

ABSTRACT
Together with ATP, the C-terminal region of the essential streptococcal FtsA protein acts as an intramolecular switch to promote its polymerization and attachment to the membrane. During septation, FtsA is known to anchor the constricting FtsZ ring and, subsequently, the divisome to the membrane. Truncation of the C terminus of the streptococcal FtsA (FtsAΔCt) facilitates a more rapid ATP-dependent polymerization in solution than is seen with the full-length protein (FtsA+). The FtsAΔCt polymers are more organized and compact than those formed in solution by FtsA+, resembling the shape of the membrane-associated FtsA+ polymers. We find that ATP, besides being needed for polymerization, is required for the attachment of FtsA+ to lipid monolayers and to vesicle membranes. We propose a model in which the binding of ATP activates a switch favoring the polymerization of FtsA and at the same time driving the amphipathic helix at its C terminus to become attached to the membrane. Conversely, when FtsA is in the cytoplasm, the C terminus is not engaged in the attachment to the membrane, and it obstructs polymerization. ATP-dependent polymerization of FtsA inside membrane vesicles causes vesicle shrinkage, suggesting that, besides providing a membrane attachment for FtsZ, the FtsA C terminus may also introduce local alterations in the membrane to facilitate septation.
IMPORTANCE
FtsA is a protein needed in many bacteria to construct a septum that divides one fully grown cell, producing two daughters. We show that the region located at the C-terminal end of the Streptococcus pneumoniae FtsA protein works as a switch triggered by ATP, a molecule that stores energy. This region contains an amphipathic helix that obstructs the assembly of FtsA into polymers in the cytoplasm. In the presence of ATP, the obstruction is removed by switching the position of the helix. The switch directs the helix to the membrane and simultaneously facilitates the polymerization of the protein. The accumulation of FtsA molecules at the membrane causes distortions, an effect produced also by proteins such as MinD, MreB, and SepF that also contain amphipathic helixes as membrane attachment devices. In the case of FtsA, these distortions may also facilitate the initial events that lead to the division of bacteria.
INTRODUCTION
At the earliest stage of bacterial division, the FtsA protein connects FtsZ, the principal component of the division machinery, to the cell membrane and forms a structure called the proto-ring at the division site (reviewed in reference 1). Correct assembly of proto-ring components enables progression of bacterial division by recruiting in successive steps the remainder of the essential divisome proteins involved in septum synthesis and cell pole formation. FtsA is a multiple connector protein with various regions able to interact independently with several of the early- and late-assembling division proteins, including itself.
FtsA belongs structurally to the actin/Hsp70/hexokinase superfamily (2–4). In the presence of ATP, FtsA monomers polymerize through a head-to-tail assembly mechanism that involves its 1C domain and the S12 and S13 β-strands (5–8). These regions are essential for cell division in Escherichia coli (9), and the residues involved in the self-interaction have been mapped in the crystal model from the Thermotoga maritima FtsA dimer (8). The 1C domain is also the main factor in recruitment of late-assembling division proteins (9, 10); dissociation of the FtsA oligomer to release this domain is thus proposed to help to recruit the late-assembling division proteins (11). In addition to the head-to-tail interactions between monomers, the polymers also establish lateral interactions to form bundles. The H1 alpha helix in domain 1A is involved in these interactions, and it is needed for the integrity of the Z ring (11). Consistent with their negligible ATP hydrolytic activity and the apparently insignificant differences between the ATP-bound and unbound crystal forms (4), FtsA polymers remain stable in vitro (6–8).
At its C terminus, FtsA has a flexible, disordered hydrophilic linker that connects the core of the protein to a membrane-targeting sequence (MTS). In E. coli FtsA, the MTS is an essential, conserved amphipathic helix which participates in anchoring the protein to the cell membrane (12) and in regulating FtsA self-interaction (13).
In this work, we have investigated the role of the streptococcal FtsA C-terminal region in its ATP-dependent polymerization and explored the relationship between the two activities previously assigned to this region, i.e., FtsA self-interaction and attachment to the cytoplasmic membrane. We used FtsA from Streptococcus pneumoniae produced heterologously in E. coli (5), since this protein is more stable than the E. coli FtsA and shows ATP-dependent polymerization activity (6). Our results suggest that, upon ATP binding, the position of the MTS acts as a switch to modify simultaneously the polymerization activity and the interaction of the protein with the membrane.
RESULTS
The FtsA C-terminal region is involved in FtsA polymerization.
The C terminus of E. coli FtsA, containing the MTS, has been described to have a role in the interaction between FtsA molecules (13). To study the role of the FtsA C-terminal region in the protein self-interaction, we took advantage of the ability of S. pneumoniae FtsA to polymerize in the presence of ATP (5, 6). Purification of FtsA from S. pneumoniae yields two distinct bands of different sizes in analysis by SDS-PAGE (6). The mobility of the larger band corresponds to the molecular weight of the full gene product, whereas the smaller one fits with a deletion comprising the last 52 residues at the C terminus of the protein as confirmed by amino acid sequencing (6). We purified a homogeneous variant of the streptococcal FtsA lacking the 52 C-terminal amino acids (FtsAΔCt) fused to a His tag after expressing a suitably deleted genetic construction (see Materials and Methods).
Measurement of polymerization by light-scattering assay showed that at a 2.5 or 5 µM concentration, the FtsAΔCt protein assembled into large structures in the presence of 2 mM ATP (Fig. 1A). The C-terminal-truncated protein assembled more rapidly than the purified full-length protein (containing a six-His tag at its N-terminal end; simplified here as FtsA+) and presented a 20% reduced lag time to initiation of polymerization. Consistently with the absence of measurable ATPase activity (6), the polymers are stable and their amount depends only on the protein concentration. Nonetheless, our results suggest that all the FtsA+ and FtsAΔCt monomers are finally incorporated into stable polymers in solution, as polymerization reaches the maximal scatter intensity level in reaction mixtures containing FtsA+ or FtsAΔCt or an equimolar mixture of FtsA+ and FtsAΔCt (Fig. 1A). From these results, we infer that the MTS causes a temporal delay of the FtsA polymerization in solution but does not affect the final amount of polymer.
FIG 1 .
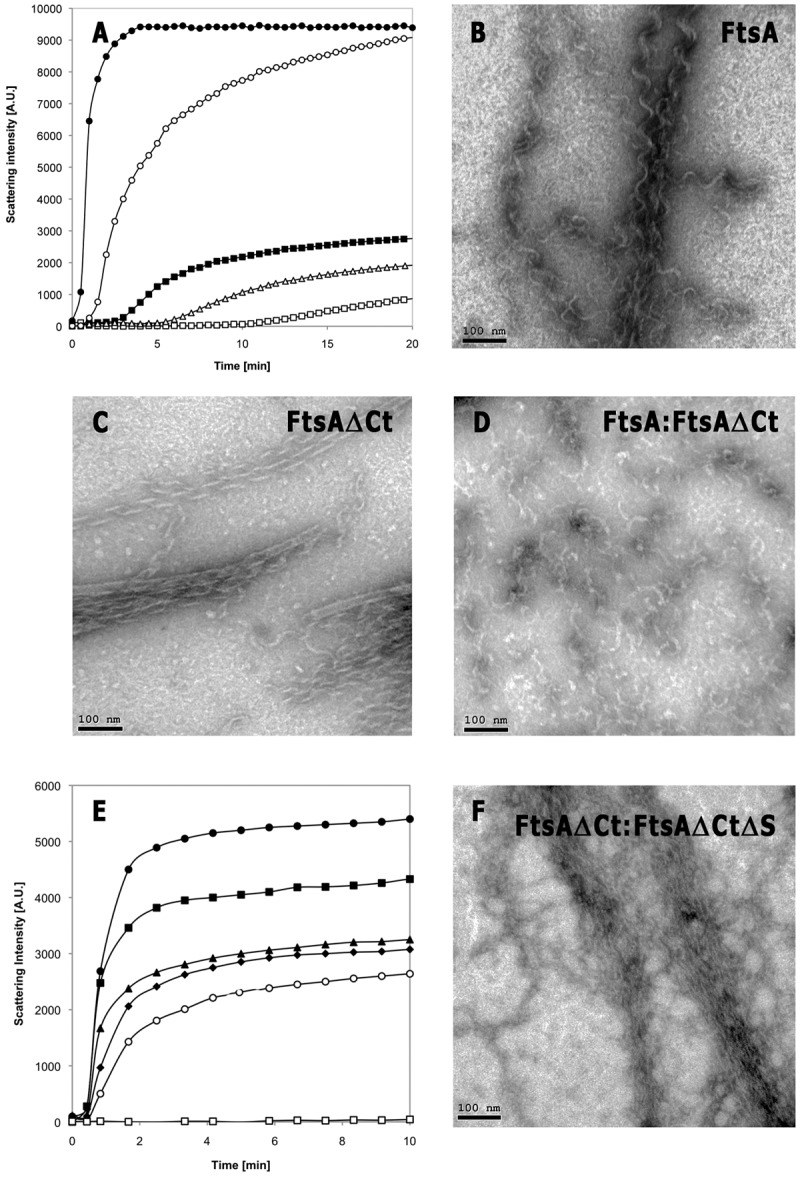
In vitro polymerization of FtsA proteins. (A) Light-scattering measurements of 5 µM FtsA+ (○), 5 µM FtsAΔCt (●), 2.5 µM FtsA+ (□), 2.5 µM FtsAΔCt (■), and a mixture of 2.5 µM FtsA+ with 2.5 µM FtsAΔCt (△) after the addition of 2 mM ATP. A.U., arbitrary units. (B to D) Electron microscopy of polymers formed by 5 µM FtsA+ (B), 5 µM FtsAΔCt (C), and a mixture of 2.5 µM FtsA+ and 2.5 µM FtsAΔCt (D). Bars, 100 nm. (E) Light-scattering measurements of 5 µM FtsAΔCt (●), 5 µM FtsAΔCtΔS (□), or 5 µM FtsAΔCt in the presence of 1.25, 2.5, 3.75, and 5 µM FtsAΔCtΔS (■, ▲, ♦, and ○, respectively). (F) Electron microscopy images of polymers formed by a mixture of 5 µM FtsAΔCt and 2.5 µM FtsAΔCtΔS. Bar, 100 nm.
Images of the polymers formed by FtsAΔCt showed bundles of helical protofilaments morphologically similar to those formed by FtsA+ (Fig. 1B and C). This suggests that the two proteins may establish similar head-to-tail interactions and lateral contacts when polymerizing. FtsAΔCt polymers nevertheless had a more regular and compact organization than FtsA+ polymers, frequently forming tubular structures (18 ± 2 nm in diameter), suggesting a role of the C terminus as a steric hindrance for polymerization.
Since FtsA+ and FtsAΔCt assembled in bundles of helical protofilaments when assayed separately, we reasoned that similar bundles would result from the simultaneous polymerization of the two proteins in a mixture at equimolar concentrations. The lag time and the molar mass values obtained for a polymerization mixture containing FtsA+ and FtsAΔCt were within the ranges individually shown by each species (Fig. 1A). However, transmission electron microscopy (EM) images nonetheless showed that the polymers formed by the mixture of the two proteins were a disordered network of short filaments and protein clumps (Fig. 1D). In contrast to the results seen with the FtsA+ or FtsAΔCt polymers, the filaments formed when the two proteins were assayed together barely bundled, suggesting that the lateral contacts among protofilaments were impaired. Given the tendency to self-associate shown by the hydrophobic faces within amphipathic helixes, we suggest that MTS-MTS interactions may partially counteract the negative effect of this region, as the FtsA+ polymers were more ordered than those formed by the mixture.
These results indicate that the C-terminal region might interfere with FtsA polymerization in solution, probably by disturbing the initial nucleation event proposed for the FtsA+ polymerization (6) and/or the subsequent addition of the monomers to the protofilaments.
The C-terminal region of FtsA affects polymerization but not the head-to-tail interactions.
The interference of the C-terminal ends in polymerization could be exerted through the distortion of the surfaces involved in the head-to-tail interaction or, alternatively, by masking other, unknown interaction sites in the FtsA+ molecule. To discriminate between the two possibilities, we measured the ability of FtsAΔCt to polymerize in the presence of truncated FtsA proteins from S. pneumoniae that lacked S12-to-S13 β strands or the 1C domain (at the top or bottom of the crystallographic model, respectively). These proteins inhibit FtsA+ head-to-tail bidirectional polymerization by capping the opposite ends and cannot polymerize (5). We designed FtsAΔCt constructs in which these regions were additionally deleted. Deletion of the 1C domain from FtsAΔCt resulted in a protein (FtsAΔCtΔ1C) with a high tendency to aggregate, making analysis in solution unreliable (not shown).
We have measured whether the double-deletion FtsAΔCtΔS protein, in which both the C-terminal end and the S12-to-S13 region were lacking, affected the polymerization and morphology of the FtsAΔCt polymers. FtsAΔCtΔS itself did not polymerize, and, when added to FtsAΔCt, it reduced both the maximum level and rate of polymerization (Fig. 1E). In addition, the polymers obtained from a mixture containing both proteins (Fig. 1F) were shorter, less abundant, less compact, and less defined than polymers formed by FtsAΔCt alone (Fig. 1C). This effect is similar to that exerted by FtsAΔS on FtsA+ polymerization where both proteins contain their C termini (5). These results indicate that, in addition to the head-to-tail interaction, the C terminus has a distinct role in FtsA polymerization.
Association of the C-terminal region of Streptococcus pneumoniae FtsA (SnFtsA) with the E. coli cytoplasmic membrane prevents the initiation of constrictions and distorts the membrane.
The FtsA proteins from E. coli, T. maritima, and Bacillus subtilis, which do not form polymers in solution, can polymerize inside the cytoplasm of E. coli cells (8, 12). Given that the streptococcal FtsA+ and FtsAΔCt proteins polymerize in vitro, we tested whether they could also form polymers inside E. coli. For this purpose, these two streptococcal proteins were overproduced separately in E. coli as fusions to the fluorescent mCherry protein. Data in Fig. 2A and B show that mCherry FtsA+ appeared in close proximity to the membrane and inhibited septation. This effect may have resulted from an excess of C-terminal amphipathic FtsA helices attached to the membrane. Indeed, our results show a high density of labeled proteins in the vicinity of the cytoplasmic membrane that appears distorted (Fig. 2A to C).
FIG 2 .
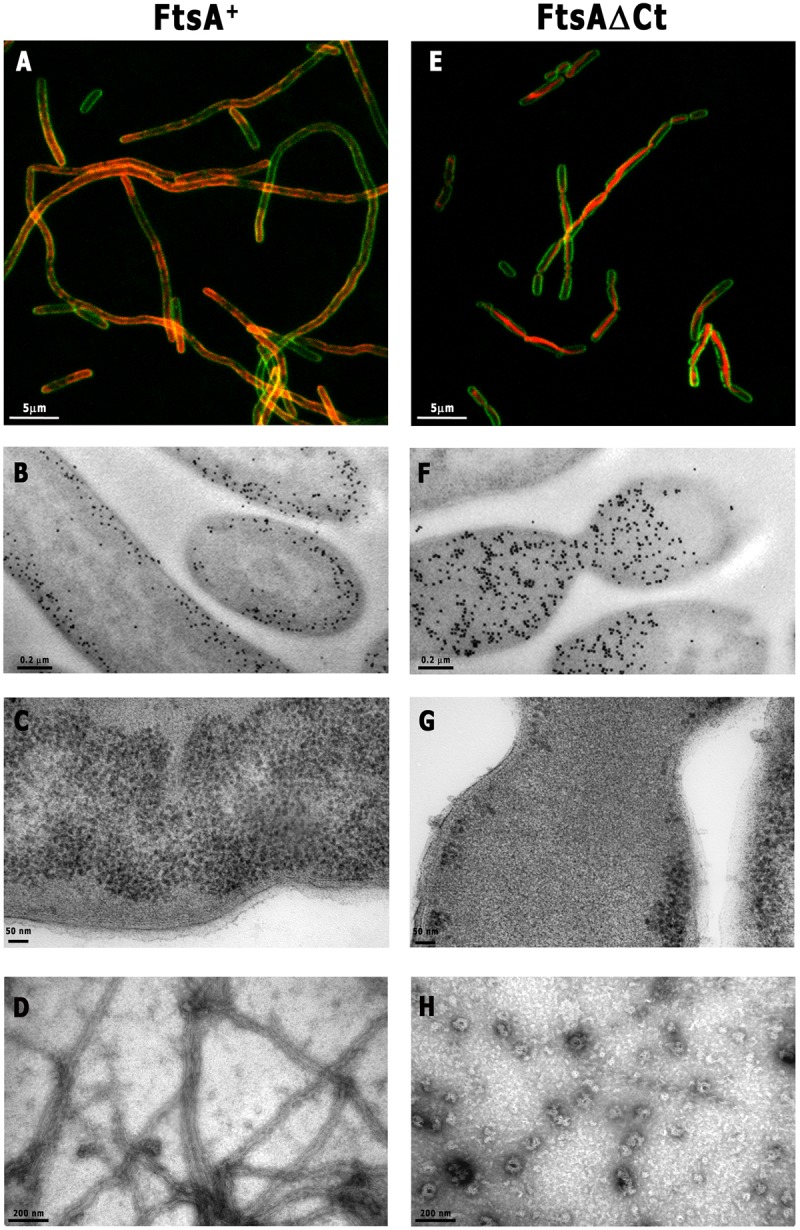
Localization of streptococcal FtsA+ and FtsAΔCt overproduced in E. coli and on lipid monolayers. FtsA (A to C) and FtsAΔCt (E to G) were fused with mCherry (red) at the N terminus and overproduced in E. coli (3 h). Membranes were stained with FM1-43FX (green). Samples were analyzed by confocal microscopy (A and E) and electron microscopy (B, C, F, and G). Panels B and F show the gold immunolocalization of FtsA and FtsAΔCt, respectively. For monolayer assay, a lipid-coated electron microscopy grid was incubated with FtsA+ or FtsAΔCt, followed by the addition of 2 mM ATP (D or H, respectively). Samples were washed extensively with polymerization buffer and fixed with 2% uranyl acetate (see Materials and Methods). (H) Note that polymers formed by FtsAΔCt occurred very occasionally on the lipid monolayer. Scale bars are as indicated.
As expected, the mCherry FtsAΔCt fusion, in which the membrane-targeting helix was absent, failed to localize near the cytoplasmic membrane. Its overproduction did not prevent the initiation of constrictions and caused no apparent distortions in the membrane (Fig. 2E to G). These constrictions failed to complete division, perhaps because the bulk of the mCherry FtsAΔCt observed at the constriction may block them.
To examine if the accumulated FtsA+ proteins could be arranged as polymers on the cytoplasmic membrane, we further analyzed the in vitro polymerization of FtsA+ on a lipid monolayer. Purified FtsA+ formed long, tubular polymers densely covering a lipid monolayer 40 min after the addition of ATP. They appear as nested tubes similar to those found for FtsAΔCt when polymerized in solution (Fig. 1B and 2D). In contrast to FtsA+, only occasional FtsAΔCt polymers were found on the lipid monolayer in the presence of ATP, as exemplified by the one shown in Fig. 2H. As FtsAΔCt lacks the membrane-targeting sequence, its binding to the lipid monolayer after washing was likely unspecific.
Fusions of mCherry to the amino terminus of the nonpolymerizing FtsAΔS or FtsAΔ1C protein resulted in proteins that, as expected, did not polymerize in solution. These proteins nevertheless contained an intact C terminus (5), and they consequently localized in the proximity of the cytoplasmic membrane similarly to the FtsA+ protein (see Fig. S1 in the supplemental material). However, these nonpolymerizing proteins failed to distort the membrane as FtsA+ does. On the other hand, a similar mCherry fusion to the double-deletion FtsAΔCtΔS protein was distributed all over the cytoplasm, as would be expected given the absence of a membrane-attaching amphipathic helix and its inability to polymerize. When overproduced, the products of this FtsAΔCtΔ1C fusion localized as inclusion bodies, as would be expected from the tendency of the double mutant to form aggregates (see Fig. S1). Together, these results suggest that the FtsA C terminus, besides mediating the association of the protein with the membrane, might produce an alteration in its structure when attached in an orderly manner in large amounts, as would occur in the FtsA polymers.
The attachment of FtsA+ polymers to the inner surface of GUVs induces shrinkage.
FtsA+ associates with cell membranes in vivo, and, with the addition of ATP, it forms lipid-anchored polymers in vitro. We have analyzed the localization of FtsA+ inside giant unilamellar vesicles (GUVs) and the effects of its polymerization. The use of GUVs provides a membrane-confined environment, intermediate between the cell and the lipid monolayer, in which the effects of polymerization can be followed over time.
Purified FtsA+ and its variants were labeled with Alexa 488 and incorporated separately into vesicles. In the absence of ATP, all the FtsA variants were distributed homogenously in the vesicle lumen (Fig. 3). After addition of ATP, the bulk of FtsA+ localized at the vesicle membrane and only a residual amount could be observed in the lumen. This protein attached more efficiently to lipids than the nonpolymerizing FtsAΔS variant (Fig. 3), which conserves the C terminus and the ATP-binding pocket (5), suggesting that a weak membrane affinity of FtsA monomers should be compensated for by the protein multimerization. The FtsAΔS could be detected on the membrane only in the presence of ATP (Fig. 3), suggesting that the ATP may shift the protein from a state in which it hinders membrane attachment to a membrane-bound state even as a monomer. On the other hand, the majority of the FtsAΔCt protein, lacking an MTS, failed to localize at the vesicle membrane upon addition of ATP (Fig. 3) even if it had a higher tendency to polymerize (Fig. 1). Therefore, we propose that the shift promoted by ATP involves the movement of the C-terminal region and, consequently, of the MTS. However, we then tested the ability of a double-deletion FtsAΔCtΔS protein to interact with vesicles and found that it was not totally abolished (not shown). This result suggests that FtsA may contain secondary membrane-interacting regions additional to the C terminus that could be exposed upon the ATP addition.
FIG 3 .

Spatial distribution of Alexa 488-labeled FtsA+, FtsAΔCt, and FtsAΔS inside GUVs. The indicated proteins (green) were encapsulated in GUVs (see Materials and Methods). Representative equatorial cross-sectional images for each protein taken shortly after addition of 2 mM ATP (3 to 7 min) and after prolonged (>10-min) incubation are shown. The percentages of vesicles showing similar localization patterns are indicated in the upper right corner of the respective images. The total number of vesicles observed in each case is indicated in brackets. Bar, 10 µm.
These results suggest that, to efficiently associate to the membrane, FtsA needs both the presence of the MTS and the formation of polymers triggered by ATP. The fact that polymerization and membrane attachment are synergistically enhanced in the presence of ATP would also explain why E. coli and T. maritima FtsA polymers cannot be detected in vitro unless they are membrane associated (7, 8).
After 10 min of incubation with ATP, over 70% of the vesicles containing FtsA+ shrank, while those containing any of the truncated FtsA variants showed no apparent deformation. After further incubation periods (over 45 min) or at higher protein concentrations (over 13 µM), the truncated proteins were also able to promote deformation in 70% of the observed vesicles (not shown). We postulate that vesicle deformation may result from the progression of polymerization, leading to the formation of large assemblies near the membrane and thus to the accumulation of membrane-anchored amphipathic helices. In the case of FtsAΔCt, we observed brilliant protein clusters in the lumen of vesicles, in accordance with its ability to polymerize. Consistently with our in vivo results with the mCherry-tagged FtsAΔCt, these structures were not attached to the vesicle membrane, and therefore the vesicles showed no deformation after prolonged observation times (Fig. 2E to G and 3).
When FtsA+ was added to the buffer outside the vesicles, it formed polymers that could be found decorating their outer surface in part upon the addition of ATP (see Fig. S2 in the supplemental material), which confirmed the affinity of this protein for lipid membranes. We conclude that the recruitment of FtsA+ to the membrane is predominantly mediated by the C terminus and that it requires the polymerization induced by ATP.
DISCUSSION
We propose that an intramolecular switch, driven by ATP binding and involving the displacement of its C terminus, operates in the essential FtsA cell division protein from S. pneumoniae to allow its polymerization and its attachment to the membrane (Fig. 4).
FIG 4 .

The role of the C terminus in FtsA ATP-dependent polymerization and attachment to the membrane. (A) FtsA monomers (blue) are not anchored to the membrane in the absence of nucleotide, and free C-terminal ends temporarily disturb the polymerization process in the cytoplasm (bottom). The FtsA molecule domains (1A, 1C, 2A, and 2B), the MTS (blue rectangles), and the flexible linker (blue lines) that connects the MTS to the rest of the protein are represented. ATP (green triangles) triggers the FtsA polymerization and membrane attachment mediated by a shift of the position of the MTS (top). Local perturbations produced by the abundance of MTS in the membrane (gray) are represented in red. (B) The lack of C termini in FtsAΔCt facilitates the ATP-dependent polymerization of the protein in solution, but it does not interact specifically with the membrane.
Our proposal is based on data showing the different behavior of the FtsAΔCt protein, in which the C terminus is absent, relative to that of FtsA+. Differences can be observed in three properties: the ability to polymerize in solution, the shape of the polymers, and the localization in the cytoplasmic membrane or in lipid surfaces. In the presence of ATP, FtsAΔCt polymerizes in solution faster than FtsA+. Whereas polymers of FtsA+ in solution appear loose, the polymers formed both by FtsAΔCt in solution and by FtsA+ when associated to lipid surfaces are more compact. Finally, FtsAΔCt cannot localize, even in the presence of ATP, either in the cytoplasmic membrane of E. coli cells when expressed heterologously or in the membranes of GUVs when artificially included in them. Moreover, FtsAΔCt forms organized structures in the cytoplasm or in the lumen of the vesicles, whereas under similar conditions the FtsA+ structures are readily associated to the membrane both in cells and vesicles.
A similar increased polymerization when associated to the membrane has been reported for other proteins such as MinD, MreB, and SepF in which their attachment to the membrane is mediated by an amphipathic helix (MTS) (14–16). In the case of MinD, ATP binding causes the displacement of the MTS, allowing both the interaction between monomers and association to the membrane (17).
A possible mechanism to facilitate FtsA polymerization in the presence of ATP may be based on the different position occupied by the 1C domain in the nucleotide-free dimer structure relative to the one adopted in the ATP-bound one. Upon overlaying the structure of the ATP-free monomer on the top monomer of the ATP-bound form, it became evident that the 1C domain in the top monomer did not contact the 1A domain of the bottom one (see Fig. S3 in the supplemental material). It is possible, then, that the binding of ATP works by bringing into closer contact the 1C and 1A domains of monomers and therefore promoting polymerization.
Our results favoring the operation of an intramolecular switch acting on both polymerization and membrane attachment do not offer specific clues on which of the two processes occurs earlier during the assembly of the proto-ring. Pichoff and Lutkenhaus (12, 18) have suggested that the interaction between FtsA and FtsZ is likely preceded by both ATP binding and attachment of FtsA to the membrane, an interpretation compatible with our results. In E. coli, FtsA initially works together with ZipA to assemble FtsZ into the proto-ring; in addition, a role for the MTS FtsA in the recruitment of the late E. coli divisome proteins (FtsN) has been suggested (19, 20). It is possible as well that the local enrichment in amphipathic helixes created at the membrane regions in which FtsA becomes associated may weaken the membrane at the site of insertion (21). This effect has been observed to happen in the attachment of SepF (14), MreB (16), and MinD (17, 22) as well. Our results favor this possibility, as the attachment of FtsA+ to the membrane of vesicles upon ATP addition results in their shrinking. Besides promoting polymerization and association to the membrane, the ATP-driven switch would initiate the differentiation of a potential septation site in which first a proto-ring and later a divisome are assembled.
Note that, while the E. coli proto-ring contains an additional protein, ZipA, to provide attachment to the membrane, the proto-ring of S. pneumoniae does not contain ZipA. In this case, the role of ZipA may be replaced by those of other membrane proteins recruiting FtsZ, such as EzrA or SepF (reviewed in reference 23), or by the higher number of FtsA molecules, such as the nearly 2,200 in S. pneumoniae (6), instead of the approximately 740 present in E. coli (24). Consistent with the role of both proteins being membrane anchors for FtsZ, a large amount of ZipA produces invaginations in the cytoplasmic membrane of E. coli (25), a behavior reminiscent of the deformations caused in vesicles by the presence of the ATP-bound streptococcal FtsA protein.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
E. coli strain DH5α (26) was used as a host for gene cloning and E. coli strain C41 (27) for overproduction and purification of streptococcal proteins. E. coli strains were routinely grown at 37°C in Luria-Bertani (LB) agar or broth supplemented with ampicillin (100 µg ml−1) when necessary.
Plasmid constructions and DNA manipulation.
Standard protocols for molecular cloning, transformation, and DNA analysis were performed as previously described (28). Oligonucleotides were purchased from Sigma-Aldrich and restriction enzymes and DNA polymerase from Roche Diagnostics.
Primers MK1 (5′ CGGGATCCATGGCTAGAGAAGGCTT 3′) and MK3 (5′ CGGAATTC TTAAGCTGTTTTTTGCAG 3′) were used to amplify the pRSETA_ftsA sequence (6), except the sequence encoding the last 52 amino acids of the FtsA C-terminal region from Gln406 to Glu457. The plasmid named pMKV1 bears the desired deletion to produce truncated FtsA lacking the C terminus, termed FtsAΔCt. A similar procedure was used to amplify the ftsAΔCt genes additionally lacking the sequence encoding domain 1C (ftsAΔCtΔ1C) or the S12 and S13 β sheets (ftsAΔCtΔS) from the pARV48 and pARV49 plasmids encoding FtsAΔ1C and FtsAΔS (5) to yield the plasmids pMKV12 and pMKV2, respectively.
Plasmids from pMKV29 to pMKV34 encoding FtsA and its deletion mutants (FtsAΔ1C, FtsAΔS, FtsAΔCt, FtsAΔCtΔS, and FtsAΔCtΔ1C, respectively), fused with mCherry at the N-terminal end, were obtained by inserting the mCherry gene into the pRSETA_ftsA, pARV48, pARV49, pMKV1, pMKV2 and pMKV12 plasmids, respectively, to span the His tag-encoding sequence derived from the pRSETA vector (Invitrogen). All mutations were confirmed by DNA sequencing.
Purification of streptococcal His-tagged proteins.
The FtsA protein and its variants were overproduced in the C41 strain and purified from the inclusion bodies by affinity chromatography in denaturing conditions, as previously described (5). Samples were renaturalized by sequential dialysis in polymerization buffer (20 mM Tris-HCl [pH 7.5], 50 mM NaCl, 5 mM MgCl2, 10% glycerol) supplemented with decreasing concentrations (2.5, 1.25, 0.5, and 0 M) of guanidine chloride and stored at −80°C.
FtsA polymerization assays in solution and visualization of polymer morphology.
For standard polymerization assays, thawed purified proteins were centrifuged (13,000 × g, 15 min) to remove protein aggregates and then diluted to a 5 µM final concentration in polymerization buffer. After purified protein was tempered in the reaction mix (room temperature, 5 min), 2 mM ATP (buffered at pH 8) was added to initiate polymerization. The polymerization process was followed by 90° light-scattering measurements using a Hitachi F-2500 fluorescence spectrophotometer. Excitation and emission wavelengths were set to 320 nm with 5-nm slit widths (29).
The morphology of polymers containing FtsA or its variants was observed using transmission electron microscopy 20 min after ATP addition. Samples were applied to a 400-mesh collodion-coated glow-discharged copper grid, negatively stained with 2% uranyl acetate, inspected with a Jeol JEM1011 transmission electron microscope, and photographed with an 11-megapixel charge-coupled Gatan Erlanghsen ES1000W camera. The software used for image capture was Gatan Digital Micrograph 1.8.0, and the software used for processing was Adobe Photoshop CS5.
FtsA overproduction in vivo.
Overnight cultures of E. coli C41 cells transformed with plasmids from pMKV29 to pMKV34 encoding mCherry-tagged FtsA or its deletion proteins (FtsAΔ1C, FtsAΔS, FtsAΔCt, FtsAΔCtΔS, and FtsAΔCtΔ1C, respectively) were diluted 1:100 and grown at 37°C in ampicillin-supplemented LB medium. Cultures were maintained by sequential dilution under exponentially balanced growth conditions at below an optical density at 600 nm (OD600) of 0.2 for at least 3 h; they were induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) (3 h), and samples were collected for visualization.
For confocal microscopy imaging, cells were subjected to membrane staining with FM1-43FX (Molecular Probes) and fixed according to methods described in reference 30. Images were collected with a Leica TCS SP5 AOBS inverted confocal microscope with a 63× (numerical aperture [NA], 1.4 to 0.6; oil HCX PL APO lambda-blue) immersion objective (Leica, Mannheim, Germany). Argon ion lasers were used for laser lines at 488 nm, which excited FM1-43FX; a diode-pumped solid-state (DPSS) laser was used to excite mCherry at 561 nm.
To examine the organization of these C41 cells, cryo-transmission electron microscopy was performed as previously described (25). For FtsA and FtsAΔCt immunolabeling, ultrathin sections of the samples were first blocked with TBST (30 mM Tris-HCl [pH 8], 150 mM NaCl, 1% bovine serum albumin [BSA], 0.02% Tween 20) followed by a 1-h incubation with MVA1 anti-FtsA antibody (6). Samples were incubated with EM goat F(ab')2 anti-rabbit immunogold conjugate (BBInternational, Cardiff, United Kingdom). All images were processed with Adobe Photoshop CS5.
Lipid monolayer assay.
Monolayer assays were performed as previously described (8) with some modifications. Based on the previously estimated streptococcal polar lipid membrane composition (31, 32), 0.2 µg phosphatidylglycerol and cardiolipin (Avanti) at a 1:1 ratio were dissolved in chloroform and floated on polymerization buffer in Teflon wells. After the chloroform evaporated, electron microscopy grids were placed onto the lipid monolayer and FtsA+ or FtsAΔCt proteins were injected underneath, into the polymerization buffer, to achieve a final concentration of 2 µM and incubated (40 min), and 2 mM ATP was added to trigger polymerization. The reaction was allowed to proceed for 20 min, followed by standard grid staining with 2% uranyl acetate, as described for the polymer visualization.
Polymerization of proteins encapsulated in giant unilamellar vesicles.
FtsA+ and its truncation mutants (0.75 mg/ml) were labeled with Alexa 488 (Molecular Probes) (1:5 M ratio)–20 mM HEPES-HCl [pH 8]–50 mM NaCl–5 mM MgCl2 at 4°C for 30 min. The reaction was terminated by addition of Tris-HCl (pH 8) to achieve a final concentration of 100 mM. Excess free probe was removed by gel filtration (HiTrap Desalting; GE Healthcare), and the protein was divided into aliquots and stored at −80°C. Labeling efficiency was 0.1 to 0.2 mol Alexa 488 per mol of protein. Polymerization of labeled proteins was confirmed by 90° light-scattering assays and electron microscopy.
Giant unilamellar vesicles (GUVs) containing FtsA+ or its deletion forms (7 µM) were prepared by the droplet-transfer method (25), except that the lipid mixture contained 40% egg phosphatidylcholine (ePC), 10% dimyristoylphosphatidylcholine (DMPC), 25% phosphatidylglycerol (PG), and 25% cardiolipin (CL). This composition allows vesicle formation and maintains the 1 PG/1 CL lipid ratio (31, 32). DMPC increases membrane fluidity, allowing diffusion of external ATP into GUVs (33). Membranes were stained with 8% rhodamine (Avanti). A high concentration of inert macromolecules (50 mg/ml Ficoll) was added to mimic the conditions of the cytosol (34). ATP (2 mM) was added to the vesicles to trigger FtsA polymerization. Images were collected with a Leica TCS SP5 AOBS inverted confocal microscope with a 63× immersion objective (NA, 1.4 to 0.6; oil HCX PL APO lambda-blue). Argon ion lasers were used for laser lines at 488 nm to excite Alexa 488. Images were processed with Adobe Photoshop CS5.
SUPPLEMENTAL MATERIAL
In vivo localization of FtsAΔS and FtsAΔ1C and their C-terminal-truncated forms, FtsAΔCtΔS and FtsAΔCtΔ1C. Confocal microscopy images show mCherry-FtsA variants (red) overproduced in E. coli (3 h). Membranes were stained with FM1-43FX (green). Bar, 5 µm. Download
Spatial distribution of Alexa 488-labeled FtsA+ proteins when not encapsulated inside GUVs. Empty GUVs were prepared without proteins by the droplet-transfer method. Lipids were stained with rhodamine (red), and Alexa 488-labeled FtsA+ (green) was added externally. Merged images of representative equatorial cross sections were taken using a confocal microscope before and 2 min after the addition of ATP (2 mM). Bar, 10 µm. Download
Overlay analysis of the crystal structures of FtsA reveals that a slight difference in the position of the 1C domain caused by ATP binding can promote the establishment of the FtsA-FtsA interaction. The apo-bound (orange; PDB code 1E4F) and ATP-bound (blue; PDB code 1E4G) surface plots of T. maritima FtsA (4) have been matched to the upper monomer of the FtsA dimer (gray; PDB code 4A2A) (8). ATP of 1E4G is shown in red. Cartoons were created in UCSF Chimera version 1.8.1 (35). A detailed view of the 1C domain and its contacts is shown at the bottom. Download
ACKNOWLEDGMENTS
This work was funded by grant BIO2008-04478-C03 from Spanish Ministerio de Educación y Ciencia and by contract HEALTH-F3-2009-223431 (DIVINOCELL) from the European Commission (to both M.V. and G.R.). M.K. was supported by La Caixa Foundation International Fellowship Program (La Caixa/CNB).
We thank Orietta Massidda for critical reading of the manuscript. We also thank the Confocal Microscopy Facility of CNB-CSIC and CIB-CSIC and the Electron Microscopy Facility of CNB-CSIC for their technical support.
Footnotes
Citation Krupka M, Cabré EJ, Jiménez M, Rivas G, Rico AI, Vicente M. 2014. Role of the FtsA C terminus as a switch for polymerization and membrane association. mBio 5(6):e02221-14. doi:10.1128/mBio.02221-14.
REFERENCES
- 1. Rico AI, Krupka M, Vicente M. 2013. In the beginning, Escherichia coli assembled the proto-ring: an initial phase of division. J. Biol. Chem. 288:20830–20836. 10.1074/jbc.R113.479519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bork P, Sander C, Valencia A. 1992. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc. Natl. Acad. Sci. U. S. A. 89:7290–7294. 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sánchez M, Valencia A, Ferrándiz MJ, Sander C, Vicente M. 1994. Correlation between the structure and biochemical activities of FtsA, an essential cell division protein of the actin family. EMBO J. 13:4919–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van den Ent F, Löwe J. 2000. Crystal structure of the cell division protein FtsA from Thermotoga maritima. EMBO J. 19:5300–5307. 10.1093/emboj/19.20.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krupka M, Rivas G, Rico AI, Vicente M. 2012. Key role of two terminal domains in the bidirectional polymerization of FtsA protein. J. Biol. Chem. 287:7756–7765. 10.1074/jbc.M111.311563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lara B, Rico AI, Petruzzelli S, Santona A, Dumas J, Biton J, Vicente M, Mingorance J, Massidda O. 2005. Cell division in cocci: localization and properties of the Streptococcus pneumoniae FtsA protein. Mol. Microbiol. 55:699–711. 10.1111/j.1365-2958.2004.04432.x. [DOI] [PubMed] [Google Scholar]
- 7. Martos A, Monterroso B, Zorrilla S, Reija B, Alfonso C, Mingorance J, Rivas G, Jiménez M. 2012. Isolation, characterization and lipid-binding properties of the recalcitrant FtsA division protein from Escherichia coli. PLoS One 7:e39829. 10.1371/journal.pone.0039829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Szwedziak P, Wang Q, Freund SM, Löwe J. 2012. FtsA forms actin-like protofilaments. EMBO J. 31:2249–2260. 10.1038/emboj.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rico AI, García-Ovalle M, Mingorance J, Vicente M. 2004. Role of two essential domains of Escherichia coli FtsA in localization and progression of the division ring. Mol. Microbiol. 53:1359–1371. 10.1111/j.1365-2958.2004.04245.x. [DOI] [PubMed] [Google Scholar]
- 10. Corbin BD, Geissler B, Sadasivam M, Margolin W. 2004. Z-ring-independent interaction between a subdomain of FtsA and late septation proteins as revealed by a polar recruitment assay. J. Bacteriol. 186:7736–7744. 10.1128/JB.186.22.7736-7744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pichoff S, Shen B, Sullivan B, Lutkenhaus J. 2012. FtsA mutants impaired for self-interaction bypass ZipA suggesting a model in which FtsA’s self-interaction competes with its ability to recruit downstream division proteins. Mol. Microbiol. 83:151–167. 10.1111/j.1365-2958.2011.07923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pichoff S, Lutkenhaus J. 2005. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol. Microbiol. 55:1722–1734. 10.1111/j.1365-2958.2005.04522.x. [DOI] [PubMed] [Google Scholar]
- 13. Yim L, Vandenbussche G, Mingorance J, Rueda S, Casanova M, Ruysschaert JM, Vicente M. 2000. Role of the carboxy terminus of Escherichia coli FtsA in self-interaction and cell division. J. Bacteriol. 182:6366–6373. 10.1128/JB.182.22.6366-6373.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duman R, Ishikawa S, Celik I, Strahl H, Ogasawara N, Troc P, Löwe J, Hamoen LW. 2013. Structural and genetic analyses reveal the protein SepF as a new membrane anchor for the Z ring. Proc. Natl. Acad. Sci. U. S. A. 110:E4601–E4610. 10.1073/pnas.1313978110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu Z, Lutkenhaus J. 2003. A conserved sequence at the C-terminus of MinD is required for binding to the membrane and targeting MinC to the septum. Mol. Microbiol. 47:345–355. 10.1046/j.1365-2958.2003.03321.x. [DOI] [PubMed] [Google Scholar]
- 16. Salje J, van den Ent F, de Boer P, Löwe J. 2011. Direct membrane binding by bacterial actin MreB. Mol. Cell 43:478–487. 10.1016/j.molcel.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu Z, Gogol EP, Lutkenhaus J. 2002. Dynamic assembly of MinD on phospholipid vesicles regulated by ATP and MinE. Proc. Natl. Acad. Sci. U. S. A. 99:6761–6766. 10.1073/pnas.102059099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pichoff S, Lutkenhaus J. 2007. Identification of a region of FtsA required for interaction with FtsZ. Mol. Microbiol. 64:1129–1138. 10.1111/j.1365-2958.2007.05735.x. [DOI] [PubMed] [Google Scholar]
- 19. Shiomi D, Margolin W. 2007. Dimerization or oligomerization of the actin-like FtsA protein enhances the integrity of the cytokinetic Z ring. Mol. Microbiol. 66:1396–1415. 10.1111/j.1365-2958.2007.05998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shiomi D, Margolin W. 2008. Compensation for the loss of the conserved membrane targeting sequence of FtsA provides new insights into its function. Mol. Microbiol. 67:558–569. 10.1111/j.1365-2958.2007.06085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zimmerberg J, Kozlov MM. 2006. How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 7:9–19. 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
- 22. Suefuji K, Valluzzi R, RayChaudhuri D. 2002. Dynamic assembly of MinD into filament bundles modulated by ATP, phospholipids, and MinE. Proc. Natl. Acad. Sci. U. S. A. 99:16776–16781. 10.1073/pnas.262671699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Massidda O, Nováková L, Vollmer W. 2013. From models to pathogens: how much have we learned about Streptococcus pneumoniae cell division? Environ. Microbiol. 15:3133–3157. 10.1111/1462-2920.12189. [DOI] [PubMed] [Google Scholar]
- 24. Rueda S, Vicente M, Mingorance J. 2003. Concentration and assembly of the division ring proteins FtsZ, FtsA, and ZipA during the Escherichia coli cell cycle. J. Bacteriol. 185:3344–3351. 10.1128/JB.185.11.3344-3351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cabré EJ, Sánchez-Gorostiaga A, Carrara P, Ropero N, Casanova M, Palacios P, Stano P, Jiménez M, Rivas G, Vicente M. 2013. Bacterial division proteins FtsZ and ZipA induce vesicle shrinkage and cell membrane invagination. J. Biol. Chem. 288:26625–26634. 10.1074/jbc.M113.491688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580. 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 27. Miroux B, Walker JE. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260:289–298. 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 28. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, New York, NY. [Google Scholar]
- 29. Mingorance J, Rueda S, Gómez-Puertas P, Valencia A, Vicente M. 2001. Escherichia coli FtsZ polymers contain mostly GTP and have a high nucleotide turnover. Mol. Microbiol. 41:83–91. 10.1046/j.1365-2958.2001.02498.x. [DOI] [PubMed] [Google Scholar]
- 30. Uehara T, Dinh T, Bernhardt TG. 2009. LytM-domain factors are required for daughter cell separation and rapid ampicillin-induced lysis in Escherichia coli. J. Bacteriol. 191:5094–5107. 10.1128/JB.00505-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Epand RF, Savage PB, Epand RM. 2007. Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (ceragenins). Biochim. Biophys. Acta 1768:2500–2509. 10.1016/j.bbamem.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 32. Trombe MC, Lanéelle MA, Lanéelle G. 1979. Lipid composition of aminopterin-resistant and sensitive strains of Streptococcus pneumoniae. Effect of aminopterin inhibition. Biochim. Biophys. Acta 574:290–300. 10.1016/0005-2760(79)90010-9. [DOI] [PubMed] [Google Scholar]
- 33. Bessonov K, Harauz G. 2010. Molecular dynamics investigation of myelin basic protein stability on lipid membranes. Studies by Undergraduate Researchers at Guelph 4:79–86. [Google Scholar]
- 34. Jiménez M, Martos A, Vicente M, Rivas G. 2011. Reconstitution and organization of Escherichia coli proto-ring elements (FtsZ and FtsA) inside giant unilamellar vesicles obtained from bacterial inner membranes. J. Biol. Chem. 286:11236–11241. 10.1074/jbc.M110.194365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In vivo localization of FtsAΔS and FtsAΔ1C and their C-terminal-truncated forms, FtsAΔCtΔS and FtsAΔCtΔ1C. Confocal microscopy images show mCherry-FtsA variants (red) overproduced in E. coli (3 h). Membranes were stained with FM1-43FX (green). Bar, 5 µm. Download
Spatial distribution of Alexa 488-labeled FtsA+ proteins when not encapsulated inside GUVs. Empty GUVs were prepared without proteins by the droplet-transfer method. Lipids were stained with rhodamine (red), and Alexa 488-labeled FtsA+ (green) was added externally. Merged images of representative equatorial cross sections were taken using a confocal microscope before and 2 min after the addition of ATP (2 mM). Bar, 10 µm. Download
Overlay analysis of the crystal structures of FtsA reveals that a slight difference in the position of the 1C domain caused by ATP binding can promote the establishment of the FtsA-FtsA interaction. The apo-bound (orange; PDB code 1E4F) and ATP-bound (blue; PDB code 1E4G) surface plots of T. maritima FtsA (4) have been matched to the upper monomer of the FtsA dimer (gray; PDB code 4A2A) (8). ATP of 1E4G is shown in red. Cartoons were created in UCSF Chimera version 1.8.1 (35). A detailed view of the 1C domain and its contacts is shown at the bottom. Download