

Abstract
Reductive methylation of lysyl side-chain amines has been a successful tool in the advancement of high resolution structural biology. The utility of this method has continuously gained ground as a protein chemical modification; first, as a tool to aid protein crystallization and later, as a probe in protein nuclear magnetic resonance (NMR) spectroscopy. As an isotope-labeling strategy for NMR studies, reductive methylation has contributed to the study of protein-protein interactions and global conformational changes. While more detailed structural studies using this labeling strategy are possible, the hurdle of assigning the NMR peaks to the corresponding reductively methylated amine hinders its use. In this review, we discuss and compare strategies used to assign the NMR peaks of reductively methylated protein-amines.
Keywords: reductive methylation, lysozyme, chemical modification, dimethyllysine, isotopic labeling
Introduction
Reductive methylation is an effective tool for studying protein structure and function and has been used since 1968 to study the biological functions of specific lysines [1]. This sparse labelling method allows for lysines to be chemically modified under slightly basic conditions in the presence of a reducing agent and formaldehyde. The protein N-terminal α-amine and the lysyl side-chain ε-amines undergo methylation to produce monomethylamines, which have a slightly higher acid dissociation constant (pKa) compared to the unmodified primary amines [2]. In the presence of excess formaldehyde, the monomethylamine readily reacts to produce a dimethylamine (Figure 1). Proteins can be isotopically labeled in this manner with the use of labeled formaldehyde (13C and 14C) and/or reducing agent (2H and 3H) [2-5]. Since lysines are polar and the side-chains are typically solvent-exposed, it is easy to homogeneously modify most lysines by reductive methylation. Some exceptions are when the lysine is involved in a salt bridge or buried in tertiary or quaternary structures. The reductive methylation reaction conditions are mild, so proteins can be modified without denaturation. Reductive methylation of proteins is usually performed at pH 7.5-10.0; but, in an unpublished study, we found that reductive methylation is achievable as low as pH 4.0, extending the applicability of the method. Another attractive feature is the retained charge of the amines with reductive methylation, closely maintaining the isoelectric point (pI) of the protein. Due to the small size of the methyl groups, the modification rarely introduces global changes to the protein structure or interferes with its activity, the most important feature of reductive methylation.
Figure 1.
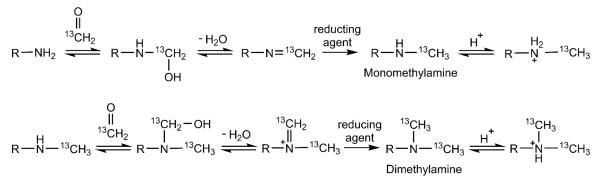
The reductive methylation reaction with 13C-formaldehyde. The top row shows the reaction to form the monomethylamine, and the bottom row shows the continuing reaction to form the dimethylamine.
Reductive methylation of proteins has been used to advance many techniques in the study of protein structure and function. Reductive methylation can facilitate protein crystallization by altering the dynamics of the lysyl side-chains through steric and hydrophobic interactions. Aiding protein crystallization is one of the early applications of the reductive methylation reaction and is still a major application today [6-7]. Rypniewski et al. demonstrated the first high-resolution three-dimensional (3D) structure of a reductively methylated protein [8]. In this work, the structures of lysozyme and reductively methylated lysozyme were compared, revealing highly similar structures with a root mean squared deviation of 0.40 Å in the backbone atoms (Figure 2). As a result, reductive methylation has had a major impact on studying the biophysical properties of proteins, especially structure.
Figure 2.

Comparison of the α-carbon positions for native and reductively methylated hen egg white lysozyme. Reprinted with permission from [8]. Copyright 1993 American Chemical Society.
Other techniques, such as bioactivity assays, electrophoresis, and circular dichroism (CD), have also benefited from reductive methylation [4,9-12]. Electrophoresis is a useful technique for assessing biophysical properties, such as the molecular weight and the pI, of medium to large proteins. Reductive methylation has been used in electrophoresis to radiolabel protein samples with 14C and 3H, improving the detection sensitivity over the traditional Coomassie Blue staining method [13]. CD is a technique that can probe the secondary structure of a protein by monitoring the differential absorption of circularly polarized light at ultraviolet wavelengths. Each unique combination of amino acids and secondary structure gives a distinct absorption fingerprint. Reductively methylated proteins have been shown to conserve the structural fingerprint of their native counters, which allows CD to be used to analyze the effects of methylation on protein binding [14-15]. Mass spectrometry (MS) is a powerful technique for analyzing isotopic composition, mass, and chemical structures of proteins and peptides. Reductive methylation has provided many advantages in MS proteomics studies for peptide enrichment [16], separations [17], quantitation [18], detection [19], and signal enhancement [20]. In addition, reductive methylation has been used to identify and associate protein biomarkers with breast cancer using a proteomics MS approach [21]. MS is also being used to determine tertiary and quaternary protein structure [22-23], and the reductive methylation reaction could be used in this way to identify structurally protected lysines or the N-terminus.
Reductive methylation has also positively impacted the study of proteins by NMR. NMR can be used to study the high-resolution 3D structure, ligand interactions, and dynamics of proteins. The technique relies on NMR observable nuclei, such as 1H, 15N, and 13C. Due to the low natural abundance of 15N and 13C, these isotopes must be incorporated into the protein. Metabolic labeling using bacterial hosts and recombinant DNA is the most common approach to produce fully 15N- and 13C-labeled proteins [24]. This labeling technique is not amenable to all proteins, especially those requiring post-translational modifications. Reductive methylation provides an alternative labeling strategy where 13C-formaldehyde is used to incorporate 13C-methyl groups. This labeling method introduces sparse 13C-labels that can be used as sensitive probes to determine lysyl pKa values [25-29], discover the roles of lysines in enzyme activity [30-32], study protein-protein interactions [33-34], and observe pseudo-contact shifts and paramagnetic relaxation enhancement (PRE) effects [2,25,35].
Despite the significant advantages reductive methylation provides NMR-studies of proteins, the utility is mitigated by the difficulty in assigning the 13C-methyl peaks to their respective amines. Traditional NMR assignment strategies for fully 15N- and 13C-labeled proteins rely on through-bond, scalar couplings or through-space, nuclear Overhauser effects. Because the reductive methylation reaction produces sparse labeling, these assignment methods are not applicable. Here, we review alternative assignment strategies for reductively methylated proteins and compare the results for the assignment of reductively 13C-dimethylated hen egg white lysozyme.
NMR Assignment Methods
Assignment of the dimethyllysyl NMR peaks was pioneered in 1973 by Bradbury and Brown [25]. This work established the utility of the dimethylamino group to probe the microenvironment of lysines by studying the dimethylamino 1H-NMR peaks of lysozyme as a function of gadolinium (III) (Gd3+) concentration. Through PRE, Gd3+ has a broadening effect on NMR peaks, which is inversely related to the distance to the sixth power between Gd3+ and the nucleus of interest. Broader lines were observed as the concentration of bound Gd3+ increased, with the group closest to the paramagnetic ion broadening the most. Assignments made with this method were based on a single Gd3+ binding site observed in the X-ray crystal structure determined by Blake and Rabstein (not published). Corroboration of the results with experimental pKa values and the 3D structure allowed for the assignment of the six ε-dimethyllysyl, 1H-NMR peaks. It should be noted that lysozyme has six lysines, so seven dimethylamino peaks are expected; however, inadequate modification of the N-terminal α-amino group, confirmed by experimental pKa values, resulted in six peaks. While absolute assignment of the ε-dimethyllysyl peaks was accomplished, this assignment method is limited to proteins that bind paramagnetic ions and have known structures with ion-to-methyl distances in the range of 15 – 24 Å. In 1975, Brown and Bradbury published an addendum to this procedure using reductively methylated ribonuclease A (RNase-A) [26]. In this work, the authors compared the pKa values obtained from pH titrations of the dimethylamines to those of model compounds to make assignments. The pKa of the N-terminal α-dimethylamine of a protein is distinct and typically around 7.0. The lysyl ε-dimethylamines fall within the range of 9.5 – 11.0, depending on their microenvironment. When the dimethylamines are involved in a salt bridge, the pKa is typically lower. Because of their distinct pKa values, the authors were able to assign the α-dimethylamino peak (pKa = 6.6) as well as the ε-dimethylamino peak of Lys41 (pKa = 8.6), with the aid of the crystal structure, which shows Lys41 in a salt bridge. The remaining dimethylamines had pKa values in the range of 10.6 – 11.2 and could not be assigned by titration alone.
In 1979, Jentoft and Dearborn described new methods for reductive methylation and the study of dimethylamines on proteins [36]. Reductive methylation of proteins with sodium borohydride (NaBH4) suffers from low efficiency due to the strength of this reducing agent. In addition to reducing the Schiff base, NaBH4 also reduces aldehydes and ketones; thus, the reduction of formaldehyde to methanol competes with the desired, forward reaction. Other problems associated with NaBH4 are its strong pH dependence (pH 9.0) and the reduction of disulfide bonds. The reductive methylation method presented by Jentoft and Dearborn uses sodium cyanoborohydride (NaCNBH3), a milder reducing agent that does not reduce aldehydes or ketones at neutral pH. Additional benefits of NaCNBH3 are the larger pH range (pH 7.0 – 9.0) and a higher degree of lysine modification. Another difference between the methods is that Jentoft and Dearborn used 13C-NMR to study the dimethylamino peaks instead of 1H-NMR. Reductive methylation of the lysyl side-chains can yield monomethylamines and dimethylamines depending on the efficiency of the reaction and limiting reagents. The 1H-chemical shifts of both mono- and dimethyl peaks are similar and often overlap each other as well as other 1H-signals from the protein. Titration (pH) experiments are often used to differentiate the α- and ε-dimethylamino peaks; however, significant 1H-signal overlap can render this method ineffective as signals shift in the same direction with pH. On the other hand, the 13C-chemical shifts of methylamines are sensitive to the “amine order.” Monomethylamines (Δ ~32 – 34) and dimethylamines (Δ ~41 – 44) are isolated from each other as well as other signals from the protein. Jentoft and Dearborn demonstrated that α-dimethylamino peaks shift upfield while the ε-dimethylamino peaks shift downfield with increasing pH. Using the new reductive methylation method and 13C-NMR, Jentoft et al. studied RNase-A. Peak assignments of RNase-A were made by comparing the 13C-spectra of dimethylated RNase-A to the spectra of model compounds and verifying both the α-dimethylamino and Lys41 ε-dimethylamino peaks by pH titrations. This work demonstrated the benefits of using the 13C-NMR peaks and became the basis for ensuing experiments.
Variations of Jentoft and Dearborn’s method were later used by Gerken et al. to study lysozyme and Sparks et al. to study apolipoprotein A-1 (apoA-1) [28,31,37]. Instead of using model compounds to verify assignments made from experimental pKa values, Gerken et al. used the X-ray structure of lysozyme and Sparks et al. used the predicted secondary structure of apoA-1 to make and verify assignments. Like their predecessors, each of the methods is limited by the number of distinct pKa values. Additionally, the assignment methods used by Gerken et al. and Sparks et al. are limited by their reliance on determined and predicted structural data, respectively. Using pH titrations for assigning dimethylamino peaks is proven to be a useful tool; however, supplementing with a secondary technique is necessary to achieve absolute assignment. In order to use reductive methylation as a strategy to study protein structure by NMR, it is important that the assignment techniques not rely on prior knowledge of the protein other than the amino acid sequence.
In 1988, Dick et al. assigned 13C-NMR peaks of reductively methylated fd gene 5 protein (G5P) using pH titration and peptide sequencing of partially radiolabeled (3H and 14C) protein [30]. This method advanced the development of an ideal assignment strategy and was the first to utilize partial methylation for assignment. In this method, G5P was reductively 13C-methylated both free (control) and bound to a ligand followed by subsequent 14C-methylation to completion (unbound) to incorporate radiolabels. The protein was enzymatically digested and sequenced by Edman degradation. Radioactivity was monitored during degradation to assess the level of protection by bound ligand of each lysyl residue. Those lysyl residues with higher radioactivity exhibited lower peak intensities in the 13C-NMR spectrum. One of the lysines of G5P is the C-terminal residue, which Dick et al. cleaved using carboxypeptidase B. Subsequent difference spectra of methylated G5P and the carboxypeptidase-modified G5P were used to assign the C-terminal lysyl residue. Degenerate levels of radioactive isotope-incorporation led to group-wise assignments; however, absolute assignments were possible by comparing the data to the X-ray crystal structure.
The assignment of dimethylamino NMR peaks soon progressed with the introduction of two dimensional (2D) NMR. The 2D experiments improved the resolution of the dimethylamino peaks by utilizing the chemical shift dispersion of the dimethylamino peaks in the 13C-dimension with the sensitivity of 1H-detection. Zhang and Vogel used site-directed mutagenesis to assign the dimethylamino peaks in a 1H-13C heteronuclear multiple quantum coherence (HMQC) spectrum of reductively methylated mammalian calmodulin (CaM) [29]. Individual mutants (Lys → Gln) of CaM were generated for each of the 7 lysl residues, followed by reductive methylation with 13C-formaldehyde. Each peak was assigned by its absence in the spectrum of the corresponding mutant. This assignment method is the first to achieve absolute assignment without prior knowledge of the target protein’s structure and has significant implications towards protein structural studies. Although technically sound and successful, site-directed mutagenesis can require extensive work for those proteins containing many lysines.
In 2000, Ashfield et al. presented an assignment method that utilized matrix assisted laser desorption ionization – time of flight (MALDI-TOF) MS to identify partially 13C-methylated lysines on tryptic peptides [34]. The use of MS to probe the extent of methylation at each lysine is an alternative approach to the peptide sequencing (Edman degradation) method used by Dick et al. and does not require radio-labeling. Dimethylamino NMR peaks of human MIP-1α D26A (hMIP-1α), which contains 3 lysines, were assigned by correlating the disappearance of unmodified peptide masses in the mass spectra with the appearance of mono- and dimethylamino peaks in 13C-NMR and 2D 1H-13C heteronuclear single quantum coherence (HSQC) NMR spectra. Reductive methylation of dimeric hMIP-1α revealed that one of the lysyl residues was completely protected from modification. The authors were able to make absolute assignments of the 3 reductively methylated lysines of hMIP-1α with only the amino acid sequence as prior knowledge.
The MS-assisted assignment method, developed by Macnaughtan et al., expands on the Ashfield method with the introduction of using MS to measure the amount of 13C incorporated at each dimethylamine [38]. This method utilized partial labeling for assignment based on the varying rates of the reductive methylation reaction at each site. Partial labeling with 13C-formaldehyde, followed by labeling with excess natural abundance formaldehyde, resulted in varying levels of 13C at each dimethylamine. The amount of 13C-incorporation at each site was measured by NMR, using the peak volumes in a 2D 1H-13C HSQC spectrum, and MS, using the shift in the isotopic profile of peptides produced by trypsin digestion. MS analysis also provides the identity of the dimethylamine through the unique mass-to-charge ratio (m/z) of the peptide, which can also be verified by tandem MS experiments. By correlating the NMR and MS data for 13C-incorporation, the dimethylamino peaks can be assigned knowing only the amino acid sequence of the target protein. Assignments of the dimethylamino NMR peaks of hen egg white lysozyme were made through the correlation of 13C-incorporation percent values calculated for both tryptic peptides with MS and NMR peaks of 2D 1H-13C HSQC NMR experiments. While the range of partial labeling was good (30.2 – 71.4 %), the accuracy of the NMR and/or MS measurements of 13C-incorporation did not allow for complete assignment. In addition, the MS data were incomplete because individual isotopic profile measurements could not be made for the two N-terminal dimethylamines or for the tandem lysines, Lys96 and Lys97. The inherent difficulty in assigning the N-terminal lysine led to the supplemental methods by Roberson et al. [39]. This work presented two techniques to identify and assign the N-terminal lysine α- and ε-dimethylamino NMR peaks. The first method used aminopeptidase to identify the two peaks belonging to the N-terminal lysine, and the second method used pH-induced chemo-selectivity to modify either the α-amine or the lysine side-chain ε-amines. Assignments were made by the loss of peaks in 2D 1H-13C HSQC spectra and by using the MS-assisted assignment method on selectively labeled samples. The pH-selectivity technique provides advantages over the titration approach in assigning the α-dimethylamino peak because it does not require a supplemental technique or structural knowledge to confirm the assignments. It also has the added benefit of assigning the N-terminal ε-dimethyllysyl peak, which has a pKa similar to other ε-dimethyllysines. Establishment of these methods to supplement the original work by Macnaughtan et al. aided in further assignments of lysozyme; however, limited accuracy and degenerate labeling did not allow for absolute assignments, despite intense efforts to vary the extent of labeling [40].
Recently, Larda et al. published a new approach for the assignment of ε-methyllysyl peaks for protein structural studies [2]. Instead of focusing on dimethylamines, the authors presented a protocol that favors monomethylation of the amino groups to take advantage of the improved resolution and overcome exchange broadening. In addition, deuterium tags allowed for the use of pulse sequences that filter out natural abundance 13C-signals. Assignments of methylamino peaks were made by comparing aromatic nuclear Overhauser effects (NOEs) and line broadening effects from dissolved nitroxide spin-labels to that of predicted effects from the X-ray crystal structure. Although absolute assignments were made, the general applicability of the method is limited because of the dependence on the assignments made by Macnaughtan et al. and the crystal structure.
Assignments of Reductively Methylated Lysozyme
Lysozyme has been the model protein most often used in developing an assignment strategy for dimethyllysyl NMR peaks. With six lysines and the N-terminal amine, seven sites of methylation are possible and will be referred to as αLys1, εLys1, Lys13, Lys33, Lys96, Lys97, and Lys116. Depending on the pH, ionic strength, and temperature of the reductively methylated lysozyme sample, the methyl groups of a single dimethylamine can be equivalent and exhibit one NMR peak or they can be non-equivalent or in slow exchange and exhibit two NMR peaks. The number of peaks and the peak widths depend on the dynamics associated with the methyl groups, which are influenced by bond rotations, amine-inversion, and pKa. Assigning the NMR peaks of the dimethylamino groups to the lysines and N-terminal amine is usually performed within the pH range of 8 – 9 at room temperature, when each site has equivalent methyl groups and seven peaks are observed as shown in Figure 3. Assignments of the dimethylamino peaks have been made in part or in absolution by five methods, which we refer to as Bradbury, Gerken, Macnaughtan, Roberson, and Larda. Each assignment strategy is discussed and compared in detail and summarized in Table 1.
Figure 3.

1H-13C HSQC spectrum of 13C-dimethylated hen egg white lysozyme (120 μM) at pH 8.1 collected on a 600 MHz spectrometer in approximately 5 min. The peak numbering scheme is the same as in Table 1. Reprinted with permission from [38]. Copyright 2005 American Chemical Society.
Table 1.
Summary of the strategies to assign the dimethylamino NMR peaks of reductively 13C-methylated hen egg white lysozyme.
| Name | Assignment Strategy | Advantages | Disadvantages |
|---|---|---|---|
| Bradbury [25] | Compare experimental pKa values to model compounds and predicted electrostatic interactions from the X-ray crystal structure |
Assignment of the α-amine and ε-amines involved in electrostatic interactions |
Relies on the 3D protein structure; pKa values were not sufficient to assign all ε- amines |
| Compare selective broadening effects of Gd3+ to predictions from the X-ray crystal structure |
Assignment of amines within 24 Å of the paramagnetic ion |
Relies on the 3D protein structure; relies on a known paramagnetic ion-binding site |
|
| Gerken [37] | Compare experimental pKa values and chemical shift to model compounds and predicted electrostatic interactions from the X-ray crystal structure |
Assignment of the α-amine and ε-amines involved in electrostatic interactions |
Relies on the 3D protein structure; pKa values were not sufficient to assign all ε- amines |
| Macnaughtan [38] | Correlates isotope- incorporation measurements from NMR peaks and MS isotopic profiles of tryptic peptides |
Does not require a 3D protein structure |
Degenerate isotope- incorporation limits absolute assignments; the α- and ε- amines must be separated to measure the MS isotopeincorporation |
| Roberson [39] | Uses pH to alter the relative rates of the α-amine reaction compared to the ε-amines |
Does not require a 3D protein structure; absolute assignment of the α-amine; the MS isotope-incorporation of an ε-amine near the N- terminus can be measured |
Limited to assigning the α- amine and one ε-amine near the N-terminus |
| Uses aminopeptidase to assign the α-amine and an ε- amine near the N-terminus |
Does not require a 3D protein structure |
Limited to assigning the α- amine and one ε-amine near the N-terminus; relies on aminopeptidase activity toward the protein of interest |
|
| Larda [2] | Compares spin-label-induced line broadening with predictions from the X-ray crystal structure |
Assignment of amines affected differently by soluble spin-labels |
Relies on the 3D protein structure and data from another method |
In the first study, conducted by Bradbury and Brown, six resolved peaks corresponding to the six ε-dimethyllysines were titrated to obtain pKa values and then assigned using selective broadening effects with increasing concentrations of Gd3+ [25]. This method is based on the assumption that the paramagnetic ion binds in a single location between Glu35 and Asp52. Each residue was assigned based on the observed pKa values compared to those of model compounds and the side-chain interactions interpreted from the X-ray crystal structure. The authors noted that the only ambiguity in the assignments was between peaks 1 and 2, corresponding to Lys97 and Lys96, respectively, citing identical chemical shifts at pH 4.5. Paramagnetic perturbation studies have been useful in protein structure determination; however, when used to assign NMR peaks, this technique requires structural knowledge of the protein. For that reason this method is not applicable to proteins whose structures have not been solved. In addition, the assumption made in this assignment strategy was questioned by Gerken et al., citing multiple binding sites for Gd3+ at high concentrations [37].
Gerken et al. used observed pKa values and chemical shifts to make assignments of lysozyme dimethylamino NMR peaks [37]. The αLys1 peak was assigned by its distinct pKa and chemical shift. A suggested “salt bridge” between Lys13 and the C-terminal Leu129 in the X-ray crystal structure aided the assignment of Lys13’s dimethylamino peak. This assignment was confirmed with the altered pKa value of Lys13 when Leu129 was cleaved. Likewise, the εLys1 was assigned with assistance from the suggested ion pairing between εLys1 and Glu7. Lys97 was deduced to be either peak 1 or 4 (peak numbering based on Figure 3 and not on Gerken et al.); however, the remaining peaks could not be unambiguously assigned. Table 2 summarizes the peak assignments for each method for direct comparison. Gerken’s assignments agree with Bradbury’s for αLys1, εLys1, and Lys97 (peak 1), but other assignments were different or missing. Like Bradbury and Brown’s method, this method is limited to those proteins whose 3D structures have been solved. Even though both methods used suggested salt bridges and ion pairings from the crystal structure to justify their assignments, the assignments of all, except for αLys1, εLys1, and Lys97, conflict. Viable arguments were presented to rationalize the assignments from both methods, which suggest that reliance on the crystal structure introduces bias when assigning the NMR peaks.
Table 2.
Assignments of the dimethylamino NMR peaks to the lysyl residue number and N-terminal amine of reductively methylated hen egg white lysozyme with peak numbers corresponding to those in Figure 3.
| Peak number | Bradburya | Gerkenb | Macnaughtanc | Robersond | Lardae |
|---|---|---|---|---|---|
| 1 | 97 | ~97 | ε1 or 13 | 13 | 13 |
| 2 | 96 | 33 or 116 | 33 or 116 | 116 | |
| 3 | 116 | 13 | ε1 or 13 | 97 | 96 |
| 4 | 13 | ~97 | α1 | 96 | 97 |
| 5 | 33 | 33 or 116 | 33 or 116 | 33 | |
| 6 | ε1 | ε1 | 96 | ε1 | ε1 |
| 7 | α1 | α1 | 97 | α1 | α1 |
Data from reference [25].
Data from reference [37].
Data from reference [38].
Based on data from references [39] and [38].
Data from reference [2].
Assignments of lysozyme dimethylamino NMR peaks by Macnaughtan et al. relied on the correlation of isotope incorporation measurements of the intact protein using NMR and of dimethylamino, tryptic peptides using MS [38]. Definitive assignments were made for three of the seven peaks (αLys1, Lys96, and Lys97), but the remaining four peaks were assigned pairwise (εLys1 and Lys13; Lys33 and Lys116) as shown in Table 1. Absolute assignment of all dimethylamino peaks was prohibited by two problems associated with this method. First, isotope-incorporation was degenerate for two pairs of sites. The reaction rates at these sites are comparable, likely due to similar microenvironments. Various methods have been tested to break the degeneracy, including varying the reducing agent and using 18-crown-6-ether as a selective protecting agent [39]. Improving the accuracy of the NMR and/or MS measurements may reveal a difference in the level of labeling. The second problem is that individual isotope-incorporation values could not be measured for all dimethylamines with MS. For peptides that contain two dimethylamines, only the average isotope-incorporation can be measured with MS. Trypsin is commonly used to digest proteins for MS analysis, but it does not cleave at dimethylated lysines. Consecutive lysines, such as Lys96 and Lys97 in lysozyme, pose a significant challenge for digestion. One possible solution is to use high-resolution tandem MS to measure the isotopic profile of peptide fragments. On the other hand, the selectivity of trypsin can be an advantage. The assignments of Lys96 and Lys97 are based on the observation of peptide Lys97-Arg112 in the MS, which suggests that some Lys96 was not methylated, even under conditions of excess formaldehyde. Consequently, the NMR peak with the lowest peak area (peak 6) was assigned to Lys96 (Table 1). Another instance of a peptide with two dimethylamines occurs when the N-terminal amino acid is a lysine, which is the case for lysozyme. The α- and ε-dimethylamines of the N-terminal lysine cannot be separated using a protease, so other methods must be used.
Roberson et al. published two methods to distinguish, identify, and assign both the αLys1 and εLys1 NMR peaks as a supplement to the MS-assisted assignment method [39]. The NMR spectrum of lysozyme treated with aminopeptidase displayed the disappearance of peaks 6 and 7, identifying these peaks as the dimethylamino groups of the N-terminal lysine. Further investigation of the peaks with pH-induced selectivity of the reductive methylation reaction confirmed those results. At low pH, the α-amine methylated at a higher rate than any of the side-chain ε-amines, and at high pH, no methylation was observed for the α-amine. The high pH sample made it possible to distinguish the αLys1 peak from the εLys1 peak. With little to no methylation at the α-amine, the N-terminal peptide could be analyzed with MS to give a 13C-incorporation value for the ε-dimethylamine, which made the assignment of εLys1 possible (independent of the aminopeptidase method). Although peaks 6 and 7 were previously assigned to Lys96 and Lys97, respectively, data from the N-terminal methods were conclusive and these peaks are εLys1 and αLys1, respectively. Combining these assignments with Macnaughtan’s data [38] provides new assignments for peaks 1 (Lys13), 3 (Lys97), and 4 (Lys96), but peaks 2 and 5 are still ambiguous (Lys33 or Lys116) due to degenerate labeling (Table 2). The assignment of αLys1 and εLys1 may provide insight to improve the MS-assisted assignment method. The average 13C-incorporation measurements of αLys1 and εLys1 are 54.2% by NMR (Peak 6 = 30.2%, Peak 7 = 78.3%) and 44.8% by MS (peptide Lys1-Arg5). The difference between the values is very large and clearly indicates that the accuracy of the NMR and/or MS measurement must be improved for the method to be viable.
Larda et al. introduced the use of monomethylamines and deuterium-filtered NMR to improve the quality of the NMR peaks for assignment [2]. Using data from Macnaughtan et al. and predicted, spin-label-induced line broadening from the X-ray crystal structure, all monomethyl- and dimethylamino signals were assigned (Table 1). Like the Bradbury and Gerken methods, this assignment method is limited by its reliance on the crystal structure. The adjusted assignments made by combining Roberson and Macnaughtan’s data agree with Larda’s assignments, with the exception of Lys96 and Lys97. It is not surprising that the assignments of Lys96 and Lys97 are unresolved, since a pair of consecutive lysines is difficult to distinguish using spin-labels or MS.
To date, the only consensus and conclusive assignments of dimethylamino peaks of hen egg white lysozyme are αLys1 and εLys1. Although there is some overlap in the assignment of the remaining peaks, they are difficult to accept as definite. The use of crystal structures to aid in the assignments produces conflicting results and limits the applicability of the method. While proteomics MS is useful, it is not without its limitations, including the need to separate, identify, and measure every dimethylamine. Advanced tandem MS methods and more accurate modeling of dimethylated lysines from crystal structures may alleviate the current challenges to dimethylamino NMR peak assignment. Analyzing lysine mutants of lysozyme is the most direct and reliable way to definitely assign the NMR peaks, as was done with calmodulin.[29] Even though the definitive assignments of reductively 13C-methylated hen egg white lysozyme may bias future method development, a retrospective evaluation of the strategies reviewed would be possible.
Conclusion
Over the last 40 years, labeling proteins with reductive methylation has proved useful to many analytical techniques and permitted the study of proteins with limited prior knowledge of structural properties. Using isotopically labeled reagents, radio- or magnetic-isotopes can be incorporated in high yield and with little impact on the protein’s overall fold or activity. Labeling a protein with 13C-dimethylamino groups provides a means of studying proteins by NMR and is particularly useful for labeling proteins from source or purified from eukaryotic cell cultures. The 13C-dimethylamino NMR peaks can be studied in a number of ways to characterize the protein’s activity and probe its tertiary and quaternary structures. The applicability of the reductive methylation strategy to NMR is limited by the lack of a universal assignment strategy that does not require a high resolution structure or recombinant expression. When the structure of a protein is available, soluble (nitroxide) or bound (Gd3+) spin-labels and measured pKa values can be used to make assignments. This strategy produced conflicting results for hen egg white lysozyme, but improved computational modeling of reductively methylated proteins may provide more accurate predictions of paramagnetic effects and the state of salt bridges in solution. Using partial labeling of the protein (radio- or magnetic-isotopes) and a secondary method to identify and correlate the data (Edman degradation/scintillation or MS) has the advantage of not requiring a protein structure for assignment. Such an assignment strategy has the potential to be universal, but requires higher accuracy in measuring the amount of partial labeling for absolute correlation. Our review of five assignment strategies applied to reductively 13C-methylated hen egg white lysozyme exemplifies the difficulty of the task and creativity devoted to overcoming the assignment-hurdle.
Acknowledgement
The research described was supported in part by Award Number R00RR024105 from the National Center for Research Resources, National Institutes of Health.
Footnotes
Subject Category: Labeling Procedures
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Means G, Feeney R. Reductive alkylation of amino groups in proteins. Biochemistry. 1968;7:2192–2201. doi: 10.1021/bi00846a023. [DOI] [PubMed] [Google Scholar]
- [2].Larda ST, Bokoch MP, Evanics F, Prosser RS. Lysine methylation strategies for characterizing protein conformations by NMR. J. Biomol. NMR. 2012;54:199–209. doi: 10.1007/s10858-012-9664-z. [DOI] [PubMed] [Google Scholar]
- [3].Rice RH, Means GE. Radioactive labeling of proteins in-vitro. J. Biol. Chem. 1971;246:831–832. [PubMed] [Google Scholar]
- [4].Ottesen M, Svensson B. Use of reductive methylation for radioactive labeling of proteins. C. R. Trav. Lab. Carlsberg. 1971;38:445–456. [PubMed] [Google Scholar]
- [5].She YM, Rosu-Myles M, Walrond L, Cyr TD. Quantification of protein isoforms in mesenchymal stem cells by reductive dimethylation of lysines in intact proteins. Proteomics. 2012;12:369–379. doi: 10.1002/pmic.201100308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rayment I. Reductive alkylation of lysine residues to alter crystallization properties of proteins. Methods Enzymol. 1997;276:171–179. [PubMed] [Google Scholar]
- [7].Walter TS, Meier C, Assenberg R, Au KF, Ren JS, Verma A, Nettleship JE, Owens RJ, Stuart DI, Grimes JM. Lysine methylation as a routine rescue strategy for protein crystallization. Structure. 2006;14:1617–1622. doi: 10.1016/j.str.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rypniewski W, Holden H, Rayment I. Structural consequences of reductive methylation of lysine residues in hen egg white lysozyme: An X-ray analysis at 1.8-angstrom resolution. Biochemistry. 1993;32:9851–9858. doi: 10.1021/bi00088a041. [DOI] [PubMed] [Google Scholar]
- [9].Vanderwel H, Wiersma A, Brouwer JN. Tritium labeling of Thaumatin I. A sweet-tasting protein from Thaumatococcus daniellii benth, by reductive methylation, J. Label. Compd. Radiopharm. 1978;14:735–740. [Google Scholar]
- [10].Khanh NQ, Linde R, Lipecky R, Gassen HG. Inactivation of ribosomal-protein S1 by means of reductive methylation. Hoppe-Seylers Z. Physiol. Chem. 1978;359:284–284. [Google Scholar]
- [11].Guha S, Szulmajster J. Analysis of ribosomal protein conformation in Bacillus subtilis by reductive methylation: Identification of proteins with different conformation in monosomes and polysomes. FEBS J. 1980;118:103–108. doi: 10.1016/0014-5793(80)81228-2. [DOI] [PubMed] [Google Scholar]
- [12].Heacock CS, Bernstein BW, Duhaiman AS, Amorese DA, Bamburg JR. In vitro labeling of proteins by reductive methylation: Application to proteins involved in supramolecular structures. J. Cell. Biochem. 1982;19:77–91. doi: 10.1002/jcb.240190107. [DOI] [PubMed] [Google Scholar]
- [13].Finger JM, Choo KH. Double-label reductive methylation of tissue proteins for precision two-dimensional polyacrylamide-gel electrophoretic analysis. Biochem. J. 1981;193:371–374. doi: 10.1042/bj1930371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gray DM, Sherry AD, Teherani J, Kansy JW. Reductive methylation of the lysyl residues in the fd gene 5 DNA-binding protein: CD and 13C NMR results on the modified protein. J. Biomol. Struct. Dyn. 1984;2:77–91. doi: 10.1080/07391102.1984.10507548. [DOI] [PubMed] [Google Scholar]
- [15].Fretheim K, Egelandsdal B, Harbitz O. Effect of alkylation with different sized substituents on the conformation of ovomucoid, lysozyme and ovotransferrin. Int. J. Peptide Protein Res. 1985;25:601–607. doi: 10.1111/j.1399-3011.1985.tb02216.x. [DOI] [PubMed] [Google Scholar]
- [16].Prokai-Tatrai K, Guo J, Prokai L. Selective chemoprecipitation and subsequent release of tagged species for the analysis of nitropeptides by liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics. 2011;10:1–10. doi: 10.1074/mcp.M110.002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hsu JL, Huang SY, Shiea JT, Huang WY, Chen SH. Beyond quantitative proteomics: Signal enhancement of the a(1) ion as a mass tag for peptide sequencing using dimethyl labeling. J. Proteome Res. 2005;4:101–108. doi: 10.1021/pr049837+. [DOI] [PubMed] [Google Scholar]
- [18].Guo J, Prokai-Tatrai K, Prokai L. Relative quantitation of protein nitration by liquid chromatography-mass spectrometry using isotope-coded dimethyl labeling and chemoprecipitation. J. Chromatogr. A. 2012;1232:266–275. doi: 10.1016/j.chroma.2011.12.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chicooree N, Connolly Y, Tan C-T, Malliri A, Li Y, Smith DL, Griffiths JR. Enhanced detection of ubiquitin isopeptides using reductive methylation. J. Am. Soc. Mass Spectrom. 2013;24:421–430. doi: 10.1007/s13361-012-0538-0. [DOI] [PubMed] [Google Scholar]
- [20].Hsu JL, Huang SY, Chen SH. Dimethyl multiplexed labeling combined with microcolumn separation and MS analysis for time course study in proteomics. Electrophoresis. 2006;27:3652–3660. doi: 10.1002/elps.200600147. [DOI] [PubMed] [Google Scholar]
- [21].Cohen A, Wang E, Chisholm KA, Kostyleva R, O’Connor-McCourt M, Pinto DM. A mass spectrometry-based plasma protein panel targeting the tumor microenvironment in patients with breast cancer. J. Proteomics. 2013;81:135–147. doi: 10.1016/j.jprot.2012.11.004. [DOI] [PubMed] [Google Scholar]
- [22].Kriwacki R, Reisdorph N, Siuzdak G. Protein structure characterization with mass spectrometry. Spectr.-Int. J. 2004;18:37–47. [Google Scholar]
- [23].Kussmann M, Roepstorff P. Characterisation of the covalent structure of proteins from biological material by MALDI mass spectrometry – possibilities and limitations. Spectr.-Int. J. 1998;14:1–27. [Google Scholar]
- [24].Nettleship JE, Assenberg R, Diprose JM, Rahman-Huq N, Owens RJ. Recent advances in the production of proteins in insect and mammalian cells for structural biology. J. Struct. Biol. 2010;172:55–65. doi: 10.1016/j.jsb.2010.02.006. [DOI] [PubMed] [Google Scholar]
- [25].Bradbury JH, Brown LR. Determination of dissociation constants of lysine residues of lysozyme by proton-magnetic-resonance spectroscopy. Eur. J. Biochem. 1973;40:565–576. doi: 10.1111/j.1432-1033.1973.tb03228.x. [DOI] [PubMed] [Google Scholar]
- [26].Brown L, Bradbury J. Proton-magnetic-resonance studies of lysine residues of ribonuclease A. Eur. J. Biochem. 1975;54:219–227. doi: 10.1111/j.1432-1033.1975.tb04131.x. [DOI] [PubMed] [Google Scholar]
- [27].Huque M, Vogel H. 13C NMR studies of the lysine side chains of calmodulin and its proteolytic fragments. J. Protein Chem. 1993;12:695–707. doi: 10.1007/BF01024928. [DOI] [PubMed] [Google Scholar]
- [28].Sparks D, Phillips M, Lundkatz S. The conformation of apolipoprotein A-I in discoidal and spherical recombinant high density lipoprotein particles: 13C NMR studies of lysine ionization behavior. J. Biol. Chem. 1992;267:25830–25838. [PubMed] [Google Scholar]
- [29].Zhang M, Vogel H. Determination of the side chain pKa values of the lysine residues in calmodulin. J. Biol. Chem. 1993;268:22420–22428. [PubMed] [Google Scholar]
- [30].Dick L, Sherry A, Newkirk M, Gray D. Reductive methylation and 13C NMR studies of the lysyl residues of fd gene 5 protein: Lysines 24, 46, and 69 may be involved in nucleic acid binding. J. Biol. Chem. 1988;263:18864–18872. [PubMed] [Google Scholar]
- [31].Jentoft JE, Jentoft N, Gerken TA, Dearborn DG. 13C NMR studies of ribonuclease A methylated with [13C]formaldehyde. J. Biol. Chem. 1979;254:4366–4370. [PubMed] [Google Scholar]
- [32].Jentoft J, Gerken T, Jentoft N, Dearborn D. [13C]Methylated ribonuclease A: 13C NMR studies of the interaction of lysine 41 with active site ligands. J. Biol. Chem. 1981;256:231–236. [PubMed] [Google Scholar]
- [33].Hattori Y, Furuita K, Ohki I, Ikegami T, Fukada H, Shirakawa M, Fujiwara T, Kojima C. Utilization of lysine 13C-methylation NMR for protein–protein interaction studies. J. Biomol. NMR. 2013;55:19–31. doi: 10.1007/s10858-012-9675-9. [DOI] [PubMed] [Google Scholar]
- [34].Ashfield J, Meyers T, Lowne D, Varley P, Arnold J, Tan P, Yang J, Czaplewski L, Dudgeon T, Fisher J. Chemical modification of a variant of human MIP-1 alpha; Implications for dimer structure. Protein Sci. 2000;9:2047–2053. doi: 10.1110/ps.9.10.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dick LR, Geraldes C, Sherry AD, Gray CW, Gray DM. 13C NMR of methylated lysines of fd gene-5 protein: Evidence for a conformational change involving lysine-24 upon binding of a negatively charged lanthanide chelate. Biochemistry. 1989;28:7896–7904. doi: 10.1021/bi00445a052. [DOI] [PubMed] [Google Scholar]
- [36].Jentoft N, Dearborn DG. Labeling of proteins by reductive methylation using sodium cyanoborohydride. J. Biol. Chem. 1979;254:4359–4365. [PubMed] [Google Scholar]
- [37].Gerken T, Jentoft J, Jentoft N, Dearborn D. Intramolecular interactions of amino groups in 13C reductively methylated hen egg-white lysozyme. J. Biol. Chem. 1982;257:2894–2900. [PubMed] [Google Scholar]
- [38].Macnaughtan MA, Kane AM, Prestegard JH. Mass spectrometry assisted assignment of NMR resonances in reductively 13C-methylated proteins. J. Am. Chem. Soc. 2005;127:17626–17627. doi: 10.1021/ja056977r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Roberson KJ, Brady PN, Sweeney MM, Macnaughtan MA. Methods to identify the NMR resonances of the 13C-dimethyl N-terminal amine on reductively methylated proteins. J. Vis. Exp. 2013;82:e50875. doi: 10.3791/50875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Roberson KJ, Macnaughtan MA. Attempts towards unambiguously assigning 13C-dimethylamine NMR resonances. The All Results Journal: Chem. 2013;4:10–16. [Google Scholar]