

Abstract
Background
Allosteric HIV-1 integrase (IN) inhibitors (ALLINIs) are an important new class of anti-HIV-1 agents. ALLINIs bind at the IN catalytic core domain (CCD) dimer interface occupying the principal binding pocket of its cellular cofactor LEDGF/p75. Consequently, ALLINIs inhibit HIV-1 IN interaction with LEDGF/p75 as well as promote aberrant IN multimerization. Selection of viral strains emerging under the inhibitor pressure has revealed mutations at the IN dimer interface near the inhibitor binding site.
Results
We have investigated the effects of one of the most prevalent substitutions, H171T IN, selected under increasing pressure of ALLINI BI-D. Virus containing the H171T IN substitution exhibited an ~68-fold resistance to BI-D treatment in infected cells. These results correlated with ~84-fold reduced affinity for BI-D binding to recombinant H171T IN CCD protein compared to its wild type (WT) counterpart. However, the H171T IN substitution only modestly affected IN-LEDGF/p75 binding and allowed HIV-1 containing this substitution to replicate at near WT levels. The x-ray crystal structures of BI-D binding to WT and H171T IN CCD dimers coupled with binding free energy calculations revealed the importance of the Nδ- protonated imidazole group of His171 for hydrogen bonding to the BI-D tert-butoxy ether oxygen and establishing electrostatic interactions with the inhibitor carboxylic acid, whereas these interactions were compromised upon substitution to Thr171.
Conclusions
Our findings reveal a distinct mechanism of resistance for the H171T IN mutation to ALLINI BI-D and indicate a previously undescribed role of the His171 side chain for binding the inhibitor.
Electronic supplementary material
The online version of this article (doi:10.1186/s12977-014-0100-1) contains supplementary material, which is available to authorized users.
Keywords: HIV-1 integrase, Allosteric inhibitors, Aberrant multimerization, Drug resistance
Background
Rapid evolution of HIV-1 phenotypes conferring resistance to current antiretroviral therapies is a major clinical problem. The multifunctional nature of HIV-1 integrase (IN) provides attractive and unexploited targets for developing complementary antiretroviral compounds to enhance the treatment options for HIV-1 infected patients. During the early stage of HIV-1 replication, IN mediates integration of the reverse transcribed viral genome into human chromatin. This activity proceeds in two steps with the first step, termed 3’ processing, occurring when IN cleaves a GT dinucleotide from the 3’ ends of the viral DNA. The second step, a transesterification reaction termed strand transfer, inserts the processed viral DNA ends into host chromosomal DNA [1]. Three clinically approved antiretroviral drugs raltegravir (RAL), elvitegravir (EVG) and dolutegravir (DTG) inhibit IN strand transfer activity and are collectively referred to as IN strand transfer inhibitors or INSTIs [2]. Importantly, HIV-1 mutations that confer cross-resistance to both RAL and EVG have been identified in patients [3-5]. While the second generation INSTI, DTG, appears to exhibit a higher genetic barrier to resistance, substitutions in IN that confer low-level resistance to DTG have been identified [6].
IN catalytic activities depend on the correct assembly of the stable synaptic complex (SSC) or intasome, where individual IN subunits engage the viral DNA ends to form the fully functional IN tetramer [7]. Each of the three IN domains, the N-terminal domain (NTD), the catalytic core domain (CCD) and the C-terminal domain (CTD), contribute to the assembly of the SSC through protein-protein and protein-DNA interactions [8-12]. Unliganded IN subunits exhibit highly dynamic interplay with the inhibition of this exchange through the stabilization of subunit-subunit interactions prior to their binding to viral DNA results in the loss of enzymatic function [11,13]. Initial studies with the small molecule inhibitor tetra-acetylated-chicoric acid have shown that the inhibitor binds at the IN dimer interface and promotes the incorrect multimerization of IN, which in turn compromises IN catalytic activity in vitro [14]. These findings have provided important proof-of-concept for a new mechanism for inhibition of IN activity through the modulation of its multimeric state.
Integration in infected cells is significantly enhanced by the cellular chromatin associated protein LEDGF/p75 which acts as a bimodal tether to link the lentiviral preintegration complex to active genes [15-20]. LEDGF/p75 association with chromatin is mediated through its N-terminal segment containing the PWWP domain, which selectively recognizes the H3K36me3 histone mark as well as non-specifically engages nucleosomal DNA [21-23]. LEDGF/p75 also binds the IN tetramer through its C-terminal integrase binding domain (IBD) by inserting a small loop into a V-shaped cavity located at the HIV-1 IN CCD dimer interface [20,24-26]. LEDGF/p75 Asp366 establishes a pair of hydrogen bonds with IN Glu170 and His171 backbone amides, whereas LEDGF/p75 Ile365 and Leu368 engage in hydrophobic interactions with both IN subunits [20,24]. In addition, the LEDGF/IBD α-helix 4 forms electrostatic interactions with α-helix 1 of the IN NTD [26]. Antagonism of HIV-1 IN interaction with LEDGF/p75 through knockout (KO) of the cellular Psip1 gene, which encodes for LEDGF/p75 protein, resulted in marked decrease of HIV-1 infectivity [18,27,28]. Additionally, overexpression of the LEDGF/IBD, which is capable of both competing with endogenous LEDGF/p75 as well as inhibiting the formation of the SSC by stabilizing incorrect IN multimers [13], was able to potently inhibit HIV-1 replication [17,29]. These studies have established the importance and molecular basis of the interaction of HIV-1 IN with LEDGF/p75 and have highlighted the primary LEDGF/p75 binding pocket at the IN CCD-CCD dimer interface for anti-HIV-1 drug development.
Multifunctional allosteric IN inhibitors (ALLINIs) have been discovered that potently inhibit HIV-1 replication (reviewed in [30-33]). These compounds were identified through two separate methods, including a high throughput screen for 3’-processing inhibitors or through the rational design of inhibitors that block the IN-LEDGF/p75 interaction [33-36]. ALLINIs bind to HIV-1 IN in the principal LEDGF/p75 binding pocket and bridge between two IN subunits [36-42]. ALLINIs share several common structural features including a central quinoline ring and the carboxylic acid moiety on a modifiable one-carbon linker attached to position 3 of the central ring. Similar to LEDGF/p75 residue Asp366, the ALLINI carboxylic acid forms hydrogen bonds with Glu170 and His171 backbone amides of one IN subunit. Additionally, the quinoline ring extends to form hydrophobic contacts with the second IN subunit akin to LEDGF/p75 residue Leu368. However, unlike LEDGF/p75, potent ALLINIs also contain a tert-butoxy ether oxygen at the modifiable carbon, which forms an additional hydrogen bond with the side chain of IN residue Thr174 [36,37,39,40].
ALLINIs inhibited both IN-LEDGF/p75 binding and LEDGF/p75 independent assembly of functional SSCs in vitro [36,37,40,43]. The latter inhibitory activity has been attributed to the ability of ALLINIs to prematurely stabilize interacting IN subunits and promote aberrant higher order protein multimerization [37,40,43]. Consistent with these observations, in infected cells ALLINIs impaired a step at or prior to 3’-processing and could reduce LEDGF/p75 mediated integration into active transcription units [40,44]. Unexpectedly though, the primary activity of ALLINIs occurs during the late stage of HIV-1 replication [39,41,42,45,46]. Virions produced in the presence of ALLINIs exhibited an eccentric morphology characterized by the electron dense material being mislocalized outside of the capsid core and were furthermore defective for reverse transcription during the subsequent round of infection [39,45,46]. This phenotype is similar to the one caused by certain HIV-1 IN mutations, which are typed as class II, suggesting that IN structure may play a yet unidentified role during HIV-1 maturation [12,39,47-51]. Potential contributions of LEDGF/p75 and its interactions with IN during the late stage of HIV-1 replication are unlikely due to the observations that fully infectious virus particles were formed in LEDGF/p75 KO or knockdown cells [27,39,45]. Consistent with this view, LEDGF/p75 over expression did not affect ALLINI potencies in virus producer cells [45]. Instead, ALLINI induced aberrant IN multimerization has been shown to correlate with the inhibition of correct particle assembly. Further support for this notion has been provided by the recent development of multimerization selective IN inhibitors or MINIs. These compounds are not effective inhibitors of IN-LEDGF/p75 binding and instead potently induce aberrant IN multimerization during virus particle production and result in eccentric, non-infectious particles [44].
Genotyping HIV-1 in cell culture under the selective pressure of the archetypal ALLINI BI-1001 and its several analogs have identified substitutions in the IN coding sequence near the inhibitor binding sites [36,40,52]. Of these, the A128T IN substitution was the most prevalent mutation with HIV-1NL4-3(A128T IN) displaying marked resistance to respective ALLINI compounds [36,38,40,52]. Interestingly, crystallographic studies have revealed that BI-1001 is still able to bind A128T IN CCD by maintaining all hydrogen bonding interactions, but that the quinoline ring bridging the two IN subunits was slightly shifted compared with the wild type (WT) protein [38]. Consequently, BI-1001 was unable to promote aberrant multimerization of recombinant A128T IN, whereas it maintained its ability to inhibit IN-LEDGF/p75 binding in vitro [38]. The results of these studies are consistent with the interpretation that aberrant IN multimerization rather than IN-LEDGF/p75 binding is the primary target of this inhibitor in infected cells.
Selection of HIV-1 variants in the presence of ALLINI BI-D (Additional file 1: Figure S1), a more potent analog of BI-1001, did not result in the A128T mutation but instead revealed several amino acid changes near the inhibitor binding sites including Y99H, L102F, A/T124D, and H171T [52]. Of these, HIV-1 bearing the single amino acid H171T substitution was found to be one of the most prominent mutations persisting at the highest concentration of BI-D tested [52]. Here, we have investigated the mechanism of resistance for the H171T IN mutation. Our findings show that unlike A128T, the H171T IN substitution causes resistance to BI-D by reducing the binding affinity of the inhibitor to IN. Our structural studies have elucidated a previously undescribed role of the His171 side chain for hydrogen bonding with BI-D tert-butoxy ether oxygen, which is compromised upon the H171T substitution. Since LEDGF/p75 lacks a tert-butoxy moiety, the H171T substitution has minimal effects on IN-LEDGF/p75 binding and accordingly, HIV-1NL4-3(H171T IN), replicated in cells at WT levels. These findings have uncovered the structural and mechanistic basis for H171T IN resistance to BI-D and are expected to facilitate in the development of second generation ALLINIs with increased potency and decreased potential to evolve drug resistance.
Results
To assess the functional significance of the H171T IN substitution we introduced the mutation into both HIV-1NL4.3 and recombinant IN. As indicated in Figure 1A and B, the H171T IN substitution did not significantly alter virus release from transfected cells as measured by p24 production or affect the infectivity of the mutant virus. We next examined the biochemical properties of purified recombinant H171T IN. Size exclusion chromatography (SEC) experiments revealed that WT and H171T INs similarly formed tetramers and monomers (Figure 1C). In a homogeneous time-resolved fluorescence-based (HTRF) IN activity assay [38], the mutant IN protein exhibited near WT levels of catalytic function in the absence of LEDGF/p75 (Figure 1D). Furthermore, LEDGF/p75 was able to stimulate the strand transfer activity of mutant IN similarly to WT IN (Figure 1E). This suggested that even though the H171T IN substitution is within the IN-LEDGF/p75 binding pocket, IN retains the ability to effectively bind LEDGF/p75 in vitro and during virus infection. To test this directly, we compared LEDGF/p75 binding to WT and H171T INs utilizing a HTRF-based binding assay [38]. WT IN bound LEDGF/p75 with a Kd of 3.3 ± 0.3 nM, whereas H171T IN bound LEDGF/p75 with a Kd of ~10.5 ± 0.3 nM, a 3.2-fold decrease in affinity (Figure 1F).
Figure 1.
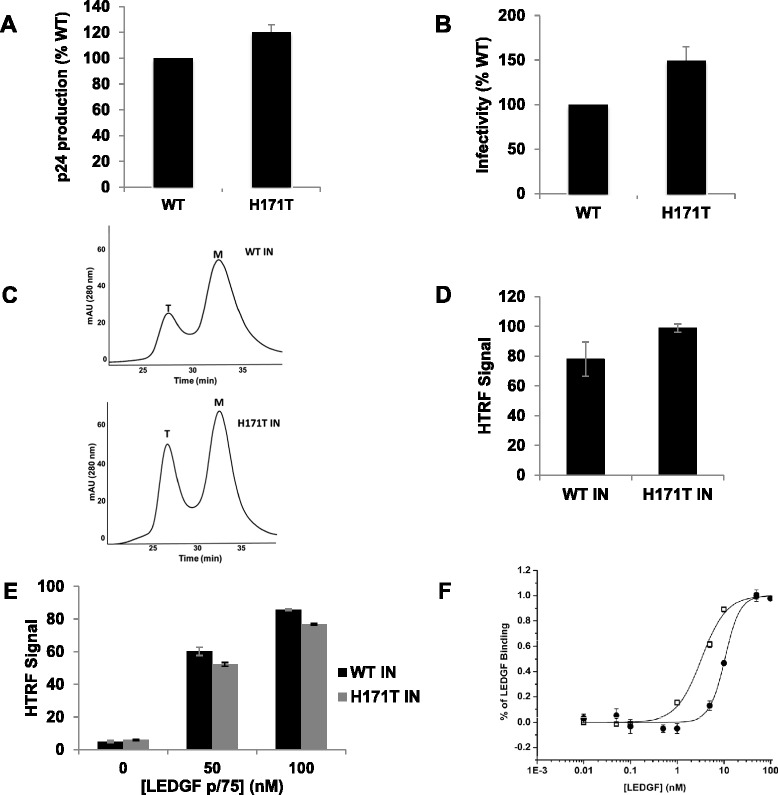
Effects of the H171T IN substitution on HIV-1 replication and recombinant IN activities. (A) p24 production of HIV-1NL4-3 and HIV-1NL4-3(H171T IN) plotted as percent WT with standard deviations shown for n = 3 independent experiments. (B) Single round infection of HIV-1NL4-3 and HIV-1NL4-3(H171T IN) determined by luciferase expression and plotted as percent WT infectivity with standard deviations shown for three independent experiments. (C) SEC of WT and H171T INs. The peaks corresponding to tetrameric (T) and monomeric (M) forms of IN are indicated. (D) HTRF-based LEDGF/p75 independent integration assay and (E) HTRF-based LEDGF/p75 dependent integration assays showing stimulation of WT and H171T IN activities at indicated LEDGF/p75 concentrations. Total HTRF signal is plotted with standard deviation for three independent experiments shown. (F) HTRF-based assays to determine LEDGF/p75 binding affinities for WT (opened boxes) or H171T (closed circles) INs. Error bars indicate standard deviation for three independent experiments.
Although the inhibition of particle maturation determines BI-D potency, the inhibitor displays a second, substantially weaker activity during the early phase of HIV-1 replication ([39], Table 1). To gain insight into the mechanism of BI-D action and the mode of H171T resistance, HIV-1NL4-3(H171T IN) was evaluated during both the early and late stages of the replication lifecycle. When drug exposure was limited to the acute phase of infection, BI-D EC50 values increased from 1.17 μM for VSV-G pseudotyped HIV-1NL4-3 to 12.4 μM for the VSV-G pseudotyped HIV-1NL4-3(H171T IN), ~11-fold resistance (Table 1). However, the H171T IN substitution resulted in significantly higher levels of resistance (~68-fold) when drug exposure was limited to the late stage of replication. To examine the apparent differences in resistance levels between the early and late stages of replication, anti-viral potencies of BI-D were determined in LEDGF/p75 KO cells. The complete removal of endogenous LEDGF/p75 increased BI-D potency during the acute phase of HIV-1NL4-3 infection, but had minimal effect on the inhibitor activity when exposure was limited to the late stage. These results are consistent with other studies and indicate that during the early stage of replication, LEDGF/p75 is able to compete with ALLINIs for binding to IN and hence reduce inhibitor potency [39,53]. However, LEDGF/p75 expression levels did not detectably alter ALLINI potencies during the late stage of viral replication ([27,39,41,42,45,46], also see Table 1). The virus containing the H171T IN substitution still conferred resistance to BI-D in the absence of endogenous LEDGF/p75, with 28- and 45-fold resistance observed in the KO cells during the early and late stages, respectively.
Table 1.
Effects of the H171T IN substitution on antiviral activities of BI-D
| Producer cells | Target cells | LEDGF KO producer cells | LEDGF KO target cells | |||||
|---|---|---|---|---|---|---|---|---|
| EC 50 (μM) | Fold change | EC 50 (μM) | Fold change | EC 50 (μM) | Fold change | EC 50 (μM) | Fold change | |
| WT IN | 0.090 ± 0.031a | -- | 1.17 ± 0.1a | -- | 0.080 ± 0.01 | -- | 0.067 ± 0.02 | -- |
| H171T IN | 6.11 ± 0.63 | 67.9x | 12.4 ± 0.85 | 10.6x | 2.20 ± 0.64 | 28x | 2.98 ± 0.7 | 44.5x |
Means ± S.D. are shown for at least 3 independent experiments.
aData from Ref [39].
The vast majority of virions produced in the presence of BI-D display eccentric core morphology, where the electron dense material normally situated within the conical core is mislocalized adjacent to a translucent capsid core and the viral membrane [39]. Consistent with these findings, when WT HIV-1NL4-3 virions were produced in the presence of 0.18 μM BI-D (a dose equivalent to 2×EC50; Table 1), 77% of the virions displayed an eccentric morphology (Figure 2). However, at the same concentration of BI-D the H171T IN mutant virus resulted in only 27% of the virions with eccentric core morphologies (Figure 2). When BI-D concentrations were increased to 12 μM, which corresponds to 2×EC50 for HIV-1NL4-3 bearing the H171T IN substitution, the eccentric virion morphology for the mutant virus increased to 82% (Figure 2).
Figure 2.
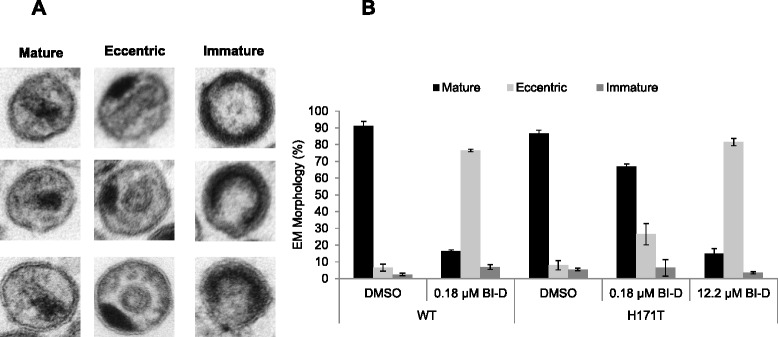
Concentration dependent effects of BI-D on viral core morphology for HIV-1 NL4-3 and HIV-1 NL4-3(H171T IN) . (A) Representative images of mature, eccentric and immature virion morphologies as visualized by electron microscopy. (B) Quantitation of counted virions (100 for WT or H171T per experiment). Virions were produced in the presence of DMSO, 0.18 μM BI-D or 12.2 μM BI-D as indicated. Graphed are averages and standard deviation for n = 2 independent experiments.
We next wished to dissect the mechanism of the HIV-1NL4-3(H171T IN) resistance to BI-D. Surface plasmon resonance (SPR) was used to compare BI-D binding to recombinant WT and H171T IN CCD proteins. Figure 3 shows that BI-D bound WT IN CCD with a Kd of 0.123 μM, which is in good agreement with the antiviral activities of BI-D measured in cell culture (Table 1). BI-D binding to H171T IN CCD resulted in a significantly higher Kd of 10.3 μM. Significantly, the observed ~84-fold decrease in the binding affinity of BI-D to the mutant IN CCD (Figure 3) roughly correlated with the ~68-fold decrease in the EC50 value seen for antiviral activity against HIV-1NL4-3 bearing the H171T IN substitution (Table 1).
Figure 3.
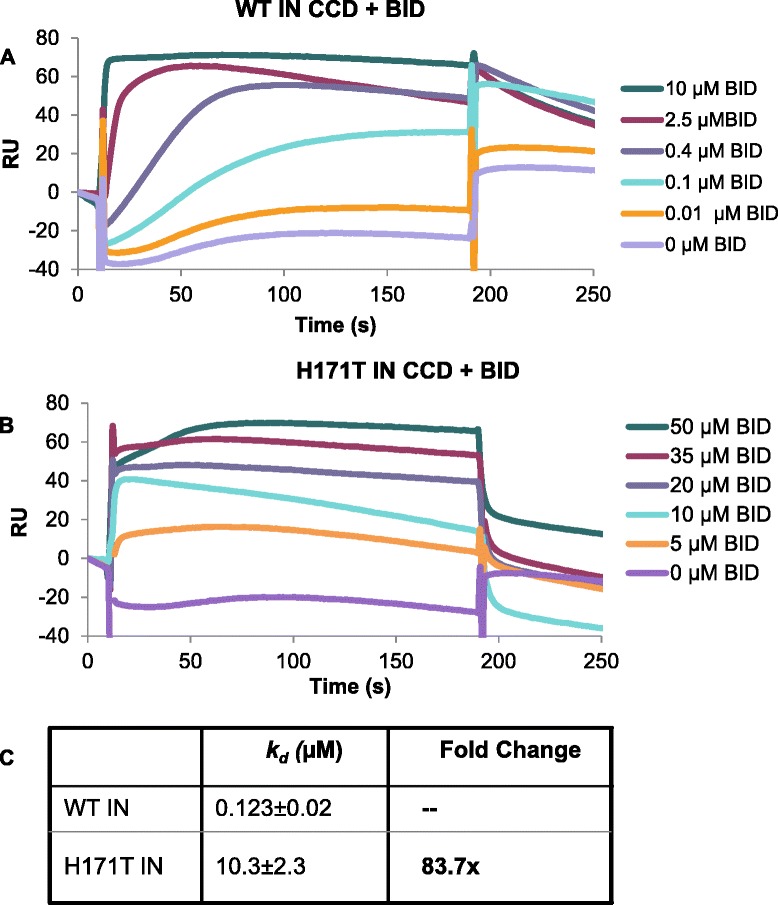
SPR analysis of BI-D interactions with WT and H171T mutant IN CCDs. SPR binding kinetics for BI-D interactions with (A) WT IN CCD and (B) H171T IN CCD at indicated inhibitor concentrations. Binding affinities are summarized in C.
Previous studies have indicated that the primary mechanism of action of ALLINIs is through the promotion of aberrant IN multimerization [38-41,43]. Therefore, we compared the effects of BI-D on aberrant multimerization of WT and H171T INs using dynamic light scattering (DLS). In the absence of inhibitor (DMSO control), peaks for soluble WT or mutant IN were not observed by this technique (Figure 4). Instead, a background signal corresponding to <1 nm diameter was detected both in the presence of IN and in the buffer alone sample, indicating that the reaction buffer contained small size particles. For inhibitor experiments, we examined two concentrations of BI-D: one (~0.120 μM) that correlated to the Kd value of the inhibitor binding to WT IN CCD (~0.123 μM) and the other (10 μM) that correlated with the Kd value for inhibitor binding to H171T IN CCD (~10.3 μM). Incubation of 0.12 μM BI-D with WT IN for 15 min resulted in a peak corresponding to particles with a diameter of 51 nm, which significantly exceeds an estimated diameter of 7.5 nm for the IN tetramer in the SSC [54]. The size of the oligomer continued to increase further to 106 nm and 142 nm diameter at 20 and 30 minutes, respectively (Figure 4A). In contrast, the same BI-D concentration (0.12 μM) failed to elicit higher order H171T IN oligomers even after 30 min incubation (Figure 4B). However, when the concentration of BI-D was increased to 10 μM, higher order oligomerization of H171T IN was detected in a time dependent manner (Figure 4D). As expected, 10 μM BI-D also induced higher order oligomers of WT IN (Figure 4C). This suggests that at lower concentrations, BI-D is unable to promote higher order IN oligomerization of H171T IN likely due to the decreased affinity of the inhibitor binding to the mutant protein (Figure 3). However, under conditions of increased inhibitor, BI-D is able to bind H171T IN (Figure 3) and promote aberrant IN multimerization (Figure 4D). This indicates that the H171T IN substitution confers resistance to BI-D by decreasing inhibitor binding affinity and hence correspondingly decreasing aberrant IN multimerization.
Figure 4.
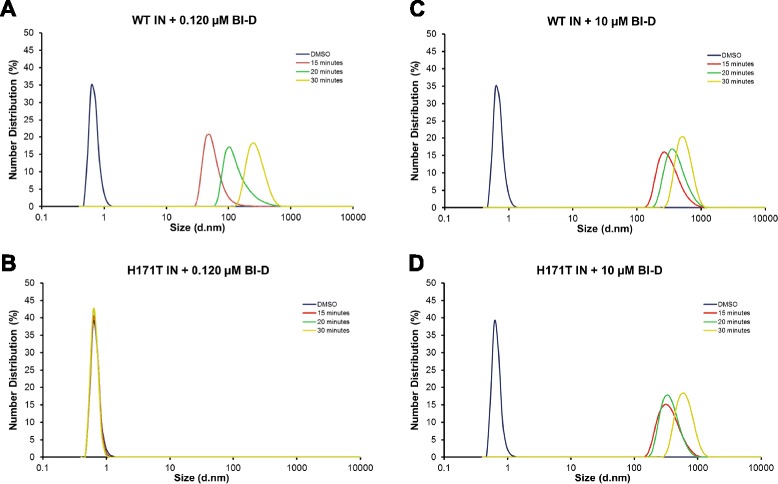
DLS analysis of BI-D induced oligomerization of recombinant WT and the H171T INs. Shown are the size distributions (%) of IN after DMSO treatment (blue) or BI-D treatment after 15 minutes (red), 20 minutes (green) and 30 minutes (yellow) incubation. BI-D treatments include (A) WT IN +0.120 μM BI-D, (B) H171T IN +0.120 μM BI-D, (C) WT IN +10 μM BI-D and (D) H171T IN +10 μM BI-D. The peak with the diameter size of <1 nm detected in these samples has also been observed for the buffer alone sample indicating that small size particles unrelated to IN or BI-D were present in our preparations.
To understand the structural basis for the reduced binding affinity of BI-D to H171T IN, we solved the crystal structure of BI-D in complex with H171T CCD dimer (Figure 5A) and compared it to the complex of inhibitor bound to WT CCD dimer ([39], also see Figure 5B). As expected (Figure 3) with the high concentration of BI-D (~5 mM) used in the crystallographic experiments, the inhibitor bound to H171T CCD dimers. Furthermore, the H171T substitution did not detectably affect the inhibitor position within the binding pocket (compare Figure 5A and B). BI-D hydrophobic interactions with IN CCD subunit 2 as well as hydrogen bonding between the inhibitor carboxylic acid, and backbone amides of subunit 1 were fully preserved in both crystal structures. Furthermore, the Thr174 side chain similarly hydrogen bonded to the tert-butoxy ester oxygen in both the WT and mutant IN structures.
Figure 5.
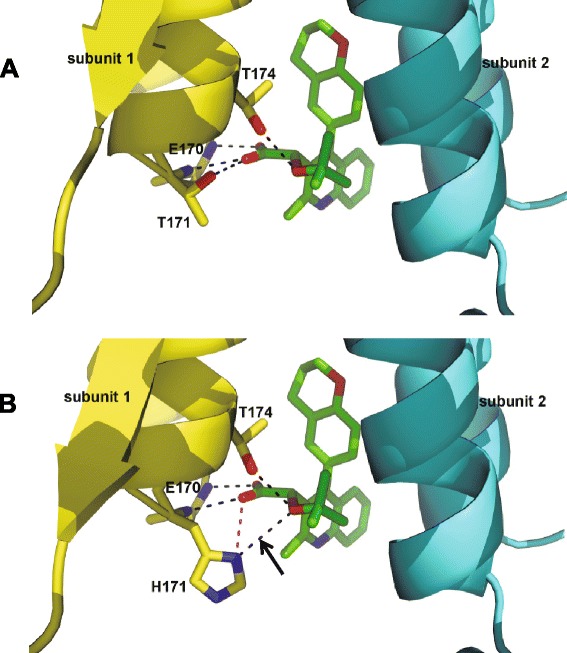
Crystal structures of BI-D bound to WT and H171T CCD dimers. Panel A is the H171T CCD dimer and panel B is the WT CCD dimer. BI-D is colored green and individual IN subunits are colored yellow and cyan. Oxygen atoms are shown in red and nitrogen atoms are in blue. Black dash-lines indicate hydrogen bonding interactions, whereas the magenta dash-line shows the electrostatic interaction between the protonated Nδ- on His171 and the carboxylic acid of BI-D. The arrow indicates the hydrogen bond between the protonated Nδ- on His171 and the ether oxygen on the tert-butoxy (B), which is absent in the H171T IN CCD structure (A).
Importantly, though, we observed differential interactions of His171 and Thr171 side chains with the inhibitor. The imidazole group of His171 formed both an electrostatic interaction with the carboxylic acid and a hydrogen bond with the tert-butoxy oxygen of BI-D (Figure 5B). However, the side chain of Thr171 which establishes a novel hydrogen bond with the carboxylic acid was unable to form a hydrogen bond with the tert-butoxy moiety of BI-D (compare Figure 5A and B).
To understand how these structural differences contributed to the markedly reduced ability of the inhibitor to bind H171T IN, we performed absolute binding free energy calculations for the interactions between WT and H171T IN CCD dimers with BI-D (Table 2 and Additional file 1: Figure S2). Nδ-, Nε- and doubly (Nδ- and Nε-) protonated forms of His171 were considered in our calculations. Table 2 shows that the doubly-protonated form of His171 and the Nδ-protonated form of His171 have similar calculated ΔGbind (cal) of -11.2 kcal/mol and -10.1 kcal/mol respectively; whereas a significantly weaker ΔGbind(cal) value of -5.9 kcal/mol was obtained for the Nε-protonated form of His171. The calculated ΔGbind (cal) for the IN containing Thr171 was -6.7 kcal/mol (Table 2). For comparison, Table 2 also shows the ΔGbind (exp) values determined using experimental Kd values for BI-D binding to WT and H171T IN CCDs (Figure 3). Comparison of ΔGbind (cal) and ΔGbind (exp) suggests that BI-D binding to WT IN preferentially stabilizes the doubly protonated form and/or the Nδ-protonated tautomer states of His171, while the Nε-protonated tautomer is a relatively minor species in the inhibitor-bound complex. We also decomposed the computed binding free energy ΔGbind (cal) into contributions from electrostatic and non-polar interactions. We compared the results for both BI-D binding to the H171T IN CCD and its WT counterpart (data not shown), which suggests that the less favorable electrostatic interactions between BI-D and Thr171 primarily contribute to the lower Kd value for the inhibitor binding to the mutant protein. Significantly, comparisons between the molecular dynamics (MD) simulated structures obtained with different protonation states of His171 and the crystal structure of BI-D in complex with the WT IN CCD dimer are consistent with the trend in the computed binding free energies (see Additional file 1: Table S1 and Figure S2).
Table 2.
Binding free energy calculations for BI-D interactions with WT and the H171T mutant IN CCDs
| Receptor | ΔG bind (cal) | ΔG bind (exp) |
|---|---|---|
| WT IN (His171-doubly-protonated) | -11.2 | -9.5 |
| WT IN (His171-Nδ-protonated) | -10.1 | |
| WT IN (His171-Nε-protonated) | -5.9 | |
| His171T IN | -6.7 | -6.8 |
Unit: kcal/mol. ∆Gbind (cal) represent calculated values for BI-D binding to His171 containing either Nδ-, Nε- or doubly protonated tautomer states and H171T IN CCDs. ∆Gbind (exp) values have been calculated based on the experimental data.
The simulations also revealed the importance of hydrogen bonding interactions between the ether oxygen on the tert-butoxy moiety on BI-D and the protonated Nδ- on His171. In contrast, to the mutant IN CCD, the hydroxyl group of the Thr171 side chain does not form such interactions with the ether oxygen on the inhibitor (Figure 5). This result is supported by the published observations that ALLINIs which contain an oxygen ether linkage are considerably more potent inhibitors of HIV-1NL4.3 with WT IN than those lacking such an oxygen atom (reviewed in [31]). To further test this notion we have examined the effects of the H171T IN substitution on the antiviral activities of LEDGIN-6, which lacks the tert-butoxy group ([36,37], also see Additional file 1: Figure S1). LEDGIN-6 was only ~3.4-fold less potent with respect to HIV-1NL4.3 (H171T IN) (EC50 of 41.4 ± 6.4 μM) (data not shown) versus WT HIV-1NL4.3 (EC50 of 12.2 ± 2.9 μM) [37]. For comparison, BI-D, which unlike LEDGIN-6 contains the tert-butoxy group, was significantly more sensitive (~68-fold, Table 1) to the H171T IN substitution in HIV-1NL4.3 likely due to the disruption of the hydrogen bonding between the tert-butoxy ether oxygen and Nδ-protonated His171.
Next, we wanted to understand the structural basis as to why the H171T substitution had significantly less effect on LEDGF/p75 binding (~3.2-fold) compared with BI-D binding (~84-fold) to recombinant HIV-1 IN. Comparison of available crystal structures of BI-D or LEDGF/IBD bound to WT HIV-1 IN CCDs [20,39] revealed that the hydrogen bonding between the protonated Nδ on His171 and the tert-butoxy ether oxygen of BI-D is unique to the inhibitor because LEDGF/IBD does not similarly contact IN. To examine LEDGF/IBD interactions with the mutant IN we simulated the H171T change in the available crystal structure [20], which revealed that unlike BI-D, LEDGF/IBD made the same number of hydrogen bonds with WT and H171T mutant INs (Additional file 1: Figure S3). In particular, LEDGF/p75 D366 can hydrogen bond with the side chain of Thr171 replacing the lost electrostatic interaction that occurred with His171. To further test these observations, we performed relative binding free energy calculations for LEDGF/IBD binding to WT and H171T IN CCDs. Free energy perturbation analysis allowed us to calculate that ΔΔGb = ΔGb(H171T) − ΔGb(WT) = 1.08 kcal/mol. Since Kd = eΔGb/RT, , the calculated ΔΔGbof 1.08 kcal/mol translates into a relative binding affinity ratio . Experimentally, the Kd of LEDGF/p75 binding to wild type and H171T mutant INs are ~3.3 nM and 10.5 nM respectively, i.e. , which is in good agreement with the calculated relative affinity ratio of 6.2.
Discussion
Published studies have shown that ALLINIs are anchored to the IN dimer interface through their key pharmacophore, the carboxylic acid, hydrogen bonding with the backbone amides of IN residues Glu170 and His171. Furthermore, the Thr174 side chain has been implicated in hydrogen bonding with the ALLINI tert-butoxy ether oxygen [37,38,40]. However, the contributions of the His171 side chain for interacting with this class of inhibitors have not been previously elucidated. Yet, genotyping of HIV-1NL4-3 variants under increasing selective pressure of ALLINI BI-D has revealed the H171T IN substitution as a key amino acid substitution. In addition, this variant persists at the highest inhibitor concentration tested suggesting that the amino acid side chain change at position 171 contributes to the evolved resistance to BI-D [52]. The present studies provide mechanistic and structural clues for these observations. We show that HIV-1NL4-3 containing the H171T IN substitution confers ~68-fold resistance to BI-D. Significantly, this level of resistance in infected cells correlates closely with ~84-fold reduced binding affinity of the inhibitor to recombinant H171T IN CCD as compared with its WT counterpart. Crystallographic experiments and binding free energy calculations have indicated that Nδ-protonated or doubly protonated forms of the imidazole ring of His171 can engage in both electrostatic interactions with BI-D carboxylic acid as well as hydrogen bonding with the tert-butoxy ether oxygen of the inhibitor. These interactions are compromised by the H171T substitution, with the Thr171 side chain forming a less electrostatically favorable hydrogen bond with the BI-D carboxylic acid and lacking any additional interactions with the tert-butoxy ether oxygen (Figure 5).
In contrast with the significant reduction in BI-D binding affinity, the H171T IN substitution only minimally reduced LEDGF/p75 binding affinity to recombinant H171T IN. In infected cells, where endogenous LEDGF/p75 levels significantly exceed what is needed for HIV-1 integration, HIV-1NL4-3 with the H171T substitution was not compromised for HIV-1 replication. The MD simulations and binding free energy calculation have revealed important differences between BI-D and LEDGF/IBD for their binding to HIV-1 IN. The Nδ protonated His171 hydrogen bonds the tert-butoxy ether oxygen of BI-D, which is compromised upon the H171T IN substitution. In contrast, such interactions are not formed between WT IN and LEDGF/IBD. Accordingly, the H171T IN change minimally affects the IN-LEDGF/p75 binding. Furthermore, unlike BI-D which engages only a small pocket at the CCD-CCD dimer interface, LEDGF/p75 establishes additional extensive interactions with HIV-1 IN, which extend beyond the CCD-CCD dimer interface and include strong electrostatic interactions between positively charged residues along LEDGF/IBD α-helix 4 and a number of acidic residues of α-helix 1 of IN [26]. HIV-1 seems to exploit these structural differences between BI-D and LEDGF/p75 interactions with IN during the process of evolution of the H171T IN escape mutation.
The mechanism for H171T IN resistance is distinct from the previously described mechanism of resistance for the A128T IN escape mutation under the selective pressure of related, archetypal inhibitor BI-1001 [38]. The A128T IN substitution does not significantly reduce BI-1001 binding to HIV-1 IN CCD with all hydrogen bonding and electrostatic interactions of BI-1001 with HIV-1 IN being fully preserved in WT and A128T INs. Instead, the substitution of Ala with bulkier and polar Thr repositioned BI-1001 at the IN CCD dimer interface and reduced its ability to effectively bridge between two IN subunits. Consequently, bound BI-1001 failed to induce aberrant multimerization of recombinant A128T IN and accordingly HIV-1NL4-3 containing the A128T IN substitution exhibited marked resistance to BI-1001 [38]. In contrast, the H171T IN substitution was able to resist BI-D through decreasing the ability of the inhibitor to bind IN. However, at high BI-D concentrations, BI-D is able to bind, effectively bridging two IN subunits, and inducing aberrant IN multimerization. Collectively these findings provide important structural and mechanistic details for a novel mechanism of resistance.
Analysis of our crystal structure of BI-D bound to HIV-1 IN CCDs has revealed 13 residues (Gln95, Tyr99, Leu102, Thr124, Thr125, Trp132, Ala128, Ala129, Ala169, Glu170, His171, Lys173 and Met178) that are within 5 Å of the inhibitor. Of these only IN amino acids 124 and 125 are polymorphic with Thr predominating at both positions in clade B, whereas the majority of clade C strains contain Ala124 and Ala125 [55]. Recent studies [33] have shown that these polymorphic substitutions only modestly affected the antiviral potencies of various ALLINIs. We also note close structural similarity between BI-D and BI-224436, the first ALLINI to advance into phase 1a clinical trials ([33], also see Additional file 1: Figure S3). These two compounds exhibit similar antiviral activities in cell culture with an EC50 range of 51-90 nM for BI-D and 11-27 nM for BI-224436 with respect to different viral strains [33,39,56]. However, the latter compound has been chosen for clinical trials due to its excellent pharmacokinetic profile in rats [33]. These two compounds differ only in the substituted ring system, with BI-D and BI-224436 containing bicyclic and tricyclic arenes, respectively ([33], also see Additional file 1: Figure S3). While the crystal structure for BI-224436 bound to HIV-1 IN CCDs has not been published, based on its close structural similarity with BI-D we predict that both bi- and tricyclic arenes would similarly dock in the hydrophobic pocket that encompasses Leu102, Ala128, Ala129, Trp132 and Met178. At the same time the key interactions between the Nδ- protonated imidazole group of His171 with the tert-butoxy ether oxygen and electrostatic interactions with the inhibitor carboxylic acid are likely to be equally important for both BI-D and BI-224436 binding to HIV-1 IN. Therefore, our findings are expected to facilitate in the development of improved IN inhibitors for potential clinical use.
Conclusions
We have elucidated a distinct mechanism of resistance for the H171T IN mutation to the potent ALLINI BI-D. Our findings indicate the importance of the His171 side chain for binding the inhibitor and that its substitution to Thr171 markedly reduces the binding affinity and corresponding inhibitory activity of BI-D.
Methods
Antiviral compounds, plasmids, and DNA constructs
BI-D and LEDGIN-6 were synthesized as previously described [37,53]. Plasmid pNL4.3/Xmal [57] encodes for replication competent HIV-1NL4-3. Plasmids pNLX.Luc.R- [58] or pNL4-3.Luc.Env- [59] encodes for single-round HIV-1NL4-3 carrying the luciferase reporter gene. Vesicular stomatitis virus G glycoprotein was encoded by pCG-VSV-G [57]. The H171T substitution was introduced into the IN coding region of pNLX.Luc.R-, pNL4-3.Luc.Env- and pNL4-3/Xmal using PCR-site directed mutagenesis (Agilent) and verified by dideoxy sequencing.
Cells, viruses and antiviral assays
Parental HEK293T and HEK293T LEDGF/p75 KO cells [60] were grown in Dulbecco’s modified Eagle medium (Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum (FBS) (Invitrogen), 100 IU/mL penicillin, and 100 μg/mL streptomycin (Gibco). SupT1 cells were maintained in RPMI medium 1640 containing 10% FBS, 100 IU/mL penicillin and 100 μg/mL streptomycin.
HIV-Luc was pseudotyped by cotransfecting HEK293T cells with either pNLX.Luc.R- or pNL4-3.Luc.Env- and with pCG-VSV-G using PolyJet DNA transfection reagent (SignaGen Laboratories).
To determine antiviral activity during virus production, DMSO or the inhibitor was added at indicated concentrations during media exchange at 18-20 hrs post-transfection. Cell-free supernatants were measured for p24 content utilizing a commercial p24 ELISA kit (Advanced Biosciences Laboratories). SupT1 cells were infected in triplicate with HIV-Luc normalized for p24 content (5 ng/mL p24). Luciferase values were determined 48 hrs post-infection.
To assess antiviral activity during the early stage of HIV-1 replication, DMSO or the inhibitor was added at the indicated concentrations to the target cells 30 min before or at the time of infection. Luciferase values, expressed as relative light units were determined 48 hrs post-infection.
Electron microscopy
Cell-free HIV-1NL4-3 from transfected HEK293T cell supernatants was concentrated via ultracentrifugation at 4°C for 2 hrs in a Beckman SW41 rotor at 32,000 rpm prior to fixation in 2% paraformaldehyde and submission to the Harvard Medical School Electron Microscopy core facility. Images were taken with a JEOL 1200EX microscope equipped with an AMT 2 k charge-coupled device camera. Virus particles (100 per experimental sample) were counted by eye.
Expression and purification of recombinant proteins
WT HIV-1 IN, H171T HIV-1 IN and LEDGF/p75 recombinant proteins with 6xHis or FLAG tags were expressed in E. coli and purified as described previously [37]. WT and H171T HIV-1 IN CCD (residues 50-212) containing the F185K mutation were expressed in E. coli and purified as described [37].
Crystallization and X-ray crystal structure determination
Recombinant H171T IN CCD (50-212) containing the F185K solubilizing substitution was prepared to ~8 mg/ml and grown at 4°C using hanging drop vapor diffusion method. The crystallization buffer contains 8% PEG 8 K, 0.1 M Na cacodylate, pH 6.5, 0.1 M ammonium sulfate and 5 mM DTT. Protein (1 μl) was mixed with the equal volume of the crystallization buffer. Within 4 weeks the cubic shape crystals reached 0.1- 0.2 mm in size. The soaking buffer containing 5 mM BI-D was prepared by dissolving the compound in crystallization buffer supplemented with 10% DMSO. The protein crystal was soaked in the buffer for 12 hrs at 4°C before it was flash-frozen with liquid nitrogen. Diffraction data from the crystals were collected at 100 F on a Rigaku Raxis 4++ image plate detector in OSU Crystallography Facility. The intensity data integration and reduction were performed with HKL2000 program [61]. Molecular Replacement program Phaser [62] in CCP4 package method was used to solve the structure. Coot [63] was used for the subsequent refinement and building of the structure. Refmac5 [64] of the CCP4 package was used for the restraint refinement. The crystal belong to space group P3121 with cell dimensions of a = b =72.2 Å, c =66 Å; One 18 kDa monomer resides in the asymmetric unit. The structure was refined to 1.94 Å with Rcryst/Rfree =0.190/0.221. Coordinates have been deposited in the Protein Data Bank with accession number 4TSX (Additional file 1: Table S2).
Binding free energy calculations
The absolute binding free energies between the various forms of IN CCD dimer and BI-D were calculated using the double decoupling method (DDM) [65-68] in explicit solvent (TIP3P water model [69] plus counterions) at 300 K. The protein molecules are modeled by the Amber ff99sb-ILDN force field [70], and the ligand BI-D is described by the Amber GAFF [71] parameters set. The partial charges of the ligands are obtained using the AM1-BCC method [72]. A DDM calculation involves two legs of simulation, in which a restrained ligand is gradually decoupled from the receptor binding pocket or from the aqueous solution. In each leg of the decoupling simulations, the Coulomb interaction is turned off first using 11 lambda windows, and the Lennard-Jones interactions are then turned off in 17 lambda windows. The two decoupling free energies ΔGgas*→complex and ΔGgas→water associated with the two legs of the DDM cycle were determined using thermodynamic integration (TI). The Hamiltonian derivative 〈∂U/∂λ〉λ at a series of l from 0 to 1 were collected and integrated to obtain the free energy difference. For absolute binding free energy calculations, the MD simulation at each λ was performed using the GROMACS [73,74] version 4.6.4 for 15 ns; the last 10 ns was used for the calculation of binding free energy.
SEC
A Superdex 200 10/300 GL column (GE Healthcare) was used to analyze multimeric forms of recombinant WT and H171T INs in buffer containing 20 mM HEPES (pH 6.8), 750 mM NaCl, 10 mM MgSO4, 0.2 mM EDTA, 5 mM BME, 5% glycerol and 200 μM ZnCl2. The following proteins were used to calibrate the column: bovine thyroglobulin (670,000 Da), bovine gamma-globulin (158,000 Da), chicken ovalbumin (44,000 Da), horse myoglobin (17,000 Da) and vitamin B12 (1,350 Da). Proteins were detected by absorbance at 280 nm. All the procedures were performed at 4°C.
DLS
DLS experiments were carried out as previously reported [44]. Briefly, DMSO, 0.12 μM BI-D, or 10 μM BI-D was incubated with 200 nM WT or H171T IN. DLS signals were recorded at room temperature after 15, 20 and 30 minutes using a Malvern Nano series Zetasizer instrument.
SPR
SPR was performed using a Biacore T100 (GE Healthcare). A series s sensor chip NTA (GE Healthcare) was conditioned with 0.5 mM NiCl2 at a flow rate of 10 μl/min for 1 min followed by a 1 min wash of 3 mM EDTA at a flow rate of 10 μl/min. WT or H171T 6xHis-CCD 4 μg/mL in HBS-P, (GE Healthcare) was immobilized on the chip to 4400 response units (RU). Indicated BI-D concentrations in HBS-P with 10% DMSO was flowed over the cell for 180 s at a flow rate of 40 μl/min followed by a 5 min dissociation. The chip was regenerated with 500 mM imidazole.
Catalytic activities of recombinant INs
LEDGF/p75 dependent integration and LEDGF/p75 independent integration assays were performed using HTRF-based assays as described [38] with minor modifications. LEDGF/p75 independent assays were executed by incubating 400 nM WT or H171T IN with 50 nM Cy-5 labeled donor DNA and 10 nM biotinylated target DNA. For LEDGF/p75 dependent integration assays, 100 nM WT or H171T IN was incubated with 50 nM Cy-5 labeled donor, 10 nM biotinylated target DNA and 50 nM or 100 nM LEDGF/p75. After the addition of europium-streptavidin, the HTRF signal was recorded using a Perkin Elmer EnSpire multimode plate reader.
HTRF-based assay for IN-LEDGF/p75 interactions
A previously described HTRF-based assay [37] was modified to monitor LEDGF/p75 binding to WT and H171T INs. Briefly, C-terminally FLAG-tagged LEDGF/p75 (0.01 – 100 nM) was titrated in the binding buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 2 mM MgCl2, 0.1% Nonidet P-40, 1 mg/ml BSA) containing 10 nM N-terminally 6XHis WT or H171T HIV-1 IN. Anti-6xHis-XL665 and anti-FLAG-EuCryptate antibodies (Cisbio, Inc., Bedford, MA) were added to the reaction and the HTRF signal vs LEDGF/p75 concentration curves were fitted to Hill Equation to identify the Kd values for IN-LEDGF/p75 binding.
Acknowledgments
We thank Dr. Dennis Bong (OSU, Chemistry Department) for the use of the DLS instrument and Dr. Drew Dangel for technical assistance with SPR experiments. The present study was supported by the National Institutes of Health (grants AI062520 to M.K., GM103368 to M.K., A.E., R.M.L. and J.R.F., AI039394 to A.E., AI110310 to M.K. and J.R.F., AI110270 to J.J.K., GM30580 to R.M.L., AI77344 and the John H. Tietze Foundation Trust to E.M.P.).
Additional file
Structural analysis of the free energy simulations of BI-D binding to HIV-1 IN CCD; Table S1. Heavy RMS deviation between the simulated bound ligand and the crystal structures; Table S2. Data collection and structure refinement statistics; Figure S1. Chemical structures of BI-D, LEDGIN-6 and BI-224436; Figure S2. The MD simulated structures obtained using different His171 protonation states; Figure S3. The MD simulations reveal similar hydrogen bonding between LEDGF/p75 Asp366 with the WT (left) and the H171T mutant (right) IN CCDs.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MK, AE, RML, EP and JRF designed the study. AS, KJ, LF, JJK and RCL carried out the experiments. ND performed binding free energy calculations. HF, PAP, and NJ provided reagents. AS and MK wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Alison Slaughter, Email: slaughter.89@osu.edu.
Kellie A Jurado, Email: kelliejurado@fas.harvard.edu.
Nanjie Deng, Email: nanjie.deng@gmail.com.
Lei Feng, Email: feng.203@osu.edu.
Jacques J Kessl, Email: kessl.1@osu.edu.
Nikoloz Shkriabai, Email: shkriabai.1@osu.edu.
Ross C Larue, Email: larue.22@osu.edu.
Hind J Fadel, Email: fadel.hind@mayo.edu.
Pratiq A Patel, Email: pratiq.a.patel@vanderbilt.edu.
Nivedita Jena, Email: jena.2@osu.edu.
James R Fuchs, Email: fuchs.42@osu.edu.
Eric Poeschla, Email: poeschla.eric@mayo.edu.
Ronald M Levy, Email: tuf28314@temple.edu.
Alan Engelman, Email: alan_engelman@dfci.harvard.ed.
Mamuka Kvaratskhelia, Email: kvaratskhelia.1@osu.edu.
References
- 1.Brown PO. Integration. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Plainview, NY: Cold Spring Harbor Laboratory; 1997. pp. 161–204. [PubMed] [Google Scholar]
- 2.Hazuda DJ. HIV integrase as a target for antiretroviral therapy. Curr Opin HIV AIDS. 2012;7:383–389. doi: 10.1097/COH.0b013e3283567309. [DOI] [PubMed] [Google Scholar]
- 3.Sichtig N, Sierra S, Kaiser R, Daumer M, Reuter S, Schulter E, Altmann A, Fatkenheuer G, Dittmer U, Pfister H, Esser S. Evolution of raltegravir resistance during therapy. J Antimicrob Chemother. 2009;64:25–32. doi: 10.1093/jac/dkp153. [DOI] [PubMed] [Google Scholar]
- 4.Steigbigel RT, Cooper DA, Kumar PN, Eron JE, Schechter M, Markowitz M, Loutfy MR, Lennox JL, Gatell JM, Rockstroh JK, Katlama C, Yeni P, Lazzarin A, Clotet B, Zhao J, Chen J, Ryan DM, Rhodes RR, Killar JA, Gilde LR, Strohmaier KM, Meibohm AR, Miller MD, Hazuda DJ, Nessly ML, DiNubile MJ, Isaacs RD, Nguyen BY, Teppler H. Raltegravir with optimized background therapy for resistant HIV-1 infection. N Engl J Med. 2008;359:339–354. doi: 10.1056/NEJMoa0708975. [DOI] [PubMed] [Google Scholar]
- 5.Metifiot M, Vandegraaff N, Maddali K, Naumova A, Zhang X, Rhodes D, Marchand C, Pommier Y. Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. AIDS. 2011;25:1175–1178. doi: 10.1097/QAD.0b013e3283473599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wares M, Mesplede T, Quashie PK, Osman N, Han Y, Wainberg MA. The M50I polymorphic substitution in association with the R263K mutation in HIV-1 subtype B integrase increases drug resistance but does not restore viral replicative fitness. Retrovirology. 2014;11:7. doi: 10.1186/1742-4690-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li M, Mizuuchi M, Burke TR, Jr, Craigie R. Retroviral DNA integration: reaction pathway and critical intermediates. EMBO J. 2006;25:1295–1304. doi: 10.1038/sj.emboj.7601005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maertens GN, Hare S, Cherepanov P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature. 2010;468:326–329. doi: 10.1038/nature09517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krishnan L, Li X, Naraharisetty HL, Hare S, Cherepanov P, Engelman A. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc Natl Acad Sci U S A. 2010;107:15910–15915. doi: 10.1073/pnas.1002346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kessl JJ, Li M, Ignatov M, Shkriabai N, Eidahl JO, Feng L, Musier-Forsyth K, Craigie R, Kvaratskhelia M. FRET analysis reveals distinct conformations of IN tetramers in the presence of viral DNA or LEDGF/p75. Nucleic Acids Res. 2011;39:9009–9022. doi: 10.1093/nar/gkr581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson BC, Metifiot M, Ferris A, Pommier Y, Hughes SH. A homology model of HIV-1 integrase and analysis of mutations designed to test the model. J Mol Biol. 2013;425:2133–2146. doi: 10.1016/j.jmb.2013.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKee CJ, Kessl JJ, Shkriabai N, Dar MJ, Engelman A, Kvaratskhelia M. Dynamic modulation of HIV-1 integrase structure and function by cellular lens epithelium-derived growth factor (LEDGF) protein. J Biol Chem. 2008;283:31802–31812. doi: 10.1074/jbc.M805843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kessl JJ, Eidahl JO, Shkriabai N, Zhao Z, McKee CJ, Hess S, Burke TR, Jr, Kvaratskhelia M. An allosteric mechanism for inhibiting HIV-1 integrase with a small molecule. Mol Pharmacol. 2009;76:824–832. doi: 10.1124/mol.109.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J Biol Chem. 2003;278:372–381. doi: 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- 16.Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med. 2005;11:1287–1289. doi: 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- 17.Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. An essential role for LEDGF/p75 in HIV integration. Science. 2006;314:461–464. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- 18.Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007;21:1767–1778. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Busschots K, Vercammen J, Emiliani S, Benarous R, Engelborghs Y, Christ F, Debyser Z. The interaction of LEDGF/p75 with integrase is lentivirus-specific and promotes DNA binding. J Biol Chem. 2005;280:17841–17847. doi: 10.1074/jbc.M411681200. [DOI] [PubMed] [Google Scholar]
- 20.Cherepanov P, Ambrosio AL, Rahman S, Ellenberger T, Engelman A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc Natl Acad Sci U S A. 2005;102:17308–17313. doi: 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eidahl JO, Crowe BL, North JA, McKee CJ, Shkriabai N, Feng L, Plumb M, Graham RL, Gorelick RJ, Hess S, Poirier MG, Foster MP, Kvaratskhelia M. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 2013;41:3924–3936. doi: 10.1093/nar/gkt074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8:e1002717. doi: 10.1371/journal.pgen.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Nuland R, van Schaik FM, Simonis M, van Heesch S, Cuppen E, Boelens R, Timmers HM, van Ingen H. Nucleosomal DNA binding drives the recognition of H3K36-methylated nucleosomes by the PSIP1-PWWP domain. Epigenetics Chromatin. 2013;6:12. doi: 10.1186/1756-8935-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cherepanov P, Sun ZY, Rahman S, Maertens G, Wagner G, Engelman A. Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat Struct Mol Biol. 2005;12:526–532. doi: 10.1038/nsmb937. [DOI] [PubMed] [Google Scholar]
- 25.Hare S, Di Nunzio F, Labeja A, Wang J, Engelman A, Cherepanov P. Structural basis for functional tetramerization of lentiviral integrase. PLoS Pathog. 2009;5:e1000515. doi: 10.1371/journal.ppat.1000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hare S, Shun MC, Gupta SS, Valkov E, Engelman A, Cherepanov P. A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog. 2009;5:e1000259. doi: 10.1371/journal.ppat.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fadel HJ, Morrison JH, Saenz DT, Fuchs JR, Kvaratskhelia M, Ekker SC, Poeschla EM. TALEN knockout of the PSIP1 gene in human cells: analyses of HIV-1 replication and allosteric integrase inhibitor mechanism. J Virol. 2014;88:9704–9717. doi: 10.1128/JVI.01397-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall HM, Ronen K, Berry C, Llano M, Sutherland H, Saenz D, Bickmore W, Poeschla E, Bushman FD. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One. 2007;2:e1340. doi: 10.1371/journal.pone.0001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Rijck J, Vandekerckhove L, Gijsbers R, Hombrouck A, Hendrix J, Vercammen J, Engelborghs Y, Christ F, Debyser Z. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J Virol. 2006;80:11498–11509. doi: 10.1128/JVI.00801-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jurado KA, Engelman A. Multimodal mechanism of action of allosteric HIV-1 integrase inhibitors. Expert Rev Mol Med. 2013;15:e14. doi: 10.1017/erm.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engelman A, Kessl JJ, Kvaratskhelia M. Allosteric inhibition of HIV-1 integrase activity. Curr Opin Chem Biol. 2013;17:339–345. doi: 10.1016/j.cbpa.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demeulemeester J, Chaltin P, Marchand A, De Maeyer M, Debyser Z, Christ F. LEDGINs, non-catalytic site inhibitors of HIV-1 integrase: a patent review (2006–2014) Expert Opin Ther Pat. 2014;24:609–632. doi: 10.1517/13543776.2014.898753. [DOI] [PubMed] [Google Scholar]
- 33.Fader LD, Malenfant E, Parisien M, Carson R, Bilodeau F, Landry S, Pesant M, Brochu C, Morin S, Chabot C, Halmos T, Bousquet Y, Bailey MD, Kawai SH, Coulombe R, LaPlante S, Jakalian A, Bhardwaj PK, Wernic D, Schroeder P, Amad M, Edwards P, Garneau M, Duan J, Cordingley M, Bethell R, Mason SW, Bös M, Bonneau P, Poupart MA, et al. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med Chem Lett. 2014;5:422–427. doi: 10.1021/ml500002n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fenwick C, Lamorte L, Bethell R, Bonneau P, Cordingly M, Coulombe R, Duan J, Edwards P, Mason S, Poupart M-A, Simoneau B, Tsantrizos Y, Yoakim C: Discovery of a novel HIV-1 non-catalytic site integrase inhibitor [abstract]. In 4th International Conference on Retroviral Integration. 047th edition. Siena: 2011:s5.
- 35.Tsantrizos YS, Boes M, Brochu C, Fenwick C, Malenfant E, Mason S, Pesant M. Inhibitors of Human Immunodeficency Virus Repllication. Germany: Boehringer Ingelheim International GmbH; 2007. [Google Scholar]
- 36.Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyser Z. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat Chem Biol. 2010;6:442–448. doi: 10.1038/nchembio.370. [DOI] [PubMed] [Google Scholar]
- 37.Kessl JJ, Jena N, Koh Y, Taskent-Sezgin H, Slaughter A, Feng L, de Silva S, Wu L, Le Grice SF, Engelman A, Fuchs JR, Kvaratskhelia M. A multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J Biol Chem. 2012;287:16801–16811. doi: 10.1074/jbc.M112.354373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng L, Sharma A, Slaughter A, Jena N, Koh Y, Shkriabai N, Larue RC, Patel PA, Mitsuya H, Kessl JJ, Engelman A, Fuchs JR, Kvaratskhelia M. The A128T resistance mutation reveals aberrant protein multimerization as the primary mechanism of action of allosteric HIV-1 integrase inhibitors. J Biol Chem. 2013;288:15813–15820. doi: 10.1074/jbc.M112.443390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras-Colas A, Patel PA, Fuchs JR, Kvaratskhelia M, Engelman A. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc Natl Acad Sci U S A. 2013;110:8690–8695. doi: 10.1073/pnas.1300703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsiang M, Jones GS, Niedziela-Majka A, Kan E, Lansdon EB, Huang W, Hung M, Samuel D, Novikov N, Xu Y, Mitchell M, Guo H, Babaoglu K, Liu X, Geleziunas R, Sakowicz R. New class of HIV-1 integrase (IN) inhibitors with a dual mode of action. J Biol Chem. 2012;287:21189–21203. doi: 10.1074/jbc.M112.347534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gupta K, Brady T, Dyer BM, Malani N, Hwang Y, Male F, Nolte RT, Wang L, Velthuisen E, Jeffrey J, Van Duyne GD, Bushman FD. Allosteric inhibition of human immunodeficiency virus integrase: late block during viral replication and abnormal multimerization involving specific protein domains. J Biol Chem. 2014;289:20477–20488. doi: 10.1074/jbc.M114.551119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Rouzic E, Bonnard D, Chasset S, Bruneau JM, Chevreuil F, Le Strat F, Nguyen J, Beauvoir R, Amadori C, Brias J, Vomscheid S, Eiler S, Lévy N, Delelis O, Deprez E, Saïb A, Zamborlini A, Emiliani S, Ruff M, Ledoussal B, Moreau F, Benarous R. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology. 2013;10:144. doi: 10.1186/1742-4690-10-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christ F, Shaw S, Demeulemeester J, Desimmie BA, Marchand A, Butler S, Smets W, Chaltin P, Westby M, Debyser Z, Pickford C. Small-molecule inhibitors of the LEDGF/p75 binding site of integrase block HIV replication and modulate integrase multimerization. Antimicrob Agents Chemother. 2012;56:4365–4374. doi: 10.1128/AAC.00717-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma A, Slaughter A, Jena N, Feng L, Kessl JJ, Fadel HJ, Malani N, Male F, Wu L, Poeschla E, Bushman FD, Fuchs JR, Kvaratskhelia M. A new class of multimerization selective inhibitors of HIV-1 Integrase. PLoS Pathog. 2014;10:e1004171. doi: 10.1371/journal.ppat.1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balakrishnan M, Yant SR, Tsai L, O'Sullivan C, Bam RA, Tsai A, Niedziela-Majka A, Stray KM, Sakowicz R, Cihlar T. Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PLoS One. 2013;8:e74163. doi: 10.1371/journal.pone.0074163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Desimmie BA, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Van Remoortel B, Hofkens J, De Rijck J, Hendrix J, Bannert N, Gijsbers R, Christ F, Debyser Z. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology. 2013;10:57. doi: 10.1186/1742-4690-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engelman A, Englund G, Orenstein JM, Martin MA, Craigie R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J Virol. 1995;69:2729–2736. doi: 10.1128/jvi.69.5.2729-2736.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Engelman A. In vivo analysis of retroviral integrase structure and function. Adv Virus Res. 1999;52:411–426. doi: 10.1016/S0065-3527(08)60309-7. [DOI] [PubMed] [Google Scholar]
- 49.Engelman A. Pleiotropic nature of HIV-1 integrase mutations. In: Neamati N, editor. HIV-1 Integrase: Mechanism and Inhibitor Design. Hoboken: John Wiley & Sons; 2011. pp. 67–81. [Google Scholar]
- 50.Bukovsky A, Gottlinger H. Lack of integrase can markedly affect human immunodeficiency virus type 1 particle production in the presence of an active viral protease. J Virol. 1996;70:6820–6825. doi: 10.1128/jvi.70.10.6820-6825.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cannon PM, Wilson W, Byles E, Kingsman SM, Kingsman AJ. Human immunodeficiency virus type 1 integrase: effect on viral replication of mutations at highly conserved residues. J Virol. 1994;68:4768–4775. doi: 10.1128/jvi.68.8.4768-4775.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fenwick CW, Tremblay S, Wardrop E, Bethell R, Coulomb R, Elston R, Faucher AM, Mason S, Simoneau B, Tsantrizos Y, Yoakim C. Resistance studies with HIV-1 non-catalytic site integrase inhibitors [abstract] Antivir Ther. 2011;16(Suppl 1):A9. [Google Scholar]
- 53.Wang H, Jurado KA, Wu X, Shun MC, Li X, Ferris AL, Smith SJ, Patel PA, Fuchs JR, Cherepanov P, Kvaratskhelia M, Hughes SH, Engelman A. HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 2012;40:11518–11530. doi: 10.1093/nar/gks913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kotova S, Li M, Dimitriadis EK, Craigie R. Nucleoprotein intermediates in HIV-1 DNA integration visualized by atomic force microscopy. J Mol Biol. 2010;399:491–500. doi: 10.1016/j.jmb.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rhee SY, Liu TF, Kiuchi M, Zioni R, Gifford RJ, Holmes SP, Shafer RW. Natural variation of HIV-1 group M integrase: implications for a new class of antiretroviral inhibitors. Retrovirology. 2008;5:74. doi: 10.1186/1742-4690-5-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fader LD, Carson R, Morin S, Bilodeau F, Chabot C, Halmos T, Bailey MD, Kawai SH, Coulombe R, Laplante S, Mekhssian K, Jakalian A, Garneau M, Duan J, Mason SW, Simoneau B, Fenwick C, Tsantrizos Y, Yoakim C. Minimizing the contribution of enterohepatic recirculation to clearance in rat for the NCINI class of inhibitors of HIV. ACS Med Chem Lett. 2014;5:711–716. doi: 10.1021/ml500110j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown HE, Chen H, Engelman A. Structure-based mutagenesis of the human immunodeficiency virus type 1 DNA attachment site: effects on integration and cDNA synthesis. J Virol. 1999;73:9011–9020. doi: 10.1128/jvi.73.11.9011-9020.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu R, Limon A, Devroe E, Silver PA, Cherepanov P, Engelman A. Class II integrase mutants with changes in putative nuclear localization signals are primarily blocked at a postnuclear entry step of human immunodeficiency virus type 1 replication. J Virol. 2004;78:12735–12746. doi: 10.1128/JVI.78.23.12735-12746.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 60.Fadel HJ, Morrison J, Saenz D, Ekker SC, Fuchs JR, Kvaratskhelia M, Poeschla EM. Conference on Retroviruses and Opportunistic Infections; March 3-6. Boston: IAS-USA/CROI Foundation; 2014. p. 177. [Google Scholar]
- 61.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW, Sweet RM, editors. Methods in Enzymology. New York: Academic; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 62.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 65.Gilson MK, Given JA, Bush BL, McCammon JA. The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys J. 1997;72:1047–1069. doi: 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boresch S, Tettinger F, Leitgeb M, Karplus M. Absolute binding free energies: a quantitative approach for their calculation. J Phys Chem B. 2003;107:9535–9551. doi: 10.1021/jp0217839. [DOI] [Google Scholar]
- 67.Wang J, Deng Y, Roux B. Absolute binding free energy calculations using molecular dynamics simulations with restraining potentials. Biophys J. 2006;91:2798–2814. doi: 10.1529/biophysj.106.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deng NJ, Zhang P, Cieplak P, Lai L. Elucidating the energetics of entropically driven protein-ligand association: calculations of absolute binding free energy and entropy. J Phys Chem B. 2011;115:11902–11910. doi: 10.1021/jp204047b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 70.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 72.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 73.Hess B, Kutzner C, Van Der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theor Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 74.Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics (Oxford, England) 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]