with HCV genotype 1 or 4 infection achieve SVR (summarized in68). Indeed, HCV genotypes 1 and 4 induce high levels of hepatic ISG expression in the infected patient liver before therapy resulting in an IFN-insensitive phenotype that attenuates treatment responses69-71.
Figure 3. Factors that influence the host response to IFN therapy during HCV infection.
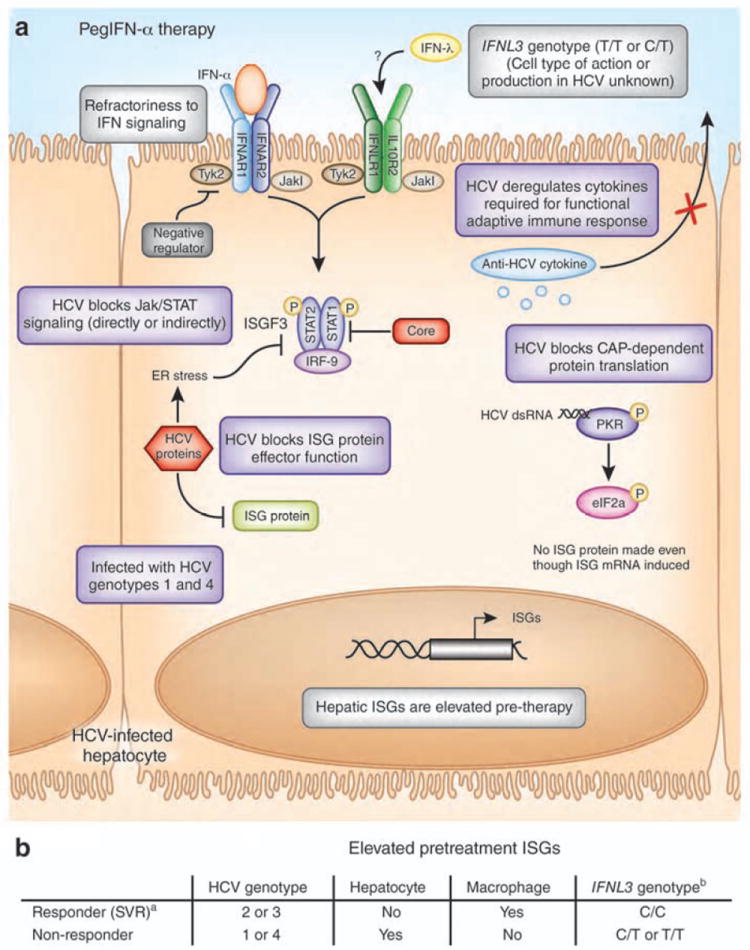
(A) In the HCV-infected hepatocyte, a number of possible factors influence therapy responses to pegylated IFN-α. These factors include the patient IFNL3 genotype, the viral genotype, and the activation status of hepatic ISGs prior to therapy, including ISGs that are negative regulators of IFNAR that make the cells refractory to IFN signaling. HCV itself could regulate the host response in the infected cell to prevent IFN therapy action by directly or indirectly blocking Jak/STAT signaling, ISG protein effector function, CAP-dependent protein translation, and cytokines required for functional adaptive immune responses (see purple boxes). (B) Table of key factors that determine HCV therapy responses.[a(SVR: sustained virologic release); b(the nucleotide SNP at rs12979860)]
What explains the viral genotypic differences in therapy responses? Further, why does HCV persist in the liver in spite of high levels of IFN signaling and elevated hepatic ISGs, many with known antiviral actions? Within the HCV-infected hepatocyte, HCV proteins, including Core, could be antagonizing IFN signaling through Jak-STAT pathway inactivation (reviewed in42) or suppression of specific ISGs that could otherwise limit HCV infection (Fig. 3). Additionally, there are known genotypic differences in HCV-mediated immune regulation that likely contribute to the observed differences in therapy responses. For example, NS5A and E2 from different genotypes differentially bind and regulate PKR, with the IFN-resistant viral genotypes being better able to block PKR function62-64. Importantly, the viral sequences encoding E2 (PePHD/PKR inhibitory domain) and NS5A (PKR binding domain) show the most evolution between HCV genotypes, supporting the idea that E2 and NS5A are potential determinants of genotype-specific clinical outcome68. In addition, differential levels of MAVS cleavage among HCV genotypes were found in the livers of HCV patients in which those with fully cleaved MAVS (genotypes 2 and 3) exhibited lower levels of pre-therapy ISG expression59. Interestingly, higher pre-therapy genetic variability of HCV within the major genotypes, especially within the sequences of Core, NS3, and NS5A, correlates with a more successful therapy outcome72. Taken together, this data suggests that alterations in the sequences of these viral regulatory factors lead to increased immune regulation during HCV infection. This increased immune regulation, while beneficial to the virus, would be predicted to prevent pre-therapy induction of ISGs and thereby make this virus more susceptible to HCV therapy.
While IFN therapy induces many anti-HCV ISGs (see Table 1)69, it is unlikely that any single ISG or its regulation by HCV completely governs the outcome of IFN therapy during HCV infection73. Further within the liver, there is no single ISG among all of those expressed prior to therapy that is associated with poor response to treatment but it is rather the overall composition of induced ISGs that associates with poor therapy responses69,74-75. Indeed, it is likely a combination of factors mediating control of innate immune and IFN signaling, along with viral and host genetic variability (to be discussed below), that all contribute to the differential responses to IFN-based therapy in HCV patients.
Host genetic components that determine the outcome of HCV infection and response to therapy
While viral genotype differences influence clinical outcome to IFN-based therapies, host genetic differences also play a role in this outcome. Genome-wide association studies (GWAS) have identified single-nucleotide polymorphisms (SNPs) upstream of the IFNL3 locus that can predict both successful clinical outcome to HCV therapy6-7,9-10 and spontaneous HCV clearance8,10. These SNPs associate with altered mRNA expression of IFNL3, which encodes the antiviral cytokine IFNL3, suggesting that IFNL3 expression levels are likely associated with HCV clearance and response to therapy6-7,70. We note that while several early studies failed to find altered mRNA expression of IFNL3 associated with these SNPs74,76, as the specificity of real-time PCR primers for IFNL3 (that differentiate between IFNL3 and the closely related IFNL2) has increased, it is now clear that these identified SNPs in the IFNL3 locus do impact the expression of IFNL3 within the liver, peripheral blood mononuclear cells, and whole blood6-7,70,77-78, with the minor/unfavorable allele/haplotype resulting in less in IFNL3 expression.
Detailed analyses of the SNPs within the haplotype/genomic block at the IFNL3 gene locus have revealed candidate functional SNPs involved in both HCV natural and treatment-based clearance (Fig. 4)79-81. However, studies of the function of these genetic elements have not yet defined the single causal variants and how these variations regulate IFNL3 mRNA expression. The locations of the current candidate casual SNPs within the IFNL3 gene suggest possible regulatory mechanisms (see Fig. 4). As the SNPs in IFNL3 do appear to impact IFNL3 expression6-7,70,77-78,82, it is unlikely that these SNPs are simply in linkage disequilibrium with some other gene involved in HCV pathogenesis. Defining how IFNL3 polymorphisms enhance HCV therapy response rates will be important for the development of new therapeutic strategies to suppress HCV through treatment with recombinant IFN-λ and to better understand the role of IFNL3 in the host response to HCV infection. A recent study showed that patients with the unfavorable IFNL3 genotype have depressed innate immune function, particularly with respect to NK cells83, suggesting the decreased expression of IFNL3 affects immunity and therefore clearance of HCV.
Figure 4. Single nucleotide polymorphisms (SNPs) in the IFNL gene locus.
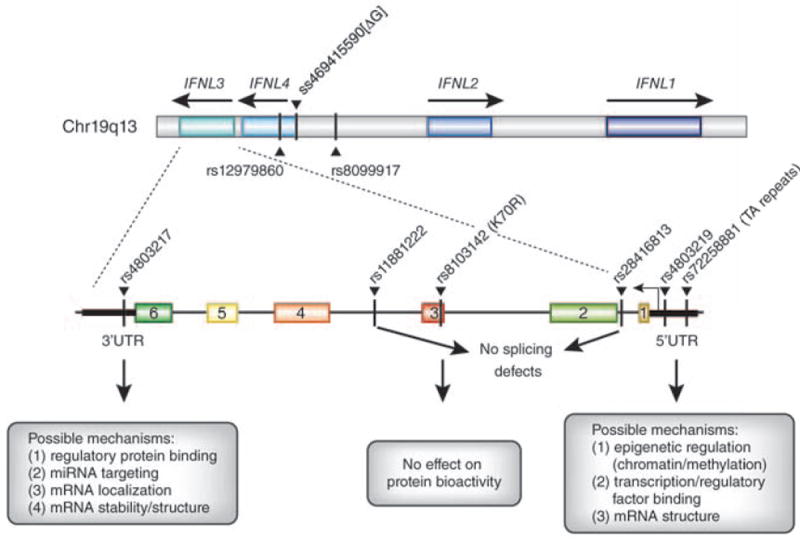
A schematic of the IFNL1-4 gene locus in Chromosome 19q13 is depicted, based on the determined sequence of the 5’ and 3’ ends of the IFNL3 mRNA81. The significant proximal SNPs around IFNL3 associated with response to IFN therapy are shown in the map, as indicated by the red dash. Candidate functional SNPs near the IFNL3 and IFNL4 locus, along with possible regulatory mechanisms at these sites, are depicted. Several of these SNPs have been tested as candidate functional SNPs (rs4803219 and rs11881222, role in splicing; rs8103142 (K70R), role in protein bioactivity) and found to have no affect on IFNL381.
Additionally, a dinucleotide polymorphism that can create or disrupt an open reading frame for a new gene, IFNL4, has been identified84-85 (Fig. 4). In this case, it has been proposed that loss of expression of IFNL4 is protective for HCV. Based on the current data, it is still not clear what the role, if any, is for IFNL4 in the differential responses to HCV therapy, but it is clear that the IFN-λ locus is important for immune control of HCV infection.
The type III IFNs (IFN-λ) now include IFNL3 and the closely related cytokines IFNL1, IFNL2, as well as IFNL4. As IFNL4 only shares 29% homology with the other IFN-λ genes and functions in a slightly different manner84, we will limit our discussion here to IFNL1-3. These cytokines have antiviral and immunomodulatory activity, similar to the type I IFNs. IFN-λ signals through a receptor consisting of the IL10R2 and IFNLR1 subunits that converges on the Jak/STAT pathway, induces similar ISG profiles as type I IFN, and can inhibit HCV replication in vitro (reviewed in86). However, the cellular producers and receptor proteins for type III IFN have a more limited distribution than type I IFN87, suggesting a more localized role for type III IFN in immunity. Interestingly, the kinetics of ISG induction varies between these cytokines, with IFN-λ inducing a more sustained expression of ISGs than IFN-α88. Further, unlike IFN-α IFN-λ signaling does not become refractory to repeated applications89, and it can function cooperatively with type I IFN to impart enhanced ISG induction and antiviral activity90. Therefore, we could speculate that in the innate immune response to HCV, IFN-λ provides a localized, sustained activation of ISGs in specific cell types in the infected liver, and primes an effective immune response, both at the innate and adaptive levels, for HCV clearance. In fact, IFN-λ has emerged as new candidate for HCV therapy with lower side effects than IFN-α treatment86, likely due to its restricted receptor distribution. It is still not clear why the genetic polymorphisms associated with natural and therapy-induced control of HCV are found specifically in IFNL3 (and now IFNL4), and not within the IFNL1 or IFNL2 loci, and why IFNL3 specifically would play such a key role in the host response to HCV. More studies aimed at understanding how these very similar genes are activated and regulated during HCV infection, as well as determining both the cell types of production and the mechanism of induction of the IFN-λ cytokines during HCV infection are needed. In particular, new studies need to continue to carefully discriminate between the virtually indistinguishable IFNL2 and IFNL3 genes, both at the protein and mRNA levels. Moreover, there is a need to actually demonstrate the expression IFNL4 (both protein and mRNA levels) and its regulation in patients, as well to understand the relationship of IFNL4 on impacting levels of IFNL3. Only then will we have a foundation of understanding of the role of the IFN-λ cytokines in HCV immune control and response to therapy in the liver.
The polymorphisms in IFNL3 associated with response to treatment are not wholly predictive of HCV therapy and infection outcome, as between 20-40% of those patients with the favorable IFNL3 genotype do not respond to current IFN-based therapy6,9-10. Therefore, other factors must contribute to a successful outcome of therapy. In fact, the strongest predictor of response to therapy is dictated by the pretreatment expression pattern of ISG mRNA within the liver69-70,91. Patients with pre-therapy induction of ISG mRNA in hepatocytes achieve the lowest therapy response rates. While we do not know why this correlates to lower therapy response rates, it is possible that the pre-therapy induction of ISGs prevents further induction of ISG levels by IFN-α during therapy due to activation of negative regulators leading to refractoriness to signaling89, or that PKR translational suppression of ISGs prevents their effector function, precluding effective therapy responses37,89(see Fig. 2). Interestingly, in Kupffer cells, the resident liver macrophages, the pre-therapy induction level of ISGs is also a strong predictor of therapy response71. In Kupffer cells, the phenotype is opposite of that seen in hepatocytes, with virtually all non-responders lacking pre-therapy induction of ISGs, while responders have strongly-induced ISGs91. This suggests that during HCV infection, Kupffer cells (activated to induce ISGs, either by cytokine signaling or through phagocytosis and sensing of viral products75,92-93; Fig. 5) may play a key protective role for the host in both therapy responses and HCV clearance. Understanding how macrophages are activated and interact with hepatocytes for immune control during HCV infection and treatment will play a key role in determining the mechanisms driving this phenotype, as well as HCV pathogenesis.
Figure 5. IFN induction by HCV in the liver.
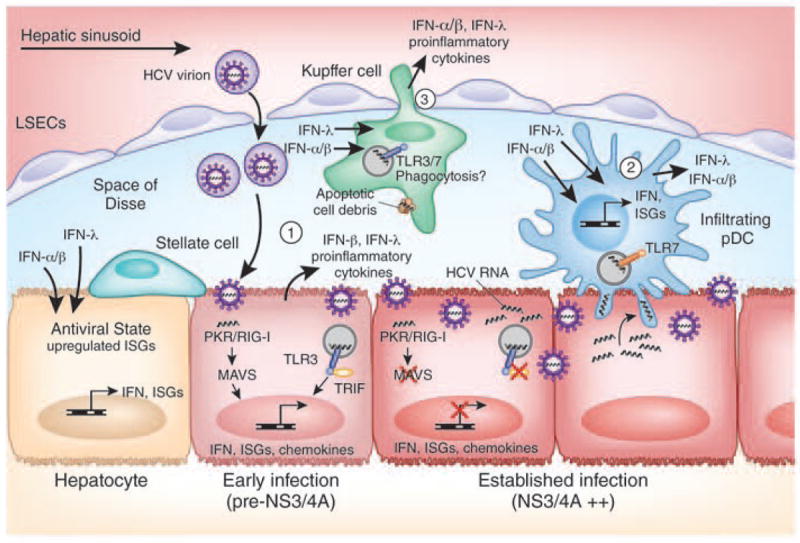
The liver contains different cell types that can secrete IFN upon stimulation by HCV infection, including (1) hepatocytes, (2) infiltrating plasmacytoid dendritic cells (pDCs), and (3) Kupffer cells. The activation of these cells is required to prime the adaptive immune response to HCV, which plays a major role in eventual viral clearance16. Both type I and type III IFN can act on uninfected hepatocytes to induce an antiviral state that limits virus spread. In the infected cell, HCV can evade IFN induction through viral-mediated processes, including NS3/4A-mediated cleavage of MAVS and TRIF, impacting HCV outcome of infection and disease progression. Cross-talk between hepatocytes and hepatic stellate cells during HCV infection has also been shown to induce inflammatory cytokines and chemokines (not depicted here)100.
Why do some patients have pre-therapy induction of ISGs in hepatocytes while others do not? It is known that host genetic differences, as well as HCV-induced ISG expression profiles in liver cell subtypes can predict therapy responses. However, they are often independent predictors of therapy responses meaning that the patient IFNL3 genotype cannot definitively predict the hepatic ISG activation state70,91,94. Thus, the ISG expression profile of hepatocytes and macrophages is still the single best predictor of therapy response, where virtually all patients that lack pre-induced hepatic ISGs respond to therapy while those who lack pre-induced macrophage ISGs fail therapy91. Therefore, the host genotype at IFNL3 or other host loci and viral genotype likely predispose the HCV infected patient to a particular therapy response outcome but it is a combination of factors, including cross-talk between liver cell subtypes and environmental factors (such as age, race, sex), which ultimately dictates the response to therapy (Fig. 3B).
Endogenous hepatic IFN regulates innate immunity to HCV
The hepatic pre-therapy ISG mRNA expression profiles of some patients with chronic HCV infection indicates that IFN (either Type I or Type III) is being produced endogenously during infection. The cell type of Type I or Type III IFN production could be the infected cell as well as bystander cells responding to virus exposure33,75,95. Importantly, the fact that patients with the favorable IFNL3 SNPs generally have higher levels of IFNL3 and lower pre-therapy hepatic ISG levels, but higher macrophage ISG levels91, suggest that other cell types within the liver besides the infected hepatocytes could be producing the IFN. Further, production of Type I and/or Type III IFN or response to the IFN at the level of the receptor or downstream signaling in the infected hepatocyte is likely regulated by HCV infection itself. We know this is the case for Type 1 IFN, and it is quite possible that this is also true for IFNL3 (see Figs. 2 and 3). Here, we propose a number of virus/host interactions in the context of the liver that could drive the hepatic production of IFN and proinflammatory cytokines (Fig. 5). Before viral proteins accumulate during infection, HCV activation of RIG-I and/or PKR actions in hepatocytes can stimulate IRF-3 activation, leading to an early, albeit transient, induction of IFN-β and ISG expression35-36,96. HCV can also induce type III IFN in primary liver cultures97-98. Resident or infiltrating liver myeloid cells, including plasmacytoid dendritic cells (pDC) and Kupffer cells, could produce type I or III IFN after stimulation by viral products from neighboring infected hepatocytes. In particular, exosomal transfer of HCV RNA from hepatocytes to neighboring pDCs can stimulate high level type I and type III IFN production through TLR7, as well as through RIG-I signaling95,99, while Kupffer cells may be able to phagocytose HCV92 to stimulate local type I and III IFN production. While this local production of IFN could serve an obvious antiviral function for suppression of acute HCV infection, the chronic production of IFN (particularly IFN-α) also regulates ISG expression and antiviral function that limits the efficacy of both the local hepatic IFN and IFN therapy69,89. During HCV infection, cytokines produced by pDCs, Kupffer cells, and infected hepatocytes within the liver could also impact the recruitment of immune cells to the liver, including myeloid cells, NK cells, and T cells. HCV regulation of this cytokine induction could play a key regulatory role in dictating activation of these immune cells for an effective adaptive immune response to HCV, resulting in changes in HCV-induced pathogenesis and varying outcomes of IFN therapy14. This cross talk between liver cell subtypes would be expected to be influenced by the patient IFNL3 genotype, with those with the favorable genotype having increased immune cell function and better viral clearance, as has been suggested83. Understanding the cross talk between liver cell subtypes is now key to defining the complex nature of both type I and type III IFN actions, priming of the adaptive immune response, especially generation of virus-specific CTLs, therapy outcome, and HCV suppression.
Summary and insights
While the immune response can clear HCV43, virus exposure often carries forward into a chronic infection. Chronic HCV is linked to dysregulation of innate and adaptive immune signaling and viral-induced cytotoxicity/apoptosis in the hepatic environment which together affect infection outcome, response to therapy, and likely contribute to liver fibrosis and cirrhosis. HCV accomplishes this immune dysregulation by evading the host innate immune response at multiple points, to prevent the initial coordination of innate immunity that primes the entire immune response, including adaptive immunity, leading to increased viral replication and contributing to viral persistence. Within the liver microenvironment, multiple cell types contribute to the immune response towards HCV, and interruption of any aspect of the innate response by HCV could lead to an unbalanced, ineffective immune response, both in natural and treatment-induced HCV clearance, that could drive the progression to liver disease. A full understanding of how viral immune modulation imparts infection outcome requires us to define the spectrum of antiviral and immunomodulatory cytokines that govern the cross-talk between the various cell types within the complex liver tissue. For example, how do IFN-α and IFN-λ responses crosstalk to one another and with hepatic responses to proinflammatory cytokines, such as IL-1β, IP10, and others, implicated in hepatic inflammation? What are the key producer cells for these cytokines in the infected liver, and how is production of these cytokines stimulated and/or regulated during HCV infection? There may also be a role for hepatic stellate cells in mediating some of these processes as they have recently been implicated in driving inflammatory responses during HCV-infection of hepatocytes100.
Clinical trials with IFN-λ are already showing great promise for its role as an effective treatment antiviral cytokine, with fewer side effects than observed in patients undergoing IFN-α-based therapy. While genetic studies have revealed insights of IFNL3 polymorphisms and their linkage with an effective immune response that clears HCV, we note that IFNL3 genotype is only one of the factors that predict therapy responses (Fig. 2B). While knowledge of one’s IFNL3 genotype could be used to guide therapy decisions, further genetic studies to define additional host factors that impact HCV clearance and/or disease progression will help us understand what dictates successful immune responses and treatment outcomes against HCV.
It is quite likely that an upcoming treatment era for HCV may fully embrace direct acting antivirals alone, such as NS3 protease, NS5A replication complex, or NS5B polymerase inhibitors, in the absence of IFN combination therapy. While these therapies are providing a major step forward in treating HCV infected patients, viral breakthrough and drug resistance will need to be considered and monitored carefully in these situations101. In addition, efficacy of these drugs for HCV genotypes other than genotype 1 still needs to be determined.
Research to continue onward in these exciting discoveries and developments will no doubt reveal the key factors required for successful immunity to HCV. In the coming age of personalized medicine, this knowledge could lead to individually tailored HCV therapies, based on knowledge of both one’s host and viral genotype. An understanding of these detailed components of an effective immune response to HCV both at the innate and adaptive levels will be useful to guide the development of the long desired HCV vaccine.
Acknowledgments
We thank Gale laboratory members, Adelle McFarland, and Ram Savan for helpful discussion and comments on this manuscript. Supported by NIH grants AI060389, AI88778, DA024563 (M.G.) and the Irvington Institute Fellowship Program of the Cancer Research Institute (S.M.H.)
References
- 1.Lavanchy D. The global burden of hepatitis C. Liver Int. 2009;29(Suppl 1):74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 2.Seeff LB. The history of the “natural history” of hepatitis C (1968-2009) Liver Int. 2009;29(Suppl 1):89–99. doi: 10.1111/j.1478-3231.2008.01927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soriano V, Peters MG, Zeuzem S. New therapies for hepatitis C virus infection. Clin Infect Dis. 2009;48:313–320. doi: 10.1086/595848. [DOI] [PubMed] [Google Scholar]
- 4.Hofmann WP, Zeuzem S. A new standard of care for the treatment of chronic HCV infection. Nat Rev Gastroenterol Hepatol. 2011;8:257–264. doi: 10.1038/nrgastro.2011.49. [DOI] [PubMed] [Google Scholar]
- 5.Sarrazin C, Hezode C, Zeuzem S, Pawlotsky JM. Antiviral strategies in hepatitis C virus infection. J Hepatol. 2012;56(Suppl):S88–S100. doi: 10.1016/S0168-8278(12)60010-5. [DOI] [PubMed] [Google Scholar]
- 6.Suppiah V, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka Y, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 8.Thomas DL, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ge D, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 10.Rauch A, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138:1338–1345. 1345 e1331–1337. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 11.Andre P, Perlemuter G, Budkowska A, Brechot C, Lotteau V. Hepatitis C virus particles and lipoprotein metabolism. Semin Liver Dis. 2005;25:93–104. doi: 10.1055/s-2005-864785. [DOI] [PubMed] [Google Scholar]
- 12.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–952. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 13.Su AI, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A. 2002;99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rehermann B. Pathogenesis of Chronic Viral Hepatitis: A New Role for Natural Killer Cells. Nat Med. 2013 doi: 10.1038/nm.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suthar MS, et al. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 2010;6:e1000757. doi: 10.1371/journal.ppat.1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thimme R, Binder M, Bartenschlager R. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol Rev. 2011 doi: 10.1111/j.1574-6976.2011.00319.x. [DOI] [PubMed] [Google Scholar]
- 17.Kumar A, et al. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-kappaB. EMBO J. 1997;16:406–416. doi: 10.1093/emboj/16.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McAllister CS, Samuel CE. The RNA-activated protein kinase enhances the induction of interferon-beta and apoptosis mediated by cytoplasmic RNA sensors. J Biol Chem. 2009;284:1644–1651. doi: 10.1074/jbc.M807888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loo YM, et al. Viral and therapeutic control of IFN-Beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M., Jr Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uzri D, Gehrke L. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J Virol. 2009;83:4174–4184. doi: 10.1128/JVI.02449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.You S, Rice CM. 3’ RNA elements in hepatitis C virus replication: kissing partners and long poly(U) J Virol. 2008;82:184–195. doi: 10.1128/JVI.01796-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saito T, et al. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A. 2007;104:582–587. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu HM, et al. The Mitochondrial Targeting Chaperone 14-3-3epsilon Regulates a RIG-I Translocon that Mediates Membrane Association and Innate Antiviral Immunity. Cell Host Microbe. 2012;11:528–537. doi: 10.1016/j.chom.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang F, et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479:423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gack MU, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 27.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang N, et al. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J Virol. 2009;83:9824–9834. doi: 10.1128/JVI.01125-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 30.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. 2006;176:4894–4901. doi: 10.4049/jimmunol.176.8.4894. [DOI] [PubMed] [Google Scholar]
- 32.Li K, et al. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology. 2012;55:666–675. doi: 10.1002/hep.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dansako H, et al. Class A Scavenger Receptor 1 (MSR1) Restricts Hepatitis C Virus Replication by Mediating Toll-like Receptor 3 Recognition of Viral RNAs Produced in Neighboring Cells. PLoS Pathog. 2013;9:e1003345. doi: 10.1371/journal.ppat.1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A. 2009;106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnaud N, et al. Hepatitis C virus reveals a novel early control in acute immune response. PLoS Pathog. 2011;7:e1002289. doi: 10.1371/journal.ppat.1002289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnaud N, et al. Hepatitis C virus controls interferon production through PKR activation. PLoS ONE. 2010;5:e10575. doi: 10.1371/journal.pone.0010575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe. 2009;6:513–522. doi: 10.1016/j.chom.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koev G, Duncan RF, Lai MM. Hepatitis C virus IRES-dependent translation is insensitive to an eIF2alpha-independent mechanism of inhibition by interferon in hepatocyte cell lines. Virology. 2002;297:195–202. doi: 10.1006/viro.2002.1455. [DOI] [PubMed] [Google Scholar]
- 39.Shimoike T, McKenna SA, Lindhout DA, Puglisi JD. Translational insensitivity to potent activation of PKR by HCV IRES RNA. Antiviral Res. 2009;83:228–237. doi: 10.1016/j.antiviral.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 40.Schoggins JW, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horner SM, Gale M., Jr Intracellular innate immune cascades and interferon defenses that control hepatitis C virus. J Interferon Cytokine Res. 2009;29:489–498. doi: 10.1089/jir.2009.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu L, Fisher BE, Thomas DL, Cox AL, Ray SC. Spontaneous clearance of primary acute hepatitis C virus infection correlated with high initial viral RNA level and rapid HVR1 evolution. Hepatology. 2012 doi: 10.1002/hep.25575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morikawa K, et al. Nonstructural protein 3-4A: the Swiss army knife of hepatitis C virus. J Viral Hepat. 2011;18:305–315. doi: 10.1111/j.1365-2893.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- 45.Brass V, et al. Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc Natl Acad Sci U S A. 2008;105:14545–14550. doi: 10.1073/pnas.0807298105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horner SM, Park HS, Gale M., Jr Control of innate immune signaling and membrane targeting by the Hepatitis C virus NS3/4A protease are governed by the NS3 helix alpha0. J Virol. 2012;86:3112–3120. doi: 10.1128/JVI.06727-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foy E, et al. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 48.Foy E, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A. 2005;102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meylan E, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 50.Baril M, Racine ME, Penin F, Lamarre D. MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J Virol. 2009;83:1299–1311. doi: 10.1128/JVI.01659-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Z, et al. GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. J Virol. 2007;81:964–976. doi: 10.1128/JVI.02076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Y, et al. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci U S A. 2007;104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iwasaki Y, et al. Long-Term Persistent GBV-B Infection and Development of a Chronic and Progressive Hepatitis C-Like Disease in Marmosets. Front Microbiol. 2011;2:240. doi: 10.3389/fmicb.2011.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin R, et al. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J Virol. 2006;80:6072–6083. doi: 10.1128/JVI.02495-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 57.Dixit E, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horner SM, Liu HM, Park HS, Briley J, Gale M., Jr Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bellecave P, et al. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology. 2010;51:1127–1136. doi: 10.1002/hep.23426. [DOI] [PubMed] [Google Scholar]
- 60.Li K, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Z, et al. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity. 2011;34:866–878. doi: 10.1016/j.immuni.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gale MJ, Jr, et al. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 63.Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 64.Noguchi T, et al. Effects of mutation in hepatitis C virus nonstructural protein 5A on interferon resistance mediated by inhibition of PKR kinase activity in mammalian cells. Microbiol Immunol. 2001;45:829–840. doi: 10.1111/j.1348-0421.2001.tb01322.x. [DOI] [PubMed] [Google Scholar]
- 65.de Veer MJ, et al. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- 66.Jaeckel E, et al. Treatment of acute hepatitis C with interferon alfa-2b. N Engl J Med. 2001;345:1452–1457. doi: 10.1056/NEJMoa011232. [DOI] [PubMed] [Google Scholar]
- 67.Simmonds P. Genetic diversity and evolution of hepatitis C virus--15 years on. J Gen Virol. 2004;85:3173–3188. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- 68.Pang PS, Planet PJ, Glenn JS. The evolution of the major hepatitis C genotypes correlates with clinical response to interferon therapy. PLoS ONE. 2009;4:e6579. doi: 10.1371/journal.pone.0006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sarasin-Filipowicz M, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A. 2008;105:7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dill MT, et al. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology. 2011;140:1021–1031. doi: 10.1053/j.gastro.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 71.Chen L, et al. Cell-type specific gene expression signature in liver underlies response to interferon therapy in chronic hepatitis C infection. Gastroenterology. 2010;138:1123–1133. e1121–1123. doi: 10.1053/j.gastro.2009.10.046. [DOI] [PubMed] [Google Scholar]
- 72.Donlin MJ, et al. Pretreatment sequence diversity differences in the full-length hepatitis C virus open reading frame correlate with early response to therapy. J Virol. 2007;81:8211–8224. doi: 10.1128/JVI.00487-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Metz P, et al. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology. 2012 doi: 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- 74.Honda M, et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology. 2010;139:499–509. doi: 10.1053/j.gastro.2010.04.049. [DOI] [PubMed] [Google Scholar]
- 75.Lau DT, et al. Innate Immune Tolerance and the Role of Kupffer Cells in Differential Responses to Interferon Therapy Among Patients With HCV Genotype 1 Infection. Gastroenterology. 2012 doi: 10.1053/j.gastro.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Urban TJ, et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology. 2010;52:1888–1896. doi: 10.1002/hep.23912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fukuhara T, et al. Variants in IL28B in liver recipients and donors correlate with response to peg-interferon and ribavirin therapy for recurrent hepatitis C. Gastroenterology. 2010;139:1577–1585. 1585 e1571–1573. doi: 10.1053/j.gastro.2010.07.058. [DOI] [PubMed] [Google Scholar]
- 78.Langhans B, et al. Interferon-lambda serum levels in hepatitis C. J Hepatol. 2011;54:859–865. doi: 10.1016/j.jhep.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 79.de Castellarnau M, et al. Deciphering the interleukin 28B variants that better predict response to pegylated interferon-alpha and ribavirin therapy in HCV/HIV-1 coinfected patients. PLoS ONE. 2012;7:e31016. doi: 10.1371/journal.pone.0031016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.di Iulio J, et al. Estimating the net contribution of interleukin-28B variation to spontaneous hepatitis C virus clearance. Hepatology. 2011;53:1446–1454. doi: 10.1002/hep.24263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sugiyama M, Tanaka Y, Wakita T, Nakanishi M, Mizokami M. Genetic variation of the IL-28B promoter affecting gene expression. PLoS ONE. 2011;6:e26620. doi: 10.1371/journal.pone.0026620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int. 2013 doi: 10.1111/liv.12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Naggie S, et al. Dysregulation of innate immunity in hepatitis C virus genotype 1 IL28B-unfavorable genotype patients: impaired viral kinetics and therapeutic response. Hepatology. 2012;56:444–454. doi: 10.1002/hep.25647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Prokunina-Olsson L, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45:164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bibert S, et al. IL28B expression depends on a novel TT/-G polymorphism which improves HCV clearance prediction. The Journal of Experimental Medicine. 2013 doi: 10.1084/jem.20130012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kelly C, Klenerman P, Barnes E. Interferon lambdas: the next cytokine storm. Gut. 2011;60:1284–1293. doi: 10.1136/gut.2010.222976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marcello T, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131:1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 89.Makowska Z, Duong FH, Trincucci G, Tough DF, Heim MH. Interferon-beta and interferon-lambda signaling is not affected by interferon-induced refractoriness to interferon-alpha in vivo. Hepatology. 2011;53:1154–1163. doi: 10.1002/hep.24189. [DOI] [PubMed] [Google Scholar]
- 90.Pagliaccetti NE, et al. Interleukin-29 functions cooperatively with interferon to induce antiviral gene expression and inhibit hepatitis C virus replication. J Biol Chem. 2008;283:30079–30089. doi: 10.1074/jbc.M804296200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McGilvray I, et al. Hepatic Cell-Type Specific Gene Expression Better Predicts HCV Treatment Outcome Than IL28B Genotype. Gastroenterology. 2012;142:1122–1131 e1121. doi: 10.1053/j.gastro.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 92.Negash A, et al. IL-1β Production through the NLRP3 Inflammasome by Hepatic Macrophages Links Hepatitis C Virus Infection with Liver Inflammation and Disease. PLoS Pathog. doi: 10.1371/journal.ppat.1003330. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Negash AA, et al. IL-1beta Production through the NLRP3 Inflammasome by Hepatic Macrophages Links Hepatitis C Virus Infection with Liver Inflammation and Disease. PLoS Pathog. 2013;9:e1003330. doi: 10.1371/journal.ppat.1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Asahina Y, et al. Association of gene expression involving innate immunity and genetic variation in interleukin 28B with antiviral response. Hepatology. 2012;55:20–29. doi: 10.1002/hep.24623. [DOI] [PubMed] [Google Scholar]
- 95.Takahashi K, et al. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci U S A. 2010;107:7431–7436. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lau DT, et al. Interferon regulatory factor-3 activation, hepatic interferon-stimulated gene expression, and immune cell infiltration in hepatitis C virus patients. Hepatology. 2008;47:799–809. doi: 10.1002/hep.22076. [DOI] [PubMed] [Google Scholar]
- 97.Marukian S, et al. Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology. 2011;54:1913–1923. doi: 10.1002/hep.24580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thomas E, et al. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology. 2012;142:978–988. doi: 10.1053/j.gastro.2011.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stone AE, et al. Hepatitis C Virus Pathogen Associated Molecular Pattern (PAMP) Triggers Production of Lambda-Interferons by Human Plasmacytoid Dendritic Cells. PLoS Pathog. 2013;9:e1003316. doi: 10.1371/journal.ppat.1003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nishitsuji H, et al. HCV infection induces inflammatory cytokines and chemokines mediated by the cross-talk between hepatocytes and stellate cells. J Virol. 2013 doi: 10.1128/JVI.00974-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lange CM, Zeuzem S. Perspectives and challenges of interferon-free therapy for chronic hepatitis C. J Hepatol. 2013;58:583–592. doi: 10.1016/j.jhep.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 102.Sumpter R, Jr, et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jiang D, et al. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J Virol. 2008;82:1665–1678. doi: 10.1128/JVI.02113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kanazawa N, et al. Regulation of hepatitis C virus replication by interferon regulatory factor 1. J Virol. 2004;78:9713–9720. doi: 10.1128/JVI.78.18.9713-9720.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gale M, Jr, et al. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol Cell Biol. 1998;18:5208–5218. doi: 10.1128/mcb.18.9.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Itsui Y, et al. Expressional screening of interferon-stimulated genes for antiviral activity against hepatitis C virus replication. J Viral Hepat. 2006;13:690–700. doi: 10.1111/j.1365-2893.2006.00732.x. [DOI] [PubMed] [Google Scholar]
- 107.Pichlmair A, et al. IFIT1 is an antiviral protein that recognizes 5’-triphosphate RNA. Nat Immunol. 2011;12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- 108.Raychoudhuri A, et al. ISG56 and IFITM1 proteins inhibit hepatitis C virus replication. J Virol. 2011;85:12881–12889. doi: 10.1128/JVI.05633-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang C, et al. Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J Virol. 2003;77:3898–3912. doi: 10.1128/JVI.77.7.3898-3912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu XY, Chen W, Wei B, Shan YF, Wang C. IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J Immunol. 2011;187:2559–2568. doi: 10.4049/jimmunol.1100963. [DOI] [PubMed] [Google Scholar]
- 111.Wilkins C, Woodward J, Lau DTY, Tyrrell DL, Gale M., Jr IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology. 2012 doi: 10.1002/hep.26066. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang IC, et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 2011;7:e1001258. doi: 10.1371/journal.ppat.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yao L, et al. Identification of the IFITM3 gene as an inhibitor of hepatitis C viral translation in a stable STAT1 cell line. J Viral Hepat. 2011;18:e523–529. doi: 10.1111/j.1365-2893.2011.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ishibashi M, Wakita T, Esumi M. 2’,5’-Oligoadenylate synthetase-like gene highly induced by hepatitis C virus infection in human liver is inhibitory to viral replication in vitro. Biochem Biophys Res Commun. 2010;392:397–402. doi: 10.1016/j.bbrc.2010.01.034. [DOI] [PubMed] [Google Scholar]
- 115.Han JQ, Barton DJ. Activation and evasion of the antiviral 2’-5’ oligoadenylate synthetase/ribonuclease L pathway by hepatitis C virus mRNA. RNA. 2002;8:512–525. doi: 10.1017/s1355838202020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Helbig KJ, et al. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology. 2011;54:1506–1517. doi: 10.1002/hep.24542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Helbig KJ, Lau DT, Semendric L, Harley HA, Beard MR. Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology. 2005;42:702–710. doi: 10.1002/hep.20844. [DOI] [PubMed] [Google Scholar]