

Abstract
Th17 cells play an important role in multiple sclerosis (MS) and its autoimmune model, experimental autoimmune encephalomyelitis (EAE). However, studies have not addressed how enhanced Th17 immune responses can affect demyelinating diseases. We induced EAE with MOG in RORγt transgenic C57BL/6 mice that overexpress a Th17 inducing transcription factor. RORγt transgenic mice developed more severe EAE than wild-type mice with more robust anti-MOG Th17 immune responses. In contrast, mice overexpressing T-bet, a Th1-inducing transcription factor, were resistant to EAE. Therefore, a genetic bias toward Th17 immune responses could contribute to CNS immunopathology.
1. Introduction
Experimental autoimmune encephalomyelitis (EAE) is widely used as an autoimmune model of multiple sclerosis (MS) (Tsunoda and Fujinami, 1996). Mice with EAE have inflammatory demyelinating lesions in the central nervous system (CNS) that are histologically similar to MS. Active EAE is mediated by myelin-specific T cells, which are activated in the periphery by sensitization with CNS antigen and recruited into the CNS. Once in the CNS, they re-encounter myelin antigen and start the inflammatory process resulting in inflammatory demyelination. In C57BL/6 mice, EAE can be induced by sensitization with myelin oligodendrocyte glycoprotein (MOG), which results in tail and hind limb paralysis (Tsunoda and Fujinami, 1996). C57BL/6 mice with EAE develop clinical signs that appear around 2 weeks post-induction (p.i.) and begin to subside about 1 month p.i.
Previously, in EAE, the most prominent immune effector cells that have been demonstrated to influence the outcome of disease are T helper (Th)1 and Th2 cells (Martinez et al., 2013). Th1 cells require the transcription factor T-bet for differentiation, secrete proinflammatory cytokines such as interferon (IFN)-γ, and are thought to play a pathogenic role in EAE (Sato et al., 2011). Th2 cells can antagonize Th1 cells, secrete anti-inflammatory cytokines such as interleukin (IL)-4 and IL-10, and play a regulatory role in most types of EAE.
Currently, a newly discovered Th subtype, Th17, has also been implicated to play a pathogenic role in MS and EAE. Th17 cells express the transcription factor retinoic acid-related orphan receptor (ROR)γt and secrete the proinflammatory cytokines IL-17, IL-21, IL-22 and tumor necrosis factor (TNF)-α (Harrington et al., 2006). In mice, naive CD4+ T cells are differentiated into Th17 cells by priming in the presence of transforming growth factor (TGF)-β and IL-6, which induces their hallmark transcription factor RORγt (Bettelli et al., 2006), while IL-23 promotes the survival of Th17 cells (Stritesky et al., 2008). Since the IL-17 receptor and IL-22 receptor are present on a broad range of cell types, Th17 cells can promote a widespread reaction that includes the production of IL-6 and other pro-inflammatory cytokines. The release of inflammatory cytokines from Th17 cells can cause immunopathology; dysregulation of Th17 cells has been implicated in many immune-mediated diseases ranging from MS to inflammatory bowel disease (IBD) (Ichiyama et al., 2008).
The increased frequency of IL-17-secreting cells in EAE led to the theory that they could be a critical effector cell population of disease. Komiyama et al first reported attenuation of EAE in IL-17 knockout (KO) mice: the onset of disease was delayed and both the clinical and pathological severity of disease were reduced (Komiyama et al., 2006). Experimentally, the roles of Th cells have been investigated in animal models of MS mainly by suppression of each Th response using blocking monoclonal antibodies (mAbs) directed against different Th cell-derived cytokines as well as gene knockout mice of these specific cytokines and mediators (Cua et al., 2003, Gran et al., 2002, Liblau et al., 1997). Although these “loss-of-function” studies have been informative, they have not addressed how increased Th17 immune responses, which have been found in MS patients, can affect the induction and clinical and pathological outcomes in EAE (Lovett-Racke et al., 2011). We have developed transgenic (Tg) mice that overexpress RORγt in T cells (Yoh et al., 2012). Compared with wild-type mice, the RORγt Tg mice have significantly higher amounts of IL-17 in the sera and after stimulation a higher percent of T cells convert to Th17 cells in vitro. We used these mice to investigate how a bias toward a Th17 immune response could affect EAE, since, in theory the incidence and clinical course of EAE can affected by a preexisting bias (“gain-of-function”) towards the Th17 subset prior to the induction of disease.
In this report, we have investigated how overexpression of RORγt or T-bet in the T cells of mice could affect EAE, induced with MOG. We demonstrated that the severity of EAE was enhanced in RORγt Tg mice, both clinically and histologically, while the T-bet Tg mice showed no signs of disease. The RORγt Tg mice have a significantly more robust anti-MOG immune response and secrete higher levels of inflammatory cytokines compared with wild-type mice.
2. Materials and Methods
2.1 Mice
To generate RORγt Tg and T-bet Tg mice, a full-length cDNA encoding the murine RORγt or T-bet protein was inserted into a VA CD2 transgene cassette that contained the upstream gene regulatory region and locus control region of the human CD2 gene (Kiwamoto et al., 2006, Yoh, Morito, 2012). RORγt or T-bet were preferentially expressed in T cells in RORγt Tg and T-bet Tg mice, respectively (Harrington, Mangan, 2006, Sato, Omura, 2011, Yoh, Morito, 2012). The Tg mice were maintained as heterozygotes for the transgene by breeding them with wild-type C57BL/6 mice. Animals were maintained on 12/12-hour (hr) light/dark cycles in standard animal cages with filter tops under specific pathogen-free conditions in our animal care facility at Louisiana State University Health Sciences Center (LSUHSC)-Shreveport and given standard laboratory rodent chow and water ad libitum. All experimental procedures involving the use of animals were reviewed and approved by the Institutional Animal Care and Use Committee of LSUHSC and performed according to the criteria outlined by the National Institutes of Health.
2.2 EAE induction
Four to six-week-old wild-type littermates, RORγt Tg, or T-bet Tg C57BL/6 mice were sensitized subcutaneously in the base of the tail with 100 nmol of MOG35-55 peptide (United Peptide Corporation, Rockville, MD) emulsified in complete Freund’s adjuvant (CFA) composed of Imject® Freund’s incomplete adjuvant (Pierce Biotechnology, Rockford, IL) and Mycobacterium tuberculosis H37 Ra (Difco Laboratories, Detroit, MI) (Sato et al., 2013). The final concentration of M. tuberculosis in the MOG/CFA solution was 2 mg/ml (200 μl/mouse). Mice were also injected intraperitoneally with 400 ng of pertussis toxin (List Biological Laboratories Inc., Campbell, California) on days 0 and 2. Clinical scores of EAE were evaluated as follows: 0, no signs; 1, paralyzed tail; 2, mild hind limb paresis; 3, moderate hind limb paralysis; 4, complete hind limb paraplegia; 5, fore limb paralysis or moribund (Fernando et al., 2014).
2.3 Neuropathology
Mice were perfused with phosphate-buffered saline (PBS) followed by a 4% paraformaldehyde solution (Sigma-Aldrich) in PBS. The spinal cords were harvested and fixed with 4% paraformaldehyde. The spinal cords were divided into 10 to 12 transversal segments and embedded in paraffin. Four-μm-thick sections were stained with Luxol fast blue (Solvent blue 38; Sigma-Aldrich) for myelin visualization. Histological scoring of the spinal cords was performed as previously described (Tsunoda et al., 2008, Tsunoda et al., 2001). For scoring of spinal cord sections, each spinal cord section was divided into four quadrants: the ventral funiculus, the dorsal funiculus, and each lateral funiculus. Any quadrant containing demyelination, meningitis, or perivascular cuffing was given a score of 1 in that pathological class. The total number of positive quadrants for each pathological class was determined and then divided by the total number of quadrants present on the slide and multiplied by 100 to give the percent involvement for each pathological class. An overall pathology score was also determined by giving a positive score if any pathology was present in the quadrant, and presented as the percent involvement.
2.4 Lymphoproliferation assay
Mononuclear cells (MNCs) were isolated from the inguinal lymph nodes of mice using Histopaque 1083 (Sigma-Aldrich) (Sato, Martinez, 2013). MNCs were cultured in RPMI 1640 medium (Mediatech) supplemented with 10% fetal bovine serum (FBS) (Mediatech), 2 mM L-glutamine (Mediatech), 50 mM β-mercaptoethanol (Sigma-Aldrich), and 1% antibiotic-antimycotic solution (Mediatech), at 2 × 105 cells/well in 96-well plates (Corning) and stimulated with 50 μg/ml MOG35-55 peptide or purified protein derivative (PPD) from Mycobacterium Tuberculosis (World Health Organization International Lab for Biological Standards, Copenhagen, Denmark) for 5 days. For CD4 and CD8 blocking, the cells were cultured with unlabeled GK1.5 and Lyt-2.43, respectively, for the 5 day period (Dialynas et al., 1983). To assess the lymphoproliferation, [3H]thymidine (PerkinElmer, Inc., Waltham, MA) was added in the culture system at the concentration of 1 μCi/well for the last 24 hr. MNCs were harvested on Reeves Angel 934AH filters (Brandel, Gaithersburg, MD) using PHD™ Harvester (Brandel). The incorporated radioactivity was measured by Wallac 1409 Liquid Scintillation Counter (PerkinElmer). All cultures were performed in triplicate and the data were expressed as the change in counts per minute (cpm) (Δcpm = experimental cpm - no stimulation cpm).
2.5 Cytokine enzyme linked immunosorbent assay (ELISA)
MNCs isolated from inguinal lymph nodes of mice were cultured at 8 × 106 cells/well in 6-well plates (Corning Inc., Corning, NY) and stimulated with 4 ml of 5 μg/ml of concanavalin A (ConA) or 50 μg/ml of MOG35-55 for 48 hr. Culture supernatants were harvested and stored at −80 °C until examined. The levels of IL-4, IL-6, IL-10, and IFN-γ concentrations were detected with BD OptEIA kits (BD Biosciences, San Diego, CA) and IL-17A was detected with a Mouse IL-17A ELISA MAX™ kit (Biolegend, Inc., San Diego, CA) according to the manufacturer’s instructions (Tsunoda et al., 2005). ELISAs were performed in duplicate in 96-well plates.
2.6 Flow cytometry
MNCs in the CNS tissue were separated on discontinuous Percoll gradients, as described previously (Tsunoda et al., 1996). Brain and spinal cord tissues were removed en bloc and were dissociated by passing them through a stainless steel mesh. The dissociated tissues were pelleted at 1,000 rpm for 10 minutes (min). The pellets were suspended in 5 ml of 70% Percoll (GE Healthcare Bio-Sciences, Pittsburgh, PA) in PBS. Each suspension was overlaid with 5 ml of 30% Percoll, and centrifuged at 1,200 g for 30 min at room temperature. MNCs were removed from the 30%/70% Percoll interface.
Fc receptors of cells were blocked with anti-CD16/32 (Biolegend, San Diego, CA) (Martinez et al., 2014). Cells were stained with antibodies against CD4 (Biolegend), IFN-γ (Biolegend), and IL-17A (Biolegend). Cells were permeabilized and fixed using the BD Cytofix/Cytoperm™ Plus Fixation/Permeabilization Kit (BD Biosciences, San Jose, CA). The flow cytometry data was acquired on a FACSCalibur (BD Biosciences) and analyzed using Cellquest Pro (BD Biosciences). For intracellular cytokine staining, cells were incubated with MOG35-55 at 50 μg/ml for 48 hr, and 5 ng/ml phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich), 500 ng/ml ionomycin (Sigma-Aldrich), and 1 μg/ml of brefeldin A (GolgiPlug™, BD Biosciences) for 6 hr before staining.
3. Results
3.1 RORγt Tg mice develop more severe EAE
We sensitized both C57BL/6 wild-type littermates and RORγt Tg mice with MOG 35-55 to induce EAE and found that both strains developed classical EAE with flaccid ascending tail and hind limb paralysis. The RORγt Tg mice developed significantly more severe clinical disease compared with wild-type mice [mean maximum clinical score ± standard error of mean (SEM): wild-type, 2.3 ± 0.3; RORγt Tg, 3.3 ± 0.3; P < 0.05, Mann-Whitney U test], despite a slightly delayed onset (average day of disease onset ± SEM: wild-type, 13.5 ± 0.5; RORγt Tg, 15 ± 0.5; P < 0.05, t test) (Figure 1). The incidence was slightly higher in the RORγt Tg mice (19/21, 90.5%) compared with the wild-type mice (18/23, 78.3%), but did not reach a statistical difference. The RORγt Tg mice did not fully recover from disease and had lasting hind limb paralysis during the 2-month observation period, while most wild-type mice fully recovered from disease around 4 weeks p.i., clinically. The cumulative score during the 2-month observation period was significantly higher in RORγt Tg mice than in wild-type mice (mean cumulative score ± SEM: wild-type, 36.6 ± 10; RORγt Tg, 97 ± 23.9; P < 0.05, Mann-Whitney U test).
Figure 1.

Clinical scores for wild-type (closed boxes) and RORγt Tg (open circles) mice with experimental autoimmune encephalomyelitis (EAE) induced with myelin oligodendrocyte glycoprotein (MOG)35-55. RORγt Tg mice developed significantly more severe disease with lasting neurological deficits (*, P < 0.05, compared with wild-type mice, Mann-Whitney U test). Clinical scores of EAE were evaluated as follows: 0, no signs; 1, paralyzed tail; 2, mild hind limb paresis; 3, moderate hind limb paralysis; 4, complete hind limb paralysis; 5, fore limb paralysis or moribund. The results are representative of four experiments, consisting of 7–15 mice per experiment, for a total 21 wild-type and 19 RORγt Tg mice.
Histologically, we found that the severity in clinical disease was associated with enhanced demyelination in the spinal cord (Figures 2A and B). All symptomatic mice had demyelinating lesions in their dorsal funiculus; the RORγt Tg mice had demyelinating lesions in their lateral funiculi leading to a higher pathology score in the RORγt Tg mice compared with the wild-type mice, which rarely had lesions in their lateral funiculi (overall score ± SEM: wild-type, 24 ± 6; RORγt Tg, 52 ± 3; P < 0.05, t test) (Figure 2C). The distribution of lesions along the spinal cord axis was similar between the wild-type and RORγt Tg mice.
Figure 2.
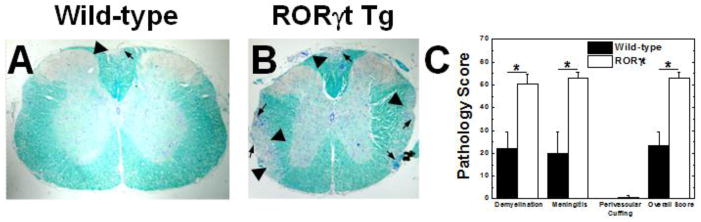
Spinal cord pathology of EAE mice. A and B. RORγt Tg mice developed more inflammation (arrows) and demyelination (arrow heads) compared with wild-type mice. Spinal cord segments stained with Luxol fast blue. C. Quantification of spinal cord pathology showed that the RORγt Tg mice had more demyelination, meningitis, and overall pathology (*, P < 0.05 compared with wild-type mice, t test). The results are representative of four independent experiments, consisting of 7–15 mice per experiment, for a total 21 wild-type and 19 RORγt Tg mice. Spinal cords were harvested 2 months post induction. Magnification A and B: ×32.
3.2 RORγt Tg mice have more robust immune responses to MOG
To test whether the levels of MOG-specific immune responses correlated with neuropathology, we isolated MNCs from wild-type and RORγt Tg mice with EAE during the early stage of disease (19 days p.i.), or during the late stage of disease (2 months p.i.), and compared lymphoproliferative responses to MOG between the two mouse strains. We found that the lymphoproliferative response to MOG was significantly higher in RORγt Tg mice compared with wild-type mice at both the early and late stages of EAE (P < 0.05, t test) (Figure 3). When the MNCs from the RORγt Tg mice or wild-type mice were incubated with MOG35-55, in the presence of CD4 blocking Ab, the response was suppressed in both mouse strains, while CD8 blocking antibody did not significantly suppress MOG-specific lymphoproliferation. This indicates that in both mouse strains CD4+ T cells were the predominant MOG35-55-specific effector cells in EAE, and that the difference in disease severity was likely due to an increase in MOG-specific CD4+ T cells, but not CD8+ T cells in the RORγt Tg mice.
Figure 3.

Cellular immune responses to MOG during the early (A) and late (B) stages of EAE. RORγt Tg mice (open bars) had a significantly more robust immune response to MOG compared with wild-type mice (closed bars) (*, P < 0.05, t test), which was suppressed by CD4 blocking antibody (Ab), but not CD8 blocking Ab (A and B). Mononuclear cells (MNCs) were isolated from the inguinal lymph nodes of wild-type or RORγt Tg mice during the peak of the early stage (19 days p.i.) or the late (2 months p.i.) stage of EAE and stimulated with MOG35-55. For CD4 and CD8 blocking, the cells were cultured with unlabeled anti-CD4 or CD8 antibody, respectively, during the 5 day incubation period. Lymphoproliferation was quantified by a [3H]thymidine incorporation assay. Δcpm = [counts per minute (cpm) of treatment group] - (cpm of no treatment group). The results are representative of two experiments (early stage) and four experiments (late stage) consisting of 9 or 11 mice for a total of 9 wild-type and 11 RORγt Tg mic (early stage) and 7–15 mice for a total 21 wild-type and 19 RORγt Tg mice (late stage). Results are mean + SEM from three pools of inguinal lymph nodes from two mice per pool.
Since the RORγt Tg mice had a more robust immune response to MOG, we determined the cytokine profile of the MNCs by stimulation with MOG or ConA, a T-cell mitogen, to further characterize the immune response using ELISAs. As expected, MNCs from the RORγt Tg mice produced significantly more IL-17 in response to MOG and ConA at both the early stage (Figure 4A) and late stage (Figure 5A). During the early stage, we found that MNCs from RORγt Tg mice also produced significantly higher levels of IL-10, and IL-6, while there was no significant difference in IFN-γ production (Figures 4B–D). During the late stage, MNCs from the RORγt Tg mice produced significantly higher levels of IL-10, IL-6, and IFN-γ than wild-type mice (Figures 5B–D). IL-4 production was undetectable in response to MOG or ConA during the early or late stage of EAE.
Figure 4.

Cytokine responses during the early stage of EAE. MNCs from RORγt Tg mice (open bars) produced significantly interleukin (IL)-17 (A) and IL-10 (B) IL-6 (C) and interferon (IFN)-γ (D) in response to MOG compared with control mice (closed bars) (*, P < 0.05, t test). Concanavalin A (ConA) also induced higher IL-17, IL-10 and IL-6 but not IFN-γ production in RORγt Tg mice than wild-type mice. Mononuclear cells (MNCs) were isolated from the inguinal lymph nodes of RORγt or wild-type mice (19 days p.i.) and stimulated with ConA or MOG35-55 for 48 hours (hr). The levels of cytokine production in the culture supernatants were measured by ELISA. The results are representative of two independent experiments consisting of 9 or 11 mice, for a total of 9 wild-type and 11 RORγt Tg mice. Results are mean + SEM from three pools of inguinal lymph nodes from two mice per pool. ND: Not detectable.
Figure 5.

Cytokine responses during the late stage of EAE. MNCs from the RORγt Tg mice (open bars) produced significantly more IL-17 (A), IL-10 (B), IL-6 (C), and IFN-γ (D) in response to MOG35-55 compared with wild-type mice (closed bars) (*, P < 0.05, t test). ConA also induced higher production of all cytokines in RORγt Tg mice compared with wild-type mice. MNCs were isolated from the inguinal lymph nodes of wild-type or RORγt Tg mice 2 months p.i. and stimulated with ConA or MOG35-55 for 48 hr. The levels of cytokine production in the culture supernatants were measured by ELISA. The results are representative of 4 experiments, consisting of 7–15 mice, for a total 21 wild-type and 19 RORγt Tg mice. Results are mean + SEM from three pools of inguinal lymph nodes from two mice per pool.
To determine the cell types that produce IL-17 and IFN-γ, we have isolated MNCs from the CNS and regional lymph nodes of the wild-type and RORγt Tg mice during the peak of EAE. After in vitro incubation with MOG35-55, we conducted intracellular cytokine staining of the MNCs. In both the CNS and lymph nodes, we found that the percentages of CD4+ were similar between wild-type and RORγt Tg mice (mean percentage of CD4+ cells ± SEM, CNS: wild-type, 37.4 ± 3.0; RORγt Tg, 42.3 ± 9.5; lymph node: wild-type, 13.1 ± 0.3; RORγt Tg, 16.5 ± 0.4) and that IL-17+ and IFN-γ+ cells were predominantly CD4+ cells (data not shown). RORγt Tg mice had higher percentages of IL-17 single positive and IL-17+/IFN-γ+ double positive CD4+ cells and lower percentages of IFN-γ single positive CD4+ cells both in the CNS and lymph nodes, compared with wild-type mice (Figure 6).
Figure 6.

Flow cytometric analysis of MNCs from mice 19 days after MOG sensitization. Both wild-type and RORγt Tg mice were at the peak of EAE, while T-bet Tg mice were asymptomatic. RORγt Tg mice and T-bet Tg mice had higher percentages of IL-17+ and IFN-γ+ CD4+ cells, compared with wild-type mice, respectively. MNCs from the CNS or inguinal lymph nodes were stimulated with MOG35-55, PMA, ionomycin and brefeldin A and stained for the cell surface marker CD4 and intracellular cytokines IL-17 and IFN-γ. Representative dot plots after gating on CD4+ cells (four mice/group). CNS: central nervous system; LN: lymph node.
3.3 T-bet overexpression reduces the severity of EAE
Since T-bet Tg mice have previously been shown to develop altered susceptibilities to immune mediated diseases (Ishizaki et al., 2007, Shimohata et al., 2009), we sensitized both C57BL/6 wild-type littermates and T-bet Tg mice with MOG 35-55 to induce EAE. Interestingly, we found that most wild-type mice developed EAE, while the T-bet Tg mice did not develop any clinical signs during the 2-month observation period (Figures 7A). Histologically, the T-bet Tg mice had little or no inflammation in the spinal cord (Figure 7B). Consistent with the clinical and neuropathological data, the T-bet mice had a significantly less lymphoproliferation in response to MOG (Figure 7C).
Figure 7.
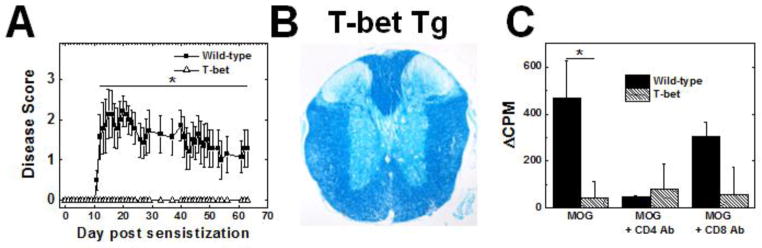
EAE in T-bet Tg mice. Wild-type mice (closed squares) and T-bet Tg (open triangles) mice were sensitized with MOG. T-bet Tg mice did not develop clinical signs (A), spinal cord pathology (B), or substantial immune responses to MOG35-55 (C) 2 months p.i. [*, P < 0.05, compared with wild-type mice, Mann-Whitney U test (A) and t test(C)]. A. Clinical scores of EAE. B. Luxol fast blue staining of the spinal cord. C. Lymphoproliferative responses to MOG35-55. The results are representative of two experiments, consisting of 10-15 mice, for a total of 9 wild-type and 11 T-bet Tg mice. Magnification B: × 28.
When wild-type mice with EAE were at the peak of disease, we isolated MNCs from the CNS and regional lymph nodes from MOG-sensitized T-bet Tg mice, and conducted intracellular cytokine staining after stimulation with MOG. In both the CNS and lymph nodes, we found that the percentages of CD4+ cells were similar between wild-type and T-bet Tg mice (mean percentage of CD4+ cells ± SEM, CNS: wild-type, 37.4 ± 3.0; T-bet Tg, 44.3 ± 2.0; lymph node: wild-type, 13.1 ± 0.3; T-bet Tg, 15.2 ± 0.1) and that IL-17+ and IFN-γ+ cells were predominately CD4+ cells (data not shown). Compared with wild-type mice, T-bet Tg mice had higher percentages of IFN-γ+ CD4+ cells in both the CNS and lymph nodes, but lower percentages of IL-17+ cells (Figure 6).
4. Discussion
In this report, we have demonstrated that RORγt Tg mice developed more severe EAE than wild-type mice. This is likely due to a genetic bias toward Th17 immune responses. The deleterious role of Th17 cells has been reported in many studies, but these have generally involved KO animals, where Th17 immune responses have been impaired (Komiyama, Nakae, 2006, Langrish et al., 2005). To our knowledge this is the first study to demonstrate how genetically enhanced Th17 immune responses could worsen EAE. These observations are in agreement with previous reports that Th17 cells are a major pathogenic effector cells in EAE.
We did not observe any abnormal immune responses during the 2-month observation period, except that they had higher serum IL-17 levels compared with wild-type mice [mean serum IL-17 (pg/ml) ± SEM: wild-type, 132.7 ± 18.8; RORγt Tg, 217.5 ± 18.5; P < 0.05, t test]. Unlike BALB/c × C57BL/6 RORγt Tg F1 mice that develop splenomegaly and enlarged lymph nodes (Yoh, Morito, 2012), we did not observe any differences in size of spleens or lymph nodes (data not shown). Additionally, if the MNCs from either mouse strain were incubated with PPD from Mycobacterium Tuberculosis, which is a component of Freund’s adjuvant, the lymphoproliferation was similar between the wild-type and RORγt Tg mice (Δcpm at 50μg/ml PPD ± SEM, 2 months p.i.: wild-type, 2019.6 ± 678.3; RORγt Tg, 1900.5 ± 305.8; P < 0.05, t test).
While the RORγt Tg mice developed more severe EAE, the day of disease onset was delayed, compared with wild-type mice. This was unexpected, since IL-17 KO mice have been reported to show a delayed onset of EAE (Komiyama, Nakae, 2006). It is possible that Th17 cells by themselves may not be able to cross the blood-brain barrier to initiate disease as efficiently as other cells types or that they are not the primary initiators of disease and act as an effector cell population once another cell population has initiated disease. Alternatively, the coordination of different effector cell types may be important in disease onset.
As expected MOG-sensitized RORγt Tg mice secreted more IL-17 compared with the wild-type mice. However, interestingly, the RORγt Tg mice also secreted significantly more IFN-γ compared with the wild-type mice, despite having a lower percentage of CD4+/IFN-γ+ cells. This discrepancy can be accredited to a percentage of IL-17+/IFN-γ+ double positive cells and the robust proliferation of the MNCs from the RORγt Tg mice in response to MOG35-55. Although Th17 and Th1 immune responses suppress each other, clinically and experimentally, the presence of both Th1 and Th17 cells have been demonstrated in many immune-mediated disease and their animal models, including MS and EAE, suggesting that Th1 and Th17 cells play pathogenic roles in certain immune mediated diseases, synergistically. The delay in the onset of disease in the RORγt Tg mice may reflect such synergistic effects. IL-17+/IFN-γ+ double-positive cells have been demonstrated to be present in inflamed tissue or the peripheral blood of individuals with chronic inflammatory conditions, including MS, and its animal models (Kebir et al., 2009, Momcilovic et al., 2008, Suryani and Sutton, 2007). The authors suggested that these double-positive cells exhibited a Th17 phenotype and eventually converted to IL-17+/IFN-γ− cells. Although, we found that the secretion of IL-17 and IFN-γ in the RORγt Tg mice remained high over 2 months p.i., the ratio of IL-17/IFN-γ production was higher during the early stage of disease than in the late stage of disease, suggesting a relative decrease in IL-17 production (e.g. IL-17 to IFN-γ ratio after MOG stimulation in RORγt Tg mice: early, 33.8; late, 0.21). Here, Th1 cells may antagonize Th17 cells and be beneficial in Th17 cell mediated disease, and may also explain why the T-bet Tg mice did not develop EAE.
We also found that the RORγt Tg mice produced higher levels of IL-6 and IL-10. The increased in IL-6 production in response to MOG could be a secondary effect of the enhanced IL-17 production, since it has been shown that IL-17 can induce IL-6 in other cell types (Hwang et al., 2004). Although, Th17 cells have been shown to secrete IL-10 in response to certain stimuli (Zielinski et al., 2012), it is unknown if Th17 cells produce IL-10 in response to MOG or other self-antigens. Alternatively, a large population of IL-10 producing anti-inflammatory cells could be generated in response to MOG to counteract the inflammatory responses in the RORγt Tg mice and they may be responsible for the increased IL-10 response. The greater cytokine responses could also be due to a higher percentage of MOG-specific cells in the T cell population. In theory, the greater amount of damage in the RORγt Tg mice would release more myelin antigen, leading to a greater expansion of MOG-specific T cells; this would agree with the greater lymphoproliferative response in the RORγt Tg mice.
Although a lack of T-bet has been shown to reduce the severity of EAE (Nath et al., 2006, Yang et al., 2009), interestingly, we found that overexpression of T-bet also suppressed EAE. It may be necessary for T-bet to be expressed within a certain range for it to be effective in causing disease, where no expression or expression beyond a certain level both could prevent disease. Previously, we (KY and ST) demonstrated that T-bet mice developed spontaneous dermatitis and accelerated autoimmune glomerulonephritis (Ishizaki, Yamada, 2007, Shimohata, Yamada, 2009). In the current experiment, we also found that the T-bet mice develop spontaneous dermatitis, regardless of MOG-sensitization, while we did not observe CNS involvement in the T-bet Tg mice. Since it has been shown that these T-bet Tg mice have suppressed Th17 immune responses, the Th1 cells might infiltrated the CNS, but need help from Th17 cells to initiate immune-mediated disease in the CNS (Kondo et al., 2012). Future experiments will require the exact influence of T-bet on the severity of EAE, such as crossing the RORγt and T-bet Tg mice.
The alteration of disease course and severity shown here in RORγt and T-bet Tg mice demonstrate how genetic factors could influence the susceptibility and severity of autoimmune diseases, particularly how a bias to Th17 immune responses enhances the severity of disease. Thus, a genetic bias toward a Th17 immune response could render individuals more susceptible to autoimmune disease; clinically, it may cause nonsymptomatic inflammation to evolve into clinical disease. For example, an infection could result in the release of sequestered auto-antigens from immune-privileged tissue, which can induce autoimmunity by epitope (or determinant) spreading (McCarthy et al., 2012). This could lead to autoimmune inflammatory diseases among a certain population of people with a Th17 bias, even in what would be an innocuous infection in the general population (Martinez et al., 2012). “Gain-of-function” mutations have been shown to cause auto-inflammatory diseases and change Th immune responses in humans (Masters et al., 2009). Here, we demonstrated how a gain-of-function can change susceptibility and severity of an autoimmune model of MS. Translational application of information from our findings will be clinically useful in the future, for example, where if cytokine profiles are found that are biased toward Th17 immune responses in an individual whose family members with MS, the individual may be advised to be monitored for disease development and might take prophylactic medicine to block Th17 immune responses.
Highlights.
RORγt Tg mice mice overexpressing RORγt favored Th17 immune responses
RORγt Tg mice developed more severe EAE, clinically and histologically
RORγt Tg mice had a more robust anti-myelin immune response
A Th17 biased immune response exacerbated the severity of an autoimmune model for MS
T-bet Tg mice did not develop EAE
Acknowledgments
This work was supported by the fellowships (F. Sato and S. Omura) from the Malcolm Feist Cardiovascular Research Endowment, LSU Health Sciences Center, Shreveport, and grants from the National Institute of General Medical Sciences COBRE Grant (P30-GM110703). We thank Elaine A. Cliburn for excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Dialynas DP, Quan ZS, Wall KA, Pierres A, Quintans J, Loken MR, et al. Characterization of the murine T cell surface molecule, designated L3T4, identified by monoclonal antibody GK1.5: similarity of L3T4 to the human Leu-3/T4 molecule. Journal of immunology. 1983;131:2445–51. [PubMed] [Google Scholar]
- Fernando V, Omura S, Martinez NE, Sato F, Kawai E, Elliott SF, Takahashi S, Yoh K, Tsunoda I. Regulation of an autoimmune model for multiple sclerosis in Th2-biased GATA3 transgenic mice. International Journal of Molecular Sciences. 2014;15:1700–18. doi: 10.3390/ijms15021700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, et al. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. Journal of immunology. 2002;169:7104–10. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Current opinion in immunology. 2006;18:349–56. doi: 10.1016/j.coi.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Hwang SY, Kim JY, Kim KW, Park MK, Moon Y, Kim WU, et al. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis research & therapy. 2004;6:R120–8. doi: 10.1186/ar1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyama K, Yoshida H, Wakabayashi Y, Chinen T, Saeki K, Nakaya M, et al. Foxp3 inhibits RORgammat-mediated IL-17A mRNA transcription through direct interaction with RORgammat. The Journal of biological chemistry. 2008;283:17003–8. doi: 10.1074/jbc.M801286200. [DOI] [PubMed] [Google Scholar]
- Ishizaki K, Yamada A, Yoh K, Nakano T, Shimohata H, Maeda A, et al. Th1 and type 1 cytotoxic T cells dominate responses in T-bet overexpression transgenic mice that develop contact dermatitis. Journal of immunology. 2007;178:605–12. doi: 10.4049/jimmunol.178.1.605. [DOI] [PubMed] [Google Scholar]
- Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, et al. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Annals of neurology. 2009;66:390–402. doi: 10.1002/ana.21748. [DOI] [PubMed] [Google Scholar]
- Kiwamoto T, Ishii Y, Morishima Y, Yoh K, Maeda A, Ishizaki K, et al. Transcription factors T-bet and GATA-3 regulate development of airway remodeling. American journal of respiratory and critical care medicine. 2006;174:142–51. doi: 10.1164/rccm.200601-079OC. [DOI] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. Journal of immunology. 2006;177:566–73. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Iizuka M, Wakamatsu E, Yao Z, Tahara M, Tsuboi H, et al. Overexpression of T-bet gene regulates murine autoimmune arthritis. Arthritis and rheumatism. 2012;64:162–72. doi: 10.1002/art.33335. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. The Journal of experimental medicine. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liblau R, Steinman L, Brocke S. Experimental autoimmune encephalomyelitis in IL-4-deficient mice. International immunology. 1997;9:799–803. doi: 10.1093/intimm/9.5.799. [DOI] [PubMed] [Google Scholar]
- Lovett-Racke AE, Yang Y, Racke MK. Th1 versus Th17: are T cell cytokines relevant in multiple sclerosis? Biochimica et biophysica acta. 2011;1812:246–51. doi: 10.1016/j.bbadis.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NE, Sato F, Kawai E, Omura S, Chervenak RP, Tsunoda I. Regulatory T cells and Th17 cells in viral infections: implications for multiple sclerosis and myocarditis. Future virology. 2012;7:593–608. doi: 10.2217/fvl.12.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NE, Sato F, Kawai E, Omura S, Takahashi S, Yoh K, et al. Th17-biased RORgammat transgenic mice become susceptible to a viral model for multiple sclerosis. Brain Behav Immun. 2014 doi: 10.1016/j.bbi.2014.07.008. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NE, Sato F, Omura S, Minagar A, Alexander JS, Tsunoda I. Immunopathological patterns from EAE and Theiler’s virus infection: Is multiple sclerosis a homogenous 1-stage or heterogenous 2-stage disease? Pathophysiology: the official journal of the International Society for Pathophysiology / ISP. 2013;20:71–84. doi: 10.1016/j.pathophys.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annual review of immunology. 2009;27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DP, Richards MH, Miller SD. Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods in molecular biology. 2012;900:381–401. doi: 10.1007/978-1-60761-720-4_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SD, Gerety SJ, Kennedy MK, Peterson JD, Trotter JL, Tuohy VK, et al. Class II-restricted T cell responses in Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease. III. Failure of neuroantigen-specific immune tolerance to affect the clinical course of demyelination. Journal of neuroimmunology. 1990;26:9–23. doi: 10.1016/0165-5728(90)90115-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momcilovic M, Miljkovic Z, Popadic D, Miljkovic D, Mostarica-Stojkovic M. Kinetics of IFN-gamma and IL-17 expression and production in active experimental autoimmune encephalomyelitis in Dark Agouti rats. Neuroscience letters. 2008;447:148–52. doi: 10.1016/j.neulet.2008.09.082. [DOI] [PubMed] [Google Scholar]
- Nath N, Prasad R, Giri S, Singh AK, Singh I. T-bet is essential for the progression of experimental autoimmune encephalomyelitis. Immunology. 2006;118:384–91. doi: 10.1111/j.1365-2567.2006.02385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato F, Martinez NE, Shahid M, Rose JW, Carlson NG, Tsunoda I. Resveratrol exacerbates both autoimmune and viral models of multiple sclerosis. The American journal of pathology. 2013;183:1390–6. doi: 10.1016/j.ajpath.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato F, Omura S, Martinez NE, Tsunoda I. Animal models of multiple sclerosis. In: Minagar A, editor. Neuroinflammation. London, UK: Elsevier; 2011. pp. 55–79. [Google Scholar]
- Shimohata H, Yamada A, Yoh K, Ishizaki K, Morito N, Yamagata K, et al. Overexpression of T-bet in T cells accelerates autoimmune glomerulonephritis in mice with a dominant Th1 background. Journal of nephrology. 2009;22:123–9. [PubMed] [Google Scholar]
- Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. Journal of immunology. 2008;181:5948–55. doi: 10.4049/jimmunol.181.9.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryani S, Sutton I. An interferon-gamma-producing Th1 subset is the major source of IL-17 in experimental autoimmune encephalitis. Journal of neuroimmunology. 2007;183:96–103. doi: 10.1016/j.jneuroim.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Tsunoda I, Fujinami RS. Two models for multiple sclerosis: experimental allergic encephalomyelitis and Theiler’s murine encephalomyelitis virus. Journal of neuropathology and experimental neurology. 1996;55:673–86. doi: 10.1097/00005072-199606000-00001. [DOI] [PubMed] [Google Scholar]
- Tsunoda I, Iwasaki Y, Terunuma H, Sako K, Ohara Y. A comparative study of acute and chronic diseases induced by two subgroups of Theiler’s murine encephalomyelitis virus. Acta Neuropathol. 1996;91:595–602. doi: 10.1007/s004010050472. [DOI] [PubMed] [Google Scholar]
- Tsunoda I, Libbey JE, Kuang LQ, Terry EJ, Fujinami RS. Massive apoptosis in lymphoid organs in animal models for primary and secondary progressive multiple sclerosis. The American journal of pathology. 2005;167:1631–46. doi: 10.1016/S0002-9440(10)61247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda I, Tanaka T, Fujinami RS. Regulatory role of CD1d in neurotropic virus infection. Journal of virology. 2008;82:10279–89. doi: 10.1128/JVI.00734-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda I, Wada Y, Libbey JE, Cannon TS, Whitby FG, Fujinami RS. Prolonged gray matter disease without demyelination caused by Theiler’s murine encephalomyelitis virus with a mutation in VP2 puff B. Journal of virology. 2001;75:7494–505. doi: 10.1128/JVI.75.16.7494-7505.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, et al. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. The Journal of experimental medicine. 2009;206:1549–64. doi: 10.1084/jem.20082584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh K, Morito N, Ojima M, Shibuya K, Yamashita Y, Morishima Y, et al. Overexpression of RORgammat under control of the CD2 promoter induces polyclonal plasmacytosis and autoantibody production in transgenic mice. European journal of immunology. 2012;42:1999–2009. doi: 10.1002/eji.201142250. [DOI] [PubMed] [Google Scholar]
- Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature. 2012;484:514–8. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]