

Highlights
-
•
Large RNA domains are key regulators of various critical viral processes.
-
•
Structural determinants of RNA elements that mediate cap-independent translation.
-
•
tRNA-like structures mimic the L shape of tRNAs for host synthetase recognition.
-
•
Nuclear export and programmed frameshifting rely on structures within viral ORFs.
-
•
Technological advances allow for improved large RNA structure determination.
Keywords: Viral RNA, RNA structure, Viral translation, Genome packaging, tRNA-like structures
Abstract
The field of viral molecular biology has reached a precipice for which pioneering studies on the structure of viral RNAs are beginning to bridge the gap. It has become clear that viral genomic RNAs are not simply carriers of hereditary information, but rather are active players in many critical stages during replication. Indeed, functions such as cap-independent translation initiation mechanisms are, in some cases, primarily driven by RNA structural determinants. Other stages including reverse transcription initiation in retroviruses, nuclear export and viral packaging are specifically dependent on the proper 3-dimensional folding of multiple RNA domains to recruit necessary viral and host factors required for activity. Furthermore, a large-scale conformational change within the 5′-untranslated region of HIV-1 has been proposed to regulate the temporal switch between viral protein synthesis and packaging. These RNA-dependent functions are necessary for replication of many human disease-causing viruses such as severe acute respiratory syndrome (SARS)-associated coronavirus, West Nile virus, and HIV-1. The potential for antiviral development is currently hindered by a poor understanding of RNA-driven molecular mechanisms, resulting from a lack of structural information on large RNAs and ribonucleoprotein complexes. Herein, we describe the recent progress that has been made on characterizing these large RNAs and provide brief descriptions of the techniques that will be at the forefront of future advances. Ongoing and future work will contribute to a more complete understanding of the lifecycles of retroviruses and RNA viruses and potentially lead to novel antiviral strategies.
1. Introduction
While significant progress has been made toward understanding the complex molecular choreography involved in viral replication, during recent years a bottleneck has emerged in the form of poorly understood mechanisms involving viral RNAs. The importance of understanding viral molecular biology goes beyond development of antiviral therapies. First, the dependence and co-evolution of viruses with their hosts along with host mechanisms inherent to viral infection have given unique insights into host cellular processes such as cap-independent translation (Filbin and Kieft, 2009, Miller et al., 2007, Nicholson and White, 2011) and frameshifting (Dinman, 2012, Giedroc and Cornish, 2009), as well as other basic molecular biology (Barozzi et al., 2007, Hershey and Chase, 1952, Roulston et al., 1999). Also, many technological developments have been driven by the need for novel methods of characterizing viral function (described herein). Finally, questions about immune system function and the evolutionary race between hosts and viruses with high mutation rates, particularly RNA viruses and retroviruses, are beginning to be deciphered (Cascalho and Platt, 2007, Ding and Voinnet, 2007, Iyer et al., 2006, Sanjuan et al., 2010). However, to date viral structural biology has mostly focused on the actions of viral proteins and their interactions with host factors.
Methods of characterizing large RNA structures are constantly improving, and there are large gaps in our understanding of viral replication that such structural data can help to fill. The small size of virus particles requires that, in many cases, a single RNA element can play a role in multiple distinct functions, thus optimizing virus replication. In the case of RNA viruses and retroviruses, the RNA genome acts as both the genetic material and as an active participant in many stages of the lifecycle. For example, specific functional domains of genomic and differentially spliced viral mRNAs have shown the ability to regulate cap-independent translation initiation mechanisms (Filbin and Kieft, 2009, Miller et al., 2007, Nicholson and White, 2011), transcription/translation (Dreher, 2009, Fechter et al., 2001), reverse transcription in retroviruses (Lu et al., 2011b), cellular localization (Hammarskjold and Rekosh, 2011, Stake et al., 2013), programmed frameshifting (Dinman, 2012, Giedroc and Cornish, 2009) and viral packaging (Dreher, 2009, Fechter et al., 2001) (Fig. 1 ).
Fig. 1.
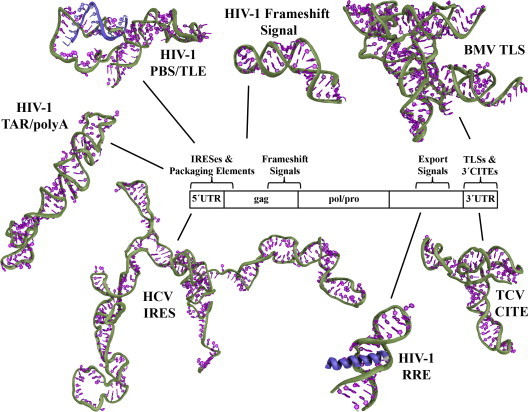
Distribution of functional domains in viral genomic RNA. The 5′UTR consists of many highly structured elements such as the TAR/polyA domain of HIV-1 (Jones et al., 2014), HIV-1 tRNA-like PBS/TLE domain (Jones et al., 2014) and the HCV IRES (Perard et al., 2013), all modeled based on SAXS data. NMR spectroscopy has yielded high-resolution structures of small sections of the HIV-1 frameshift signal (Staple and Butcher, 2005) and RRE export signal (Battiste et al., 1996), both located within coding regions. A SAXS-derived model of the full-length HIV-1 RRE has also recently been published (Fang et al., 2013). Large, structured RNAs within the 3′UTR of plant viral genomes have also been shown to be functional; for example, the BMV TLS (Felden et al., 1994) and TCV 3′CITE (Zuo et al., 2010) were modeled using probing data and a combination SAXS/NMR approach, respectively.
Viruses with RNA genomes constitute a number of human pathogens such as influenza virus, SARS-coronavirus, hepatitis C virus, West Nile virus, HIV-1 and HTLV-1. Indeed, there have been reports linking highly-structured viral RNA genomes to persistence of infection in hosts (Davis et al., 2005). Viral RNA functions, although critical to viral lifecycle progression and infectivity, have been largely neglected in terms of antiviral therapy due in large part to our lack of high quality 3D structural data. We propose that improvements in technology and methods of RNA structure determination will revolutionize our understanding of viral replication and lead to novel antiviral strategies aimed at disrupting RNA-centric functions. Knowledge of viral RNA structure has already led to improvements in potential antiviral therapies. For example, information regarding the structured regions of the HIV-1 genome was used to help design more effective shRNA inhibitors of viral replication (Low et al., 2012), and a crystal structure of a hepatitis C virus inhibitor bound to a portion of the viral internal ribosome entry site (IRES) revealed the binding pocket and has led to subsequent improvements of the inhibitor (Dibrov et al., 2012, Zhou et al., 2013). Therefore, it is important to assess the current state of the field and address potential schemes for moving the field forward. In this review, we first describe recent progress that has been made toward solving large viral RNA structures, with a focus on non-enzymatic elements. As such, we will not be discussing viral ribozymes or other catalytic RNAs (reviews of this topic include Talini et al., 2009, Talini et al., 2011, Wu et al., 2009). Secondly, we highlight common methods of characterizing large viral RNA structure and provide advantages, disadvantages and recent advances for each.
2. Current progress in characterization of large viral RNA structure
2.1. Internal ribosome entry site (IRES) elements
IRES elements are found in viral RNAs that either require, or are capable of proceeding by cap-independent mechanisms for recruiting and engaging the ribosome. IRESes are employed across many viral families and genera. While all IRESes ultimately function as cis-acting enhancers of RNA translation, there is great variety in IRES structures and number of cofactors required. Presently, no consensus IRES sequence/structure has been observed, and there appears to be an inverse correlation between degree of IRES foldedness and number of cofactors required (reviewed in Filbin and Kieft, 2009). IRESes from the viral family Dicistroviridae are highly structured and able to recruit the 80S ribosome efficiently without the aid of any eukaryotic initiation factors (eIFs) or IRES trans-acting factors (ITAFs) (Wilson et al., 2000). The IRES structures of several Dicistroviridae family members have been characterized. For example, the Cricket paralysis virus (CrPV) IRES in complex with the 80S ribosome was solved to ∼17 Å resolution using cryo-electron microscopy (cryo-EM) (Spahn et al., 2004), revealing tertiary structure characterized by an elongated, yet tightly defined shape that is able to manipulate the ribosome upon binding. A high-resolution crystal structure of the related Plautia staliintestine virus (PSIV) IRES alone was later solved, revealing that the RNA contained both rigid and flexible regions, explaining the observed order of ribosomal subunit recruitment (Pfingsten et al., 2006). A domain of the CrPV IRES containing a critical pseudoknot motif was also crystallized and revealed a striking tRNA–mRNA structural mimicry, providing a structural explanation for its role in binding the P-site of the ribosome (Costantino et al., 2008). The importance of this structural mimicry was validated by additional crystal structures of CrPV and PSIV IRES domains bound to the 70S ribosome (Zhu et al., 2011). Recently, structures of the CrPV IRES bound to the ribosome were determined to atomic-level resolution using single-particle cryo-EM (Fernandez et al., 2014). The structures showed that IRES binding initially occurs at the ribosomal A site in a pretranslocation conformation, with translocation into the P site required to move the first codon of the IRES-associated message into the A site, allowing translation to initiate.
The successful characterization of the entire Dicistroviridae IRES structures is due, in part, to their relatively high degree of foldedness. IRESes from Hepatitis C virus (HCV) and related viruses are able to initiate translation internally, but are generally less structured and require additional cellular factors not needed by CrPV or PSIV-like IRESes (Otto and Puglisi, 2004, Pineiro and Martinez-Salas, 2012). As such, high-resolution structural data only exists for subdomains of the HCV IRES. The focus of this review is large viral RNA structures, and so these works will not be extensively discussed (see also Filbin and Kieft, 2009, Lukavsky, 2009). Early probing and small-angle X-ray scattering (SAXS) characterization of the full-length HCV IRES confirmed that it folded into a defined tertiary structure of a non-globular nature (Kieft et al., 1999). Cryo-EM structures of the full-length HCV IRES bound to the 40S (Spahn et al., 2001) and 80S (Boehringer et al., 2005) ribosomes have been solved to ∼20 Å and ∼25 Å resolution, respectively. The 40S-IRES structure reveals that addition of the IRES is sufficient to alter the conformation of the 40S subunit, underscoring the active role that the RNA plays in initiating its own translation. The IRES-80S complex shows that upon 60S association, both the IRES and the ribosome structures are significantly altered. Siridechadilok and coworkers determined a cryo-EM structure of the HCV IRES in complex with eIF3 (a required cofactor for 40S recruitment by this class of IRES), revealing that the IRES interacts with eIF3 at the same position as the cap-dependent pathway cofactor eIF4G (Siridechadilok et al., 2005). Furthermore, modeling this structure with the 40S suggests that the HCV IRES effectively mimics the positioning of eIF4G, essentially acting as a functional replacement in translation initiation. Recently, the final subdomain of the HCV IRES was solved at high resolution by X-ray crystallography (XRC), and the structures were assembled into the available cryo-EM envelope of the full-length IRES revealing that they all fit well (Berry et al., 2011). This structure further supported the notion that the HCV IRES functions by specific positioning of the start codon in the ribosomal P-site. The 40S-eIF3-CSFV IRES (Classical swine fever virus, a close HCV relative) ternary complex was solved to ∼9 Å resolution by cryo-EM, showing that, in this system, the IRES binds the 40S subunit in the same position as eIF3, relegating eIF3 to binding only with the IRES itself (Hashem et al., 2013). This structure helps rationalize how CSFV infection efficiently hijacks cellular translation in favor of viral translation; it not only competitively displaces a necessary cellular translation cofactor in favor of a viral IRES, it provides a novel site for the cofactor to dock and become sequestered. In all of the HCV IRES-complex structures, the RNA appears to adopt a single, elongated conformation. However, a recent study employing SAXS in conjunction with molecular dynamics suggests that the HCV apoIRES is composed of rigid parts that move independently of each other (Perard et al., 2013) (Fig. 1). Taken together, the HCV IRES demonstrates a complex and highly orchestrated mechanism of translation initiation in which structural characterization played a large role in our understanding.
Human immunodeficiency virus type 1 (HIV-1) is capable of initiating translation via both cap-dependent and independent mechanisms (reviewed in Balvay et al., 2007). While an IRES element was initially proposed to be located within the gag ORF (Buck et al., 2001), subsequent studies have focused on the 5′UTR (Brasey et al., 2003, Plank et al., 2013, Vallejos et al., 2011); the results of these studies suggest that the gag ORF in fact inhibits IRES activity (Brasey et al., 2003, Plank et al., 2013). While the matter is still controversial, the possibility remains that both IRES locations exist, and that they each play distinct roles in the viral lifecycle (Yilmaz et al., 2006). It should be noted that the HIV-2 gag ORF has been unambiguously identified as an IRES, actively recruiting translation-initiation factors to the RNA (Herbreteau et al., 2005, Locker et al., 2011). Different splice variants of HIV-1 mRNAs containing the 5′UTR have all been shown to possess the same IRES secondary structure, suggestive of a common mechanism for translational control (Charnay et al., 2009, Plank et al., 2014). In contrast to the HCV IRES, which specifically positions the start codon in the P-site of the ribosome, the HIV-1 IRES appears to function by first recruiting initiation factors to the RNA, after which they can scan until finding a start site (Plank et al., 2014). HIV-1 IRES activity is enhanced by still unidentified cellular cofactors, present only in the G2/M phase of the cell cycle, suggesting a specific timing when HIV-1 IRES activity is required (Vallejos et al., 2011). A clear picture has yet to emerge as to which mutations the HIV-1 IRES can tolerate while still maintaining functionality (Brasey et al., 2003, Plank et al., 2013, Plank et al., 2014, Vallejos et al., 2011), but overall it appears as though the RNA plays a more passive role than other viral IRESes. Consistent with this, HIV IRES activity requires more cellular factors than HCV, for example. Although the precise sequence that constitutes the HIV-1 IRES is not clearly defined, 3D structures of elements within the 5′UTR have been elucidated (reviewed in Lu et al., 2011b). Recently, our lab used SAXS in conjunction with MD and simulated annealing to generate models of three ∼100-nucleotide (nt) domains of the 5′UTR, revealing that the primer-binding site (PBS) region mimics the 3D structure of a tRNA (Jones et al., 2014) (Fig. 1). It is possible that this tRNA-like element (TLE) interacts with the ribosome, although it has also been shown to bind lysyl-tRNA synthetase, which is co-packaged into viral particles along with the HIV-1 reverse transcription primer, tRNALys (Jones et al., 2013). Further studies will be required to fully define the role(s) that the TLE plays in the viral lifecycle. Future efforts will likely be aimed at precisely determining the necessary cofactors for HIV-1 IRES activity, and further characterizing the whole 5′UTR structure alone and in complexes. A number of studies have shown that translation initiation can also occur via ribosomal scanning in HIV-1 (Berkhout et al., 2011, Miele et al., 1996), and which mechanism is dominant is not entirely clear. It remains possible that HIV-1 uses both mechanisms, and additional conditions such as cell cycle progression or stress may factor into the choice.
The Picornaviridae family comprises the final class of viral IRESes, of which Foot-and-mouth disease virus (FMDV) is an exemplar. This class is the least folded, requires the greatest number of cellular cofactors, and cannot even bind the 40S subunit on its own (reviewed in Filbin and Kieft, 2009). The tertiary structure of this class of IRESes alone or in relevant complexes is unknown; however, it has been suggested that tertiary contacts are indeed formed and are important for FMDV IRES activity (Fernandez-Miragall et al., 2006).
2.2. 3′ Cap-independent translation elements (CITEs)
The 3′ cap-independent translation element (CITE) is an RNA structure found within 3′UTRs of plant viruses. These RNA motifs enhance translation of the viral genome, but are considered distinct from IRESes based on their location in the 3′UTR. The mechanisms by which CITEs act to promote translation remain poorly understood, but interactions with 5′UTRs, ribosomal subunits, and translation initiation factors have been described and are thought to be involved in enhancing translation (reviewed in Miller et al., 2007, Nicholson and White, 2011). To date, structural analysis of CITEs has largely been limited to examination of their secondary structures, with a few notable exceptions. The 3′UTR of the Turnip crinkle virus (TCV) contains a kissing-loop-T-shaped structure class of CITE that interacts with the 60S ribosomal subunit. Through a combination of SAXS and nuclear magnetic resonance (NMR) spectroscopy, a low-resolution structure was determined for a 102-nt RNA corresponding to the TCV CITE. This element was found to mimic the overall shape and certain nt motifs of tRNA (Zuo et al., 2010) (Fig. 1). This experimental structural study largely confirmed previous molecular modeling work that predicted that the tertiary structure would resemble tRNA (McCormack et al., 2008). These results also support data suggesting that the TCV CITE interacts with the 60S subunit via the P-site in a manner that mimics tRNA, and that this interaction is partly responsible for the observed translational enhancement (Stupina et al., 2008). In contrast, the Panicum mosaic virus-like (PMV) CITE class has been shown to interact with the initiation factor eIF4F via its eIF4E subunit, an interaction whose affinity correlates positively with the CITEs translational enhancement capabilities (Wang et al., 2009b). Furthermore, besides the TCV CITE, the PMV CITE is the only other CITE for which a 3D structure has also been elucidated (Wang et al., 2011). In this study, Wang and coworkers determined the secondary structures of CITEs from several viral genera using selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) probing and RNA footprinting experiments, both in the presence and absence of eIF4E. The authors then built computational models of the apoCITE and docking models of CITE–eIF4E complexes using constraints derived from the probing experiments and previous literature. Interestingly, the PMV CITE interacts with eIF4E in a manner that resembles the interaction between eIF4E and the 5′-m7GpppN cap. Efforts are underway to examine the Barley yellow dwarf virus CITE (representative of another class) by X-ray crystallography (Kraft et al., 2011). Future structural work in this field promises not only to improve our understanding of how this diverse set of RNA elements function, but also to reveal what makes them similar/different from the closely related IRES element (Nicholson and White, 2011).
2.3. Plant viral tRNA-like structures (TLSs)
The extreme 3′end of a number of (+) strand RNA plant viruses have been shown to mimic tRNA structures. These so called tRNA-like structures (TLSs) are aminoacylated in vivo, and this activity has been linked to regulating translation vs. transcription of the viral genome, maintenance of its 3′end, and encapsidation of the RNA (reviewed in Dreher, 2009, Fechter et al., 2001). A majority of structural studies have focused on the Turnip yellow mosaic virus (TYMV) TLS, and through various probing studies combined with computational modeling found that it bares a striking resemblance to tRNA in terms of overall fold and tertiary interactions, despite differences in base pairing (de Smit et al., 2002, Dumas et al., 1987, Giegé et al., 1993). An NMR structure revealed that the 3′ half of the TLS folds into a coaxially-stacked H-type pseudoknot that mimics the overall tRNA acceptor stem/T arm structure (Kolk et al., 1998). SAXS analysis confirmed that the RNA element mimics the overall shape of tRNA (Hammond et al., 2009). Further SAXS structures of the TYMV TLS showed that an upstream RNA element required for efficient aminoacylation folds as an independent domain, and suggested a role for the upstream domain in acting as a molecular switch, modulating TLS interactions with its protein partners (Hammond et al., 2010). A recent crystal structure confirmed many of the previous observations, and showed that the TYMV TLS strikingly resembles tRNA, despite being composed of distinct tertiary interactions (Colussi et al., 2014). Interestingly, the TLS contains one face that precisely mimics tRNA structure, while the other face is more divergent, providing a structural explanation for the ability of this RNA to serve as a substrate for aminoacylation, as well as in other non-tRNA roles.
The related Tobacco mosaic virus (TMV) and Brome mosaic virus (BMV) TLS tertiary structures have also been modeled. Early 3D models of the BMV TLS (Felden et al., 1994) (Fig. 1) and TMV TLS (Felden et al., 1996) generated based on RNA structure-probing suggested that both TLSs mimicked the canonical L shape of a tRNA and contained pseudoknot interactions reminiscent of the D–T interaction in tRNA. A more recent examination using SAXS and probing showed that, while both BMV and TMV TLSs retained some tRNA-like tertiary contacts, they did not fold into a static conformation like the TYMV TLS, but instead remained relatively dynamic in solution (Hammond et al., 2009). This result is interesting in light of the fact that all three TLSs are competent to be aminoacylated; further studies will be required to determine if this difference in RNA dynamics bears any significance on the viral lifecycle.
2.4. Nuclear export signals
A hallmark of retroviruses is reverse transcription of viral genomic RNA (vRNA) and integration into the host genome. Retroviruses hijack host transcriptional machinery to generate transcripts coding for viral proteins. Messages encoding longer retroviral proteins (such as Gag, Pol, and Env) must be exported from the nucleus containing some or all of their introns. A number of retroviruses bypass this hurdle by encoding cis-acting RNA elements that interact with nuclear export factors either directly, or indirectly via viral proteins (reviewed in Hammarskjold and Rekosh, 2011, Stake et al., 2013). In the case of HIV-1, the virally encoded Rev protein oligomerizes on the Rev Response Element (RRE), subsequently engaging the Crm1 nuclear export pathway and localizing the unspliced or partially spliced viral RNAs to the cytoplasm (Stake et al., 2013). Efforts to characterize the HIV-1 RRE and Rev-RRE structures in their entirety have been hindered by the size of the RRE (∼350 nt) and the propensity for Rev to aggregate. Early efforts were limited to characterization of short Rev-derived peptides bound to the high affinity RRE stem loop IIB solved by NMR (Battiste et al., 1996, Gosser et al., 2001, Peterson and Feigon, 1996, Ye et al., 1996) (Fig. 1), while the apo stem loop IIB structure was elucidated using X-ray crystallography (Ippolito and Steitz, 2000) and NMR (Peterson and Feigon, 1996). While there are minor discrepancies between the various studies, they all provide a clear structural rationale for Rev's critical interaction with a non-canonical G:G/G:A base pair motif in the RRE, in what is thought to be the nucleating event of Rev oligomerization. However, it is known that the entire RRE is required for functionality in vivo (Hammarskjold and Rekosh, 2011). A more recent study using atomic force microscopy (AFM) characterized the full-length RRE by itself and bound to Rev protein (Pallesen et al., 2009), showing that multiple copies of Rev coated the entire RRE. Protein engineering efforts by the Frankel laboratory yielded Rev mutants whose oligomeric state was controlled, leading to a SAXS structure of a full-length Rev dimer bound to RRE stem loop IIB (Daugherty et al., 2010a). In the same study, the authors examined the full-length RRE:Rev complex by electron microscopy (EM), showing that the RNP assembly was consistent with a single hexameric Rev formed on one RRE. The finding is significant as it suggests an active role of RRE in determining the extent of Rev oligomerization; indeed, in the absence of RRE, Rev forms heterogeneous oligomeric complexes in solution (Daugherty et al., 2010a). Recently, SAXS was used to characterize a ∼230-nt RRE construct including stem loops IIB and IA, generating molecular envelopes and an ensemble of atomic models showing that the RRE fluctuates between “A” and “H” shaped conformations (Fang et al., 2013). Furthermore, the SAXS structure is consistent with the distance between the RNA binding domains of the Rev dimer (Daugherty et al., 2010b, DiMattia et al., 2010), suggesting a model where a Rev dimer interacts with both helices of the RRE to nucleate a controlled oligomerization.
Much of the work toward determining the tertiary structure of viral nuclear export signals has focused on HIV-1, but a few other viruses have been studied. The only 3D structure of the HIV-2 RRE comes from a homology model based on the SHAPE-probed secondary structure (Lusvarghi et al., 2013a); the structure resembles the “A”- and “H”-shaped folds observed for HIV-1 RRE (Fang et al., 2013). This result is consistent with the observation that HIV-1 Rev is capable of exporting RNA containing either the HIV-1 or HIV-2 RRE (Dillon et al., 1990). Additional 3D characterization of nuclear export elements from other viruses will be needed to understand the structural basis for why certain viruses are able to interact directly with host export machinery, while others require viral specific adaptor molecules.
2.5. Frameshift signals
Programmed ribosomal frameshifting is a common mechanism employed by viruses to expand their typically limited genetic information. For example, many retroviruses encode their pol or pro ORFs without a start codon. These genes must be translated as the product of a highly controlled-1 ribosomal-frameshifting event that leads to a certain percentage of the gag gene stop codon being shifted out-of-frame, so that the ribosome may continue and produce gag–pol or gag–pro proteins. Furthermore, precise control of frameshift gene product levels has been linked to optimal viral function. These kinds of controlled-frameshift events occur in a number of other viruses as well. Frameshift signals generally require two elements: a heptanucleotide “slippery sequence”, where the actual frameshifting occurs, and a structured RNA element 6–8 nt downstream that can stall the ribosome (the overall topic is reviewed in Dinman, 2012, Giedroc and Cornish, 2009). Here, we will focus on work characterizing the 3D structures of the RNA elements that induce frameshifting of the ribosome. Given their relatively small size (∼25–40 nt), there has been a great deal of success in high-resolution characterization of these elements.
Viral ribosome pausing elements generally fall into one of two structural categories: pseudoknot-containing elements and hyperstable stem loop structures. The Mouse mammary tumor virus (MMTV) was the first frameshift signal whose structure was determined at high resolution by NMR (Shen and Tinoco, 1995). This pseudoknot-containing frameshift signal was found to form two distinct coaxially-stacked helices punctuated by an intercalated adenosine that causes a distinct bend in the structure. Within the context of the MMTV system, this bend was shown to be critical for proper function, as mutations causing the pseudoknot to adopt a linear conformation (as confirmed by NMR) abrogated frameshifting activity (Chen et al., 1996). A crystal structure of the Beet western yellow virus (BWYV) frameshift pseudoknot solved to 1.6 Å resolution showed conservation of many of the structural elements observed in MMTV; the distinct bend and the characteristic triple-helix interactions were also present in the BWYV system (Egli et al., 2002, Su et al., 1999). A number of other viral frameshift signals have also been studied: NMR structures of the Pea enation mosaic virus (Nixon et al., 2002) and Sugarcane yellow leaf virus pseudoknots (Cornish et al., 2005), a crystal structure of the Potato leaf roll virus pseudoknot (Pallan et al., 2005), and a mass spectrometric three-dimensional (MS3D)-derived structure of the Feline immunodeficiency virus pseudoknot (Yu et al., 2005) have all been determined. While some differences exist in the nt composition and the specific interactions made, all of these models (determined by a variety of techniques) converge on a common overall fold; that is, one characterized by a distinctly bent conformation, and the presence of a triple helix formed by triple and quadruple base-pairing interactions. However, the bent pseudoknot motif is not universal, and several subsequent studies demonstrated that functional frameshift elements may adopt a linear conformation as well. NMR structures of the T2 bacteriophage pseudoknot (Du et al., 1996) and the Simian retrovirus-1 (SRV-1) frameshift signal (Du et al., 1997, Michiels et al., 2001) show this clearly. Nonetheless, these linear pseudoknots share many of the same features as their bent counterparts, such as a triple-helix motif.
The second class of frameshift elements is the hyperstable stem loop. HIV-1 is an example of this class and NMR structures of this frameshift signal have been solved (Gaudin et al., 2005, Staple and Butcher, 2005) (Fig. 1). While there was initially some controversy as to whether the HIV-1 frameshift element folded into a pseudoknot or discrete stem loop, these NMR structures unambiguously showed that only the stem loop conformation formed, at least in vitro in the absence of (viral) proteins. The stem loop folds into two distinct stems of roughly equal size that are bent by an internal bulge in a manner reminiscent of the bent-pseudoknot motif. An NMR structure of the Simian immunodeficiency virus (SIV) frameshift element was also reported, showing that while it too folds into a stem loop, it is not bent as is the case for HIV-1 (Marcheschi et al., 2007). Rather, the stem is coaxially stacked throughout and the apical-loop residues contain a number of H-bonding contacts, adding further stability.
Given the extremely diverse structural nature of frameshift elements, the question has naturally arisen as to what features govern the capability of these RNAs to cause the ribosome to stall and frameshifting to occur. The main hypotheses that have been favored are that structural stability of the frameshifting element, specific contacts with the ribosome by the frameshift element, or some combination of the two is critical for productive pausing of the ribosome. A consensus has not yet been reached (for more detailed discussion of this topic, see Firth and Brierley, 2012, Giedroc and Cornish, 2009). Clearly more structural data of frameshift elements bound to the ribosome would shed light on the debate. A low-resolution cryo-EM structure of an 80S ribosome paused at the Coronavirus frameshift pseudoknot contained bound elongation factor eEF2 in the A-site and an uncharacteristically bent tRNA at the P-site (Namy et al., 2006). This led the authors to propose a frameshifting mechanism based on the apparent strain that the pseudoknot was inducing within the ribosome. Higher-resolution structures of frameshift elements interacting with ribosomes are needed to establish whether there are any specific contacts made, or if the pausing is caused purely by intrinsic RNA stability.
2.6. Viral packaging signals
During the late stages of infection, viruses must assemble all of the required viral and host cell components and then egress from the cell. A variety of mechanisms exist to ensure that the correct proteins and nucleic acids are packaged into the nascent virions. Specific interactions occur between viral proteins and RNAs/DNAs in the assembly process, leading to packaging of the vRNA. In retroviruses, interactions occur between the Gag polyprotein and a structured element termed the Psi (Ψ)-packaging signal within the vRNA that lead to selective incorporation of vRNA into virions. Retroviruses are exquisitely capable of packaging only a single unspliced and dimerized vRNA despite the presence of a vast excess of cellular RNAs and spliced vRNAs; understanding the nature of this recognition has been intensely investigated but is still incompletely understood (reviewed in D'Souza and Summers, 2005, Kuzembayeva et al., 2014). The Moloney murine leukemia virus (MLV) core-packaging signal has been defined, and has been shown to direct encapsidation of heterologous vRNAs into which it is transplanted (D'Souza and Summers, 2005). Employing a segmental isotopic-labeling strategy, NMR structures of the ∼100-nt MLV Ψ-packaging signal in the apo (D'Souza et al., 2004) (Fig. 2 ) and NC-bound forms (D'Souza and Summers, 2004) have been determined. The apo structure revealed that two of the stem loops coaxially stacked with one another, forming an extended stem loop; the third stem loop formed independently and bifurcated the other two (D'Souza et al., 2004). The structure of the NC-RNA complex showed that a single NC molecule bound to a single-stranded region near the junction of the three stem loops that is only available if the packaging element is in the dimeric conformation, thus providing a structural explanation for the observation that only dimeric RNA is encapsidated (D'Souza and Summers, 2004). An NMR and cryo-ET characterization of dimeric MLV Ψ confirmed this result (Miyazaki et al., 2010). The minimal Rous sarcoma virus (RSV) Ψ element has also been identified, and an NMR structure of the 80-nt RNA bound to NC was reported (Zhou et al., 2007). Like MLV, the RSV-packaging element is composed of three stem loops, two of which are coaxially stacked, with the third forming independently. However, RSV NC was found to interact with two stem loops of the packaging signal via each of its zinc knuckles rather than at one site as in MLV.
Fig. 2.
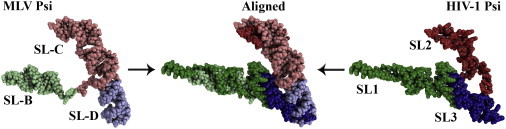
Comparison of HIV-1 and MLV Ψ packaging elements. The HIV-1 Ψ element determined by SAXS (darker colors) (Jones et al., 2014) closely resembles that of the MLV Ψ element solved using NMR (lighter colors) (D'Souza et al., 2004). The only significant deviation is located in the SL1/SL-B stem loops where HIV-1 has a longer arm. SL1/SL-B is shown in green shades, SL2/SL-C in blue shades and SL3/SL-D in red shades. HIV-1 stem loops are numbered and MLV stem loops are denoted with letters as per convention.
In contrast to both MLV and RSV, the minimal packaging signal of HIV-1 has been more difficult to identify. Conflicting reports exist as to whether the entire 5′UTR plus part of the gag ORF is required to encapsidate a heterologous RNA, or if a smaller region is sufficient (reviewed in D'Souza and Summers, 2005, Kuzembayeva et al., 2014). The general consensus seems to be that the entire 5′UTR region is necessary, but efforts to determine the minimal signal are still underway (Heng et al., 2012). Within the 5′UTR, four stem loop (SL) elements (SL1–SL4) are generally believed to be necessary for HIV-1 vRNA packaging. The structure of the individual stem loops of HIV-1 Ψ (SL1–SL3) in both the apo and NC-bound states have been extensively characterized by NMR (as reviewed in Lu et al., 2011b). A variety of studies using different techniques have attempted to elucidate the structure of the entire Ψ domain. An MS3D-based model of Ψ showed that the RNA adopts a compact conformation with tertiary contacts occurring between the apical loop of SL4 and an internal bulge within SL1 and the loops of SL2 and SL3 (Yu et al., 2008a). However, subsequent studies using Förster resonance energy transfer (FRET)-derived distance restraints and molecular dynamics (MD) (Stephenson et al., 2013), as well as SAXS in conjunction with MD and simulated annealing (Jones et al., 2014), suggested that Ψ lacked tertiary contacts, and that SL1 and SL3 coaxially stack on each other with SL2 bifurcating. Interestingly, there is a high degree of similarity between the overall structures of MLV and HIV-1 Ψ (Fig. 2), suggesting that packaging elements from a diverse set of retroviruses may share a conserved 3D shape. A structure of NC bound to the larger HIV-1 Ψ element has yet to be determined, so it is possible that the apoRNA fluctuates dynamically between a compact and open conformation, and that NC binding stabilizes one or the other.
Similar to the molecular switch observed in MLV Ψ, it has been proposed that the larger HIV-1 5′UTR alternates between a packaging-competent and non-packaging-competent conformation (Abbink and Berkhout, 2003, Abbink et al., 2005, Berkhout et al., 2002, Huthoff and Berkhout, 2001, Kenyon et al., 2013, Lu et al., 2011a, Ooms et al., 2004a, Ooms et al., 2004b, Seif et al., 2013). In the long-distance interaction (LDI) conformation, SL1 of Ψ interacts with an upstream region within the 5′UTR, preventing dimerization, while in the branched multiple-helix (BMH) conformation, SL1 is exposed. Efforts are underway to determine the structure of the 5′UTR in both conformations, and these results should further our understanding of why only dimeric vRNAs are packaged in HIV-1. AFM images of dimeric 5′UTRs suggest the formation of two intermolecular contacts, presumably between the Ψ SL1 dimerization initiation site (DIS) loops, and between the trans-activation region (TAR) loops (Andersen et al., 2004, Pallesen, 2011). A SAXS-derived 3D structure of the TAR and polyA hairpins (Fig. 1) is consistent with this possibility, as the stem loops coaxially stack and the TAR loop is available for intermolecular interactions (Jones et al., 2014). The same study showed that the SL1 hairpin loop of an HIV-1 Ψ variant that was mutated to prevent dimerization, is solvent exposed and available for intermolecular contacts. However, higher-resolution structures of the dimeric RNA will be required to confirm the presence and exact nature of these interactions.
3. Techniques for structural characterization of large functional viral RNAs and RNPs
Our understanding of the molecular mechanisms underlying viral replication is currently incomplete due to limited knowledge of the tertiary structure of genomic RNA elements and their complexes with host and viral proteins. The large size and inherent conformational flexibility of these functional RNAs and ribonucleoprotein complexes (RNPs) significantly reduces the effectiveness of conventional methods such as NMR and XRC. However, recent advances in methods and technology have made structural characterization of large RNAs and RNPs feasible. Comprehensive descriptions of the technologies have been reviewed extensively and will be referred to in the text. Here, we will focus on applications to large RNA structure determination. Each method is characterized by inherent size and resolution limitations (Fig. 3 ), as well as specific advantages, disadvantages, challenges and recent advances that will also be discussed.
Fig. 3.

General size and resolution limitations of common approaches to RNA structure determination. NMR spectroscopy can yield atomic-resolution (∼1–5 Å) structures, but has the most restrictive size limitations for RNA (∼3–35 kDa). XRC is also capable of atomic-resolution (∼1–5 Å) and can be readily applied to RNP structures up to 10 MDa with the caveat that they must be capable of forming large, well-ordered crystals. Cryo-EM has not been used specifically for RNA structure determination but has demonstrated the ability to achieve sub-nanometer resolution (up to 3 Å) in certain circumstances, however structures must be large enough to provide ample phase contrast (∼200 kDa to 20,000 MDa). SAXS bridges the gap between NMR and cryo-EM in size limitations (∼10 kDa to 3500 MDa) at the cost of resolution (currently ∼10–50 Å).
All of the methods reviewed here suffer from a few common challenges that must be overcome. First, sufficient quantities of the RNA of interest must be obtained in a pure form. In vitro transcription using T7 RNA polymerase generally provides efficient synthesis of RNAs up to ∼400–500 nt; however, since the polymerase commonly adds one (n + 1) or two (n + 2) extra nt to the 3′ end, yields can be significantly reduced by virtue of the requirement for RNAs with homogeneous ends. Denaturing polyacrylamide gel electrophoresis generally affords single-nt resolution of RNAs up to ∼50 nt (Lu et al., 2010, Reyes et al., 2009). Alternative tactics must be used for RNAs of larger size. Such approaches include the use of ribozyme cassettes flanking the sequence of interest (Batey and Kieft, 2007, Di Tomasso et al., 2012, Di Tomasso et al., 2011, Ferre-D’Amare and Doudna, 1996, Salvail-Lacoste et al., 2013, Walker et al., 2003), trans-acting ribozymes (Ferre-D’Amare and Doudna, 1996, Perrotta and Been, 1992), RNase H (Inoue et al., 1987, Lapham and Crothers, 1996), RNase P (Ziehler and Engelke, 1996) or trans-acting DNAzymes (Santoro and Joyce, 1997). Additionally, it has recently been shown that Marine cyanophage Syn5 RNA polymerase exhibits significantly reduced number of n + 1/n + 2 products during run-off transcription (Zhu et al., 2014). Second, proper folding of the RNA into a functional structure must be verified and alternative conformations rigorously removed from the sample. RNAs have a propensity to become trapped in local energy minima during folding, resulting in multiple misfolded forms, which has been referred to as “alternative conformer hell” (Fedor and Westhof, 2002, Uhlenbeck, 1995). In some cases, primarily with large RNAs, thermal denaturation/renaturation strategies may prove ineffective, requiring use of native purification (Batey and Kieft, 2007, Lukavsky and Puglisi, 2004, Toor et al., 2008) or even recombinant methods of overexpressing and purifying RNA from Escherichia coli (Nelissen et al., 2012). Mutations may be introduced to aid in sample preparation, but these changes should be vetted for function. Proper function of viral RNAs often hinges on their ability to bind viral and host proteins. Therefore, binding or other activity assays should be performed with all constructs prior to structural characterization. Also, when possible, mutations should be incorporated into viral infectivity assays. In some cases, nature has already performed this experiment through genetic diversity, highlighting the importance of comprehensive sequence analysis of viral isolates prior to structural experimentation.
3.1. Chemical modification, probing and comparative sequence analysis
While a detailed description of chemical-probing techniques is outside the scope of this review (for comprehensive reviews, see Weeks, 2010 and Lu et al., 2011b), such studies contribute important details needed for efficient tertiary structure determination. Chemical probing has been used to identify backbone flexibility and base-pairing interactions at single-nt resolution (Merino et al., 2005), leading to determination of secondary structure. However, although standardized protocols have been published over the past three decades (Low and Weeks, 2010, Lusvarghi et al., 2013b, McGinnis et al., 2009, Mortimer and Weeks, 2009, Steen et al., 2011, Wilkinson et al., 2006), unambiguous secondary structure assignment remains elusive for many RNAs largely due to RNA conformational heterogeneity and dynamics, with the 5′UTR of HIV-1 as a prime example (Lu et al., 2011b).
Recent successes in deriving the secondary structures and conformational changes of viral RNA genomes illustrates the usefulness of SHAPE probing (Archer et al., 2013, Grohman et al., 2013, Watts et al., 2009, Wilkinson et al., 2006). An initial estimate suggested that the addition of SHAPE-derived pseudo-free energy parameters to nearest neighbor free energy terms improved secondary structure prediction to 96–100% accuracy (Deigan et al., 2009). However, more recent rigorous benchmarking has revealed more modest improvements (false negative rates near 17% and false discovery rate near 20%) (Kladwang et al., 2011). Despite inconsistencies between probing data and high-resolution structures (Miyazaki et al., 2010), methods to improve accuracy are ongoing. These include the use of differential SHAPE methods, where reactivity differences between two reagents are used to correct for inherent biases of each reagent (Rice et al., 2014). Another approach has been to use dilution as a means of normalizing different data sets based on an internal standard, such as the number of full-length primer extension products (Kladwang et al., 2014). As with many other structural techniques, even low levels of sample heterogeneity can severely affect the accuracy of SHAPE-derived secondary structure models. To mitigate this effect and offer the ability to examine different conformers within a heterogeneous population, in-gel SHAPE has been developed (Kenyon et al., 2013). Briefly, a polydisperse sample is separated by native polyacrylamide electrophoresis followed by excision of each conformer, which can then be probed within the gel matrix.
Despite certain instances of uncertainty, chemical probing offers critical information toward efficient tertiary structure determination. First, knowledge of secondary structure allows for construction of RNAs that either retain or disrupt functional base-pairing networks. In instances of structural heterogeneity, mutations can also be introduced to stabilize desired structures or destabilize alternative, nonfunctional conformations. Also, regions of highly flexible residues correlate with difficulty in determining tertiary structure. These residues can be stabilized or eliminated from constructs to improve the quality of structural data. Finally, probing techniques can be used to generate data for direct use in molecular modeling of tertiary structures. All current methods of 3D structure determination require some degree of molecular modeling. For instance, XRC requires building a model into an electron density map that is further refined. Structure calculation from NMR involves generation of an energy-minimized model using NMR-derived distance and angle restraints. Chemical-probing data can similarly be used to constrain molecular models. For example, quantitative measures of solvent accessibility can be obtained from hydroxyl-radical probing (Pastor et al., 2000, Tullius and Greenbaum, 2005), greatly constraining tertiary structure models. Direct distance restraints can also be probed using bifunctional crosslinking analyzed by mass spectrometry, also known as MS3D (Fabris and Yu, 2010, Yu et al., 2008a, Yu et al., 2008b, Yu et al., 2005). Caution must be taken in analyzing and interpreting crosslinking data, as transient conformations may inadvertently be trapped, obscuring the structure of the RNA.
In addition to chemical-probing methods, nucleic acid secondary structures can also be assessed through phylogenetic and thermodynamic approaches (reviewed in Seetin and Mathews, 2012). Indeed, one of the most successful methods of secondary-structure prediction is comparative sequence analysis (CSA). CSA is predicated on the notion that structural conservation supersedes that of primary sequence conservation. Here, polymorphisms within a large number of homologous sequences can be used to differentiate between paired or unpaired residues using covariation analysis (Eddy and Durbin, 1994). Indeed, application of CSA to ribosomal RNAs was strikingly accurate (∼97%) (Gutell et al., 2002). A caveat of this technique is the need for large datasets of homologous sequences. Such large databases exist for many viral genomic RNAs such as HIV (Foley et al., 2013) and HCV (Kuiken et al., 2008). In cases where many sequence homologs are unknown or as a stand-alone method, secondary structure can be predicted based on energy minimization using the nearest neighbor model. Several tools such as MFold (Zuker, 2003) and RNA structure (Bellaousov et al., 2013, Reuter and Mathews, 2010) are freely available for automation of such calculations. As with all models, predicted secondary-structure models must be experimentally scrutinized for accuracy. For example, site-directed mutagenesis and compensatory mutation can be used to further validate predictions. Additionally, in vivo SELEX (systematic evolution of ligands by exponential enrichment), whereby specific nt in a proviral plasmid are randomized and transfected into cells followed by multiple rounds of viral replication, can also be used to identify the importance of different structural elements in viral infectivity (reviewed in Berkhout and Das, 2009). Similarly, forced-evolution experiments in which mutated viruses with replication defects are used as parents for identification of spontaneous reversions that result in partial or full rescue of replication efficiency (Abbink and Berkhout, 2008, Berkhout and Das, 2009, Berkhout et al., 1997, Klaver and Berkhout, 1994), can reveal important insights into viral RNA structure.
While it is important to consider RNA structure in order to understand how various functions are carried out, most viral RNA functions require proteins. Therefore, deciphering the mechanisms of RNA action in viral biology requires a structural rationale for how viral RNA and proteins interact to perform a specific function. While many of the methods discussed below can be applied to RNA:protein systems, additional approaches can be used to further characterize the RNPs. Here restraints based on probing of specific amino acids in the protein followed by identification using mass spectrometry can be helpful. This technique has been used in a viral context previously to identify surface lysines on HIV-1 reverse transcriptase that are protected from covalent modification upon binding to the tRNA-primed viral genome (Kvaratskhelia et al., 2002). In addition many other reagents have been identified that can be used to specifically probe carboxylic acids, cysteine, histidine, tryptophan and tyrosine residues (Mendoza and Vachet, 2009). Due to the stringency of some of these reagents and the conditions in which they are active, care should be taken to ensure that neither the modifying reagents nor the conditions appreciably affect the properties of the RNP (Kvaratskhelia et al., 2002, Shell et al., 2005, Shkriabai et al., 2006).
3.2. X-ray crystallography
In honor of the centenary anniversary of the derivation of Bragg's Law (Bragg, 1913), detailed reviews discussing current and proposed technological advances in XRC are abundant (Garman, 2014, Howard and Probert, 2014, Miller, 2014, Zhang and Ferre-D’Amare, 2014). For large RNA-only structures, XRC has been a workhorse, accounting for 138 of the 142 entries in the protein data bank (PDB) larger than 100 nt (Fig. 3). In fact, XRC is the only atomic-resolution method that has been used independently to solve an RNA-only structure larger than 101 nt. However, due to complications in crystallizing RNA such as high intrinsic flexibility and poor packing tendencies arising from the repulsive nature of polyanionic molecules (Holbrook and Kim, 1997), many RNAs are difficult to crystallize and resolution tends to wane as a function of RNA size (Fig. 4A). Obtaining well-ordered crystals for large RNAs can be exceedingly difficult; therefore, construct design is a critical first step. Careful inspection of phylogenetic variation aids in the design of constructs that effectively sample sequence space and sometimes brute-force trial-and-error of numerous variants is required. Such analyses were important factors that led to determination of RNase P RNA (Kazantsev et al., 2005), the glmS ribozyme (Klein and Ferre-D’Amare, 2006), and a number of riboswitches (Garst et al., 2008, Johnson et al., 2012b, Liberman et al., 2013, Montange and Batey, 2006, Spitale et al., 2009). Other considerations for construct design include defining domain boundaries (Correll et al., 1997), incorporating stable tetraloops (Pley et al., 1994, Scott et al., 1995) and addition of crystallization modules designed to make intermolecular contacts (Ferre-D’Amare et al., 1998b). Finally, favorable crystal contacts can be introduced by engineering hairpins into RNA constructs that are specifically recognized by small crystallization chaperone proteins such as U1A (Ferre-D’Amare and Doudna, 2000, Ferre-D’Amare et al., 1998a) and monoclonal antibody fragments (Koldobskaya et al., 2011, Piccirilli and Koldobskaya, 2011).
Fig. 4.
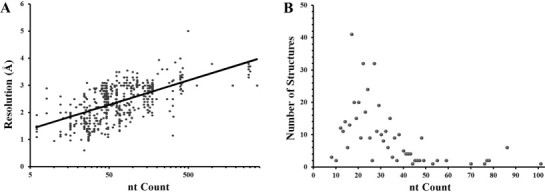
Survey of RNA-only structures from the Protein Data Bank solved using XRC and NMR. (A) XRC structure resolution as a function of the log10 of the number of nt in each structure fit to a logarithmic trendline. (B) The distribution of NMR structures by nt count reveals a grouping of structures centered near 24 nt with a scarcity of RNA structures larger than 40 residues.
Advances at synchrotron facilities are continually improving both the quality of data and the ability to solve structures of samples once thought to be intractable to XRC. An emphasis on automation, from sample preparation and data collection to image processing and model refinement, is creating a more user-friendly experience. In addition to hardware improvements, software development has led to programs (such as HKL-3000 (Minor et al., 2006), PHENIX (Adams et al., 2004, Adams et al., 2010, Adams et al., 2011, Afonine et al., 2012) and the Auto-Rickshaw web server (Panjikar et al., 2005, Panjikar et al., 2009)) with the ability to perform data reduction, phasing, generation of electron density maps and model building in a very short period of time and with minimal user intervention. On the hardware side, as improvements in technology using traditional storage ring sources continue, the advent of X-ray free-electron lasers (XFELs) promises to revolutionize the field by enabling structure determination from macromolecules unable to form large, highly-ordered crystals and monitoring dynamic molecular motions at femtosecond timescale resolution (Aquila et al., 2012, Neutze and Moffat, 2012). XFELs can produce femtosecond-duration pulses with ∼1012 photons at high energy (Emma et al., 2010). The feasibility of these types of studies has been demonstrated for known structures (Boutet et al., 2012, Chapman et al., 2011, Demirci et al., 2013, Kern et al., 2012, Kern et al., 2013, Redecke et al., 2013) and for de novo structure determination (Barends et al., 2014). The advantages of using XFELs parallel the needs of RNA XRC. For example, time-resolved diffraction is advantageous for analysis of dynamic molecules. Also, XFELs are ideally suited to analysis of micro/nanocrystals of RNA constructs for which large crystals cannot be grown.
3.3. Nuclear magnetic resonance spectroscopy
NMR-restrained modeling has produced 442 RNA-only atomic-resolution structures in the PDB ranging from 8 to 101 nt in length. However, despite this large range of sizes, the median structure is only 24 nt (Fig. 3, Fig. 4). Even for conformationally homogeneous RNAs, NMR characterization of large RNAs suffers many drawbacks (Tolbert et al., 2010). (1) Large RNAs consisting of multiple secondary-structure elements require long-range restraints on relative interhelical orientations, but interproton NOEs are only useful over very short distances (<5 Å). (2) Only four different residue types are present in RNA leading to poor chemical shift dispersion; indeed, the ribose moiety of all four residues is chemically identical. Therefore, as the size of the RNA increases, spectral overlap eventually renders unambiguous resonance assignments impossible. (3) Compared to protein, RNA is decidedly less proton rich. (4) RNAs tend to be less globular than proteins, resulting in extended structures with slower tumbling times. This further exacerbates the signal loss due to proportionally shorter transverse relaxation times as molecular mass increases.
To alleviate many of these shortcomings, new experimental strategies have been developed. A detailed description of these techniques is outside the scope of this review, but have been reviewed (Fernandez and Wider, 2003, Gao, 2013, Lukavsky and Puglisi, 2005, Tzakos et al., 2006). Briefly, isotope-labeling strategies can be used to partially alleviate spectral overlap. This can be achieved either through the combination of labeled and unlabeled NTPs during in vitro transcription reactions, or through segmental labeling and splint-mediated ligation (Lu et al., 2010). For both of these methods, commercial sources are available for uniformly-labeled NTPs. Also, selectively deuterated and 13C-enriched NTPs can be chemically synthesized (Davis et al., 2005, Scott et al., 2000). In addition to mitigating spectral overlap, residual dipolar couplings (RDCs) can be measured on selectively-labeled RNAs, yielding relative interdomain angular restraints (Mollova et al., 2000). Finally, transverse relaxation-optimized spectroscopy (TROSY) significantly reduces the line broadening resulting from short transverse relaxation times in slowly tumbling RNAs (Pervushin et al., 1997). Overall, although many large RNAs remain intractable for atomic-resolution NMR characterization, improvements are continuing to push past size barriers. For sequences up to ∼100 nt in length, NMR remains a viable option for solution-based structure determination of many RNAs.
3.4. Cryo-electron microscopy
Due to inherent size limitations, single-particle cryo-electron microscopic techniques have been applied only sparingly to RNAs. Therefore the current advantages and limitations of this technique will only be briefly described in the context of how it may be used in future studies. For more detailed reviews, see (Baker et al., 2010, Zhou, 2011). For cryo-EM, the signal-to-noise ratio increases with particle size, making it ideal for structure determination of large macromolecular complexes (Allegretti et al., 2014, Amunts et al., 2014, Fernandez et al., 2014, Liao et al., 2013). Currently, the limit of detection for sub-nanomolar resolution structure determination is ∼200 kDa (roughly equivalent to 550–600 nt), significantly larger than most of the viral RNAs described in this review (Fig. 3). However, technology is currently being pursued to improve phase contrast for smaller molecules to reduce the size barrier to 100 kDa and below (Wu et al., 2013). Despite the size limitations, many RNAs of this size would be beneficial to study. Indeed, with recent advances in cryo-EM technology, such as software for automatic post picking (Nicholson and Glaeser, 2001, Norousi et al., 2013), correction methods for beam-induced particle movement (Bai et al., 2013, Shigematsu and Sigworth, 2013), introduction of improved phase plates (Wu et al., 2013) and development of direct electron detectors (Grigorieff, 2013, McMullan et al., 2009a, McMullan et al., 2009b, McMullan et al., 2009c), near-atomic resolution can be achieved for macromolecules and complexes >200 kDa. For example, genome dimerization mediated by sequences and structures within the 5′UTR of many retroviruses are a key checkpoint regulating genomic RNA packaging; however, little is known of the structural mechanisms of dimerization. Dimers of retroviral 5′UTR constructs could, in principle, be designed to be large enough for cryo-EM analysis. Additionally, protein components known to regulate dimerization, such as Gag, could also be characterized in an RNA-bound complex.
Although cryo-EM has the capacity to achieve sub-nanomolar resolution for large macromolecules, cryo-EM and cryo-electron tomography (cryo-ET) techniques can also be used in combination with NMR spectroscopy to characterize the structures of smaller (<200 kDa) molecules. Briefly, as described in Section 3.3, a key strength of NMR spectroscopy is the ability to identify short-range distances within macromolecules; however, detection of long-range interhelical restraints such as RDCs is technically difficult and requires conformational homogeneity. Cryo-EM/ET techniques can overcome the lack of long-range distance restraints inherent to NMR. The validity of this hybrid NMR/cryo-EM/ET approach has been demonstrated for the structure of the Moloney murine leukemia virus packaging element (Miyazaki et al., 2010). For this system, alternative techniques such as measuring RDCs or SAXS were unsuccessful due to sample heterogeneity. Single-particle cryo-EM/ET methods have the powerful advantage of removing conformational subpopulations from analysis.
3.5. Small-angle X-ray scattering
As described in a recent review, the popularity of SAXS applied to biomolecules has been steadily increasing over the past decade (Graewert and Svergun, 2013). The upsurge in SAXS use can be partially attributed to key advantages of SAXS over other techniques. For instance, SAXS is a true solution technique that can be performed under biologically relevant conditions. Additionally, SAXS does not require generation of ordered crystals or incorporation of labels and does not have intrinsic size limitations (Fig. 3). In the current state of the field, however, structural information for large RNAs is limited to low resolution (∼10–50 Å), yielding only general architectural features such as orientations of helices or large conformational changes.
Also contributing to the increased popularity of SAXS (and further bolstering its usage) has been the development of improved hardware and software systems that facilitate automated, high-throughput data collection (Franke et al., 2012, Hura et al., 2009, Martel et al., 2012) and comprehensive data analysis (Blanchet et al., 2012, Forster et al., 2010, Grant et al., 2011, Konarev et al., 2006). As for XRC, the push for speed and quality in SAXS data collection has been further aided by the improved performance of single-photon pixel detectors such as the PILATUS line (Kraft et al., 2009). The recently developed successor to the PILATUS detectors, EIGER, promises to reduce pixel size and readout dead time while enhancing frame rates (Johnson et al., 2012a). Similarly, software improvements have begun to tackle key issues. A powerful application of SAXS data is in the evaluation of structural models or the validation of crystal structures in a more biologically relevant context (in solution without crystal packing influences); however, complications in evaluating the contribution of solvent has reduced the validity of theoretical scattering profiles, especially at wider angles (WAXS). This becomes important to improving the resolution limits of SAXS/WAXS experiments because incorporation of high quality wide angle data has the potential to yield sub-nanomolar resolution structural information (Kofinger and Hummer, 2013). Attempts to alleviate this issue have come in the form of three new algorithms that use all-atom explicit solvent MD simulations (Virtanen et al., 2010, Virtanen et al., 2011), Zernike expansion (Liu et al., 2012) and Poisson–Boltzmann–Langevin formalism (Poitevin et al., 2011) to improve the quality of theoretical scattering curves by incorporating solvent effects more rigorously.
The low-resolution nature of current SAXS technology and the requirement for highly homogenous samples limits its potential as a stand-alone technique. However, in combination with complementary methods, SAXS data can help to refine structural models for RNAs that are poorly suited to XRC or NMR. In a divide-and-conquer strategy, where NMR or XRC is used to determine atomic-resolution structures of short segments composing a larger RNA, SAXS can provide important long-range and global structure information for rigid-body docking to determine how the segments fit together in the context of the full macromolecule. While this has been used scarcely for large RNA complexes (Zuo et al., 2008), the feasibility of this strategy has been demonstrated for large, multidomain proteins such as CaMKII (Rosenberg et al., 2005), the hepatocyte growth factor/scattering factor (Gherardi et al., 2006), MLV Gag (Datta et al., 2011), HIV-1 gag (Datta et al., 2007) and the multi-aminoacyl-tRNA synthetase complex (Dias et al., 2013).
While rigid-body docking is applicable in certain systems, SAXS-derived de novo structure characterization requires more complex modeling techniques. Molecular modeling techniques using only SAXS data currently require the ability to produce starting structures with matching overall architectural features. Over the past decade, significant progress has been made in the field of RNA 3D structure prediction leading to freely-available software and web servers (Das and Baker, 2007, Flores et al., 2011, Jonikas et al., 2009, Parisien and Major, 2008, Popenda et al., 2012, Sharma et al., 2008). De novo models can then be evaluated by calculating theoretical scattering profiles to find the best starting structure for modeling. Depending on the amount of desired conformational space sampling, different methods of MD simulation (replica exchange, accelerated, course-grain, simulated annealing, Monte Carlo, etc.) can be used to refine the starting model by calculating theoretical SAXS data from each sampled conformation (Jones et al., 2014). Also, simulated annealing protocols can be supplemented with SAXS-derived energy restraints (Fang et al., 2013, Jones et al., 2014). Indeed, other restraints, such as secondary structure and RDCs from NMR experiments can also be used (Burke et al., 2012, Fang et al., 2013, Grishaev et al., 2008, Hennig and Sattler, 2014, Jones et al., 2014, Lipfert et al., 2008, Wang et al., 2009a, Wang et al., 2010, Zuo et al., 2010). As SAXS/WAXS technology progresses, software improvements should be directed toward more seamless incorporation of experimental restraints such as FRET distances, chemical/enzymatic probing results and NOE and RDC data from NMR experiments, thus providing a more high-throughput and user-friendly method.
4. Concluding remarks
Our understanding of viral replication has benefited from recent methodological and technological advances aimed at visualizing RNA structure. Many viral RNAs of functional importance such as IRESes, CITEs, TLSs and nuclear export, frameshifting and packaging signals have been studied; however, the field of viral RNA structural biology is still at a stage of development and our most successful high-resolution structures to date have been limited to small RNAs. Despite the limited number of RNAs studied, high-resolution structures and structural models have improved our understanding of viral replication and molecular biology. Continuing progress will hinge on expanding our abilities to solve large, functional RNA domains. In this review, we have demonstrated that such studies are possible and highlighted the techniques that are at the forefront of current capabilities. For example, studies that combine different techniques such as divide-and-conquer approaches and conflation of SAXS and NMR methods will be successful for many RNAs. Technological advances in SAXS and cryo-EM hold promise for improving resolution of large RNA structures. Careful application of new technologies, with an eye toward their limitations will provide a structural rationale for the many open-ended questions that continue to pervade viral molecular biology.
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grant R01 AI077387 (to K.M.-F. and Dr. L. Kleiman). E.D.O. is supported by The Ohio State University Chemistry-Biology Interface Program training grant and W.A.C. by the Pelotonia Postdoctoral Fellowship Program.
References
- Abbink T.E., Berkhout B. A novel long distance base-pairing interaction in human immunodeficiency virus type 1 RNA occludes the Gag start codon. J. Biol. Chem. 2003;278(13):11601–11611. doi: 10.1074/jbc.M210291200. [DOI] [PubMed] [Google Scholar]
- Abbink T.E., Berkhout B. RNA structure modulates splicing efficiency at the human immunodeficiency virus type 1 major splice donor. J. Virol. 2008;82(6):3090–3098. doi: 10.1128/JVI.01479-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbink T.E., Ooms M., Haasnoot P.C., Berkhout B. The HIV-1 leader RNA conformational switch regulates RNA dimerization but does not regulate mRNA translation. Biochemistry. 2005;44(25):9058–9066. doi: 10.1021/bi0502588. [DOI] [PubMed] [Google Scholar]
- Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C., Richardson J.S., Terwilliger T.C., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Echols N., Headd J.J., Hung L.W., Jain S., Kapral G.J., Grosse Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R.D., Read R.J., Richardson D.C., Richardson J.S., Terwilliger T.C., Zwart P.H. The Phenix software for automated determination of macromolecular structures. Methods. 2011;55(1):94–106. doi: 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P.D., Gopal K., Grosse-Kunstleve R.W., Hung L.W., Ioerger T.R., McCoy A.J., Moriarty N.W., Pai R.K., Read R.J., Romo T.D., Sacchettini J.C., Sauter N.K., Storoni L.C., Terwilliger T.C. Recent developments in the PHENIX software for automated crystallographic structure determination. J. Synchrotron Radiat. 2004;11(Pt 1):53–55. doi: 10.1107/s0909049503024130. [DOI] [PubMed] [Google Scholar]
- Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2012;68(Pt 4):352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegretti M., Mills D.J., McMullan G., Kuhlbrandt W., Vonck J. Atomic model of the F420-reducing [NiFe] hydrogenase by electron cryo-microscopy using a direct electron detector. eLife. 2014;3:e01963. doi: 10.7554/eLife.01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunts A., Brown A., Bai X.C., Llacer J.L., Hussain T., Emsley P., Long F., Murshudov G., Scheres S.H., Ramakrishnan V. Structure of the yeast mitochondrial large ribosomal subunit. Science. 2014;343(6178):1485–1489. doi: 10.1126/science.1249410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen E.S., Contera S.A., Knudsen B., Damgaard C.K., Besenbacher F., Kjems J. Role of the trans-activation response element in dimerization of HIV-1 RNA. J. Biol. Chem. 2004;279(21):22243–22249. doi: 10.1074/jbc.M314326200. [DOI] [PubMed] [Google Scholar]
- Aquila A., Hunter M.S., Doak R.B., Kirian R.A., Fromme P., White T.A., Andreasson J., Arnlund D., Bajt S., Barends T.R., Barthelmess M., Bogan M.J., Bostedt C., Bottin H., Bozek J.D., Caleman C., Coppola N., Davidsson J., DePonte D.P., Elser V., Epp S.W., Erk B., Fleckenstein H., Foucar L., Frank M., Fromme R., Graafsma H., Grotjohann I., Gumprecht L., Hajdu J., Hampton C.Y., Hartmann A., Hartmann R., Hau-Riege S., Hauser G., Hirsemann H., Holl P., Holton J.M., Homke A., Johansson L., Kimmel N., Kassemeyer S., Krasniqi F., Kuhnel K.U., Liang M., Lomb L., Malmerberg E., Marchesini S., Martin A.V., Maia F.R., Messerschmidt M., Nass K., Reich C., Neutze R., Rolles D., Rudek B., Rudenko A., Schlichting I., Schmidt C., Schmidt K.E., Schulz J., Seibert M.M., Shoeman R.L., Sierra R., Soltau H., Starodub D., Stellato F., Stern S., Struder L., Timneanu N., Ullrich J., Wang X., Williams G.J., Weidenspointner G., Weierstall U., Wunderer C., Barty A., Spence J.C., Chapman H.N. Time-resolved protein nanocrystallography using an X-ray free-electron laser. Opt. Express. 2012;20(3):2706–2716. doi: 10.1364/OE.20.002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer E.J., Simpson M.A., Watts N.J., O’Kane R., Wang B., Erie D.A., McPherson A., Weeks K.M. Long-range architecture in a viral RNA genome. Biochemistry. 2013;52(18):3182–3190. doi: 10.1021/bi4001535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X.C., Fernandez I.S., McMullan G., Scheres S.H. Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. eLife. 2013;2:e00461. doi: 10.7554/eLife.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M.L., Zhang J., Ludtke S.J., Chiu W. Cryo-EM of macromolecular assemblies at near-atomic resolution. Nat. Protoc. 2010;5(10):1697–1708. doi: 10.1038/nprot.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balvay L., Lopez Lastra M., Sargueil B., Darlix J.L., Ohlmann T. Translational control of retroviruses. Nat. Rev. Microbiol. 2007;5(2):128–140. doi: 10.1038/nrmicro1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barends T.R., Foucar L., Botha S., Doak R.B., Shoeman R.L., Nass K., Koglin J.E., Williams G.J., Boutet S., Messerschmidt M., Schlichting I. De novo protein crystal structure determination from X-ray free-electron laser data. Nature. 2014;505(7482):244–247. doi: 10.1038/nature12773. [DOI] [PubMed] [Google Scholar]
- Barozzi P., Potenza L., Riva G., Vallerini D., Quadrelli C., Bosco R., Forghieri F., Torelli G., Luppi M. B cells and herpesviruses: a model of lymphoproliferation. Autoimmun. Rev. 2007;7(2):132–136. doi: 10.1016/j.autrev.2007.02.018. [DOI] [PubMed] [Google Scholar]
- Batey R.T., Kieft J.S. Improved native affinity purification of RNA. RNA. 2007;13(8):1384–1389. doi: 10.1261/rna.528007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battiste J.L., Mao H., Rao N.S., Tan R., Muhandiram D.R., Kay L.E., Frankel A.D., Williamson J.R. Alpha helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science. 1996;273(5281):1547–1551. doi: 10.1126/science.273.5281.1547. [DOI] [PubMed] [Google Scholar]
- Bellaousov S., Reuter J.S., Seetin M.G., Mathews D.H. RNAstructure: web servers for RNA secondary structure prediction and analysis. Nucl. Acids Res. 2013;41(Web Server issue):W471–W474. doi: 10.1093/nar/gkt290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B., Arts K., Abbink T.E. Ribosomal scanning on the 5′-untranslated region of the human immunodeficiency virus RNA genome. Nucl. Acids Res. 2011;39(12):5232–5244. doi: 10.1093/nar/gkr113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B., Das A.T. Virus evolution as a tool to study HIV-1 biology. Methods Mol. Biol. 2009;485:436–451. doi: 10.1007/978-1-59745-170-3_30. [DOI] [PubMed] [Google Scholar]
- Berkhout B., Klaver B., Das A.T. Forced evolution of a regulatory RNA helix in the HIV-1 genome. Nucl. Acids Res. 1997;25(5):940–947. doi: 10.1093/nar/25.5.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B., Ooms M., Beerens N., Huthoff H., Southern E., Verhoef K. In vitro evidence that the untranslated leader of the HIV-1 genome is an RNA checkpoint that regulates multiple functions through conformational changes. J. Biol. Chem. 2002;277(22):19967–19975. doi: 10.1074/jbc.M200950200. [DOI] [PubMed] [Google Scholar]
- Berry K.E., Waghray S., Mortimer S.A., Bai Y., Doudna J.A. Crystal structure of the HCV IRES central domain reveals strategy for start-codon positioning. Structure. 2011;19(10):1456–1466. doi: 10.1016/j.str.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet C.E., Zozulya A.V., Kikhney A.G., Franke D., Konarev P.V., Shang W., Klaering R., Robrahn B., Hermes C., Cipriani F., Svergun D.I., Roessle M. Instrumental setup for high-throughput small- and wide-angle solution scattering at the X33 beamline of EMBL Hamburg. J. Appl. Crystallogr. 2012;45(3):489–495. [Google Scholar]
- Boehringer D., Thermann R., Ostareck-Lederer A., Lewis J.D., Stark H. Structure of the hepatitis C virus IRES bound to the human 80S ribosome: remodeling of the HCV IRES. Structure. 2005;13(11):1695–1706. doi: 10.1016/j.str.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Boutet S., Lomb L., Williams G.J., Barends T.R., Aquila A., Doak R.B., Weierstall U., DePonte D.P., Steinbrener J., Shoeman R.L., Messerschmidt M., Barty A., White T.A., Kassemeyer S., Kirian R.A., Seibert M.M., Montanez P.A., Kenney C., Herbst R., Hart P., Pines J., Haller G., Gruner S.M., Philipp H.T., Tate M.W., Hromalik M., Koerner L.J., van Bakel N., Morse J., Ghonsalves W., Arnlund D., Bogan M.J., Caleman C., Fromme R., Hampton C.Y., Hunter M.S., Johansson L.C., Katona G., Kupitz C., Liang M., Martin A.V., Nass K., Redecke L., Stellato F., Timneanu N., Wang D., Zatsepin N.A., Schafer D., Defever J., Neutze R., Fromme P., Spence J.C., Chapman H.N., Schlichting I. High-resolution protein structure determination by serial femtosecond crystallography. Science. 2012;337(6092):362–364. doi: 10.1126/science.1217737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg W.L. The diffraction of short electromagnetic waves by a crystal. Proc. Camb. Philos. Soc. 1913;17:43–57. [Google Scholar]
- Brasey A., Lopez-Lastra M., Ohlmann T., Beerens N., Berkhout B., Darlix J.L., Sonenberg N. The leader of human immunodeficiency virus type 1 genomic RNA harbors an internal ribosome entry segment that is active during the G2/M phase of the cell cycle. J. Virol. 2003;77(7):3939–3949. doi: 10.1128/JVI.77.7.3939-3949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck C.B., Shen X., Egan M.A., Pierson T.C., Walker C.M., Siliciano R.F. The human immunodeficiency virus type 1 gag gene encodes an internal ribosome entry site. J. Virol. 2001;75(1):181–191. doi: 10.1128/JVI.75.1.181-191.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke J.E., Sashital D.G., Zuo X., Wang Y.X., Butcher S.E. Structure of the yeast U2/U6 snRNA complex. RNA. 2012;18(4):673–683. doi: 10.1261/rna.031138.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascalho M., Platt J.L. Novel functions of B cells. Crit. Rev. Immunol. 2007;27(2):141–151. doi: 10.1615/critrevimmunol.v27.i2.20. [DOI] [PubMed] [Google Scholar]
- Chapman H.N., Fromme P., Barty A., White T.A., Kirian R.A., Aquila A., Hunter M.S., Schulz J., DePonte D.P., Weierstall U., Doak R.B., Maia F.R., Martin A.V., Schlichting I., Lomb L., Coppola N., Shoeman R.L., Epp S.W., Hartmann R., Rolles D., Rudenko A., Foucar L., Kimmel N., Weidenspointner G., Holl P., Liang M., Barthelmess M., Caleman C., Boutet S., Bogan M.J., Krzywinski J., Bostedt C., Bajt S., Gumprecht L., Rudek B., Erk B., Schmidt C., Homke A., Reich C., Pietschner D., Struder L., Hauser G., Gorke H., Ullrich J., Herrmann S., Schaller G., Schopper F., Soltau H., Kuhnel K.U., Messerschmidt M., Bozek J.D., Hau-Riege S.P., Frank M., Hampton C.Y., Sierra R.G., Starodub D., Williams G.J., Hajdu J., Timneanu N., Seibert M.M., Andreasson J., Rocker A., Jonsson O., Svenda M., Stern S., Nass K., Andritschke R., Schroter C.D., Krasniqi F., Bott M., Schmidt K.E., Wang X., Grotjohann I., Holton J.M., Barends T.R., Neutze R., Marchesini S., Fromme R., Schorb S., Rupp D., Adolph M., Gorkhover T., Andersson I., Hirsemann H., Potdevin G., Graafsma H., Nilsson B., Spence J.C. Femtosecond X-ray protein nanocrystallography. Nature. 2011;470(7332):73–77. doi: 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charnay N., Ivanyi-Nagy R., Soto-Rifo R., Ohlmann T., Lopez-Lastra M., Darlix J.L. Mechanism of HIV-1 Tat RNA translation and its activation by the Tat protein. Retrovirology. 2009;6:74. doi: 10.1186/1742-4690-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Kang H., Shen L.X., Chamorro M., Varmus H.E., Tinoco I., Jr. A characteristic bent conformation of RNA pseudoknots promotes-1 frameshifting during translation of retroviral RNA. J. Mol. Biol. 1996;260(4):479–483. doi: 10.1006/jmbi.1996.0415. [DOI] [PubMed] [Google Scholar]
- Colussi T.M., Costantino D.A., Hammond J.A., Ruehle G.M., Nix J.C., Kieft J.S. The structural basis of transfer RNA mimicry and conformational plasticity by a viral RNA. Nature. 2014 doi: 10.1038/nature13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish P.V., Hennig M., Giedroc D.P. A loop 2 cytidine-stem 1 minor groove interaction as a positive determinant for pseudoknot-stimulated-1 ribosomal frameshifting. Proc. Natl. Acad. Sci. U.S.A. 2005;102(36):12694–12699. doi: 10.1073/pnas.0506166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correll C.C., Freeborn B., Moore P.B., Steitz T.A. Metals, motifs, and recognition in the crystal structure of a 5S rRNA domain. Cell. 1997;91(5):705–712. doi: 10.1016/s0092-8674(00)80457-2. [DOI] [PubMed] [Google Scholar]
- Costantino D.A., Pfingsten J.S., Rambo R.P., Kieft J.S. tRNA–mRNA mimicry drives translation initiation from a viral IRES. Nat. Struct. Mol. Biol. 2008;15(1):57–64. doi: 10.1038/nsmb1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza V., Dey A., Habib D., Summers M.F. NMR structure of the 101-nucleotide core encapsidation signal of the Moloney Murine Leukemia virus. J. Mol. Biol. 2004;337(2):427–442. doi: 10.1016/j.jmb.2004.01.037. [DOI] [PubMed] [Google Scholar]
- D'Souza V., Summers M.F. Structural basis for packaging the dimeric genome of Moloney Murine Leukaemia virus. Nature. 2004;431(7008):586–590. doi: 10.1038/nature02944. [DOI] [PubMed] [Google Scholar]
- D'Souza V., Summers M.F. How retroviruses select their genomes. Nat. Rev. Microbiol. 2005;3(8):643–655. doi: 10.1038/nrmicro1210. [DOI] [PubMed] [Google Scholar]
- Das R., Baker D. Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl. Acad. Sci. U.S.A. 2007;104(37):14664–14669. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S.A., Curtis J.E., Ratcliff W., Clark P.K., Crist R.M., Lebowitz J., Krueger S., Rein A. Conformation of the HIV-1 Gag protein in solution. J. Mol. Biol. 2007;365(3):812–824. doi: 10.1016/j.jmb.2006.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S.A., Zuo X., Clark P.K., Campbell S.J., Wang Y.X., Rein A. Solution properties of Murine Leukemia virus gag protein: differences from HIV-1 gag. J. Virol. 2011;85(23):12733–12741. doi: 10.1128/JVI.05889-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty M.D., Booth D.S., Jayaraman B., Cheng Y., Frankel A.D. HIV Rev response element (RRE) directs assembly of the Rev homooligomer into discrete asymmetric complexes. Proc. Natl. Acad. Sci. U.S.A. 2010;107(28):12481–12486. doi: 10.1073/pnas.1007022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty M.D., Liu B., Frankel A.D. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat. Struct. Mol. Biol. 2010;17(11):1337–1342. doi: 10.1038/nsmb.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.H., Tonelli M., Scott L.G., Jaeger L., Williamson J.R., Butcher S.E. RNA helical packing in solution: NMR structure of a 30 kDa GAAA tetraloop-receptor complex. J. Mol. Biol. 2005;351(2):371–382. doi: 10.1016/j.jmb.2005.05.069. [DOI] [PubMed] [Google Scholar]
- de Smit M.H., Gultyaev A.P., Hilge M., Bink H.H., Barends S., Kraal B., Pleij C.W. Structural variation and functional importance of a D-loop-T-loop interaction in valine-accepting tRNA-like structures of plant viral RNAs. Nucl. Acids Res. 2002;30(19):4232–4240. doi: 10.1093/nar/gkf539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deigan K.E., Li T.W., Mathews D.H., Weeks K.M. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. U.S.A. 2009;106(1):97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirci H., Sierra R.G., Laksmono H., Shoeman R.L., Botha S., Barends T.R., Nass K., Schlichting I., Doak R.B., Gati C., Williams G.J., Boutet S., Messerschmidt M., Jogl G., Dahlberg A.E., Gregory S.T., Bogan M.J. Serial femtosecond X-ray diffraction of 30S ribosomal subunit microcrystals in liquid suspension at ambient temperature using an X-ray free-electron laser. Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun. 2013;69(Pt 9):1066–1069. doi: 10.1107/S174430911302099X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tomasso G., Dagenais P., Desjardins A., Rompre-Brodeur A., Delfosse V., Legault P. Affinity purification of RNA using an ARiBo tag. Methods Mol. Biol. 2012;941:137–155. doi: 10.1007/978-1-62703-113-4_11. [DOI] [PubMed] [Google Scholar]
- Di Tomasso G., Lampron P., Dagenais P., Omichinski J.G., Legault P. The ARiBo tag: a reliable tool for affinity purification of RNAs under native conditions. Nucl. Acids Res. 2011;39(3):e18. doi: 10.1093/nar/gkq1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias J., Renault L., Perez J., Mirande M. Small-angle X-ray solution scattering study of the multi-aminoacyl-tRNA synthetase complex reveals an elongated and multi-armed particle. J. Biol. Chem. 2013;288(33):23979–23989. doi: 10.1074/jbc.M113.489922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibrov S.M., Ding K., Brunn N.D., Parker M.A., Bergdahl B.M., Wyles D.L., Hermann T. Structure of a hepatitis C virus RNA domain in complex with a translation inhibitor reveals a binding mode reminiscent of riboswitches. Proc. Natl. Acad. Sci. U.S.A. 2012;109(14):5223–5228. doi: 10.1073/pnas.1118699109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon P.J., Nelbock P., Perkins A., Rosen C.A. Function of the human immunodeficiency virus types 1 and 2 Rev proteins is dependent on their ability to interact with a structured region present in env gene mRNA. J. Virol. 1990;64(9):4428–4437. doi: 10.1128/jvi.64.9.4428-4437.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMattia M.A., Watts N.R., Stahl S.J., Rader C., Wingfield P.T., Stuart D.I., Steven A.C., Grimes J.M. Implications of the HIV-1 Rev dimer structure at 3.2 A resolution for multimeric binding to the Rev response element. Proc. Natl. Acad. Sci. U.S.A. 2010;107(13):5810–5814. doi: 10.1073/pnas.0914946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S.W., Voinnet O. Antiviral immunity directed by small RNAs. Cell. 2007;130(3):413–426. doi: 10.1016/j.cell.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinman J.D. Mechanisms and implications of programmed translational frameshifting. Wiley Interdiscip. Rev. RNA. 2012;3(5):661–673. doi: 10.1002/wrna.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher T.W. Role of tRNA-like structures in controlling plant virus replication. Virus Res. 2009;139(2):217–229. doi: 10.1016/j.virusres.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z., Giedroc D.P., Hoffman D.W. Structure of the autoregulatory pseudoknot within the gene 32 messenger RNA of bacteriophages T2 and T6: a model for a possible family of structurally related RNA pseudoknots. Biochemistry. 1996;35(13):4187–4198. doi: 10.1021/bi9527350. [DOI] [PubMed] [Google Scholar]
- Du Z., Holland J.A., Hansen M.R., Giedroc D.P., Hoffman D.W. Base-pairings within the RNA pseudoknot associated with the simian retrovirus-1 gag-pro frameshift site. J. Mol. Biol. 1997;270(3):464–470. doi: 10.1006/jmbi.1997.1127. [DOI] [PubMed] [Google Scholar]
- Dumas P., Moras D., Florentz C., Giege R., Verlaan P., Van Belkum A., Pleij C.W. 3-D graphics modelling of the tRNA-like 3′-end of turnip yellow mosaic virus RNA: structural and functional implications. J. Biomol. Struct. Dyn. 1987;4(5):707–728. doi: 10.1080/07391102.1987.10507674. [DOI] [PubMed] [Google Scholar]
- Eddy S.R., Durbin R. RNA sequence analysis using covariance models. Nucl. Acids Res. 1994;22(11):2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli M., Minasov G., Su L., Rich A. Metal ions and flexibility in a viral RNA pseudoknot at atomic resolution. Proc. Natl. Acad. Sci. U.S.A. 2002;99(7):4302–4307. doi: 10.1073/pnas.062055599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emma P., Akre J.A., Bionta R., Bostedt C., Bozek J., Brachmann A., Bucksbaum P., Coffee R., Decker F.-J., Ding Y., Dowell D., Edstrom S., Fisher A., Frisch J., Gilevich S., Hastings J., Hays G., Hering P., Huang Z., Iversion R., Loos H., Messerschmidt M., Miahnahri A., Moeller S., Nuhn H.D., Pile G., Ratner D., Rzepiela J., Schultz D., Smith T., Stefan P., Tompkins H., Turner J., Welch J., White W., Wu J., Yocky G., Galayda J. First lasing and operation of an Ångstrom-wavelength free-electron laser. Nat. Photonics. 2010;4:641–647. [Google Scholar]
- Fabris D., Yu E.T. Elucidating the higher-order structure of biopolymers by structural probing and mass spectrometry: MS3D. J. Mass Spectrom. 2010;45(8):841–860. doi: 10.1002/jms.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X., Wang J., O’Carroll I.P., Mitchell M., Zuo X., Wang Y., Yu P., Liu Y., Rausch J.W., Dyba M.A., Kjems J., Schwieters C.D., Seifert S., Winans R.E., Watts N.R., Stahl S.J., Wingfield P.T., Byrd R.A., Le Grice S.F., Rein A., Wang Y.X. An unusual topological structure of the HIV-1 Rev response element. Cell. 2013;155(3):594–605. doi: 10.1016/j.cell.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fechter P., Rudinger-Thirion J., Florentz C., Giege R. Novel features in the tRNA-like world of plant viral RNAs. Cell. Mol. Life Sci. 2001;58(11):1547–1561. doi: 10.1007/PL00000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedor M.J., Westhof E. Ribozymes: the first 20 years. Mol. Cell. 2002;10(4):703–704. doi: 10.1016/s1097-2765(02)00700-1. [DOI] [PubMed] [Google Scholar]
- Felden B., Florentz C., Giege R., Westhof E. Solution structure of the 3′-end of brome mosaic virus genomic RNAs. Conformational mimicry with canonical tRNAs. J. Mol. Biol. 1994;235(2):508–531. doi: 10.1006/jmbi.1994.1010. [DOI] [PubMed] [Google Scholar]
- Felden B., Florentz C., Giege R., Westhof E. A central pseudoknotted three-way junction imposes tRNA-like mimicry and the orientation of three 5′ upstream pseudoknots in the 3′ terminus of tobacco mosaic virus RNA. RNA. 1996;2(3):201–212. [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Miragall O., Ramos R., Ramajo J., Martinez-Salas E. Evidence of reciprocal tertiary interactions between conserved motifs involved in organizing RNA structure essential for internal initiation of translation. RNA. 2006;12(2):223–234. doi: 10.1261/rna.2153206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez C., Wider G. TROSY in NMR studies of the structure and function of large biological macromolecules. Curr. Opin. Struct. Biol. 2003;13(5):570–580. doi: 10.1016/j.sbi.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Fernandez I.S., Bai X.C., Murshudov G., Scheres S.H., Ramakrishnan V. Initiation of translation by cricket paralysis virus IRES requires its translocation in the ribosome. Cell. 2014;157(4):823–831. doi: 10.1016/j.cell.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre-D’Amare A.R., Doudna J.A. Use of cis- and trans-ribozymes to remove 5′ and 3′ heterogeneities from milligrams of in vitro transcribed RNA. Nucl. Acids Res. 1996;24(5):977–978. doi: 10.1093/nar/24.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre-D’Amare A.R., Doudna J.A. Crystallization and structure determination of a hepatitis delta virus ribozyme: use of the RNA-binding protein U1A as a crystallization module. J. Mol. Biol. 2000;295(3):541–556. doi: 10.1006/jmbi.1999.3398. [DOI] [PubMed] [Google Scholar]
- Ferre-D’Amare A.R., Zhou K., Doudna J.A. Crystal structure of a hepatitis delta virus ribozyme. Nature. 1998;395(6702):567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- Ferre-D’Amare A.R., Zhou K., Doudna J.A. A general module for RNA crystallization. J. Mol. Biol. 1998;279(3):621–631. doi: 10.1006/jmbi.1998.1789. [DOI] [PubMed] [Google Scholar]
- Filbin M.E., Kieft J.S. Toward a structural understanding of IRES RNA function. Curr. Opin. Struct. Biol. 2009;19(3):267–276. doi: 10.1016/j.sbi.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth A.E., Brierley I. Non-canonical translation in RNA viruses. J. Gen. Virol. 2012;93(Pt 7):1385–1409. doi: 10.1099/vir.0.042499-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores S.C., Sherman M.A., Bruns C.M., Eastman P., Altman R.B. Fast flexible modeling of RNA structure using internal coordinates. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011;8(5):1247–1257. doi: 10.1109/TCBB.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley B., Leitner T., Apetrei C., Hahn B., Mizrachi I., Mullins J., Rambaut A., Wolinsky S., Korber B. Theoretical Biology and Biophysics Group, L.A.N.L., NM, LA-UR 13-26007; 2013. HIV Sequence Compendium 2013. [Google Scholar]
- Forster S., Apostol L., Bras W. Scatter: software for the analysis of nano- and mesoscale small-angle scattering. J. Appl. Crystallogr. 2010;43(3):639–646. [Google Scholar]
- Franke D., Kikhney A.G., Svergun D.I. Automated acquisition and analysis of small angle X-ray scattering data. Nucl. Instrum. Methods Phys. Res. Sect. A. 2012;689:52–59. [Google Scholar]
- Gao X. Recent advances in computational methods for nuclear magnetic resonance data processing. Genomics Proteomics Bioinform. 2013;11(1):29–33. doi: 10.1016/j.gpb.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garman E.F. Developments in X-ray crystallographic structure determination of biological macromolecules. Science. 2014;343(6175):1102–1108. doi: 10.1126/science.1247829. [DOI] [PubMed] [Google Scholar]
- Garst A.D., Heroux A., Rambo R.P., Batey R.T. Crystal structure of the lysine riboswitch regulatory mRNA element. J. Biol. Chem. 2008;283(33):22347–22351. doi: 10.1074/jbc.C800120200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudin C., Mazauric M.H., Traikia M., Guittet E., Yoshizawa S., Fourmy D. Structure of the RNA signal essential for translational frameshifting in HIV-1. J. Mol. Biol. 2005;349(5):1024–1035. doi: 10.1016/j.jmb.2005.04.045. [DOI] [PubMed] [Google Scholar]
- Gherardi E., Sandin S., Petoukhov M.V., Finch J., Youles M.E., Ofverstedt L.G., Miguel R.N., Blundell T.L., Vande Woude G.F., Skoglund U., Svergun D.I. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc. Natl. Acad. Sci. U.S.A. 2006;103(11):4046–4051. doi: 10.1073/pnas.0509040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giedroc D.P., Cornish P.V. Frameshifting RNA pseudoknots: structure and mechanism. Virus Res. 2009;139(2):193–208. doi: 10.1016/j.virusres.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giegé R., Florentz C., Dreher T.W. The TYMV tRNA-like structure. Biochimie. 1993;75(7):569–582. doi: 10.1016/0300-9084(93)90063-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosser Y., Hermann T., Majumdar A., Hu W., Frederick R., Jiang F., Xu W., Patel D.J. Peptide-triggered conformational switch in HIV-1 RRE RNA complexes. Nat. Struct. Biol. 2001;8(2):146–150. doi: 10.1038/84138. [DOI] [PubMed] [Google Scholar]
- Graewert M.A., Svergun D.I. Impact and progress in small and wide angle X-ray scattering (SAXS and WAXS) Curr. Opin. Struct. Biol. 2013;23(5):748–754. doi: 10.1016/j.sbi.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Grant T.D., Luft J.R., Wolfley J.R., Tsuruta H., Martel A., Montelione G.T., Snell E.H. Small angle X-ray scattering as a complementary tool for high-throughput structural studies. Biopolymers. 2011;95(8):517–530. doi: 10.1002/bip.21630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorieff N. Direct detection pays off for electron cryo-microscopy. eLife. 2013;2:e00573. doi: 10.7554/eLife.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishaev A., Ying J., Canny M.D., Pardi A., Bax A. Solution structure of tRNAVal from refinement of homology model against residual dipolar coupling and SAXS data. J. Biomol. NMR. 2008;42(2):99–109. doi: 10.1007/s10858-008-9267-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohman J.K., Gorelick R.J., Lickwar C.R., Lieb J.D., Bower B.D., Znosko B.M., Weeks K.M. A guanosine-centric mechanism for RNA chaperone function. Science. 2013;340(6129):190–195. doi: 10.1126/science.1230715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell R.R., Lee J.C., Cannone J.J. The accuracy of ribosomal RNA comparative structure models. Curr. Opin. Struct. Biol. 2002;12(3):301–310. doi: 10.1016/s0959-440x(02)00339-1. [DOI] [PubMed] [Google Scholar]
- Hammarskjold M.H., Rekosh D. A long-awaited structure is rev-ealed. Viruses. 2011;3(5):484–492. doi: 10.3390/v3050484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond J.A., Rambo R.P., Filbin M.E., Kieft J.S. Comparison and functional implications of the 3D architectures of viral tRNA-like structures. RNA. 2009;15(2):294–307. doi: 10.1261/rna.1360709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond J.A., Rambo R.P., Kieft J.S. Multi-domain packing in the aminoacylatable 3′ end of a plant viral RNA. J. Mol. Biol. 2010;399(3):450–463. doi: 10.1016/j.jmb.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashem Y., des Georges A., Dhote V., Langlois R., Liao H.Y., Grassucci R.A., Pestova T.V., Hellen C.U., Frank J. Hepatitis-C-virus-like internal ribosome entry sites displace eIF3 to gain access to the 40S subunit. Nature. 2013;503(7477):539–543. doi: 10.1038/nature12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng X., Kharytonchyk S., Garcia E.L., Lu K., Divakaruni S.S., LaCotti C., Edme K., Telesnitsky A., Summers M.F. Identification of a minimal region of the HIV-1 5′-leader required for RNA dimerization, NC binding, and packaging. J. Mol. Biol. 2012;417(3):224–239. doi: 10.1016/j.jmb.2012.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig J., Sattler M. The dynamic duo: combining NMR and small angle scattering in structural biology. Protein Sci. 2014;23(6):669–682. doi: 10.1002/pro.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbreteau C.H., Weill L., Decimo D., Prevot D., Darlix J.L., Sargueil B., Ohlmann T. HIV-2 genomic RNA contains a novel type of IRES located downstream of its initiation codon. Nat. Struct. Mol. Biol. 2005;12(11):1001–1007. doi: 10.1038/nsmb1011. [DOI] [PubMed] [Google Scholar]
- Hershey A.D., Chase M. Independent functions of viral protein and nucleic acid in growth of bacteriophage. J. Gen. Physiol. 1952;36(1):39–56. doi: 10.1085/jgp.36.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbrook S.R., Kim S.H. RNA crystallography. Biopolymers. 1997;44(1):3–21. doi: 10.1002/(SICI)1097-0282(1997)44:1<3::AID-BIP2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Howard J.A., Probert M.R. Cutting-edge techniques used for the structural investigation of single crystals. Science. 2014;343(6175):1098–1102. doi: 10.1126/science.1247252. [DOI] [PubMed] [Google Scholar]
- Hura G.L., Menon A.L., Hammel M., Rambo R.P., Poole F.L., 2nd., Tsutakawa S.E., Jenney F.E., Jr., Classen S., Frankel K.A., Hopkins R.C., Yang S.J., Scott J.W., Dillard B.D., Adams M.W., Tainer J.A. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat. Methods. 2009;6(8):606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huthoff H., Berkhout B. Two alternating structures of the HIV-1 leader RNA. RNA. 2001;7(1):143–157. doi: 10.1017/s1355838201001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H., Hayase Y., Iwai S., Ohtsuka E. Sequence-dependent hydrolysis of RNA using modified oligonucleotide splints and RNase H. FEBS Lett. 1987;215(2):327–330. doi: 10.1016/0014-5793(87)80171-0. [DOI] [PubMed] [Google Scholar]
- Ippolito J.A., Steitz T.A. The structure of the HIV-1 RRE high affinity rev binding site at 1.6 A resolution. J. Mol. Biol. 2000;295(4):711–717. doi: 10.1006/jmbi.1999.3405. [DOI] [PubMed] [Google Scholar]
- Iyer L.M., Balaji S., Koonin E.V., Aravind L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 2006;117(1):156–184. doi: 10.1016/j.virusres.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Johnson I., Bergamaschi A., Buitenhuis J., Dinapoli R., Greiffenberg D., Henrich B., Ikonen T., Meier G., Menzel A., Mozzanica A., Radicci V., Satapathy D.K., Schmitt B., Shi X. Capturing dynamics with Eiger, a fast-framing X-ray detector. J. Synchrotron Radiat. 2012;19(Pt 6):1001–1005. doi: 10.1107/S0909049512035972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.E., Jr., Reyes F.E., Polaski J.T., Batey R.T. B12 cofactors directly stabilize an mRNA regulatory switch. Nature. 2012;492(7427):133–137. doi: 10.1038/nature11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C.P., Cantara W.A., Olson E.D., Musier-Forsyth K. Small-angle X-ray scattering-derived structure of the HIV-1 5′ UTR reveals 3D tRNA mimicry. Proc. Natl. Acad. Sci. U.S.A. 2014;111(9):3395–3400. doi: 10.1073/pnas.1319658111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C.P., Saadatmand J., Kleiman L., Musier-Forsyth K. Molecular mimicry of human tRNALys anti-codon domain by HIV-1 RNA genome facilitates tRNA primer annealing. RNA. 2013;19(2):219–229. doi: 10.1261/rna.036681.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas M.A., Radmer R.J., Laederach A., Das R., Pearlman S., Herschlag D., Altman R.B. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA. 2009;15(2):189–199. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev A.V., Krivenko A.A., Harrington D.J., Holbrook S.R., Adams P.D., Pace N.R. Crystal structure of a bacterial ribonuclease P RNA. Proc. Natl. Acad. Sci. U.S.A. 2005;102(38):13392–13397. doi: 10.1073/pnas.0506662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon J.C., Prestwood L.J., Le Grice S.F., Lever A.M. In-gel probing of individual RNA conformers within a mixed population reveals a dimerization structural switch in the HIV-1 leader. Nucl. Acids Res. 2013;41(18):e174. doi: 10.1093/nar/gkt690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern J., Alonso-Mori R., Hellmich J., Tran R., Hattne J., Laksmono H., Glockner C., Echols N., Sierra R.G., Sellberg J., Lassalle-Kaiser B., Gildea R.J., Glatzel P., Grosse-Kunstleve R.W., Latimer M.J., McQueen T.A., DiFiore D., Fry A.R., Messerschmidt M., Miahnahri A., Schafer D.W., Seibert M.M., Sokaras D., Weng T.C., Zwart P.H., White W.E., Adams P.D., Bogan M.J., Boutet S., Williams G.J., Messinger J., Sauter N.K., Zouni A., Bergmann U., Yano J., Yachandra V.K. Room temperature femtosecond X-ray diffraction of photosystem II microcrystals. Proc. Natl. Acad. Sci. U.S.A. 2012;109(25):9721–9726. doi: 10.1073/pnas.1204598109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern J., Alonso-Mori R., Tran R., Hattne J., Gildea R.J., Echols N., Glockner C., Hellmich J., Laksmono H., Sierra R.G., Lassalle-Kaiser B., Koroidov S., Lampe A., Han G., Gul S., Difiore D., Milathianaki D., Fry A.R., Miahnahri A., Schafer D.W., Messerschmidt M., Seibert M.M., Koglin J.E., Sokaras D., Weng T.C., Sellberg J., Latimer M.J., Grosse-Kunstleve R.W., Zwart P.H., White W.E., Glatzel P., Adams P.D., Bogan M.J., Williams G.J., Boutet S., Messinger J., Zouni A., Sauter N.K., Yachandra V.K., Bergmann U., Yano J. Simultaneous femtosecond X-ray spectroscopy and diffraction of photosystem II at room temperature. Science. 2013;340(6131):491–495. doi: 10.1126/science.1234273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft J.S., Zhou K., Jubin R., Murray M.G., Lau J.Y., Doudna J.A. The hepatitis C virus internal ribosome entry site adopts an ion-dependent tertiary fold. J. Mol. Biol. 1999;292(3):513–529. doi: 10.1006/jmbi.1999.3095. [DOI] [PubMed] [Google Scholar]
- Kladwang W., Mann T.H., Becka A., Tian S., Kim H., Yoon S., Das R. Standardization of RNA chemical mapping experiments. Biochemistry. 2014;53(19):3063–3065. doi: 10.1021/bi5003426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W., VanLang C.C., Cordero P., Das R. Understanding the errors of SHAPE-directed RNA structure modeling. Biochemistry. 2011;50(37):8049–8056. doi: 10.1021/bi200524n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaver B., Berkhout B. Evolution of a disrupted TAR RNA hairpin structure in the HIV-1 virus. EMBO J. 1994;13(11):2650–2659. doi: 10.1002/j.1460-2075.1994.tb06555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D.J., Ferre-D’Amare A.R. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science. 2006;313(5794):1752–1756. doi: 10.1126/science.1129666. [DOI] [PubMed] [Google Scholar]
- Kofinger J., Hummer G. Atomic-resolution structural information from scattering experiments on macromolecules in solution. Phys. Rev. E: Stat. Nonlin. Soft Matter Phys. 2013;87(5):052712. doi: 10.1103/PhysRevE.87.052712. [DOI] [PubMed] [Google Scholar]
- Koldobskaya Y., Duguid E.M., Shechner D.M., Suslov N.B., Ye J., Sidhu S.S., Bartel D.P., Koide S., Kossiakoff A.A., Piccirilli J.A. A portable RNA sequence whose recognition by a synthetic antibody facilitates structural determination. Nat. Struct. Mol. Biol. 2011;18(1):100–106. doi: 10.1038/nsmb.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolk M.H., van der Graaf M., Wijmenga S.S., Pleij C.W., Heus H.A., Hilbers C.W. NMR structure of a classical pseudoknot: interplay of single- and double-stranded RNA. Science. 1998;280(5362):434–438. doi: 10.1126/science.280.5362.434. [DOI] [PubMed] [Google Scholar]
- Konarev P.V., Petoukhov M.V., Volkov V.V., Svergun D.I. ATSAS 2.1 a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2006;39:277–286. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft J.J., Hoy J.A., Miller W.A. Crystallization and preliminary X-ray diffraction analysis of the barley yellow dwarf virus cap-independent translation element. Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun. 2011;67(Pt 5):561–564. doi: 10.1107/S1744309111007196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft P., Bergamaschi A., Broennimann C., Dinapoli R., Eikenberry E.F., Henrich B., Johnson I., Mozzanica A., Schleputz C.M., Willmott P.R., Schmitt B. Performance of single-photon-counting PILATUS detector modules. J. Synchrotron Radiat. 2009;16(Pt 3):368–375. doi: 10.1107/S0909049509009911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiken C., Hraber P., Thurmond J., Yusim K. The hepatitis C sequence database in Los Alamos. Nucl. Acids Res. 2008;36(Database issue):D512–D516. doi: 10.1093/nar/gkm962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzembayeva M., Dilley K., Sardo L., Hu W.S. Life of psi: how full-length HIV-1 RNAs become packaged genomes in the viral particles. Virology. 2014;454–455:362–370. doi: 10.1016/j.virol.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvaratskhelia M., Miller J.T., Budihas S.R., Pannell L.K., Le Grice S.F. Identification of specific HIV-1 reverse transcriptase contacts to the viral RNA:tRNA complex by mass spectrometry and a primary amine selective reagent. Proc. Natl. Acad. Sci. U.S.A. 2002;99(25):15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapham J., Crothers D.M. RNase H cleavage for processing of in vitro transcribed RNA for NMR studies and RNA ligation. RNA. 1996;2(3):289–296. [PMC free article] [PubMed] [Google Scholar]
- Liao M., Cao E., Julius D., Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504(7478):107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman J.A., Salim M., Krucinska J., Wedekind J.E. Structure of a class II preQ1 riboswitch reveals ligand recognition by a new fold. Nat. Chem. Biol. 2013;9(6):353–355. doi: 10.1038/nchembio.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J., Ouellet J., Norman D.G., Doniach S., Lilley D.M. The complete VS ribozyme in solution studied by small-angle X-ray scattering. Structure. 2008;16(9):1357–1367. doi: 10.1016/j.str.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Morris R.J., Hexemer A., Grandison S., Zwart P.H. Computation of small-angle scattering profiles with three-dimensional Zernike polynomials. Acta Crystallogr. Sect. A: Found. Crystallogr. 2012;68(Pt 2):278–285. doi: 10.1107/S010876731104788X. [DOI] [PubMed] [Google Scholar]
- Locker N., Chamond N., Sargueil B. A conserved structure within the HIV gag open reading frame that controls translation initiation directly recruits the 40S subunit and eIF3. Nucl. Acids Res. 2011;39(6):2367–2377. doi: 10.1093/nar/gkq1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low J.T., Knoepfel S.A., Watts J.M., ter Brake O., Berkhout B., Weeks K.M. SHAPE-directed discovery of potent shRNA inhibitors of HIV-1. Mol. Ther. 2012;20(4):820–828. doi: 10.1038/mt.2011.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low J.T., Weeks K.M. SHAPE-directed RNA secondary structure prediction. Methods. 2010;52(2):150–158. doi: 10.1016/j.ymeth.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K., Heng X., Garyu L., Monti S., Garcia E.L., Kharytonchyk S., Dorjsuren B., Kulandaivel G., Jones S., Hiremath A., Divakaruni S.S., LaCotti C., Barton S., Tummillo D., Hosic A., Edme K., Albrecht S., Telesnitsky A., Summers M.F. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science. 2011;334(6053):242–245. doi: 10.1126/science.1210460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K., Heng X., Summers M.F. Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 2011;410(4):609–633. doi: 10.1016/j.jmb.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K., Miyazaki Y., Summers M.F. Isotope labeling strategies for NMR studies of RNA. J. Biomol. NMR. 2010;46(1):113–125. doi: 10.1007/s10858-009-9375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukavsky P.J. Structure and function of HCV IRES domains. Virus Res. 2009;139(2):166–171. doi: 10.1016/j.virusres.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukavsky P.J., Puglisi J.D. Large-scale preparation and purification of polyacrylamide-free RNA oligonucleotides. RNA. 2004;10(5):889–893. doi: 10.1261/rna.5264804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukavsky P.J., Puglisi J.D. Structure determination of large biological RNAs. Methods Enzymol. 2005;394:399–416. doi: 10.1016/S0076-6879(05)94016-0. [DOI] [PubMed] [Google Scholar]
- Lusvarghi S., Sztuba-Solinska J., Purzycka K.J., Pauly G.T., Rausch J.W., Grice S.F. The HIV-2 Rev-response element: determining secondary structure and defining folding intermediates. Nucl. Acids Res. 2013;41(13):6637–6649. doi: 10.1093/nar/gkt353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusvarghi S., Sztuba-Solinska J., Purzycka K.J., Rausch J.W., Le Grice S.F. RNA secondary structure prediction using high-throughput SHAPE. J. Vis. Exp. 2013;75:e50243. doi: 10.3791/50243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheschi R.J., Staple D.W., Butcher S.E. Programmed ribosomal frameshifting in SIV is induced by a highly structured RNA stem-loop. J. Mol. Biol. 2007;373(3):652–663. doi: 10.1016/j.jmb.2007.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel A., Liu P., Weiss T.M., Niebuhr M., Tsuruta H. An integrated high-throughput data acquisition system for biological solution X-ray scattering studies. J. Synchrotron Radiat. 2012;19(Pt 3):431–434. doi: 10.1107/S0909049512008072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack J.C., Yuan X., Yingling Y.G., Kasprzak W., Zamora R.E., Shapiro B.A., Simon A.E. Structural domains within the 3′ untranslated region of Turnip crinkle virus. J. Virol. 2008;82(17):8706–8720. doi: 10.1128/JVI.00416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis J.L., Duncan C.D., Weeks K.M. High-throughput SHAPE and hydroxyl radical analysis of RNA structure and ribonucleoprotein assembly. Methods Enzymol. 2009;468:67–89. doi: 10.1016/S0076-6879(09)68004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullan G., Chen S., Henderson R., Faruqi A.R. Detective quantum efficiency of electron area detectors in electron microscopy. Ultramicroscopy. 2009;109(9):1126–1143. doi: 10.1016/j.ultramic.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullan G., Clark A.T., Turchetta R., Faruqi A.R. Enhanced imaging in low dose electron microscopy using electron counting. Ultramicroscopy. 2009;109(12):1411–1416. doi: 10.1016/j.ultramic.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullan G., Faruqi A.R., Henderson R., Guerrini N., Turchetta R., Jacobs A., van Hoften G. Experimental observation of the improvement in MTF from backthinning a CMOS direct electron detector. Ultramicroscopy. 2009;109(9):1144–1147. doi: 10.1016/j.ultramic.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza V.L., Vachet R.W. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom. Rev. 2009;28(5):785–815. doi: 10.1002/mas.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino E.J., Wilkinson K.A., Coughlan J.L., Weeks K.M. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J. Am. Chem. Soc. 2005;127(12):4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- Michiels P.J., Versleijen A.A., Verlaan P.W., Pleij C.W., Hilbers C.W., Heus H.A. Solution structure of the pseudoknot of SRV-1 RNA, involved in ribosomal frameshifting. J. Mol. Biol. 2001;310(5):1109–1123. doi: 10.1006/jmbi.2001.4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele G., Mouland A., Harrison G.P., Cohen E., Lever A.M. The human immunodeficiency virus type 1 5′ packaging signal structure affects translation but does not function as an internal ribosome entry site structure. J. Virol. 1996;70(2):944–951. doi: 10.1128/jvi.70.2.944-951.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R.J. Femtosecond crystallography with ultrabright electrons and X-rays: capturing chemistry in action. Science. 2014;343(6175):1108–1116. doi: 10.1126/science.1248488. [DOI] [PubMed] [Google Scholar]
- Miller W.A., Wang Z., Treder K. The amazing diversity of cap-independent translation elements in the 3′-untranslated regions of plant viral RNAs. Biochem. Soc. Trans. 2007;35(Pt 6):1629–1633. doi: 10.1042/BST0351629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W., Cymborowski M., Otwinowski Z., Chruszcz M. HKL-3000: the integration of data reduction and structure solution – from diffraction images to an initial model in minutes. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2006;62(Pt 8):859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y., Irobalieva R.N., Tolbert B.S., Smalls-Mantey A., Iyalla K., Loeliger K., D'Souza V., Khant H., Schmid M.F., Garcia E.L., Telesnitsky A., Chiu W., Summers M.F. Structure of a conserved retroviral RNA packaging element by NMR spectroscopy and cryo-electron tomography. J. Mol. Biol. 2010;404(5):751–772. doi: 10.1016/j.jmb.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollova E.T., Hansen M.R., Pardi A. Global structure of RNA determined with residual dipolar couplings. J. Am. Chem. Soc. 2000;122(46):11561–11562. [Google Scholar]
- Montange R.K., Batey R.T. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature. 2006;441(7097):1172–1175. doi: 10.1038/nature04819. [DOI] [PubMed] [Google Scholar]
- Mortimer S.A., Weeks K.M. Time-resolved RNA SHAPE chemistry: quantitative RNA structure analysis in one-second snapshots and at single-nucleotide resolution. Nat. Protoc. 2009;4(10):1413–1421. doi: 10.1038/nprot.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namy O., Moran S.J., Stuart D.I., Gilbert R.J., Brierley I. A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature. 2006;441(7090):244–247. doi: 10.1038/nature04735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelissen F.H., Leunissen E.H., van de Laar L., Tessari M., Heus H.A., Wijmenga S.S. Fast production of homogeneous recombinant RNA – towards large-scale production of RNA. Nucl. Acids Res. 2012;40(13):e102. doi: 10.1093/nar/gks292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutze R., Moffat K. Time-resolved structural studies at synchrotrons and X-ray free electron lasers: opportunities and challenges. Curr. Opin. Struct. Biol. 2012;22(5):651–659. doi: 10.1016/j.sbi.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson B.L., White K.A. 3′ Cap-independent translation enhancers of positive-strand RNA plant viruses. Curr. Opin. Virol. 2011;1(5):373–380. doi: 10.1016/j.coviro.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Nicholson W.V., Glaeser R.M. Review: automatic particle detection in electron microscopy. J. Struct. Biol. 2001;133(2–3):90–101. doi: 10.1006/jsbi.2001.4348. [DOI] [PubMed] [Google Scholar]
- Nixon P.L., Rangan A., Kim Y.G., Rich A., Hoffman D.W., Hennig M., Giedroc D.P. Solution structure of a luteoviral P1–P2 frameshifting mRNA pseudoknot. J. Mol. Biol. 2002;322(3):621–633. doi: 10.1016/s0022-2836(02)00779-9. [DOI] [PubMed] [Google Scholar]
- Norousi R., Wickles S., Leidig C., Becker T., Schmid V.J., Beckmann R., Tresch A. Automatic post-picking using MAPPOS improves particle image detection from cryo-EM micrographs. J. Struct. Biol. 2013;182(2):59–66. doi: 10.1016/j.jsb.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Ooms M., Huthoff H., Russell R., Liang C., Berkhout B. A riboswitch regulates RNA dimerization and packaging in human immunodeficiency virus type 1 virions. J. Virol. 2004;78(19):10814–10819. doi: 10.1128/JVI.78.19.10814-10819.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms M., Verhoef K., Southern E., Huthoff H., Berkhout B. Probing alternative foldings of the HIV-1 leader RNA by antisense oligonucleotide scanning arrays. Nucl. Acids Res. 2004;32(2):819–827. doi: 10.1093/nar/gkh206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto G.A., Puglisi J.D. The pathway of HCV IRES-mediated translation initiation. Cell. 2004;119(3):369–380. doi: 10.1016/j.cell.2004.09.038. [DOI] [PubMed] [Google Scholar]
- Pallan P.S., Marshall W.S., Harp J., Jewett F.C., 3rd., Wawrzak Z., Brown B.A., 2nd., Rich A., Egli M. Crystal structure of a luteoviral RNA pseudoknot and model for a minimal ribosomal frameshifting motif. Biochemistry. 2005;44(34):11315–11322. doi: 10.1021/bi051061i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallesen J. Structure of the HIV-1 5′ untranslated region dimer alone and in complex with gold nanocolloids: support of a TAR-TAR-containing 5′ dimer linkage site (DLS) and a 3′ DIS-DIS-containing DLS. Biochemistry. 2011;50(28):6170–6177. doi: 10.1021/bi200488h. [DOI] [PubMed] [Google Scholar]
- Pallesen J., Dong M., Besenbacher F., Kjems J. Structure of the HIV-1 Rev response element alone and in complex with regulator of virion (Rev) studied by atomic force microscopy. FEBS J. 2009;276(15):4223–4232. doi: 10.1111/j.1742-4658.2009.07130.x. [DOI] [PubMed] [Google Scholar]
- Panjikar S., Parthasarathy V., Lamzin V.S., Weiss M.S., Tucker P.A. Auto-rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2005;61(Pt 4):449–457. doi: 10.1107/S0907444905001307. [DOI] [PubMed] [Google Scholar]
- Panjikar S., Parthasarathy V., Lamzin V.S., Weiss M.S., Tucker P.A. On the combination of molecular replacement and single-wavelength anomalous diffraction phasing for automated structure determination. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2009;65(Pt 10):1089–1097. doi: 10.1107/S0907444909029643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M., Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452(7183):51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- Pastor N., Weinstein H., Jamison E., Brenowitz M. A detailed interpretation of OH radical footprints in a TBP-DNA complex reveals the role of dynamics in the mechanism of sequence-specific binding. J. Mol. Biol. 2000;304(1):55–68. doi: 10.1006/jmbi.2000.4173. [DOI] [PubMed] [Google Scholar]
- Perard J., Leyrat C., Baudin F., Drouet E., Jamin M. Structure of the full-length HCV IRES in solution. Nat. Commun. 2013;4:1612. doi: 10.1038/ncomms2611. [DOI] [PubMed] [Google Scholar]
- Perrotta A.T., Been M.D. Cleavage of oligoribonucleotides by a ribozyme derived from the hepatitis delta virus RNA sequence. Biochemistry. 1992;31(1):16–21. doi: 10.1021/bi00116a004. [DOI] [PubMed] [Google Scholar]
- Pervushin K., Riek R., Wider G., Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 1997;94(23):12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson R.D., Feigon J. Structural change in Rev responsive element RNA of HIV-1 on binding Rev peptide. J. Mol. Biol. 1996;264(5):863–877. doi: 10.1006/jmbi.1996.0683. [DOI] [PubMed] [Google Scholar]
- Pfingsten J.S., Costantino D.A., Kieft J.S. Structural basis for ribosome recruitment and manipulation by a viral IRES RNA. Science. 2006;314(5804):1450–1454. doi: 10.1126/science.1133281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirilli J.A., Koldobskaya Y. Crystal structure of an RNA polymerase ribozyme in complex with an antibody fragment. Philos. Trans. R. Soc. Lond. B: Biol. Sci. 2011;366(1580):2918–2928. doi: 10.1098/rstb.2011.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineiro D., Martinez-Salas E. RNA structural elements of hepatitis C virus controlling viral RNA translation and the implications for viral pathogenesis. Viruses. 2012;4(10):2233–2250. doi: 10.3390/v4102233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plank T.D., Whitehurst J.T., Cencic R., Pelletier J., Kieft J.S. Internal translation initiation from HIV-1 transcripts is conferred by a common RNA structure. Translation. 2014;2:e27694. doi: 10.4161/trla.27694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plank T.D., Whitehurst J.T., Kieft J.S. Cell type specificity and structural determinants of IRES activity from the 5′ leaders of different HIV-1 transcripts. Nucl. Acids Res. 2013;41(13):6698–6714. doi: 10.1093/nar/gkt358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pley H.W., Flaherty K.M., McKay D.B. Model for an RNA tertiary interaction from the structure of an intermolecular complex between a GAAA tetraloop and an RNA helix. Nature. 1994;372(6501):111–113. doi: 10.1038/372111a0. [DOI] [PubMed] [Google Scholar]
- Poitevin F., Orland H., Doniach S., Koehl P., Delarue M. AquaSAXS: a web server for computation and fitting of SAXS profiles with non-uniformally hydrated atomic models. Nucl. Acids Res. 2011;39(Web Server issue):W184–W189. doi: 10.1093/nar/gkr430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popenda M., Szachniuk M., Antczak M., Purzycka K.J., Lukasiak P., Bartol N., Blazewicz J., Adamiak R.W. Automated 3D structure composition for large RNAs. Nucl. Acids Res. 2012;40(14):e112. doi: 10.1093/nar/gks339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redecke L., Nass K., DePonte D.P., White T.A., Rehders D., Barty A., Stellato F., Liang M., Barends T.R., Boutet S., Williams G.J., Messerschmidt M., Seibert M.M., Aquila A., Arnlund D., Bajt S., Barth T., Bogan M.J., Caleman C., Chao T.C., Doak R.B., Fleckenstein H., Frank M., Fromme R., Galli L., Grotjohann I., Hunter M.S., Johansson L.C., Kassemeyer S., Katona G., Kirian R.A., Koopmann R., Kupitz C., Lomb L., Martin A.V., Mogk S., Neutze R., Shoeman R.L., Steinbrener J., Timneanu N., Wang D., Weierstall U., Zatsepin N.A., Spence J.C., Fromme P., Schlichting I., Duszenko M., Betzel C., Chapman H.N. Natively inhibited Trypanosoma brucei cathepsin B structure determined by using an X-ray laser. Science. 2013;339(6116):227–230. doi: 10.1126/science.1229663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter J.S., Mathews D.H. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes F.E., Garst A.D., Batey R.T. Strategies in RNA crystallography. Methods Enzymol. 2009;469:119–139. doi: 10.1016/S0076-6879(09)69006-6. [DOI] [PubMed] [Google Scholar]
- Rice G.M., Leonard C.W., Weeks K.M. RNA secondary structure modeling at consistent high accuracy using differential SHAPE. RNA. 2014 doi: 10.1261/rna.043323.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg O.S., Deindl S., Sung R.J., Nairn A.C., Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123(5):849–860. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Roulston A., Marcellus R.C., Branton P.E. Viruses and apoptosis. Annu. Rev. Microbiol. 1999;53:577–628. doi: 10.1146/annurev.micro.53.1.577. [DOI] [PubMed] [Google Scholar]
- Salvail-Lacoste A., Di Tomasso G., Piette B.L., Legault P. Affinity purification of T7 RNA transcripts with homogeneous ends using ARiBo and CRISPR tags. RNA. 2013;19(7):1003–1014. doi: 10.1261/rna.037432.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan R., Nebot M.R., Chirico N., Mansky L.M., Belshaw R. Viral mutation rates. J. Virol. 2010;84(19):9733–9748. doi: 10.1128/JVI.00694-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro S.W., Joyce G.F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. U.S.A. 1997;94(9):4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott W.G., Finch J.T., Klug A. The crystal structure of an all-RNA hammerhead ribozyme. Nucl. Acids Symp. Ser. 1995;34:214–216. doi: 10.2210/pdb1mme/pdb. [DOI] [PubMed] [Google Scholar]
- Scott L.G., Tolbert T.J., Williamson J.R. Preparation of specifically 2H- and 13C-labeled ribonucleotides. Methods Enzymol. 2000;317:18–38. doi: 10.1016/s0076-6879(00)17004-1. [DOI] [PubMed] [Google Scholar]
- Seetin M.G., Mathews D.H. RNA structure prediction: an overview of methods. Methods Mol. Biol. 2012;905:99–122. doi: 10.1007/978-1-61779-949-5_8. [DOI] [PubMed] [Google Scholar]
- Seif E., Niu M., Kleiman L. Annealing to sequences within the primer binding site loop promotes an HIV-1 RNA conformation favoring RNA dimerization and packaging. RNA. 2013;19(10):1384–1393. doi: 10.1261/rna.038497.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., Ding F., Dokholyan N.V. iFoldRNA: three-dimensional RNA structure prediction and folding. Bioinformatics. 2008;24(17):1951–1952. doi: 10.1093/bioinformatics/btn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shell S.M., Hess S., Kvaratskhelia M., Zou Y. Mass spectrometric identification of lysines involved in the interaction of human replication protein a with single-stranded DNA. Biochemistry. 2005;44(3):971–978. doi: 10.1021/bi048208a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L.X., Tinoco I., Jr. The structure of an RNA pseudoknot that causes efficient frameshifting in mouse mammary tumor virus. J. Mol. Biol. 1995;247(5):963–978. doi: 10.1006/jmbi.1995.0193. [DOI] [PubMed] [Google Scholar]
- Shigematsu H., Sigworth F.J. Noise models and cryo-EM drift correction with a direct-electron camera. Ultramicroscopy. 2013;131:61–69. doi: 10.1016/j.ultramic.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkriabai N., Datta S.A., Zhao Z., Hess S., Rein A., Kvaratskhelia M. Interactions of HIV-1 Gag with assembly cofactors. Biochemistry. 2006;45(13):4077–4083. doi: 10.1021/bi052308e. [DOI] [PubMed] [Google Scholar]
- Siridechadilok B., Fraser C.S., Hall R.J., Doudna J.A., Nogales E. Structural roles for human translation factor eIF3 in initiation of protein synthesis. Science. 2005;310(5753):1513–1515. doi: 10.1126/science.1118977. [DOI] [PubMed] [Google Scholar]
- Spahn C.M., Jan E., Mulder A., Grassucci R.A., Sarnow P., Frank J. Cryo-EM visualization of a viral internal ribosome entry site bound to human ribosomes: the IRES functions as an RNA-based translation factor. Cell. 2004;118(4):465–475. doi: 10.1016/j.cell.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Spahn C.M., Kieft J.S., Grassucci R.A., Penczek P.A., Zhou K., Doudna J.A., Frank J. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science. 2001;291(5510):1959–1962. doi: 10.1126/science.1058409. [DOI] [PubMed] [Google Scholar]
- Spitale R.C., Torelli A.T., Krucinska J., Bandarian V., Wedekind J.E. The structural basis for recognition of the PreQ0 metabolite by an unusually small riboswitch aptamer domain. J. Biol. Chem. 2009;284(17):11012–11016. doi: 10.1074/jbc.C900024200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stake M.S., Bann D.V., Kaddis R.J., Parent L.J. Nuclear trafficking of retroviral RNAs and Gag proteins during late steps of replication. Viruses. 2013;5(11):2767–2795. doi: 10.3390/v5112767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staple D.W., Butcher S.E. Solution structure and thermodynamic investigation of the HIV-1 frameshift inducing element. J. Mol. Biol. 2005;349(5):1011–1023. doi: 10.1016/j.jmb.2005.03.038. [DOI] [PubMed] [Google Scholar]
- Steen K.A., Siegfried N.A., Weeks K.M. Selective 2′-hydroxyl acylation analyzed by protection from exoribonuclease (RNase-detected SHAPE) for direct analysis of covalent adducts and of nucleotide flexibility in RNA. Nat. Protoc. 2011;6(11):1683–1694. doi: 10.1038/nprot.2011.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson J.D., Li H., Kenyon J.C., Symmons M., Klenerman D., Lever A.M. Three-dimensional RNA structure of the major HIV-1 packaging signal region. Structure. 2013;21(6):951–962. doi: 10.1016/j.str.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupina V.A., Meskauskas A., McCormack J.C., Yingling Y.G., Shapiro B.A., Dinman J.D., Simon A.E. The 3′ proximal translational enhancer of Turnip crinkle virus binds to 60S ribosomal subunits. RNA. 2008;14(11):2379–2393. doi: 10.1261/rna.1227808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L., Chen L., Egli M., Berger J.M., Rich A. Minor groove RNA triplex in the crystal structure of a ribosomal frameshifting viral pseudoknot. Nat. Struct. Biol. 1999;6(3):285–292. doi: 10.1038/6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talini G., Branciamore S., Gallori E. Ribozymes: flexible molecular devices at work. Biochimie. 2011;93(11):1998–2005. doi: 10.1016/j.biochi.2011.06.026. [DOI] [PubMed] [Google Scholar]
- Talini G., Gallori E., Maurel M.C. Natural and unnatural ribozymes: back to the primordial RNA world. Res. Microbiol. 2009;160(7):457–465. doi: 10.1016/j.resmic.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Tolbert B.S., Miyazaki Y., Barton S., Kinde B., Starck P., Singh R., Bax A., Case D.A., Summers M.F. Major groove width variations in RNA structures determined by NMR and impact of 13C residual chemical shift anisotropy and 1H-13C residual dipolar coupling on refinement. J. Biomol. NMR. 2010;47(3):205–219. doi: 10.1007/s10858-010-9424-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor N., Keating K.S., Taylor S.D., Pyle A.M. Crystal structure of a self-spliced group II intron. Science. 2008;320(5872):77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullius T.D., Greenbaum J.A. Mapping nucleic acid structure by hydroxyl radical cleavage. Curr. Opin. Chem. Biol. 2005;9(2):127–134. doi: 10.1016/j.cbpa.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Tzakos A.G., Grace C.R., Lukavsky P.J., Riek R. NMR techniques for very large proteins and rnas in solution. Annu. Rev. Biophys. Biomol. Struct. 2006;35:319–342. doi: 10.1146/annurev.biophys.35.040405.102034. [DOI] [PubMed] [Google Scholar]
- Uhlenbeck O.C. Keeping RNA happy. RNA. 1995;1(1):4–6. [PMC free article] [PubMed] [Google Scholar]
- Vallejos M., Deforges J., Plank T.D., Letelier A., Ramdohr P., Abraham C.G., Valiente-Echeverria F., Kieft J.S., Sargueil B., Lopez-Lastra M. Activity of the human immunodeficiency virus type 1 cell cycle-dependent internal ribosomal entry site is modulated by IRES trans-acting factors. Nucl. Acids Res. 2011;39(14):6186–6200. doi: 10.1093/nar/gkr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen J.J., Makowski L., Sosnick T.R., Freed K.F. Modeling the hydration layer around proteins: HyPred. Biophys. J. 2010;99(5):1611–1619. doi: 10.1016/j.bpj.2010.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen J.J., Makowski L., Sosnick T.R., Freed K.F. Modeling the hydration layer around proteins: applications to small- and wide-angle X-ray scattering. Biophys. J. 2011;101(8):2061–2069. doi: 10.1016/j.bpj.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S.C., Avis J.M., Conn G.L. General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucl. Acids Res. 2003;31(15):e82. doi: 10.1093/nar/gng082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Zuo X., Yu P., Xu H., Starich M.R., Tiede D.M., Shapiro B.A., Schwieters C.D., Wang Y.X. A method for helical RNA global structure determination in solution using small-angle X-ray scattering and NMR measurements. J. Mol. Biol. 2009;393(3):717–734. doi: 10.1016/j.jmb.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.X., Zuo X., Wang J., Yu P., Butcher S.E. Rapid global structure determination of large RNA and RNA complexes using NMR and small-angle X-ray scattering. Methods. 2010;52(2):180–191. doi: 10.1016/j.ymeth.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Parisien M., Scheets K., Miller W.A. The cap-binding translation initiation factor, eIF4E, binds a pseudoknot in a viral cap-independent translation element. Structure. 2011;19(6):868–880. doi: 10.1016/j.str.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Treder K., Miller W.A. Structure of a viral cap-independent translation element that functions via high affinity binding to the eIF4E subunit of eIF4F. J. Biol. Chem. 2009;284(21):14189–14202. doi: 10.1074/jbc.M808841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts J.M., Dang K.K., Gorelick R.J., Leonard C.W., Bess J.W., Jr., Swanstrom R., Burch C.L., Weeks K.M. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460(7256):711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks K.M. Advances in RNA structure analysis by chemical probing. Curr. Opin. Struct. Biol. 2010;20(3):295–304. doi: 10.1016/j.sbi.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson K.A., Merino E.J., Weeks K.M. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006;1(3):1610–1616. doi: 10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]
- Wilson J.E., Pestova T.V., Hellen C.U., Sarnow P. Initiation of protein synthesis from the A site of the ribosome. Cell. 2000;102(4):511–520. doi: 10.1016/s0092-8674(00)00055-6. [DOI] [PubMed] [Google Scholar]
- Wu Q., Huang L., Zhang Y. The structure and function of catalytic RNAs. Sci. China C Life Sci. 2009;52(3):232–244. doi: 10.1007/s11427-009-0038-z. [DOI] [PubMed] [Google Scholar]
- Wu Y., Wang C., Chang J., Chen Y., Miyazaki N., Murata K., Nagayama K., Chang W. Zernike phase contrast cryo-electron microscopy reveals 100 kDa component in a protein complex. J. Phys. D: Appl. Phys. 2013;46:494008–494017. [Google Scholar]
- Ye X., Gorin A., Ellington A.D., Patel D.J. Deep penetration of an alpha-helix into a widened RNA major groove in the HIV-1 rev peptide-RNA aptamer complex. Nat. Struct. Biol. 1996;3(12):1026–1033. doi: 10.1038/nsb1296-1026. [DOI] [PubMed] [Google Scholar]
- Yilmaz A., Bolinger C., Boris-Lawrie K. Retrovirus translation initiation: issues and hypotheses derived from study of HIV-1. Curr. HIV Res. 2006;4(2):131–139. doi: 10.2174/157016206776055039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu E.T., Hawkins A., Eaton J., Fabris D. MS3D structural elucidation of the HIV-1 packaging signal. Proc. Natl. Acad. Sci. U.S.A. 2008;105(34):12248–12253. doi: 10.1073/pnas.0800509105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu E.T., Hawkins A., Kuntz I.D., Rahn L.A., Rothfuss A., Sale K., Young M.M., Yang C.L., Pancerella C.M., Fabris D. The collaboratory for MS3D: a new cyberinfrastructure for the structural elucidation of biological macromolecules and their assemblies using mass spectrometry-based approaches. J. Proteome Res. 2008;7(11):4848–4857. doi: 10.1021/pr800443f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu E.T., Zhang Q., Fabris D. Untying the FIV frameshifting pseudoknot structure by MS3D. J. Mol. Biol. 2005;345(1):69–80. doi: 10.1016/j.jmb.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Zhang J., Ferre-D’Amare A.R. New molecular engineering approaches for crystallographic studies of large RNAs. Curr. Opin. Struct. Biol. 2014;26C:9–15. doi: 10.1016/j.sbi.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Bean R.L., Vogt V.M., Summers M. Solution structure of the Rous sarcoma virus nucleocapsid protein: muPsi RNA packaging signal complex. J. Mol. Biol. 2007;365(2):453–467. doi: 10.1016/j.jmb.2006.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S., Rynearson K.D., Ding K., Brunn N.D., Hermann T. Screening for inhibitors of the hepatitis C virus internal ribosome entry site RNA. Bioorg. Med. Chem. 2013;21(20):6139–6144. doi: 10.1016/j.bmc.2013.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.H. Atomic resolution cryo electron microscopy of macromolecular complexes. Adv. Protein Chem. Struct. Biol. 2011;82:1–35. doi: 10.1016/B978-0-12-386507-6.00001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B., Tabor S., Richardson C.C. Syn5 RNA polymerase synthesizes precise run-off RNA products. Nucl. Acids Res. 2014;42(5):e33. doi: 10.1093/nar/gkt1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Korostelev A., Costantino D.A., Donohue J.P., Noller H.F., Kieft J.S. Crystal structures of complexes containing domains from two viral internal ribosome entry site (IRES) RNAs bound to the 70S ribosome. Proc. Natl. Acad. Sci. U.S.A. 2011;108(5):1839–1844. doi: 10.1073/pnas.1018582108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziehler W.A., Engelke D.R. Synthesis of small RNA transcripts with discrete 5′ and 3′ ends. Biotechniques. 1996;20(4):622–624. doi: 10.2144/19962004622. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucl. Acids Res. 2003;31(13):3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X., Wang J., Foster T.R., Schwieters C.D., Tiede D.M., Butcher S.E., Wang Y.X. Global molecular structure and interfaces: refining an RNA:RNA complex structure using solution X-ray scattering data. J. Am. Chem. Soc. 2008;130(11):3292–3293. doi: 10.1021/ja7114508. [DOI] [PubMed] [Google Scholar]
- Zuo X., Wang J., Yu P., Eyler D., Xu H., Starich M.R., Tiede D.M., Simon A.E., Kasprzak W., Schwieters C.D., Shapiro B.A., Wang Y.X. Solution structure of the cap-independent translational enhancer and ribosome-binding element in the 3′ UTR of turnip crinkle virus. Proc. Natl. Acad. Sci. U.S.A. 2010;107(4):1385–1390. doi: 10.1073/pnas.0908140107. [DOI] [PMC free article] [PubMed] [Google Scholar]