

ABSTRACT
Response regulators are proteins that undergo transient phosphorylation, connecting specific signals to adaptive responses. Remarkably, the molecular mechanism of response regulator activation remains elusive, largely because of the scarcity of structural data on multidomain response regulators and histidine kinase/response regulator complexes. We now address this question by using a combination of crystallographic data and functional analyses in vitro and in vivo, studying DesR and its cognate sensor kinase DesK, a two-component system that controls membrane fluidity in Bacillus subtilis. We establish that phosphorylation of the receiver domain of DesR is allosterically coupled to two distinct exposed surfaces of the protein, controlling noncanonical dimerization/tetramerization, cooperative activation, and DesK binding. One of these surfaces is critical for both homodimerization- and kinase-triggered allosteric activations. Moreover, DesK induces a phosphorylation-independent activation of DesR in vivo, uncovering a novel and stringent level of specificity among kinases and regulators. Our results support a model that helps to explain how response regulators restrict phosphorylation by small-molecule phosphoryl donors, as well as cross talk with noncognate sensors.
IMPORTANCE
The ability to sense and respond to environmental variations is an essential property for cell survival. Two-component systems mediate key signaling pathways that allow bacteria to integrate extra- or intracellular signals. Here we focus on the DesK/DesR system, which acts as a molecular thermometer in B. subtilis, regulating the cell membrane’s fluidity. Using a combination of complementary approaches, including determination of the crystal structures of active and inactive forms of the response regulator DesR, we unveil novel molecular mechanisms of DesR’s activation switch. In particular, we show that the association of the cognate histidine kinase DesK triggers DesR activation beyond the transfer of the phosphoryl group. On the basis of sequence and structural analyses of other two-component systems, this activation mechanism appears to be used in a wide range of sensory systems, contributing a further level of specificity control among different signaling pathways.
INTRODUCTION
Two-component systems (TCS) are signaling pathways that respond to extra- and/or intracellular cues by modifying cellular behaviors (1). A phosphotransfer cascade from a sensor histidine kinase (HK) or phosphorelay system eventually results in the phosphorylation of a conserved aspartate of the response regulator (RR), which acts as the effector component of the TCS. RRs include a basic module, the receiver (REC) domain, which, upon phosphorylation, allosterically regulates protein-protein interactions such as homo-oligomerization or specific binding to other proteins or even to other domains in multidomain RRs (2).
Structural and biochemical data have been integrated into a widely accepted model of RR allosteric regulation (2, 3). According to this model, the phosphorylation of the conserved aspartate in the REC domain occurs in and stabilizes a rarely populated preexisting active conformation (4). RR structures corresponding to active and inactive states display shifts in the position of residues around the phosphorylatable Asp residue that are ultimately coupled to a structural rearrangement of the α4β5α5 (named according to the included secondary structural elements) solvent-exposed surface of the REC domain. In many inactive RRs, this surface interacts directly with the output effector domain (5). Therefore, phosphorylation-triggered changes result in the release of the effector domain (6), with greater conformational freedom to eventually select the biologically active conformation (7). The α4β5α5 region is directly involved in phosphorylation-triggered dimerization (8) and activation (9) in members of the PhoB/OmpR family. Recent crystal structures of RRs that belong to the NarL/LuxR family show that a different surface (α1α5) is key in mediating phosphorylation-triggered dimerization (10, 11), pointing to a novel regulatory mechanism of RR activation. This α1α5 surface on RRs seems also to be important in mediating the interaction with their cognate HKs (12).
Apart from their intrinsic autokinase activity, the majority of HKs catalyze two additional reactions in concert with their cognate RRs: phosphotransfer and dephosphorylation. The study of DesK from Bacillus subtilis has shown that HKs adopt distinct and defined three-dimensional (3D) conformations that correlate with their catalytic output status (13, 14), unveiling the molecular bases of those transitions (13). Crystallographic snapshots of HKs in other organisms have recently been reported (15, 16), confirming the ability of sensor HKs to sample a discrete set of 3D conformations, ultimately controlling their functional state.
Although HKs are the most relevant phosphoryl donors for RRs in vivo, RR activation mechanisms have been extensively studied at the molecular level by using small phosphodonors in vitro (3). Do HKs work just as macromolecular phosphodonors, or do they play additional roles as modulators of their cognate RR’s functional state? Different HK-RR and related phosphorelay pairs (12, 17–21) show large protein-protein interactions among the specific partners, and protein-protein interactions have been shown to be important in RR activation (7), reinforcing the pertinence of exploring the biological role of HKs beyond phosphorylation.
The model that we have chosen to address these questions is the DesK/DesR TCS from B. subtilis. DesR, a member of the NarL family of RRs, is involved in environmental temperature sensing and membrane fluidity regulation (22) (see Fig. S1 in the supplemental material). Cold shock triggers the sensor HK DesK to switch from a phosphatase to an autokinase competent state through a rotational switch in the helical dimerization and histidine phosphotransfer domain (13, 14). DesK then catalyzes phosphotransfer to Asp54 of DesR, triggering a reorganization of DesR’s quaternary structure, a key event in the physiologic induction of the transcription of the Δ5-desaturase gene (des) (23). The promoter of des (Pdes) has been studied in detail (23, 24), revealing two DNA-binding sites, the first one, farthest away from the transcription start site, displays higher affinity for DesR-P. The two binding sites are separated by a two-nucleotide spacer (lying on the same face of the DNA double helix), and occupation of both is essential for transcription activation. We now report the crystal structures of full-length DesR in the activated conformation, as well as of its REC domain in the active and inactive states. Combining biophysical, biochemical, and in vivo evidence, we show that DesR solvent-exposed surfaces mediate dimerization and tetramerization interactions, extending our current knowledge about the molecular mechanisms of RR activation. The architecture of the DesK-DesR complex is consistent with HK-triggered activity modulation mediated through the RR’s α1α5 surface. Moreover, in vivo data support a model that explains DesR activation by its cognate HK through binding (“preactivation”) and subsequent phosphorylation. This model is predicted to reflect a widely used mechanism for TCS activation control, integrating information from several systems.
RESULTS
Crystal structures of DesR reveal a phosphorylation-triggered conformational switch between functional states.
DesR is monomeric in solution. However, phosphorylation triggers DesR dimerization, as demonstrated by size exclusion chromatography (SEC) (Fig. 1A), small-angle X-ray scattering (SAXS), and SEC-coupled SAXS (see Table S1 in the supplemental material). To gain mechanistic insights into the process of DesR activation and how oligomerization is regulated, we solved the structures of full-length DesR in complex with BeF3−/Mg2+, as well as of its REC domain alone, in the presence or absence of BeF3−/Mg2+ (see Table S2 in the supplemental material). Beryllofluoride salts are commonly used as phosphomimetics to study active forms of RRs (25). The crystal structure of full-length DesR has two molecules in the asymmetric unit (Fig. 1B), both showing a molecule of BeF3− bound to phosphorylatable Asp54 (see Fig. S2A in the supplemental material). DesR comprises 199 residues showing an N-terminal REC and a C-terminal DNA-binding domain (DBD). The REC domain (residues 1 to 132) is composed of a central, parallel β-sheet (β1 to β6) surrounded by a total of six α-helices distributed on both sides (see Fig. S2B in the supplemental material). This is a variation of the canonical (βα)5 topology present in the vast majority of RRs and also seen in other NarL-like proteins (10, 11). After a short loop (amino acids 133 to 138) that links the two domains, the DBDs (residues 139 to 193) show a helix-turn-helix fold different from those described in other RRs of the NarL/LuxR family. Instead of a tetrahelical motif, DesR displays a trihelical arrangement (Fig. 1B). One DBD is completely free, and the other buries only 357 Å2 at the interface with the REC domain, suggesting that they are in a competent configuration to interact with DNA (Fig. 1B). We posit that the structure of full-length DesR in complex with BeF3−/Mg2+ represents the activated state, mimicking the phosphorylated form of the regulator.
FIG 1 .

Structure of DesR in the active state. (A) Phosphorylation-induced oligomerization of DesRwt analyzed by SEC. The apparent molecular masses of the inactive and phosphorylated species suggest a monomer-dimer transition, according to the indicated standard molecular weight (MW) markers. Abs, absorbance. (B) Structure of the active dimer shown in two orientations. The two chains in the asymmetric unit are depicted in blue and orange. Asp54 and BeF3− are displayed as spheres, colored by atom type, to highlight the phosphorylation site. term, terminus. (C) Cartoon representation of the activated REC domain (RECa, blue) compared to the inactive state (RECb, green) (D) Structural alignment of the RECa (active, blue), RECb (inactive, green), and RECc structures (phosphorylation-competent active state, orange). Key residues Asp54, Glu56, Thr81, and Phe82 are depicted as sticks, and the Mg2+ atom and BEF ligand from RECa structure are shown as spheres and sticks, respectively.
A structural alignment of the 10 independently refined DesR REC domains shows that they can be clearly grouped into two sets according to pairwise root mean square deviations, indicating two distinct structural states (Fig. 1C; see Fig. S2C in the supplemental material). The structural configurations represented by RECa and DesR most likely correspond to the active state, whereas RECb appears to reflect the inactive state. This is based not only on the presence or absence of BeF3−/Mg2+ but also on their structural resemblance to previously reported structures (10, 26, 27) (see Fig. S2D).
The phosphorylation of Asp54 induces significant rearrangements of residues Glu8, Asp9, Glu56, Thr80, and Thr81. Even though the residue at position 56 is not well conserved among different RRs, it appears to play a critical role. In the inactive state of DesR, Glu56 makes hydrogen bonds with well-conserved Thr80, Thr81, and Arg84, actually shielding phosphorylatable Asp54 from solvent access. Correlated with phosphorylation, Glu56 moves away and achieves the new stabilized position interacting with the BeF3− group and Mg2+, the cation being essential to allow for active-state RR phosphorylation. It is worth noting that a conserved aspartate (Asp9 in DesR) within the β1α1 loop also coordinates the Mg2+ cation. This β1α1 loop follows a dramatically different trace, so that, in the inactive state, the carboxylate group of Asp9 is ~13 Å from the Mg2+ site. The shifts of Thr80 and Thr81, already seen in many RRs (27–29), are observed in DesR further spreading to the α4β5α5 region, linked to the movement of Tyr99, and the rearrangement of the β4α4 loop and hydrophobic core residues Phe88, Val96, Leu101, Tyr123, and Leu127. This overall reorganization ultimately results in a shift in the position, as a well as a decrease in the flexibility, of helix α6.
A feature common to the structures of DesR, RECa, and RECb (which includes both functional states) is that the phosphorylation site is not solvent exposed, being covered either by Glu56 (in the inactive state) or by Phe82 (in the active configuration; Fig. 1D). A third crystal form of the REC domain (RECc) was determined also in the absence of [BeFx]−y and Mg2+, but it unexpectedly showed all four RECc protomers in a configuration that is closer to that of the active state, considering both quantitative figures of structural superposition and local inspection of the phosphorylation site and nearby residues. The structure of RECc shows a different arrangement of the β4α4 loop, Tyr99 does not engage in H bonds, and there is no clear electron density to confidently model the side chains of residues Glu56, Phe82, Ala83, and Arg84. RECc has probably captured the active conformation previous to actual phosphorylation, given that Asp54 is fully exposed to the solvent, and loops β1α1 and β3α3 are in the correct position for Mg2+ coordination (Fig. 1D).
DesR dimerizes through the α1α5 surface and tetramerizes through α4β6α6 upon DNA binding.
Each one of the four crystal structures of DesR displays different packing. Yet, they all reveal two major interaction surfaces between neighboring REC domains (see Fig. S3E in the supplemental material). These interfaces engage the α1α5 (burying ~800 to 1,030 Å2 of solvent-accessible surface area) and α4β6α6 (~430 to 1,000 Å2) surfaces. These interfaces are similar to those found in crystal structures of VraR (10) and spr1814 (11), illustrating high conservation among members of the NarL family. To assess the functional roles of these surfaces, we used a strategy of structure-guided point mutagenesis aiming at the disruption of potential interactions and subsequent biophysical characterization of selected mutant variants.
The α4β6α6 surface of DesR (Fig. 2A to C) differs from the α4β5α5 dimerization surface present in many RRs (essentially members of the OmpR/PhoB family), with the additional β6 and α6 elements (Fig. 2B), burying β5α5 away from bulk solvent. The major protein-protein interactions stabilizing a potential quaternary structure are mainly polar (Fig. 2B and C), including six salt bridges and three hydrogen bonds. Hydrophobic interactions seem to be restricted to the center of this motif (Fig. 2B and C).
FIG 2 .

Functional characterization of the α4β6α6 and α1α5 oligomerization surfaces. (A) Sequence conservation of DesR (according to a multiple-sequence alignment including 7,691 sequences of the NarL/LuxR family) was mapped onto the molecular surface of the REC domain. The orientation chosen highlights the presence of supplementary secondary-structure elements β6 and α6 toward the C terminus. Highly conserved residues are magenta, and variable residues are green. (B) Electrostatic potential (negative potential red, positive potential blue) mapped onto the molecular surface of the REC domain, shown in the same orientation as in panel A. (C) Close-up of the wild-type α4β6α6 interface shown as a transparent surface between two protomers depicted in green and blue. The hydrophobic cores of interacting residues are displayed as spheres and as an outer ring, and residues engaged in five salt bridges are shown as black dotted lines. Residue Arg121 was selected for site-directed mutagenesis to disrupt key ionic bonds in the α4β6α6-mediated interaction. (D) SEC comparing the behavior of phosphorylated and unphosphorylated DesRwt with DesR_Arg121Ala. Abs, absorbance. (E) Sequence conservation mapped onto the molecular surface, colored as in panel A, highlighting the position of α1. (F) Electrostatic potential mapped onto the molecular surface, colored as in panel C and in the same orientation as in panel E. (G) Close-up of the wild-type α1α5 interface. One monomer is shown in a cartoon representation (magenta, only the α1 helix is shown for clarity), and the other is shown in an accessible-surface (gray) representation. Residues in α1 involved in hydrophobic and hydrogen bond interactions are labeled. Residue Met12 was selected for site-directed mutagenesis to disrupt the α1α5-mediated interaction because of its insertion into a hydrophobic pocket of the other monomer. (H) SEC comparing phosphorylated and unphosphorylated DesRwt with DesR_Met12Ala and DesR_Met12Ala-Ala16Arg. (I) EMSAs showing the binding of DesRwt (left), DesR-Arg121Ala (middle), and α1α5 mutant variants (left) to the Pdes promoter (including the two DesR-specific DNA-binding sites). The indicated concentrations of the recombinant proteins were preincubated with 50 mM acetyl phosphate. CI and CII indicate the singly and doubly occupied DesR-DNA complexes, respectively. Lanes M contain molecular size markers.
The second interaction surface, through the α1α5 region (including the β5α5 loop) (Fig. 2E), is conserved among NarL/LuxR RRs (Fig. 2F) and mediated largely by hydrophobic contacts (Fig. 2F and G). Interestingly, α1α5-mediated oligomerization brings the phosphorylation sites of each monomer into close proximity, generating a strongly negatively charged cleft. Among the hydrophobic contacts that stabilize the α1α5-mediated interaction, the N-terminal end of α1 has a solvent-exposed protrusion, notably including the side chain of Met12, which inserts into a hydrophobic cavity delimited by the β5α5 loop and the N-terminal tip of helix α5 (Fig. 2G). The protrusion of one monomer snugly fits into the pocket of the other monomer and vice versa.
To explore the role of these interactions in α4β6α6- and α1α5-mediated oligomerization, we compared wild-type DesR (DesRwt) with variants carrying an Arg121Ala substitution (on helix α4) or Met12Ala and Met12Ala-Ala16Arg substitutions (on helix α1). Following in vitro phosphorylation with acetyl phosphate, the quaternary structure of the different DesR proteins was analyzed by SEC (Fig. 2D and H) and SAXS (see Table S1). They all revealed stable species, consistent with well-folded proteins. While phosphorylation triggered the dimerization of DesR_Arg121Ala to levels comparable to those of DesRwt (Fig. 2D), no dimeric forms of DesR_Met12Ala or DesR_Met12Ala-Ala16Arg were detectable (Fig. 2H) under identical conditions.
Given that the α4β6α6 surface does not appear to affect DesR dimerization, at least by mutating Arg121, we then asked whether the DNA-binding behavior was affected. Interestingly, electrophoretic mobility shift assays (EMSAs) revealed that DesR_Arg121Ala is defective in the ability to occupy the second DNA-binding site on the Pdes promoter (Fig. 2I). This site is essential to the generation of a functional complex II on the DNA (23) that is able to trigger transcriptional activation. That DesR_Arg121Ala is able to generate singly occupied complex I with an affinity indistinguishable from that of DesRwt is consistent with its being able to dimerize properly, ruling out the implication of the α4β6α6-mediated interface in dimerization. In contrast, the behavior of the single-mutation variant (DesR_Met12Ala) or the double-mutation variant (DesR_Met12Ala-Ala16Arg) in EMSAs is dramatically different from that of the wild-type protein, revealing no detectable DNA binding whatsoever (Fig. 2I). The latter results are consistent with the absence of a stable phosphorylated dimeric species for both α1α5 surface mutant variants (Fig. 2H).
Taking the results together and considering that all mutant variants can be phosphorylated by acetyl phosphate in vitro (see Fig. S4A in the supplemental material), we conclude that DesR dimerizes through the α1α5 surface, while it tetramerizes in a DNA-dependent way through the α4β6α6 region. Furthermore, direct-coupling analysis (DCA) (30, 31) using a set of 7,691 sequences with the same domain architecture as DesR (see the supplemental material for full details) strongly suggests that α1α5 dimerization is a common trend within the whole NarL family (see Fig. S3).
DesR is activated through a novel allosteric mechanism.
Dimerization of the RR PhoB promotes its ability to be phosphorylated with acetyl phosphate (32). Given that PhoB dimerizes though a different interaction surface (α4β5α5) than DesR, we wondered whether α1α5-mediated dimerization in DesR produces a similar outcome. We measured the ratio (ξ) of phosphorylated DesR with respect to the total amount of protein as a function of the total protein concentration by using Phos-tag gels to separate phosphorylated from nonphosphorylated species (33) at a fixed time point. A constant ξ value would rule out a dependence of phosphorylation on the DesR dimerization state because protein concentration is a variable directly linked to oligomerization. On the one hand, we observed sigmoidal positive relationships between ξ and both DesRwt and DesR_Arg121Ala concentrations (Fig. 3; see Fig. S4B). On the other hand, the variants DesR_Met12Ala and DesR_Met12Ala-Ala16Arg, with α1α5 disrupted, abrogated this ξ dependency on protein concentration, strongly supporting the notion that homodimerization favors DesR autophosphorylation. The reduced autophosphorylation capacity observed in the mutant variants affecting the α1α5 interface necessarily implies an allosteric link between this surface and the orthosteric Asp54 carboxylate group, the site of phosphorylation. Note that the mutated residues on α1 are solvent exposed and 10 to 15 Å away.
FIG 3 .
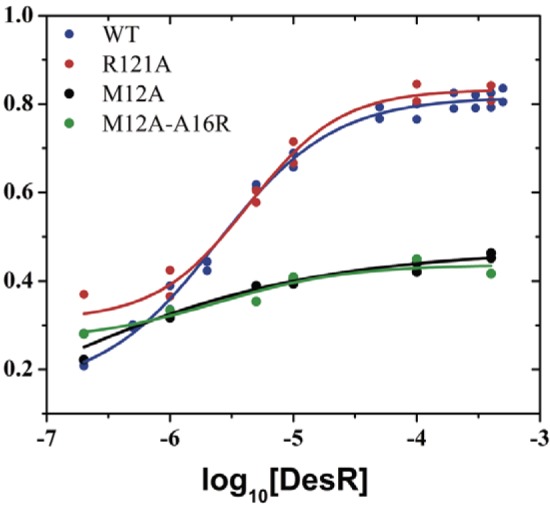
Allosteric coupling between the α1α5 dimerization surface and the phosphorylation site. Autophosphorylation in vitro with acetyl phosphate. The degree of phosphorylation (ξ) is expressed as a function of the logarithm of the protein concentration (molar). ξ was measured after 1 h of incubation with acetyl phosphate. Values from separate experiments are shown with the best-fit sigmoid curves overlaid. A representative Phos-tag SDS-PAGE analysis is shown in Fig. S4B in the supplemental material. WT, wild type.
From the dimeric structures of DesR, we predicted that dimerization could stabilize phospho-Asp by interfering with the correct positioning of a reactive water molecule, as seen in the phosphatase complex CheX-CheY structure (34). Within the β4α4 loop, Phe82 shields the BeF3− moiety and is stabilized by van der Waals contacts with Thr81, Tyr87, and Glu56 (Fig. 1D). Thr81 is in close proximity to Gln10 of the other monomer, a dimerization pair that emerges from DCA (see Fig. S3). To test this prediction, we evaluated the autodephosphorylation of DesR at different protein concentrations and found that, as expected, phospho-DesR was stabilized at higher protein levels (see Fig. S4C). Phospho-PhoB showed no detectable protein concentration dependence of its dephosphorylation (see Fig. S4C), which, together with the DCA data, strongly suggests that dimerization stabilizes the phosphorylated state of many RRs that dimerize through α1α5 within the NarL family. In the monomeric state, the β4α4 loop is expected to be more flexible, allowing for entry of the reactive water molecule.
DesR homodimerization and HK binding occur through a largely overlapping interface that involves the RR α1α5 surface.
Structural data on HK-RR complexes (12) and covariance analyses (31, 35) indicate that the RR α1α5 surface is directly involved in the interaction with its cognate HK. Given that the α1α5 surface is allosterically coupled to the phosphorylation site in DesR, we decided to analyze the relationship between DesR dimerization and DesK-DesR interaction. We generated a model by using the crystal structures of the catalytic cytoplasmic region of DesK (DesKC; Protein Data Bank [PDB] code 3GIF) and DesR (Fig. 4A; see Fig. S5A in the supplemental material), assuming that the DesK-DesR complex is similar to HK853-RR468 from Thermotoga maritima (12). We validated the model by studying disulfide cross-linking using engineered Cys mutant forms of both partners (see Fig. S5B). DesK is thus predicted to bind DesR through a surface that significantly, although not completely, overlaps the α1α5 surface engaged in DesR homodimerization. According to the modeled complex, Leu200 of DesK is expected to play a role similar to that of Met12 of DesR, occupying a cavity between helix α1 and the β5α5 loop of the REC domain. The α1 double-mutation variant DesR_Met12Ala-Ala16Arg is predicted to clash with the approaching kinase through the engineered arginine at position 16. We tested these predictions by investigating binding to the catalytic region of DesK. We determined that the single-mutation variant DesR_Met12Ala is able to form a DesK-DesR complex (Fig. 4B), whereas DesR_Met12Ala-Ala16Arg cannot. Further support for our predictions was obtained by exploring the ability of these α1α5 mutant variants to be phosphorylated by DesK and ATP in vitro, which showed that DesR_Met12Ala can be phosphorylated to levels comparable to those of DesRwt, whereas the double-mutation variant cannot (Fig. 4C). The fact that DesK associates with DesR through the RR’s α1α5 surface then opened the question of whether DesR might dimerize differently, depending on the presence or absence of the HK. The DesK-DesR complex likely leaves the α4β6α6 surface of the RR exposed to the solvent, such that DesK phosphorylation (in contrast to acetyl phosphate) could be thought to drive DesR to dimerize through the α4β6α6 interface. EMSAs performed with DesR phosphorylated by DesK in the presence of ATP (Fig. 4D) revealed a DNA shift with DesR_Arg121Ala, whereas DesR_Met12Ala resulted in no detectable complex. These results are consistent with those obtained previously with acetyl phosphate phosphorylation, confirming that DesR homodimerization always occurs through α1α5 (i.e., the α4β6α6 surface is not involved in dimerization), independently of the source of phosphorylation.
FIG 4 .

DesR forms a specific complex with DesK through the α1α5 surface. (A) The predicted binding of DesR with its cognate His-kinase DesK shows an extensive overlapping area with the α1α5-mediated dimerization of the RR. The reactive residues on the RR (Asp54 in DesR) and the HK (His188 in DesK) are shown as sticks and labeled. For clarity, only the first two helices of one monomer of DesK, predicted to interact directly with DesR, are shown in a cartoon representation (green). DesR is rendered mostly as a gray surface, except for helices α1 and α5, which are shown in cartoon (magenta). Note that helix α1 of DesK, which includes the phosphorylatable histidine, is expected to be positioned between α1 and the β5α5 loop of DesR. (B) DesR and the indicated mutant variants were evaluated for the ability to interact with DesKC_His188Val by SEC. For each run, a 50 µM concentration of DesK-H188V and/or DesR was preincubated for 10 min at room temperature. Abs, absorbance. (C) Phosphotransferase activity of 32P-labeled DesKC to DesR or the indicated mutant variants analyzed by SDS-PAGE and autoradiography. As a further control, DesR_Arg121Ala, indeed, behaves as DesRwt in DesK complexation and phosphotransfer (Fig. S5C and D in the supplemental material). WT, wild type. (D) EMSAs showing the binding of DesRwt, DesR-Arg121Ala, and DesR-Met12Ala to the Pdes promoter using DesKC and ATP for DesR phosphorylation. Lane M contains molecular size markers.
Biological relevance of the dimerization and tetramerization surfaces of DesR.
To test the biological relevance of the exposed surfaces of DesR linked to its activation state, we assayed the capacity to regulate des gene transcription in vivo by comparing DesRwt with the different point mutant variants with disrupted α1α5 or α4β6α6 interfaces. des expression was evaluated by complementing B. subtilis strain AKP21 (desk desR double knockout) (22) with a plasmid expressing wild-type desK and either wild-type desR or each of the desR surface mutant variants under the control of a xylose-inducible Pxyl promoter. AKP21 carries a lacZ reporter gene under Pdes promoter regulation. The α4β6α6 DesR_Arg121Ala mutant variant, as well as the α1α5 single-mutation variant DesR_Met12Asp (equivalent to the mutation performed by Leonard et al. [10]) and the double-mutation variant DesR_Met12Ala-Ala16Arg, lost the capacity to upregulate des transcription in response to cold shock (Fig. 5A). These results are consistent with the in vitro EMSA results showing that both surfaces are essential for DesR transcriptional activation function. Somewhat unexpectedly, the single-mutation variant DesR_Met12Ala retained in vivo regulation to extents comparable to those of DesRwt (Fig. 5A), likely because of its milder effect on the α1α5 surface than that of DesR_Met12Asp and DesR_Met12Ala-Ala16Arg.
FIG 5 .

DesK activates DesR in a phosphorylation-independent manner. (A) In vivo assays evaluating cold shock-triggered activation of des gene expression, comparing a plasmidic construct of DesRwt with those of the DesR_Asp54Ala (D54A), DesR_Asp54Asn (D54N), DesR_Met12Ala (M12A), DesR_Met12Asp (M12D), DesR_Met12Ala-Ala15Arg (M12A_A16R), and DesR_Arg121Ala (R121A) point mutant variants. (B) In vivo Pdes cold shock activation, using chromosome-integrated copies of DesRwt in the presence [K+/R+(wt)] or absence [K−/R+(wt)] of DesK, compared to DesR_Asp54Asn in the presence [K+/R+(D54N)] or absence [K−/R+(D54N)] of the kinase. B. subtilis strain AKP21 (K−/R−), which is a desK des double-knockout strain, was used as a negative control, whereas wild-type strain AKP3 was used as a positive control. WT or wt, wild type.
The cognate HK, in its phosphotransfer state, acts as an allosteric activator of the RR.
The observations that α1α5 dimerization is allosterically coupled to DesR activation (Fig. 2 and 3) and that this α1α5 surface also mediates DesK association (Fig. 4) led to the question of whether DesK binding might exert an activating effect on the regulator independently of phosphorylation. The use of the nonphosphorylatable variants DesR_Asp54Ala and DesR_Asp54Asn enabled us to differentiate between potential activation and actual phosphorylation. Strikingly, signal-dependent regulation of des was obtained with DesR_Asp54Ala and DesR_Asp54Asn to extents comparable to those obtained with DesRwt and DesR_Met12Ala (Fig. 5A). These results indicate that DesK is also able to activate its cognate RR through a phosphorylation-independent mechanism. Because the results described above were obtained with plasmid-borne genes, we investigated expressions where we inserted the desk+ des+ or desK des+ construct into the thrC locus of the chromosome under the control of the Pxyl promoter. As shown in Fig. 5B, the DesR_Asp54Asn mutant variant is able to induce des transcription at low temperatures in a DesK-dependent manner similarly to DesRwt. The strains expressing only the RRs do not exhibit des expression at low temperature, which is direct evidence of a phosphorylation-independent activating role for the cognate HK. Moreover, experiments in which the stringency of the Pxyl promoter was increased by the addition of various concentrations of glucose to the culture medium showed that larger amounts of DesR_Asp54Asn than of DesRwt are necessary to achieve the activation of Pdes (see Fig. S6 in the supplemental material). Altogether, these in vivo results unveil a phosphorylation-independent step of activation that requires a signal-triggered change in the conformation of the HK.
DISCUSSION
The crystallographic and functional studies, both in vivo and in vitro, of full-length DesR, as well as of its REC domain in the active and inactive configurations, have been instrumental in the discovery of a phosphorylation-stabilized dimeric species. The monomers interact through a “noncanonical” interface defined by helices α1 and α5 and the β5α5 loop. Furthermore, structure-guided mutant variants show that this dimerization is biologically relevant and critical for the RR activation. These results now confirm, in the context of recent reports (10, 11), that this is a widely used mechanism in TCS. A novel feature related to this noncanonical mechanism is now unveiled: the surface located in a position equivalent to that of α4β5α5 (α4β6α6 in DesR), typically involved in the dimerization of PhoB/OmpR RRs, is not engaged in DesR dimerization but is instead critical to the establishment of essential dimer-dimer interactions in DesR, enabling it to fully bind to its DNA-binding site. An additional and striking finding concerns the cognate kinase DesK, which “preactivates” DesR as a first step toward phosphotransfer, full-blown activation, and response. That homodimerization and DesK binding occur largely through the same DesR surface (α1α5) leads us to propose a shared allosteric pathway coupling this surface with the phosphorylatable aspartate. Further studies are needed to understand the derived geometric constraints during the phosphatase reaction. It can be predicted that the kinase should be able to displace the dimer-monomer equilibrium toward the phosphate-accessible monomeric species such that dephosphorylation proceeds efficiently. This scenario is likely valid for all RRs that dimerize through their α1α5 surface within an extended subset of NarL-like RRs (10, 11).
Structural bases of allosteric activation pathways.
The nonlinear behavior of DesR phosphorylation versus concentration implies oligomerization-dependent stabilization of its active conformation (Fig. 3), in agreement with recent reports (32). Disruption of the α1α5 interface impairs activation, revealing allosteric control. The comparison of DesR REC domains in the active (BeF3−-bound) versus the inactive state (Fig. 1; see Fig. S2C and S7), allows the pinpointing of key residues for this allosteric pathway. Side chains of residues in the β1α1 loop are directly involved in phosphorylation-triggered rearrangements, leading to the significant shift (maximum, ~1.8 Å) and rotation (maximum, ~30 to 40°) of helix α1, with the effect being more pronounced in the N-terminal half. The direct physical link between the α1 helix position and the preceding β1α1 loop immediately suggests a mechanistic link for α1α5-triggered activation. DesR homodimerization induces a shift of α1 (Fig. 6A) affecting the position of a conserved carboxylate side chain within the β1α1 loop (Asp9 in DesR), which is critical for cation coordination and activity. This feature, wherein the carboxylate O atoms of the conserved Asp are located too far away to coordinate the Mg2+ with a phosphorylation-competent geometry (Fig. 6B), is conserved in the inactive conformation of several RRs from different families (5, 10, 12).
FIG 6 .

Working model of DesK-DesR allosteric regulation. (A) Superposition of the active (blue) and inactive (yellow) conformations of the REC domain of DesR, illustrated in cartoon representations. BeF3− is bound to Asp54. Mg2+ is shown as a purple sphere. The direct link between the α1 shift and the competent position of Mg-coordinating Asp9 is highlighted. (B) Same orientation as in panel A, showing in solid colors the superposition between the active (cyan) and inactive (green) conformations of VraR (10). Note the side chain of Asp10, which, in concert with the movement of helix α1, shifts its side chain away from the cation coordination sphere. In a transparent representation (red), the equivalent Asp14 of inactive NarL (5) is highlighted adopting a similar distant position. RR468 (12) displays the same Asp10 rearrangement (not shown for clarity). (C) The calculated model of DesK-DesR complex is shown in the same orientation as in the previous panels. DesK is green, and the color code for the active and inactive forms of DesR are identical to those in panel A. Note the predicted favorable interactions between the active state of DesR and the kinase with contacts between the α1 helices of both partners (DesK Leu200 is predicted to occupy a position similar to that of\ DesR Met12 in the DesR homodimer). Phosphorylatable His188 is shown as a reference. (D) The cell membrane is shown at the top. Depicted is the three-state conformational transition of DesK based on the structural data we have previously reported (13); the 3D structure of its transmembrane domain is still not known. The asymmetric structure adopted by P-DesK forms a phosphotransfer complex with DesR, shifting the conformational equilibrium of the RR population toward the active state (preactivation) and transferring the phosphate. P-DesR increases the half-life of the active conformation, allowing for functional dimers to form. In the absence of phosphorylation (e.g., nonphosphorylatable DesR mutant variants), a higher DesR concentration is essential for stabilization of the activated conformation (see Fig. S6 in the supplemental material). The pathway is shut down by DesK in its phosphatase state, forming a complex with monomeric P~DesR. The DesR dimer binds first the high-affinity (RA) and then the lower-affinity (RB) DNA-binding site (23). DesR dimer-dimer interactions through the α4β6α6 interface seem crucial to the achievement of full RB occupancy. RNAP, RNA polymerase.
Our structures also indicate that when DesR is in the inactive state, Glu56 (in the β3α3 loop) is shielding phosphorylatable Asp54, stabilized by contacts with Thr80, Thr81, and Arg84. Activation of DesR implies a significant shift of Glu56, making Asp54 solvent accessible. Similar conformational changes can be identified in other RRs, such as Spo0F (36), CheY (37), NtrC (38), and RR468 (12). The residue equivalent to Glu56 in NarL (Asn61) also covers the phosphorylation site (5, 26), suggesting a common property of the inactive state. In the case of VraR, the nonphosphorylated structure does not adopt this inactive configuration, possibly because the β3α3 loop is part of the crystal packing (10).
DesR homodimerization and binding of DesK induce allosteric activation of DesR.
The association geometry of DesK and DesR evokes an activation mechanism similar to the one triggered by DesR homodimerization, with the first α helix of the HK DHp domain fulfilling the role of the RR’s α1 (Fig. 6C). DesK is thus well positioned to induce α1α5-mediated DesR activation. Alternative mechanisms cannot be ruled out. Experimental data revealing the 3D structure of DesK-DesR complexes in different functional configurations will be instrumental in this direction. Available crystallographic data from other HK-RR (12, 39) and phosphorelay (20) complexes do show a similar set of rearrangements, especially at the β1α1 and β3α3 loops and helix α1 of the RR, in support of this mechanism and its wide use among TCS. The RR’s α1α5 surface is implicated in different complexes involving RRs as partners (17–21), and this is consistent with mutual information analyses (31, 35, 40). This interface not only seems critical for specific RR recognition but, as we now show, is also key for RR activation.
We have now established that DesR’s cognate HK plays an allosteric activation effect similar to that of the one resulting from DesR dimerization, as DesK can activate the pathway independently of DesR phosphorylation. Importantly, this effect is regulated by temperature, the physiological signal in the DesK/DesR system (Fig. 6D; see Fig. S1). That Pdes activation by nonphosphorylatable DesR mutant variants occurs only after cold shock means that the functional state of DesK (in this case, its phosphotransferase-active form) is relevant for the activation mechanism. In the high-temperature regimen, DesK is mainly in its kinase-off/phosphatase-on state and the 3D structure of its cytoplasmic domain is significantly different (13), which not only explains the differential catalytic output but also predicts differences in the way it interacts with DesR. We posit that HK-RR interactions are different between the phosphatase (12) and phosphotransfer states, yet conclusive evidence awaits a high-resolution structural snapshot of an HK-RR phosphotransfer complex. We obtained a model of such a complex by using separate DesK and DesR crystal structures, resulting in an overall geometry similar to the available crystal structure (12).
A phosphorylation-independent activation mechanism of DesR can be envisaged that, although signal and DesK dependent, likely depends on DesR reaching high enough concentrations that sufficient dimer is built in the cell (see Fig. S6 and S7C in the supplemental material). At physiological DesK-DesR concentrations and ratios, DesR phosphorylation seems otherwise essential to increase the half-life of enough dimeric species that response activation stays tightly linked to signal sensing (Fig. 6D). Conclusive validation of this hypothesis awaits the quantitative determination of intracellular DesK and DesR concentrations. A working model consistent with all of the data leads us to propose a “preactivation” role for DesK, via DesR monomer binding, able to raise the concentration of active-state DesR (4, 41), a dimerization-prone state (10, 42). Enough dimeric DesR species is sufficient to exert the effector transcriptional response, and a slow active-to-inactive-state transition (43, 44) would allow sequential DesK-DesR dissociation and DesR homodimer formation. The HK-dependent allosteric activation mechanism now provides a molecular insight into reported observations such as the differential phosphorylation efficiencies of small phosphoryl donor molecules versus the cognate HK observed with the PhoB-like RR PrrA (7) or FixL-dependent in vivo signaling using nonphosphorylatable variant FixJ proteins (45). With a broader perspective, this activation role is relevant to explain why small phosphoryl donor molecules are inefficient at phosphorylating the vast majority of RRs in vivo (46), even when these phospho donors reach high intracellular concentrations (47) (Fig. 6D).
Phosphorylation-independent mechanisms have been described for a few TCS (48–52), but the regulatory mechanisms are still not fully understood. Interestingly, HKs can also be allosterically regulated by RRs: the RR DivK binds and activates the autokinase activity of HKs DivJ and PleC (53), although the mechanism is still elusive. Our evidence that DesK is able to modulate the functional state of DesR by binding through an interface expected to be used in most HK-RR pairs suggests that this allosteric mechanism is widely used, promoting RR activation in the presence of the signal and adding a further level of specificity, avoiding uncontrolled pathway activation.
MATERIALS AND METHODS
Plasmid construction and site-directed mutagenesis.
Detailed protocols used for plasmid construction and site-directed mutagenesis are included in the supplemental material (see Text S1). Lists of the primers and plasmids used are available at http://intranet.pasteur.edu.uy/pxf/Supplemental_oligos.pdf http://intranet.pasteur.edu.uy/pxf/Supplemental_plasmids.pdf, respectively.
Protein expression and purification.
Recombinant proteins were expressed as N-terminally His6-tagged fusions in Escherichia coli strain TOP10F′ (Invitrogen). Protein purifications were performed as previously described (13), except that the last SEC (HiLoad 16/60 Superdex 75 preparation grade column; GE Healthcare) for DesR (full-length construct) or DesRREC (REC domain) was preequilibrated with a mixture of 20 mM Tris HCl (pH 8), 300 mM NaCl, 10 mM MgCl2, and 0.5 mM dithiothreitol (DTT). After gel filtration, full-length DesRwt and DesRREC were concentrated to 20 and 80 mg/ml, respectively, and stored at −80°C.
Protein crystallization and structure determination.
Single crystals were grown at 20°C by using vapor diffusion techniques. Full details of the crystallization conditions and data collection protocols used are described in the supplemental material (see Table S2). Initial phases for full-length DesR were obtained by molecular replacement (54) using an RR from Staphylococcus aureus (PDB code 3B2N). The structure was rebuilt by using Coot (55) and refined with Buster (56). The three structures of the REC domain were solved by MR using the refined full-length model as the search probe.
SAXS data collection and analysis.
SAXS data from different DesR variants were collected at the BM29 (ESRF) and SWING (Soleil) synchrotron beamlines. At BM29, all samples were measured in dilution series at four different concentrations (20 to 200 µM) at 20°C (for full details, see the supplemental material).
EMSAs.
EMSAs were performed as described previously (22). Briefly, different concentrations of phosphorylated DesR or point mutant variants (previously phosphorylated at 100 µM in the presence of 50 mM acetyl phosphate) were incubated for 20 min at room temperature in a mixture of 50 mM Tris-HCl (pH 8), 5 mM MgCl2, 0.5 mM EDTA, 1.25 mM DTT, 10% glycerol, 50 mM acetyl phosphate, 5 µg/ml heparin, and 1.8 µM DNA probe. After the incubation period, glycerol was added to a final concentration of 15% and the mixture was applied to a 5% polyacrylamide gel that had been prerun for 1 h in a mixture of 45 mM Tris-borate (pH 8), 1 mM EDTA, and 4 mM MgCl2. A similar procedure was followed with DesKC instead and with 5 mM ATP as the phosphoryl donor.
Effect of protein concentration on DesR autophosphorylation.
DesR autophosphorylation reactions were performed with protein concentrations ranging from 0.2 to 500 µM at 25°C and pH 8 and incubation with a mixture of 50 mM acetyl phosphate, 300 mM NaCl, 50 mM MgCl2, and 20 mM Tris-HCl. The reaction volumes varied between 20 and 100 µl. Addition of acetyl phosphate was immediately followed by warming from 4 to 25°C. Autophosphorylation was stopped at 1 h, the mixture was cooled to 4°C, and SDS-PAGE sample buffer with 2.5 mM DTT was added. Protein samples (0.13 to 1.1 µM) were incubated with SDS-PAGE sample buffer at room temperature for 5 min and mixed with iodoacetamide (40 mM) for 10 min. Phos-tag SDS-PAGE was performed as previously described (57), with 50 µM Phos-tag acrylamide and 100 µM ZnCl2. Coomassie-stained gels were scanned with UMAX PowerLook 1120 and LabScan 5.0 (GE HealthCare) and analyzed by Image Quant TL v2005 software (GE HealthCare). The peak areas corresponding to phosphorylated (slower migration) and unphosphorylated species were integrated, and the degree of phosphorylation (ξP) was calculated according to the formula ξP = DesR − P peak area × (DesR − P peak area + DesR peak area)−1.
β-Galactosidase assays in liquid cultures.
Replicative plasmids pACRK1 (DesRwt), pACRK2 (desRM12A), pACRK3 (desRM12A,A16R), pACRK4 (desRR121A), pACRK5 (desRM12D), pACRK6 (desRM12A,A16R), and pACRK7 (desRR121A) were transformed into B. subtilis strain AKP21 (JH642 desKR::Kmr amyE::Pdes-lacZ) (22). The resulting strains were grown overnight in Spizizen minimal-salts medium supplemented with 0.5% glycerol, 0.01% tryptophan, 0.01% phenylalanine, trace elements, and 0.05% casein hydrolysate in the absence of xylose. This medium was named SMM-CAA. The cells were collected by centrifugation and diluted in SMM-CAA containing 0.1% xylose to an optical density at 525 nm (OD525) of 0.12. The xylose concentration was a critical parameter to optimize. Cultures were incubated with shaking at 37°C until they reached an OD525 of 0.3 and divided in two. One half was kept at 37°C, and the other was transferred to 25°C. Samples were taken at the times indicated and assayed for β-galactosidase activity as previously described (24). The specific activity was expressed in Miller units.
β-Galactosidase assays in plates.
Strains AKP21_ Rwt, AKP21_RD54N, AKP21_Rwt_K, and AKP21_RD54N_K carry a single copy of the desR gene (wild type or with the single point mutation D54N) integrated into the thrC locus under the control of the Pxyl (xylose-inducible) promoter. In addition, AKP21_Rwt_K and AKP21_RD54N_K contained the desK gene integrated into the same locus under the control of a second Pxyl promoter (for further details of the construction of the strains, see the supplemental material). The four strains, together with AKP3 and AKP21 (22), were streaked onto SPI-CAA medium plates containing 80 µg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal; Sigma) and different glucose (Sigma) or l-xylose (Sigma) concentrations, as indicated, and incubated at 37°C for 12 h with the corresponding antibiotics. A set of plates was kept at 37°C for an additional 48 h, and an identical set was induced by transfer to 25°C for 48 h before photography. Strains were streaked onto SPI-CAA medium plates containing 80 µg/ml X-Gal (Sigma) and different l-xylose (Sigma) concentrations, as indicated, and incubated at 37°C for 12 h. Cold shock was induced by transfer to 25°C for 48 to 72 h before were photographs taken.
Protein structure accession numbers.
The protein structures presented here have been deposited in the PDB under accession codes 4LDZ (full-length DesR in complex with BeF3−/Mg2+, activated state), 4LE0 (RECa, DesR REC domain in complex with BeF3−/Mg2+, activated state), 4LE1 (RECb, DesR apo REC domain, inactive state), and 4LE2 (RECc, DesR apo REC domain, active-state-like configuration).
SUPPLEMENTAL MATERIAL
Schematics summarizing the DesK/DesR signaling pathway. The transcriptional regulation of the Δ5-desaturase (des) is controlled by this TCS in B. subtilis. des expression is triggered by cold shock. Download
{kind=link}
Structure of the REC domain of DesR and comparison of the active and inactive states. (A) Close-up view of the phosphorylation site. The electron density map (sigmaA-weighted 2mFobs − DFcalc, contoured at 1.5σ) reveals the octahedral coordination of the Mg2+ and interactions of the BeF3− group with conserved amino acids. (B) The (βα)6 topology of the REC domain is formed by snug packing of the β6 strand on the α4β5α5 face. The (βα)6 folding of the monomer is shown with secondary-structure elements labeled. Note the central β-sheet (yellow) with surrounding α-helices (red). (C) Cartoon representation of the activated REC domain (RECa) illustrating the invariant core (pale yellow) and the shifted regions (magenta, active; green, inactive) compared to the inactive state (RECb). Two neighboring protomers according to the RECa crystal packing are shown as a transparent cartoon depicting the relative locations of the α1α5 and α4β6α6 protein-protein interfaces. Statistically significant coordinate shifts are observed in the loops that delimit the phosphorylation site (β1α1, β2α2, β3α3, and β4α4) and also in helices α2 and α6 and strand β2. (D) Structural alignment of the active and inactive conformations of DesR (active, magenta; inactive, green) and VraR (active, blue; inactive, orange). (E) Packing of RECa crystals revealing two major interaction surfaces among neighboring molecules in the upper part. These interactions are shared by all of the crystal forms of DesR and REC. Indicated by arrows toward the lower panels, the α1α5 (left) and α4β6α6 (right) interaction surfaces are shown in greater detail. Chain A is orange, and chain B is blue. The solvent-accessible surface is rendered transparent to show the extension of each buried surface, and Asp54 is represented by spheres to show the location of the phosphorylation site in each oligomer form. Download
{kind=link}
DCA analysis of α1α5 dimerization in the NarL/LuxR family. Shown is a view of the α1α5 dimerization surface of three members of this family (DesR, VraR [PDB code 4IF4], and spr1814 [PDB code 4E7P]). The two residue pairs that revealed high MI and DI values (residues 10 and 81, and 10 and 103, according to DesR residue numbering) are drawn, respectively, in the upper and lower panels, in stick representations showing their proximity in space when considering the α1α5 dimer. Download
{kind=link}
DesR in vitro autophosphorylation and autodephosphorylation assays. (A) Different proteins (as labeled on each lane) were incubated at 100 µM concentration with 50 mM acetyl phosphate for 1 h, and separated by Phos-tag SDS-PAGE. (B) One representative Phos-tag SDS-PAGE recorded with DesRwt used to generate Fig. 4A by densitometric analysis. The other reported measurements gave similar results. (C) The degree of autodephosphorylation (ξ-P) is expressed as a function of the logarithm of the protein concentration (molar). ξ-P was measured after 1 h of incubation at 25°C. Values from separate experiments are shown with the best-fit sigmoid curves overlaid. PhoB was evaluated independently by Phos-tag SDS-PAGE and tryptophan fluorescence (data not shown). Download
{kind=link}
Structural model of DesK-DesR. (A) The soluble and catalytically competent construct of the cognate HK DesK (DesKC) in the phosphotransferase state (PDB code 3GIF; Glu188 was substituted here for His) is blue, and the REC domain of DesR is green. The superimposed transparent rendering corresponds to the solvent-accessible surface. (B) SDS-PAGE stained with Coomassie blue, showing the disulfide cross-linking assay used as experimental validation of the DesK-DesR model. Leu197-Leu13 (DesK-DesR, respectively) and Leu200-Ser106 were the two pairs of residues expected to establish covalent cross-linking according to the model. The other two pairs (Ser196-Phe82 and Leu239-Gly59) were selected as negative controls, choosing residues near the interface, but expected to be far away between them. (C) Phosphotransferase activity in vitro (using 32P-labeled DesKCwt) showing indistinguishable phosphorylation of DesRwt and DesR_Arg121Ala. (D) DesKC was used to assay the formation of stable complexes with DesRwt and DesR_Arg121Ala by SEC (using the DesKC_His188Val construct locked in DesKC’s phosphatase state). Download
{kind=link}
Phosphorylation-independent activation of DesR depends on the protein concentration. Nonphosphorylatable DesR requires higher protein concentrations to activate its signaling effector response. Note the lower signal-triggered Pdes activation of the DesR_Asp54Asn mutant variant than that of DesRwt at lower expression levels (top panel, 0.5% glucose repressing the Pxyl promoter). The bottom panel shows a negative control with full repression of the xylose-inducible promoter (2% glucose). The nomenclature and plate disposition of the strains are identical to those in Fig. 5B. Download
{kind=link}
Schematic model of DesK-DesR allosteric routes. (A) The canonical allosteric route identified by comparing the active and inactive conformations of the REC domain of DesR couples the phosphorylation site to the α4β6α6 tetramerization interface. For clarity, only a subset of the residues involved is displayed as sticks. The inactive conformation is blue, and the active state is orange. The neighboring molecules, as seen in the crystals, are highlighted in green, illustrating the protein-protein interfaces. (B) The novel allosteric pathway coupling the orthosteric phosphorylation site to the α1α5 dimerization surface. The coloring scheme is the same as that in panel A. (C) Further elaboration of Fig. 6D in the situation where enough DesR is produced, allowing the detection of the signal- and DesK-dependent activation of the pathway, and unveiling the allosteric activation role of DesK by binding through the α1α5 surface of DesR. Download
{kind=link}
SAXS-derived size parameters.
X-ray diffraction data collection and structure refinement statistics.
Detailed protocols. Download
ACKNOWLEDGMENTS
We thank the staffs at synchrotron beamlines SWING (Soleil, Gif-sur-Yvette, France) and BM29 (ESRF, Grenoble, France) for assistance with SEC-coupled SAXS and SAXS, respectively. We are grateful to Pedro Alzari (Institut Pasteur) and Eduardo Groisman (Yale University) for helpful discussions. At the Institut Pasteur de Montevideo, special thanks to Matias Machado for useful input about scripting for Rosetta and Analia Lima for her assistance with gel densitometry analyses.
This work was supported by grants from the Agencia Nacional de Investigación e Innovación (ANII), Uruguay; the Agencia de Promocion Cientifica y Tecnologica (FONCYT), Argentina; and the Agence Nationale de la Recherche (ANR), France.
Footnotes
Citation Trajtenberg F, Albanesi D, Ruétalo N, Botti H, Mechaly AE, Nieves M, Aguilar PS, Cybulski L, Larrieux N, de Mendoza D, Buschiazzo A. 2014. Allosteric activation of bacterial response regulators: the role of the cognate histidine kinase beyond phosphorylation. mBio 5(6):e02105-14. doi:10.1128/mBio.02105-14.
REFERENCES
- 1. Gao R, Stock AM. 2009. Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 63:133–154. 10.1146/annurev.micro.091208.073214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bourret RB. 2010. Receiver domain structure and function in response regulator proteins. Curr. Opin. Microbiol. 13:142–149. 10.1016/j.mib.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gao R, Stock AM. 2010. Molecular strategies for phosphorylation-mediated regulation of response regulator activity. Curr. Opin. Microbiol. 13:160–167. 10.1016/j.mib.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Volkman BF, Lipson D, Wemmer DE, Kern D. 2001. Two-state allosteric behavior in a single-domain signaling protein. Science 291:2429–2433. 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 5. Baikalov I, Schröder I, Kaczor-Grzeskowiak M, Grzeskowiak K, Gunsalus RP, Dickerson RE. 1996. Structure of the Escherichia coli response regulator NarL. Biochemistry 35:11053–11061. 10.1021/bi960919o. [DOI] [PubMed] [Google Scholar]
- 6. Eldridge AM, Kang HS, Johnson E, Gunsalus R, Dahlquist FW. 2002. Effect of phosphorylation on the interdomain interaction of the response regulator, NarL. Biochemistry 41:15173–15180. 10.1021/bi026254+ PubMed. [DOI] [PubMed] [Google Scholar]
- 7. Barbieri CM, Mack TR, Robinson VL, Miller MT, Stock AM. 2010. Regulation of response regulator autophosphorylation through interdomain contacts. J. Biol. Chem. 285:32325–32335. 10.1074/jbc.M110.157164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mack TR, Gao R, Stock AM. 2009. Probing the roles of the two different dimers mediated by the receiver domain of the response regulator PhoB. J. Mol. Biol. 389:349–364. 10.1016/j.jmb.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barbieri CM, Wu T, Stock AM. 2013. Comprehensive analysis of OmpR phosphorylation, dimerization, and DNA binding supports a canonical model for activation. J. Mol. Biol. 425:1612–1626. 10.1016/j.jmb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leonard PG, Golemi-Kotra D, Stock AM. 2013. Phosphorylation-dependent conformational changes and domain rearrangements in Staphylococcus aureus VraR activation. Proc. Natl. Acad. Sci. U. S. A. 110:8525–8530. 10.1073/pnas.1302819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park AK, Moon JH, Oh JS, Lee KS, Chi YM. 2013. Crystal structure of the response regulator spr1814 from Streptococcus pneumoniae reveals unique interdomain contacts among NarL family proteins. Biochem. Biophys. Res. Commun. 434:65–69. 10.1016/j.bbrc.2013.03.065. [DOI] [PubMed] [Google Scholar]
- 12. Casino P, Rubio V, Marina A. 2009. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 139:325–336. 10.1016/j.cell.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 13. Albanesi D, Martín M, Trajtenberg F, Mansilla MC, Haouz A, Alzari PM, de Mendoza D, Buschiazzo A. 2009. Structural plasticity and catalysis regulation of a thermosensor histidine kinase. Proc. Natl. Acad. Sci. U. S. A. 106:16185–16190. 10.1073/pnas.0906699106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Trajtenberg F, Graña M, Ruétalo N, Botti H, Buschiazzo A. 2010. Structural and enzymatic insights into the ATP binding and autophosphorylation mechanism of a sensor histidine kinase. J. Biol. Chem. 285:24892–24903. 10.1074/jbc.M110.147843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Casino P, Miguel-Romero L, Marina A. 2014. Visualizing autophosphorylation in histidine kinases. Nat. Commun. 5:3258. 10.1038/ncomms4258. [DOI] [PubMed] [Google Scholar]
- 16. Mechaly AE, Sassoon N, Betton JM, Alzari PM. 2014. Segmental helical motions and dynamical asymmetry modulate histidine kinase autophosphorylation. PLoS Biol. 12:e1001776. 10.1371/journal.pbio.1001776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bell CH, Porter SL, Strawson A, Stuart DI, Armitage JP. 2010. Using structural information to change the phosphotransfer specificity of a two-component chemotaxis signalling complex. PLoS Biol. 8:e1000306. 10.1371/journal.pbio.1000306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mo G, Zhou H, Kawamura T, Dahlquist FW. 2012. Solution structure of a complex of the histidine autokinase CheA with its substrate CheY. Biochemistry 51:3786–3798. 10.1021/bi300147m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parashar V, Mirouze N, Dubnau DA, Neiditch MB. 2011. Structural basis of response regulator dephosphorylation by Rap phosphatases. PLoS Biol. 9:e1000589. 10.1371/journal.pbio.1000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zapf J, Sen U, Madhusudan, Hoch JA, Varughese KI. 2000. A transient interaction between two phosphorelay proteins trapped in a crystal lattice reveals the mechanism of molecular recognition and phosphotransfer in signal transduction. Structure 8:851–862. 10.1016/S0969-2126(00)00174-X. [DOI] [PubMed] [Google Scholar]
- 21. Zhao R, Collins EJ, Bourret RB, Silversmith RE. 2002. Structure and catalytic mechanism of the E. coli chemotaxis phosphatase CheZ. Nat. Struct. Biol. 9:570–575. 10.1038/nsb816. [DOI] [PubMed] [Google Scholar]
- 22. Aguilar PS, Hernandez-Arriaga AM, Cybulski LE, Erazo AC, de Mendoza D. 2001. Molecular basis of thermosensing: a two-component signal transduction thermometer in Bacillus subtilis. EMBO J. 20:1681–1691. 10.1093/emboj/20.7.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cybulski LE, del Solar G, Craig PO, Espinosa M, de Mendoza D. 2004. Bacillus subtilis DesR functions as a phosphorylation-activated switch to control membrane lipid fluidity. J. Biol. Chem. 279:39340–39347. 10.1074/jbc.M405150200. [DOI] [PubMed] [Google Scholar]
- 24. Najle SR, Inda ME, de Mendoza D, Cybulski LE. 2009. Oligomerization of Bacillus subtilis DesR is required for fine tuning regulation of membrane fluidity. Biochim. Biophys. Acta 1790:1238–1243. 10.1016/j.bbagen.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 25. Lee SY, Cho HS, Pelton JG, Yan D, Henderson RK, King DS, Huang L, Kustu S, Berry EA, Wemmer DE. 2001. Crystal structure of an activated response regulator bound to its target. Nat. Struct. Biol. 8:52–56. 10.1038/83053. [DOI] [PubMed] [Google Scholar]
- 26. Baikalov I, Schröder I, Kaczor-Grzeskowiak M, Cascio D, Gunsalus RP, Dickerson RE. 1998. NarL dimerization? Suggestive evidence from a new crystal form. Biochemistry 37:3665–3676. 10.1021/bi972365a. [DOI] [PubMed] [Google Scholar]
- 27. Birck C, Mourey L, Gouet P, Fabry B, Schumacher J, Rousseau P, Kahn D, Samama JP. 1999. Conformational changes induced by phosphorylation of the FixJ receiver domain. Structure 7:1505–1515. 10.1016/S0969-2126(00)88341-0. [DOI] [PubMed] [Google Scholar]
- 28. Kern D, Volkman BF, Luginbühl P, Nohaile MJ, Kustu S, Wemmer DE. 1999. Structure of a transiently phosphorylated switch in bacterial signal transduction. Nature 402:894–898. [DOI] [PubMed] [Google Scholar]
- 29. Lee SY, Cho HS, Pelton JG, Yan D, Berry EA, Wemmer DE. 2001. Crystal structure of activated CheY. Comparison with other activated receiver domains. J. Biol. Chem. 276:16425–16431. 10.1074/jbc.M101002200. [DOI] [PubMed] [Google Scholar]
- 30. Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, Zecchina R, Onuchic JN, Hwa T, Weigt M. 2011. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. U. S. A. 108:E1293–E1301. 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weigt M, White RA, Szurmant H, Hoch JA, Hwa T. 2009. Identification of direct residue contacts in protein-protein interaction by message passing. Proc. Natl. Acad. Sci. U. S. A. 106:67–72. 10.1073/pnas.0805923106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Creager-Allen RL, Silversmith RE, Bourret RB. 2013. A link between dimerization and autophosphorylation of the response regulator PhoB. J. Biol. Chem. 288:21755–21769. 10.1074/jbc.M113.471763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. 2006. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5:749–757. 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- 34. Pazy Y, Motaleb MA, Guarnieri MT, Charon NW, Zhao R, Silversmith RE. 2010. Identical phosphatase mechanisms achieved through distinct modes of binding phosphoprotein substrate. Proc. Natl. Acad. Sci. U. S. A. 107:1924–1929. 10.1073/pnas.0911185107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Skerker JM, Perchuk BS, Siryaporn A, Lubin EA, Ashenberg O, Goulian M, Laub MT. 2008. Rewiring the specificity of two-component signal transduction systems. Cell 133:1043–1054. 10.1016/j.cell.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Feher VA, Zapf JW, Hoch JA, Whiteley JM, McIntosh LP, Rance M, Skelton NJ, Dahlquist FW, Cavanagh J. 1997. High-resolution NMR structure and backbone dynamics of the Bacillus subtilis response regulator, Spo0F: implications for phosphorylation and molecular recognition. Biochemistry 36:10015–10025. 10.1021/bi970816l. [DOI] [PubMed] [Google Scholar]
- 37. Simonovic M, Volz K. 2001. A distinct meta-active conformation in the 1.1-A resolution structure of wild-type ApoCheY. J. Biol. Chem. 276:28637–28640. 10.1074/jbc.C100295200. [DOI] [PubMed] [Google Scholar]
- 38. Volkman BF, Nohaile MJ, Amy NK, Kustu S, Wemmer DE. 1995. Three-dimensional solution structure of the N-terminal receiver domain of NTRC. Biochemistry 34:1413–1424. 10.1021/bi00004a036. [DOI] [PubMed] [Google Scholar]
- 39. Podgornaia AI, Casino P, Marina A, Laub MT. 2013. Structural basis of a rationally rewired protein-protein interface critical to bacterial signaling. Structure 21:1636–1647. 10.1016/j.str.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Capra EJ, Perchuk BS, Skerker JM, Laub MT. 2012. Adaptive mutations that prevent crosstalk enable the expansion of paralogous signaling protein families. Cell 150:222–232. 10.1016/j.cell.2012.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gardino AK, Villali J, Kivenson A, Lei M, Liu CF, Steindel P, Eisenmesser EZ, Labeikovsky W, Wolf-Watz M, Clarkson MW, Kern D. 2009. Transient non-native hydrogen bonds promote activation of a signaling protein. Cell 139:1109–1118. 10.1016/j.cell.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bachhawat P, Stock AM. 2007. Crystal structures of the receiver domain of the response regulator PhoP from Escherichia coli in the absence and presence of the phosphoryl analog beryllofluoride. J. Bacteriol. 189:5987–5995. 10.1128/JB.00049-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Iynedjian PB. 2009. Molecular physiology of mammalian glucokinase. Cell. Mol. Life Sci. 66:27–42. 10.1007/s00018-008-8322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qian H. 2012. Cooperativity in cellular biochemical processes: noise-enhanced sensitivity, fluctuating enzyme, bistability with nonlinear feedback, and other mechanisms for sigmoidal responses. Annu. Rev. Biophys. 41:179–204. 10.1146/annurev-biophys-050511-102240. [DOI] [PubMed] [Google Scholar]
- 45. Reyrat JM, David M, Batut J, Boistard P. 1994. FixL of Rhizobium meliloti enhances the transcriptional activity of a mutant FixJD54N protein by phosphorylation of an alternate residue. J. Bacteriol. 176:1969–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wolfe AJ. 2005. The acetate switch. Microbiol. Mol. Biol. Rev. 69:12–50. 10.1128/MMBR.69.1.12-50.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Klein AH, Shulla A, Reimann SA, Keating DH, Wolfe AJ. 2007. The intracellular concentration of acetyl phosphate in Escherichia coli is sufficient for direct phosphorylation of two-component response regulators. J. Bacteriol. 189:5574–5581. 10.1128/JB.00564-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fraser JS, Merlie JP, Jr, Echols N, Weisfield SR, Mignot T, Wemmer DE, Zusman DR, Alber T. 2007. An atypical receiver domain controls the dynamic polar localization of the Myxococcus xanthus social motility protein FrzS. Mol. Microbiol. 65:319–332. 10.1111/j.1365-2958.2007.05785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hong E, Lee HM, Ko H, Kim DU, Jeon BY, Jung J, Shin J, Lee SA, Kim Y, Jeon YH, Cheong C, Cho HS, Lee W. 2007. Structure of an atypical orphan response regulator protein supports a new phosphorylation-independent regulatory mechanism. J. Biol. Chem. 282:20667–20675. 10.1074/jbc.M609104200. [DOI] [PubMed] [Google Scholar]
- 50. Ruiz D, Salinas P, Lopez-Redondo ML, Cayuela ML, Marina A, Contreras A. 2008. Phosphorylation-independent activation of the atypical response regulator NblR. Microbiology 154:3002–3015. 10.1099/mic.0.2008/020677-0. [DOI] [PubMed] [Google Scholar]
- 51. Schär J, Sickmann A, Beier D. 2005. Phosphorylation-independent activity of atypical response regulators of Helicobacter pylori. J. Bacteriol. 187:3100–3109. 10.1128/JB.187.9.3100-3109.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang L, Tian X, Wang J, Yang H, Fan K, Xu G, Yang K, Tan H. 2009. Autoregulation of antibiotic biosynthesis by binding of the end product to an atypical response regulator. Proc. Natl. Acad. Sci. U. S. A. 106:8617–8622. 10.1073/pnas.0900592106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paul R, Jaeger T, Abel S, Wiederkehr I, Folcher M, Biondi EG, Laub MT, Jenal U. 2008. Allosteric regulation of histidine kinases by their cognate response regulator determines cell fate. Cell 133:452–461. 10.1016/j.cell.2008.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, S SO, Vonrhein C, Womack TO. 2009. BUSTER, 2.8. 0 ed. Global Phasing Ltd., Cambridge, United Kingdom. [Google Scholar]
- 57. Barbieri CM, Stock AM. 2008. Universally applicable methods for monitoring response regulator aspartate phosphorylation both in vitro and in vivo using phos-tag-based reagents. Anal. Biochem. 376:73–82. 10.1016/j.ab.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematics summarizing the DesK/DesR signaling pathway. The transcriptional regulation of the Δ5-desaturase (des) is controlled by this TCS in B. subtilis. des expression is triggered by cold shock. Download
Structure of the REC domain of DesR and comparison of the active and inactive states. (A) Close-up view of the phosphorylation site. The electron density map (sigmaA-weighted 2mFobs − DFcalc, contoured at 1.5σ) reveals the octahedral coordination of the Mg2+ and interactions of the BeF3− group with conserved amino acids. (B) The (βα)6 topology of the REC domain is formed by snug packing of the β6 strand on the α4β5α5 face. The (βα)6 folding of the monomer is shown with secondary-structure elements labeled. Note the central β-sheet (yellow) with surrounding α-helices (red). (C) Cartoon representation of the activated REC domain (RECa) illustrating the invariant core (pale yellow) and the shifted regions (magenta, active; green, inactive) compared to the inactive state (RECb). Two neighboring protomers according to the RECa crystal packing are shown as a transparent cartoon depicting the relative locations of the α1α5 and α4β6α6 protein-protein interfaces. Statistically significant coordinate shifts are observed in the loops that delimit the phosphorylation site (β1α1, β2α2, β3α3, and β4α4) and also in helices α2 and α6 and strand β2. (D) Structural alignment of the active and inactive conformations of DesR (active, magenta; inactive, green) and VraR (active, blue; inactive, orange). (E) Packing of RECa crystals revealing two major interaction surfaces among neighboring molecules in the upper part. These interactions are shared by all of the crystal forms of DesR and REC. Indicated by arrows toward the lower panels, the α1α5 (left) and α4β6α6 (right) interaction surfaces are shown in greater detail. Chain A is orange, and chain B is blue. The solvent-accessible surface is rendered transparent to show the extension of each buried surface, and Asp54 is represented by spheres to show the location of the phosphorylation site in each oligomer form. Download
DCA analysis of α1α5 dimerization in the NarL/LuxR family. Shown is a view of the α1α5 dimerization surface of three members of this family (DesR, VraR [PDB code 4IF4], and spr1814 [PDB code 4E7P]). The two residue pairs that revealed high MI and DI values (residues 10 and 81, and 10 and 103, according to DesR residue numbering) are drawn, respectively, in the upper and lower panels, in stick representations showing their proximity in space when considering the α1α5 dimer. Download
DesR in vitro autophosphorylation and autodephosphorylation assays. (A) Different proteins (as labeled on each lane) were incubated at 100 µM concentration with 50 mM acetyl phosphate for 1 h, and separated by Phos-tag SDS-PAGE. (B) One representative Phos-tag SDS-PAGE recorded with DesRwt used to generate Fig. 4A by densitometric analysis. The other reported measurements gave similar results. (C) The degree of autodephosphorylation (ξ-P) is expressed as a function of the logarithm of the protein concentration (molar). ξ-P was measured after 1 h of incubation at 25°C. Values from separate experiments are shown with the best-fit sigmoid curves overlaid. PhoB was evaluated independently by Phos-tag SDS-PAGE and tryptophan fluorescence (data not shown). Download
Structural model of DesK-DesR. (A) The soluble and catalytically competent construct of the cognate HK DesK (DesKC) in the phosphotransferase state (PDB code 3GIF; Glu188 was substituted here for His) is blue, and the REC domain of DesR is green. The superimposed transparent rendering corresponds to the solvent-accessible surface. (B) SDS-PAGE stained with Coomassie blue, showing the disulfide cross-linking assay used as experimental validation of the DesK-DesR model. Leu197-Leu13 (DesK-DesR, respectively) and Leu200-Ser106 were the two pairs of residues expected to establish covalent cross-linking according to the model. The other two pairs (Ser196-Phe82 and Leu239-Gly59) were selected as negative controls, choosing residues near the interface, but expected to be far away between them. (C) Phosphotransferase activity in vitro (using 32P-labeled DesKCwt) showing indistinguishable phosphorylation of DesRwt and DesR_Arg121Ala. (D) DesKC was used to assay the formation of stable complexes with DesRwt and DesR_Arg121Ala by SEC (using the DesKC_His188Val construct locked in DesKC’s phosphatase state). Download
Phosphorylation-independent activation of DesR depends on the protein concentration. Nonphosphorylatable DesR requires higher protein concentrations to activate its signaling effector response. Note the lower signal-triggered Pdes activation of the DesR_Asp54Asn mutant variant than that of DesRwt at lower expression levels (top panel, 0.5% glucose repressing the Pxyl promoter). The bottom panel shows a negative control with full repression of the xylose-inducible promoter (2% glucose). The nomenclature and plate disposition of the strains are identical to those in Fig. 5B. Download
Schematic model of DesK-DesR allosteric routes. (A) The canonical allosteric route identified by comparing the active and inactive conformations of the REC domain of DesR couples the phosphorylation site to the α4β6α6 tetramerization interface. For clarity, only a subset of the residues involved is displayed as sticks. The inactive conformation is blue, and the active state is orange. The neighboring molecules, as seen in the crystals, are highlighted in green, illustrating the protein-protein interfaces. (B) The novel allosteric pathway coupling the orthosteric phosphorylation site to the α1α5 dimerization surface. The coloring scheme is the same as that in panel A. (C) Further elaboration of Fig. 6D in the situation where enough DesR is produced, allowing the detection of the signal- and DesK-dependent activation of the pathway, and unveiling the allosteric activation role of DesK by binding through the α1α5 surface of DesR. Download
SAXS-derived size parameters.
X-ray diffraction data collection and structure refinement statistics.
Detailed protocols. Download