

Abstract
HER2 is an important determinant of poor prognosis in breast cancer patients. Studies indicate that HER2 positive tumors are mostly resistant to therapy and have high metastatic potential however, the underlying mechanisms remain unknown. In this study, MDA‐MB‐231 and MCF‐7 breast cancer cells with their HER2 overexpressing syngeneic variants were used to delineate the role of HER2 in EMT and metastasis. Our results demonstrated that HER2 overexpression increased the invasive potential of cells. Our results also showed that HER2 overexpression lead to the production of TGFβ resulting in the activation of TGFβ/SMAD signaling. Furthermore, activation of SNAIL, SLUG and ZEB‐1, the transcriptional repressors of E‐cadherin and increased mesenchymal characteristics were observed in high HER2 cells. Interestingly, EMT by HER2 was mediated through TGFβ. Intravenous injection of high HER2 MDA‐MB‐231 (HH) cells in athymic nude mice showed early and substantial metastasis as compared to the parent cells establishing the direct role of HER2 in metastasis. Our results showed that inhibition of HER2 mediated EMT by cucurbitacin B a triterpenoid, resulted in the suppression of brain metastasis of breast cancer cells. Taken together, our results identify a novel mechanism of HER2 in promoting breast cancer metastasis through de novo synthesis of TGFβ leading to EMT, an initial and essential step of metastasis.
Keywords: Breast cancer, In vivo, ERBB2, EMT, TGFβ
Highlights
Study establishes the novel role of HER2 in breast cancer metastasis through TGFβ‐mediated EMT.
HER2 regulates TGFβ/SMAD pathway by inducing TGFβ do novo synthesis and activity.
HER2 overexpression enhances the rate of tumor cell invasion and metastasis in vivo.
HER2 overexpression mediates epithelial‐to‐mesenchymal transition (EMT) through TGFβ to induce metastasis.
HER2 suppression inhibits TGFβ signaling leading to reduced EMT and metastasis.
List of Abbreviations
- HER2
Human Epidermal Growth Factor Receptor 2
- TGFβ
Transforming growth factor β
- Dn TGFβR
dominant negative transforming growth factor β receptor
- HH
High HER2
- CuB
Cucurbitacin B
- MDA‐MB‐231 (HH)
High HER2 cell line MDA‐MB‐231
- MCF‐7 (HH)
High HER2 cell line MCF‐7
- PBS
Phosphate Buffered Saline
- SRB
Sulforhodamine B
- DMEM
Dulbecco's Modified Eagle's Medium
- FBS
Fetal Bovine Serum
- PSN
Penicillin Streptomycin Neomycin
- SD
Standard Deviation
- SEM
Standard Error of Means
1. Introduction
Breast cancer is the most commonly diagnosed cancer in women and a second leading cause of cancer‐related deaths worldwide (Jemal et al., 2011). Metastatic cancer is the primary cause of mortality in breast cancer patients. In spite of significantly advanced therapeutic options, about 30% of the breast cancer patients progress to metastatic disease eventually (O'Shaughnessy, 2005). In addition, about 5% patients are diagnosed with stage IV metastatic breast cancer during initial diagnosis (Irvin et al., 2011).
The oncogene HER2 is overexpressed by 2–20 fold in about 25–30% of breast cancer patients, leading to poor prognosis (Slamon et al., 1987). HER2 positive tumors are highly aggressive, difficult to treat and hence these patients have reduced survival (Irvin et al., 2011). Trastuzumab is the monoclonal antibody against HER2 and the mainline treatment for HER2 positive cancer patients (Vogel et al., 2002). However, therapy with trastuzumab is effective only in 26% of patients, i.e. almost 74% patients are primarily resistant. Amongst trastuzumab responsive tumors, only about 23% show full response indicating the development of resistance during the treatment (Vogel et al., 2002). This suggests that identification and modulation of HER2 linked signaling and mechanisms can help in the advancement of breast cancer therapeutics with better survival outcomes.
For tumors to metastasize, it is important that tumor cells acquire motility. Epithelial‐to‐mesenchymal transition (EMT) is the mechanism by which cancer cells attain an invasive phenotype (Hanahan and Weinberg, 2011). It is usually defined by the loss of E‐cadherin, a tight junction protein mediated by the transcription factor SNAIL (de Herreros et al., 2010). TGFβ is a known master regulator of EMT (Kalluri and Weinberg, 2009; Taylor et al., 2010). TGFβ promotes EMT through canonical as well as non‐canonical signaling. Few studies indicate the involvement of SNAIL in TGFβ‐induced EMT in non‐cancerous as well as pancreatic cancer cells (Brandl et al., 2010; Li et al., 2013; Yang et al., 2013).
Although HER2 positive patients present with higher rates of metastasis, direct molecular correlation between HER2 and the metastasis is not known yet. Interestingly, HER2 positive tumors that showed co‐expression of SNAIL were found to be non‐responsive to trastuzumab therapy (Oliveras‐Ferraros et al., 2012). These observations suggest a link between HER2 and EMT responsible for drug resistance and metastasis.
Based on these facts, we hypothesized that HER2 promotes EMT leading to highly metastatic tumors. In the current study we identified that stable HER2 overexpression caused enhanced expression of the transcription factors SNAIL and ZEB‐1 in breast cancer cells. Our results showed that EMT regulation by HER2 was mediated by increased production of TGFβ. Furthermore, suppression of HER2/TGFβ signaling by cucurbitacin B a triterpenoid, reversed the EMT process in breast cancer cells, leading to reduced metastasis in the mouse model of breast cancer metastasis. To the best of our knowledge this is a first report on the regulation of EMT by HER2 through TGFβ/SNAIL‐ZEB1.
2. Materials and methods
2.1. Ethics statement
Animal experiments were conducted in accordance with the ethical standards and according to approved protocol by Institutional Animal Care and Use Committee (IACUC).
2.2. Cell culture
Human breast carcinoma cell lines MDA‐MB‐231, MCF‐7, 4T‐1 and MCF‐7 and MDA‐MB‐231 cells with HER2 overexpression were maintained in DMEM supplemented with 10% FBS and 5% PSN. The MDA‐MB‐231 (HH) cells were maintained in the medium described above in the presence of 300 μg/ml zeocin. All the cells used in this study were within twenty passages after receipt or resuscitation. 4T‐1‐luc cells were obtained from PerkinElmer, Waltham, MA. MCF‐7 (HH) cells were kindly provided by Dr Huang Fei (Bristol‐Myers Squibb Co., Princeton, NJ, USA). MDA‐MB‐231 (HH) cells were kindly provided by Dr. Patricia Steeg (NIH, Bethesda, MD, USA) and Dr. Quentin Smith (Texas Tech University Health Sciences Center, Amarillo, TX, USA). The cells were maintained and passaged in culture as described by us previously (Gupta and Srivastava, 2012).
2.3. Transwell cell invasion assay
Cell invasion was performed according to manufacturer's instructions in Boyden's Transwell chamber with 8.0 μm pore size filters (BD Biosciences, San Jose, California, USA) and as described by us earlier (Boreddy et al., 2011; Gupta et al., 2013).
2.4. Western blot analysis
Whole cell lysates were prepared using 4% (w/v) CHAPS buffer. The protein was subjected to SDS‐PAGE and the segregated protein was transferred on PVDF membrane. The membrane was developed as described by us previously (Gupta and Srivastava, 2012, 2014, 2009). All the antibodies were purchased from Cell signaling (Danvers, MA), except HER2 (Abcam, Cambridge, MA), phosphorylated SMAD4 (T277) (Thermo Fisher Scientific, Rockford, IL) and E‐cadherin (BD Biosciences, Sparks, MD).
2.5. RT‐PCR analysis
Total RNA was extracted from control and treated cells using TRIzol reagent (Life Technologies, Inc., Carlsbad, CA) according to manufacturer instructions. First, cDNA synthesis was carried out using 50 U MMTV reverse transcriptase (Affymetrix, Santa Clara, CA), 0.5 mM dNTPs (R&D Systems, Minneapolis, MN), 20 U RNase inhibitor (New England Biolabs, Ipswich, MA) and 2 μg of RNA, and the final volume made to 20 μl with DEPC treated water. The reaction was allowed to proceed at 42 °C for 1 h following which the reverse transcriptase was inactivated at 95 °C for 5 min. For polymerase chain reaction (PCR), 2 μl of single‐stranded cDNA, 1XPCR buffer; 2.5 mM dNTP mix, 1.5 mM MgCl2 and 20 pmol each of forward and reverse primer, and 1U Taq DNA polymerase (Thermo Fisher, Pittsburgh, PA) in a final reaction volume of 25 μl was subjected to cycling in a thermal cycler (Thermo Fisher, Pittsburgh, PA) (Table 1). The PCR products were separated on a 1.5% agarose gel, stained with 0.5 mg/mL ethidium bromide, and visualized under UV light.
Table 1.
Primer sequence and cycling conditions.
| Target | Primer sequence | Cycling conditions |
|---|---|---|
| E‐cadherin | F: 5′‐TCC CAT CAG CTG CCCAGA AA‐3′ | 94 °C for 30 s, 58 °C for 60 s, and 72 °C for 60 s; 35 cycles |
| R: 5′‐TGA CTC CTG TGT TCC TGT TA‐3′ | ||
| Snail | F: 5′‐GGG CAG GTA TGG AGA GGA AGA‐3′ | 94 °C for 30 s, 58 °C for 60 s, and 72 °C for 60 s; 35 cycles |
| 5′‐TTC TTC TGC GCT ACT GCT GCG‐3′ | ||
| TGFβ | F: 5′‐CTC‐CGAGAAGCGG TACCTGAAC‐3′ | 94 °C for 1 min, 60 °C, 1 min, and 72 °C for 1 min); 30 cycles |
| R: 5′‐CACTTGCAGTG TGTTATCCCT‐3′ | ||
| SMAD4 | F: 5′ –GGCTGGTCGGAAAGGATT‐3′ | 94 °C for 30 s, 54 °C for 60 s, and 72 °C for 60 s; 35 cycles |
| R: 5′‐GGCTGGTCGGAAAGGATT‐3′ | ||
| SMAD2 | F: 5′‐TCACAGTCATCATGAACTCAAGG‐3′ | 94 °C for 30 s, 56 °C for 60 s, and 72 °C for 60 s; 35 cycles |
| R: 5′‐TGTGACGCATGGAAGGTCTCTC‐3′ | ||
| SMAD3 | F: 5′‐AGGAGAAATGGTGCGAGA A‐3′ | 94 °C for 30 s, 56 °C for 60 s, and 72 °C for 60 s; 35 cycles |
| R: 5′‐CCACAGGCGGCAGTAGAT‐3 | ||
| Cytokeratin‐18 | F: 5′‐GGCCACTACTTCAAGATCATC‐3′ | 94 °C for 30 s, 56 °C for 60 s, and 72 °C for 60 s; 35 cycles |
| R: 5′‐GTACTTGTCCAGTTCCTCGCG‐3 |
2.6. TGFβ ELISA
TGFβ was estimated in the media of cultured MDA‐MB‐231 and MCF‐7 cells and in the plasma of mice injected with breast cancer cells, using ELISA kit as per manufacturer's instructions (Affymetrix ebiosciences, San Diego, CA).
2.7. Immunofluorescence
Cells were plated in a 24‐well plate on a coverslip at a density of 0.1 × 106 cells/well and allowed to attach overnight. The procedure was followed as described by us previously (Gupta and Srivastava, 2012).
2.8. Immunoprecipitation assay (IP)
This assay was performed as described by us previously (Gupta and Srivastava, 2014).
2.9. Chromatin immunoprecipitation assay
CHIP assay was performed using a commercial kit (Cellsignaling, Danvers, MA). Briefly, equal numbers of MDA‐MB‐231 and HH cells were plated in 15 mm culture dishes. After 48 h incubation, the proteins were crosslinked with DNA using 37% formaldehyde and 10X glycine solution. The samples were further processed as per the manufacturer's instructions.
2.10. TGFβ dominant negative receptor transfection
The dominant negative (dn) receptor for TGFβ (TGFβR) was kindly provided by Dr. Malcolm Brenner (Bayler College of Medicine, Houston, Texas). The MDA‐MB‐231 (HH) cells were transfected with dnTGFβR using nucleofector technique (Lonza, Allendale, NJ). The cells were treated with TGFβ after transfection and the effects were compared with control cells using western blot technique.
2.11. HER2 silencing
The MDA‐MB‐231 (HH) cells were transfected with HER2 shRNA and scrambled shRNA (Genecopoeia, Rockville, MD) using nucleofection reagent as per the manufacturer's instructions. The cells were selected using puromycin selection marker and cultured for further experiments. The cells were used for experiment within initial three passages after transfection.
2.12. Wound healing assay
Wound healing assay was performed as described by us earlier (Boreddy et al., 2011; Gupta et al., 2013).
2.13. In vivo metastasis model
The MDA‐MB‐231 and HH cells with luciferase expression were collected and washed with PBS. The cells were re‐suspended in PBS, at a density of 0.5 × 106/100 μl. A 100 μl cell suspension was injected in the tail vein of athymic nude mice. Both the groups had seven mice each. The in vivo metastasis was assessed through non‐invasive live animal imaging system as described by us previously (Gupta et al., 2013). The luminescence signal from mice was used to analyze rate and extent of metastasis for both the cell types.
In another study, two experiments using 4T1‐luc cells were performed in Balb/c mice to evaluate the anti‐metastatic effects of CuB, as described by us previously (Gupta et al., 2013).
2.14. Metastasis prevention model
In this experiment, mice was administered with 1 mg/kg CuB in 100 μl PBS every third day by intraperitoneal injection (Gupta and Srivastava, 2012; Sahu et al., 2009). After 10 days of CuB treatment, intra‐cardiac injection of 4T‐1 cells was given to these mice as described above. Both control and treated group had 8 mice each. The mice were imaged after cell injection to quantitate the signal difference between control and CuB treated mice. The mice were euthanized and the brains were removed carefully, imaged for luminescence signal and fixed in 4% paraformaldehyde overnight at room temperature and processed for immunohistochemistry analysis or H& E staining.
2.15. Metastasized tumor growth suppression model
In this experiment, 4T‐1 cells were injected into the left ventricle of the heart of each mouse as described above and each mouse was imaged periodically. Twenty four hours after the tumor cell injection, mice were randomly divided into two groups with 10 mice per group. In the treated group, each mouse was given 1 mg/kg CuB and control group was administered with vehicle alone by intraperitoneal injection every third day. The mice were sacrificed at day 14 and their organs were removed carefully. The organs were imaged for luminescence signal.
2.16. Immunohistochemistry
The immunohistochemical staining was performed as described by us previously (Gupta and Srivastava, 2014).
2.17. Statistical analysis
Statistical analysis was performed using Prism 5.0 (GraphPad software Inc., San Diego, CA, USA). Results were represented as means ± SD or S.E.M with minimum value of n = 3. Data was analyzed by Student's t‐test. Differences were considered statistically significant at p < 0.05.
3. Results
3.1. HER2 promotes cellular invasion and mesenchymal phenotype
Conversion of cells from epithelial‐to‐mesenchymal phenotype is a gradual process through which cells attain spindle shape and increased motility. To visualize this process we created a wound in the monolayer of MDA‐MB‐231 cells and let the cells heal the wound. The images of these cells at different time points show a clear transformation of the cellular size and shape (Figure 1). To determine the possible role of HER2 in metastasis, we first evaluated cell invasion in high HER2 (HH) variants of MDA‐MB‐231 and MCF‐7 cells as well as in parent cell lines, using Boyden's chamber. We observed that high HER2 (HH) cells showed about 1.3–1.5 fold higher invasion relative to the parent cells in both the cell lines (Figure 2A–B). The western blot analysis of these cell lines showed about 12 fold increased HER2 expression in HH cells relative to their syngeneic controls (Figure 2C–D). Interestingly, HER2 overexpression in MDA‐MB‐231 and MCF‐7 cells was associated with increased expression of major transcription factors like SNAIL, SLUG and ZEB‐1, which are known to play role in EMT (Figure 2C–D). The MCF‐7 control cells had negligible constitutive expression of these transcription factors but their high HER2 (HH) variants showed modestly increased expression of these proteins. β‐catenin has been shown to play an important role in EMT process in cancer cells (Arend et al., 2013). We observed a significant increase in the expression of β‐catenin in HH variants of both the cell lines. The EMT was confirmed by the increase in mesenchymal marker N‐cadherin in MDA‐MB‐231 cells along with suppression of epithelial markers E‐cadherin and cytokeratin‐18 (Figure 2C–D). These observations suggest that HER2 regulates genetic expression of proteins that induce EMT.
Figure 1.
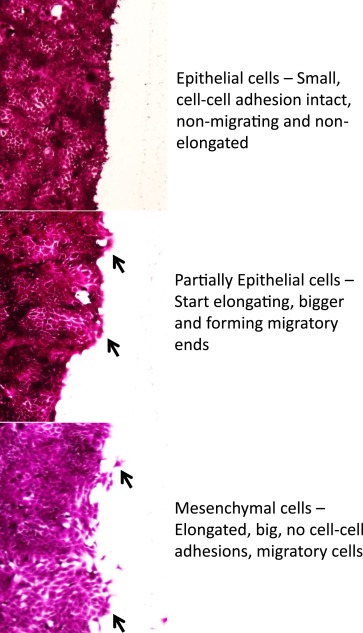
Change in cell morphology during migration: 4T‐1 cells were plated and allowed to form a monolayer. A wound was created in the monolayer. The cells were stained with Sulforrhodamine B dye and imaged using phase contrast microscope at different time points to depict the changes in cell morphology during cell migration process.
Figure 2.
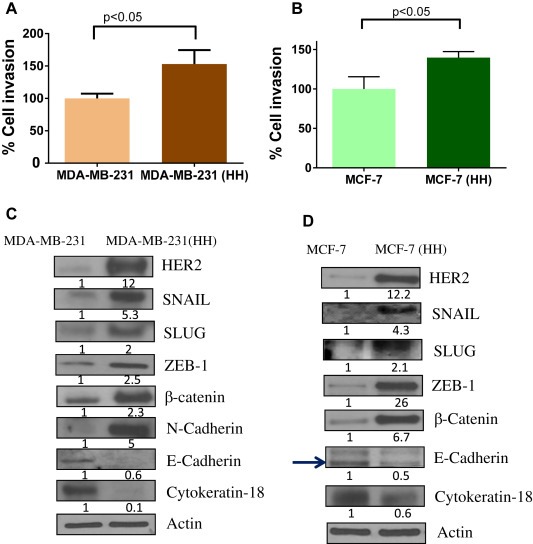
Increased invasion potential and mesenchymal characteristics of HH cells: A) Cellular invasion of parent cells was compared with syngeneic HH variants using Boyden's chamber. A) MDA‐MB‐231 vs. HH cells B) MCF‐7 vs. HH cells. The protein expression of EMT regulators was compared using western blot analysis from whole cell lysates C) MDA‐MB‐231 vs HH and D) MCF‐7 vs HH cells.
3.2. HER2 mediated TGFβ signaling regulates EMT
TGFβ is known as the master regulator of EMT (Kalluri and Weinberg, 2009). Hence we looked at TGFβ signaling in HH cells. Our results showed a 2 fold increase in TGFβ expression in MDA‐MB‐231 (HH) cells as compared to the parent cells (Figure 3A). The canonical TGFβ signaling in general is mediated by SMAD complexes. We observed a 2 fold increased expression of SMAD4, as well as phosphorylation of SMAD3 (S423/425) in HH cells (Figure 3A). However, expression of SMAD2/3 was marginally increased in MDA‐MB‐231 (HH) cells. The MCF‐7 (HH) cells showed much higher increase in TGFβ expression (Figure 3B). This was also associated with increased expressions of SMAD2/3 and phosphorylated SMAD3 (Figure 3B). To confirm whether the induction of TGFβ was at transcriptional level and not at protein level, we evaluated the mRNA levels of TGFβ and SMADs by RT‐PCR. Our results showed a significant increase in TGFβ mRNA levels in MDA‐MB‐231 (HH) as well as MCF‐7 (HH) cells as compared to parent cells (Figure 3C–D). The increased mRNA levels of TGFβ were accompanied with increased mRNA levels of SMAD4 and SMAD2 in MDA‐MB‐231 (HH) and SMAD3 in MCF‐7 (HH) cells (Figure 3C–D). In addition, a simultaneous increase in SNAIL mRNA was observed with reduction in Cytokeratin‐18 mRNA in MDA‐MB‐231 (HH) cells (Figure 3C). These results indicate that HH cells modify transcript levels of EMT regulating genes. In MCF‐7 (HH) cells, although SNAIL was undetectable, reduced mRNA of Cytokeratin‐18 and E‐cadherin were observed (Figure 3D). Furthermore, we also determined TGFβ secretion using ELISA assay. Our results showed about 7–8 fold and 3 fold increased secretion of TGFβ in the supernatant of HH cells as compared to MDA‐MB‐231 and MCF‐7 cells respectively ( Figure 3E& F).
Figure 3.
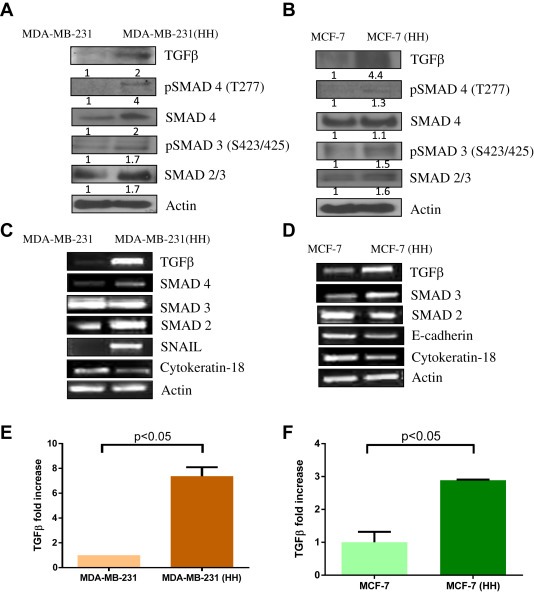
HER2 mediated increase in TGFβ: Western blot analysis from whole cell lysate for TGFβ/SMAD signaling A) MDA‐MB‐231 vs HH cells and B) MCF‐7 vs HH cells. The mRNA levels of EMT regulators were compared using RT‐PCR assay C) MDA‐MB‐231 vs HH and D) MCF‐7 vs HH cells. E) & F) TGFβ secretion was estimated in the media obtained from MDA‐MB‐231 (HH) and MCF‐7 (HH) cells and their levels compared with their respective wild type cells.
3.3. HER2 induced EMT is dependent on TGFβ signaling
Since we observed an increased expression of β‐catenin in HH cells (Figure 2C–D), we next wanted to evaluate its role in HER2 mediated EMT. In order to do that MDA‐MB‐231 and HH cells were treated with TGFβ. Our results showed a significant increase in SNAIL by TGFβ treatment in both the cell lines (Figure 4A). Expression of SNAIL was increased by 4 fold in MDA‐MB‐231 and 13.5 fold in MDA‐MB‐231 (HH) cells by TGFβ treatment (Figure 4 A). However, TGF treatment did not cause significant modulation of β‐catenin in MDA‐MB‐231 or MDA‐MB‐231 (HH) cells (Figure 4A). The role of TGFβ was further established in HH cells using a dominant negative (dn) TGFβ receptor (TGFβR). The MDA‐MB‐231 (HH) cells were transfected with dn TGFβR plasmid followed by treatment with TGFβ. Our results showed that TGFβ treatment increased the expression of SMAD4 and phosphorylation of SMAD3 (S423/425) in HH cells (Figure 4B). The TGFβ treatment also increased SNAIL and N‐cadherin expression. However, the effects of TGFβ treatment were not observed in the cells transfected with dn TGFβR plasmid, suggesting an important role of TGFβ in HER2 mediated EMT (Figure 4B). We then performed cytosolic and nuclear fractionation of MDA‐MB‐231 and MDA‐MB‐231 (HH) cells. Nuclear fraction of HH cells showed significantly increased expression of SMAD4 and phosphorylated SMAD3 (S423/425) (Figure 4C). SNAIL is a transcription factor and expected to localize in the nucleus once activated. As expected, SNAIL expression was increased in the nucleus of HH cells, whereas its level in the nucleus of MDA‐MB‐231 cells was very less (Figure 4C). However, the nuclear expression of β‐catenin did not increase in MDA‐MB‐231 cells or in HH variants. Further evidence for the involvement of SMAD signaling in HER2/TGFβ mediated EMT was provided by the immunoprecipitation (IP) studies. SMAD4 demonstrated increased binding to SMAD2/3 in HH cells as compared to parent control cells (Figure 4D). We also performed chromatin immunoprecipitation (CHIP) assay for SMAD4. Our results showed a significantly increased DNA binding of SMAD4 in HH cells as compared to MDA‐MB‐231 cells (Figure 4E). These findings clearly indicate that HER2 induced EMT was mediated by TGFβ/SMAD signaling in breast cancer cells.
Figure 4.
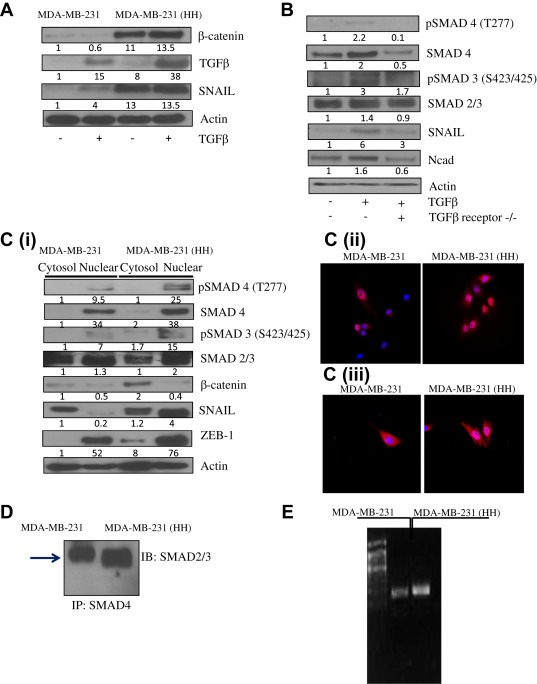
EMT in HH cells mediated by TGFβ/SMAD signaling: A) Role of β‐catenin, TGFβ and SNAIL was evaluated in MDA‐MB‐231 and HH cells by treating the cells with TGFβ. B) Role of TGFβ was evaluated using transfection of dominant negative TGFβ receptor in MDA‐MB‐231 (HH) cells. The control and transfected cells were treated with TGFβ and effects were compared by western blot technique. C) The nuclear localization of SMADs, SNAIL and β‐catenin was evaluated (i) MDA‐MB‐231 and HH cells were fractionated into cytosolic and nuclear fractions. The proteins were analyzed using western blot techqniue. MDA‐MB‐231 and HH cells were immunostained for (ii) SNAIL and (iii) pSMAD4 (T277). The red staining shows SNAIL or pSMAD4 expression and blue represents nuclear stain C) Binding of SMAD4 to SMAD2/3 was compared in MDA‐MB‐231 with HH cells using immunoprecipitation assay. D) Chromatin immunoprecipitation (CHIP) assay was performed to evaluate SMAD4 DNA binding in MDA‐MB‐231 and HH cells.
3.4. HER2 expression is required for maintenance of mesenchymal characteristics
4Although, our experiments indicated that EMT was induced by HER2, it was important to prove that suppression of HER2 could reverse EMT characteristics in breast cancer cells. Hence we used shRNA to silence HER2 expression in MDA‐MB‐231 (HH) cells. Our results showed that HER2 silencing suppressed the invasive potential of MDA‐MB‐231 (HH) cells by more than 50% (Figure 5A). Western blot results showed that about 60% HER2 was silenced by shRNA in MDA‐MB‐231 (HH) cells (Figure 5B). HER2 silencing also reduced the expression of EMT regulators such as TGFβ, SMAD4 and the transcription factor SNAIL (Figure 5B). Reversal of EMT by HER2 suppression was clearly indicated by the re‐expression of Cytokeratin‐18, an epithelial marker in MDA‐MB‐231 (HH) cells. Finally we compared the morphology of MDA‐MB‐231 (HH) cells after silencing HER2. The microscopy images clearly showed that silencing of HER2 caused reduction in cell size and cells were more flat and less elongated as compared to control HH cells (Figure 5C). The HER2 silenced cells also showed increase in the expression of Cytokeratin‐18 as observed by enhanced red staining (Figure 5C). These results confirmed the role of HER2 in EMT and cell invasion.
Figure 5.
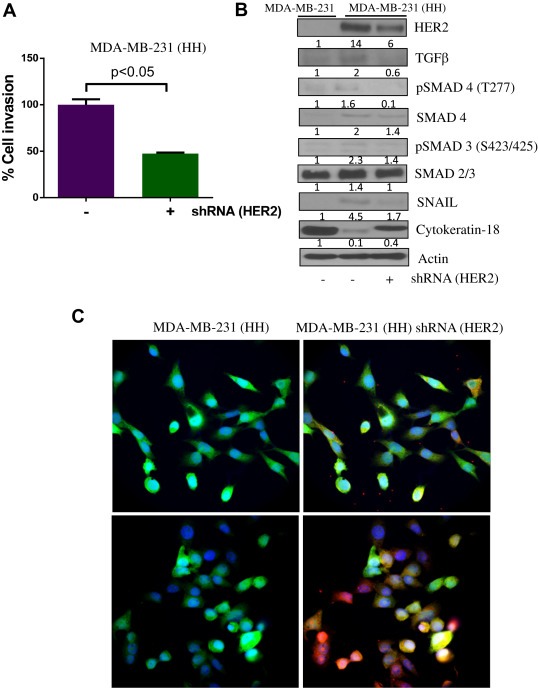
HER2 silencing reverses EMT: A) Effect of HER2 silencing was tested by invasion assay using Boyden's chamber in MDA‐MB‐231 (HH) cells. B) Effect of HER2 silencing on the TGFβ mediated EMT was evaluated using western blot analysis C) Effect of HER2 silencing on the morphology of cells was evaluated using immunofluorescent staining in MDA‐MB‐231 (HH) cells. Green staining represents GFP tagged to HER2, blue represents DAPI staining for nucleus and Red staining represents Cytokeratin‐18.
3.5. HER2 overexpressing cells show higher metastatic capability
As a proof‐of‐concept, the metastatic capability of MDA‐MB‐231 and HH cells were compared in an in vivo model. MDA‐MB‐231 and MDA‐MB‐231 (HH) cells were suspended in PBS (5 × 106/ml) and 100 μl of cell suspension was injected in the tail vein of athymic nude mice. The growth of the cells in mice was monitored in vivo by using non‐invasive imaging technique after luciferin injection. Our results showed a static growth of MDA‐MB‐231 cells, however, HH cells showed a constant increase in luminescence suggesting increase in metastatic tumor growth (Figure 6). For example, 9.5 fold increased luminescence was observed in mice injected with HH cells relative to MDA‐MB‐231 cells (Figure 6A). Moreover, luminescence was observed to be intense and widely spread in the mice injected with HH cells as compared to MDA‐MB‐231 cells (Figure 6B). After the termination of the experiment, lung and livers were removed and imaged. Our results showed 5 fold enhanced bioluminescence in the lungs of mice injected intravenously with MDA‐MB‐231 (HH) cells (Figure 6C). Although, we observed a 14 fold increased bioluminescence in the livers of mice injected with HH cells, statistical significance was not achieved due to significantly high variation (Figure 6C).
Figure 6.
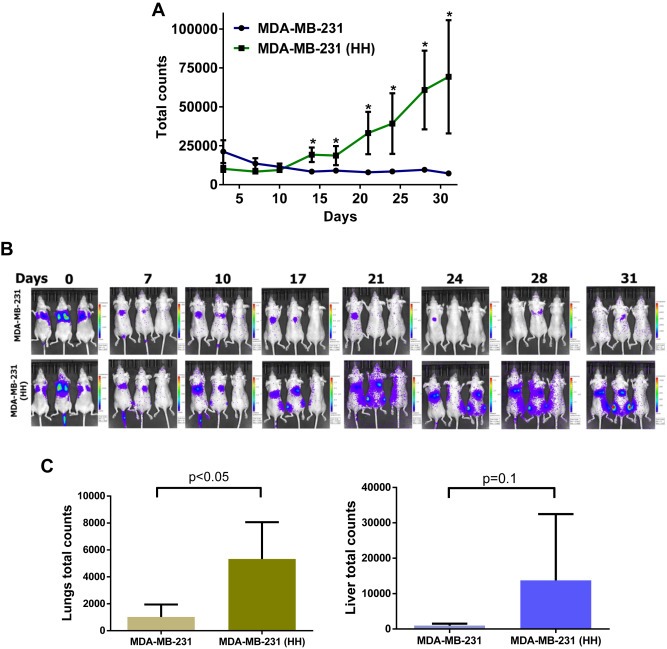
In vivo metastasis of MDA‐MB‐231 and HH cells: The MDA‐MB‐231 and HH breast cancer cells expressing luciferase were injected by tail vein. The mice were imaged using non‐invasive live animal imaging system. A) The luminescence from both the groups was quantitated and plotted against time. B) The images obtained from the imaging system and variation in luminescence with time after cell injection. C) Bioluminescence for MDA‐MB‐231 and MDA‐MB‐231 (HH) cells obtained from excised mice organs after termination of the experiment.
3.6. Inhibition of HER2 suppresses EMT
To further establish the role of HER2 in EMT and metastasis, we used cucurbitacin B (CuB), a triterpenoid compound. In our previous studies, we observed that CuB inhibited HER2 in breast cancer cells (Gupta and Srivastava, 2014). We evaluated the effects of CuB in MDA‐MB‐231, MDA‐MB‐231 (HH) and 4T‐1 cells. The western blot data showed a significant inhibition of HER2 by CuB treatment along with inhibition of TGFβ/SMAD signaling in all the three cell lines (Figure 7A–C). The CuB treatment also caused suppression of SNAIL, SLUG and ZEB‐1 as well as inhibition of mesenchymal marker N‐cadherin. Interestingly, in MDA‐MB‐231 cells, CuB treatment caused induction of Cytokeratin‐18, while in 4T‐1 cells induction of Cytokeratin‐18 as well as E‐cadherin was observed in a concentration‐dependent manner (Figure 7B–C). Furthermore, we observed that CuB treatment inhibited the mRNA levels of HER2 and TGFβ in MDA‐MB‐231 (HH) cells, suggesting the inhibition at transcriptional level (Figure 7D). These findings clearly suggested that CuB mediated HER2/TGFβ inhibition lead to suppression of EMT.
Figure 7.
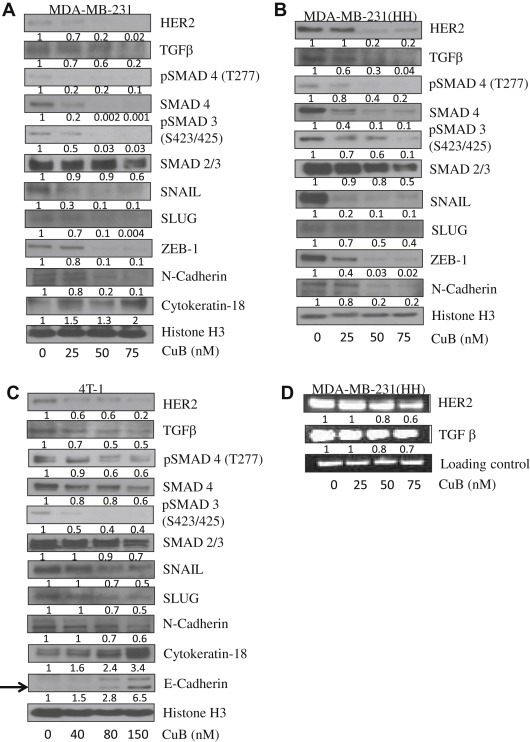
CuB mediated inhibition of EMT through HER2/TGFβ: Western blot analysis for EMT regulators in CuB treated A) MDA‐MB‐231 B) MDA‐MB‐231 (HH) and C) 4T‐1 cells. D) RT‐PCR for HER2 and TGFβ in CuB treated MDA‐MB‐231 (HH) cells.
3.7. CuB inhibits TGFβ‐mediated cell invasion and migration
Since invasion and migration are important consequences of EMT, we tested the effects of CuB in presence of TGFβ. We performed cell invasion assay in 4T‐1 cells using Boyden's chamber. CuB treatment caused about 50% reduction in cell invasion of cells (Figure 8A). TGFβ treatment increased the invasion of cells by 1.5 fold however, CuB treatment inhibited TGFβ‐mediated invasion substantially (Figure 8A).
Figure 8.
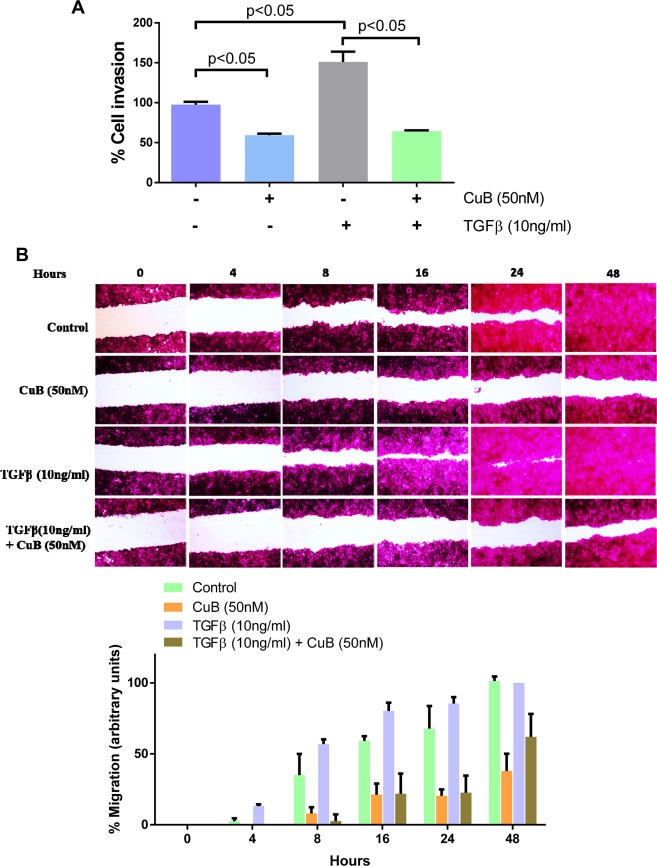
CuB inhibits TGFβ mediated invasion and migration: A) Effect of CuB on TGFβ mediated cell invasion in 4T‐1 cells. B) Effect of CuB on TGFβ mediated wound healing in 4T‐1 cells.
We then evaluated anti‐migratory properties of CuB in 4T‐1 cells. A wound was created in the monolayer of 4T‐1 cells and incubated with CuB. The wound was imaged at different time points using bright field microscope. The wound images clearly showed that CuB treatment inhibited the cell migration, whereas TGFβ promoted cell migration causing faster wound healing as compared to control cells (Figure 8B). The quantitation of wound width showed that CuB treatment inhibited wound healing by about 30–50% compared to control (Figure 8B). Interestingly, CuB treatment inhibited TGFβ‐mediated increased cell migration (Figure 8B). These results showed that CuB inhibits both cell invasion and migration.
3.8. CuB treatment inhibits breast cancer metastases
To confirm anti‐metastatic effects of CuB we used a novel in vivo model of breast cancer metastasis. The 4T‐1 luciferase expressing (Luc) breast cancer cells were injected through intracardiac route and monitored using non‐invasive live animal imaging system (PerkinElmers, Waltham, MA). We performed two different metastasis experiments to test the preventive as well as therapeutic effects of CuB.
In the first experiment, mice were treated with 1 mg/kg CuB by intraperitoneal injection every third day. The treatment started 24 h after the intracardiac cell injection as by this time 4T1 cells were stably lodged in the brain of each mouse. 4T‐1 cells are highly invasive with doubling time of about 8 h. Our results showed a steady growth of luminescence in the brain of control mice, whereas the mice from CuB treated group showed substantially reduced growth rate (Figure 9A). Our results showed that luminescence in the brain were reduced by about 86% by the end of experiment in CuB treated group (Figure 9A). The full body scanning showed about 56% reduction in the average signal of the treatment group. However, the difference was not found to be statistically significant possibly due to a large variation in control group of mice (data not shown). At the end of the experiment, brain, lungs and liver were dissected out and imaged for luminescence. The result from this experiment matched exactly with brain in vivo imaging data. We observed about 86% reduced signal in the isolated brains of CuB treated group as compared to control group (Figure 9B). The lungs and livers were also imaged for luminescence from both the groups and the treatment group showed a reduction of 50 and 64% respectively (Figure 9C&D). However, due to significant variations in the luminescence in the tissues of control group, this difference was not found to be statistically significant. The mice were weighed periodically during the experiment and also monitored closely for any visual signs of toxicity. We did not observe any difference in the body weight of mice from control group with that of treatment group, suggesting no obvious toxicity by CuB treatment (Figure 9E). We also estimated TGFβ in the plasma of mice. Our results indicated that the mice injected with HH cells exhibited about 1.3 fold higher TGFβ in plasma as compared to the mice injected with MDA‐MB‐231 cells (Figure 9F). Furthermore, the immunohistochemical staining showed a significant reduction in the expression of HER2, TGFβ and SNAIL in the lesions in the brain of CuB treated mice as compared to control mice. In addition, increased expression of E‐cadherin was observed in brain lesions from CuB treated mice (Figure 9G).
Figure 9.
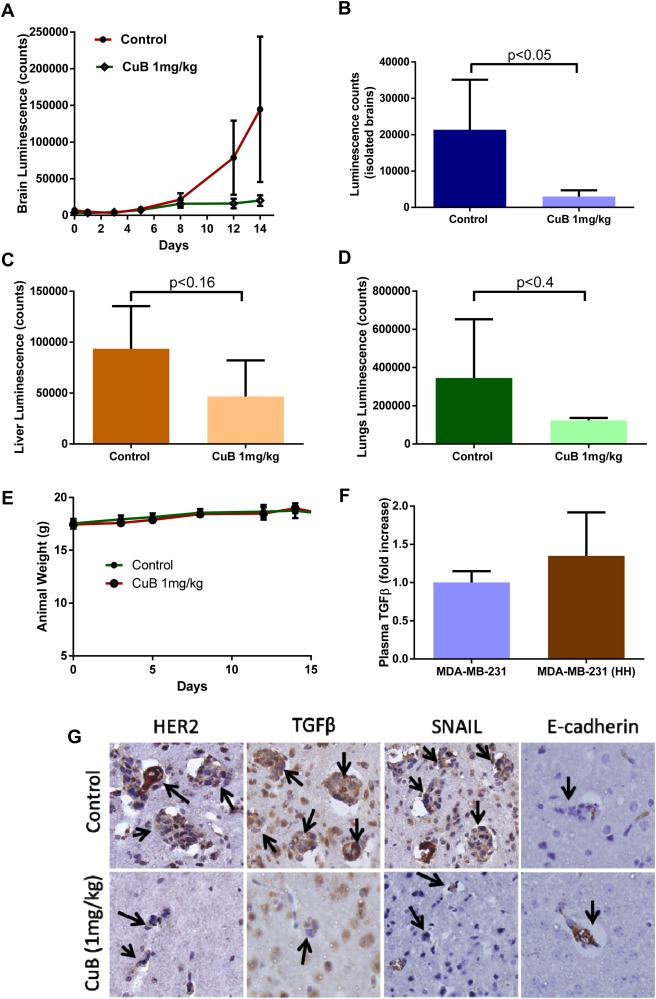
Inhibition of in vivo metastasis by CuB treatment: Luciferase expressing 4T‐1 cells were injected by intracardiac route in Balb/c mice. CuB treatment was started 24h after cell injection and the mice were imaged periodically for brain luminescence signal to analyze metastasis of breast cancer cells. A) Brain luminescence from control and CuB treated mice was quantitated and plotted against time. B) Mice brains were collected and imaged. The average luminescence was compared for control and CuB treated mice C) & D) The luminescence quantitated from liver and lungs respectively from control and CuB treated mice after termination of the experiment. E) Mice weights from control and CuB treatment groups at different time points. F) TGFβ secretion was estimated by performing ELISA in the plasma obtained from mice injected with MDA‐MB‐231 and MDA‐MB‐231 (HH) cells. G) Immunohistochemical staining for HER2, TGFβ, SNAIL and E‐cadherin in tumor lesions in brain of control and CuB treated mice.
The second experiment was performed to test if CuB pre‐treatment could reduce number of cells reaching brain after intra‐cardiac injection. Our results showed a significant reduction in whole body signal as well as in the brain of CuB treated group. We observed about 35% and 80% reduction in the signal of brain and whole body respectively in the mice treated with CuB as compared with control mice (Figure 10A and B). These findings suggest that CuB inhibits in vivo metastasis.
Figure 10.
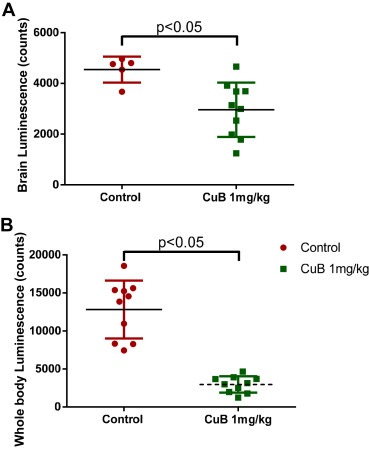
Prevention of in vivo metastasis by CuB treatment: A) & B) Luminescence signal quantitated from brain and full body respectively, of control and pre‐treatment group of mice after cell injection.
4. Discussion
About 25% of breast cancer patients overexpress HER2 making it an important target. HER2 overexpression is due to its amplification at genetic levels and associated with poor prognosis (Harari and Yarden, 2000). The exact reasons for HER2 associated poor prognosis is not known beyond that, highly aggressive tumors proliferate more rapidly. HER2 positive tumors that have been studied show metastatic patterns in patients (Kennecke et al., 2010). HER2 is implicated in increased breast cancer metastasis to brain, however no mechanistic evidence exists so far (Brufsky et al., 2011). HER2 crosstalk with other survival proteins leads to therapeutic resistance in cancer through multiple pathways (Nahta, 2012). EMT is also an important mechanism of resistance to many anti‐cancer agents (Mallini et al., 2014; Oliveras‐Ferraros et al., 2012). However, correlation between HER2 and EMT pathway has not been investigated yet. The current study provides evidence that directly correlates HER2 and metastasis through EMT.
Metastasis is a complex process with several intermediate steps (Hanahan and Weinberg, 2011). Conversion of epithelial cells into mesenchymal phenotype is one of the initial steps that promote cell migration and invasion. The mesenchymal characteristics help the cells to break cell–cell adhesions and invade circulation conduits, making it easier for cells to metastasize (Tsai and Yang, 2013). Our results show increased invasive potential of cells with HER2 overexpression, suggesting an increased metastatic potential of cells mediated by HER2. EMT is an orchestrated process, which is known to be controlled by transcription factors like SNAIL, SLUG, ZEB‐1 and TWIST. Activation of these transcription factors causes a switch between E‐cadherin and N‐cadherin, the latter provides mesenchymal properties to the cells (Gheldof and Berx, 2013). The HER2 overexpression in breast cancer cells, we used was attained through stable transfection with wild type HER2 plasmid (Huang et al., 2007; Palmieri et al., 2007). We observed that high HER2 (HH) cells expressed significantly higher levels of SNAIL and ZEB‐1 suggesting increased activation of these transcription factors. Furthermore, HH cells also showed reduced expression of E‐cadherin and Cytokeratin‐18 while increased N‐cadherin expression. These observations indicate that HER2 modulates EMT inducing proteins in breast cancer cells by enhancing the transcript levels of genes associated with EMT.
TGFβ is defined as the master regulator of EMT (Kalluri and Weinberg, 2009; Katsuno et al., 2013; Taylor et al., 2010). TGFβ generally plays a pro‐apoptotic role in normal cells or during the initial stages of cancer. However, it promotes cancer progression in advanced cancer through multiple mechanisms (Padua and Massague, 2009). One study showed an indirect crosstalk of HER2 with TGFβ signaling (Wang, 2011). This study showed that HER2 converts function of TGFβ from pro‐apoptotic to anti‐apoptotic. In line with this study, our results showed an increased expression of TGFβ with HER2 overexpression along with increased SMAD signaling. We observed increased mRNAs levels of TGFβ, SMAD3 and 4, suggesting transcriptional up‐regulation of TGFβ/SMAD signaling by HER2. These results indicate a novel mechanism of de novo synthesis of TGFβ mediated by HER2 leading to EMT in breast cancer cells. As a proof‐of‐concept, TGFβ‐mediated effects were lost when cells were transfected with dn TGFβR plasmid.
β‐catenin is an important protein known to play role in EMT and metastasis (Sanchez‐Tillo et al., 2011). In addition, β‐catenin also promotes tumor progression in HER2 positive tumors (Schade et al., 2013). We observed a significant increase in the expression of β‐catenin in HH cells, suggesting its role in HER2‐induced EMT in our model. The active form of β‐catenin translocates to the nucleus to bind with TCF/LEF complex (Behrens et al., 1996; Fagotto, 2013). In other situations, β‐catenin undergoes proteasomal degradation. Our results showed that TGFβ treatment did not alter the expression of β‐catenin. Furthermore, β‐catenin expression was not found to be increased in the nuclear fraction of HH cells. Hence even though β‐catenin expression was increased in HH cells, it was not activated. However, significant nuclear localization of SMADs was observed in HH cells along with an increased association of SMAD4 to SMAD2/3. These observations clearly indicated the activation of TGFβ/SMAD signaling in HH cells. Furthermore, increased nuclear expression of the transcription factors SNAIL and ZEB‐1 conclusively suggested that HER2‐induced EMT was mediated by TGFβ/SMAD signaling.
Confirmation of role of HER2 in cell invasion and EMT was shown by inhibition of HER2 using shRNA which reduced cell invasion as well as mesenchymal characteristics. Suppression of TGFβ/SMAD signaling was also observed with HER2 silencing. These results clearly indicated that induction of EMT in breast cancer cells was regulated by HER2 through TGFβ/SMAD signaling in a coordinated manner. Our results also suggest that TGFβ could be another target in HH cells and its inhibition combined with anti‐HER2 therapy can provide promising therapeutic outcomes. Importantly, TGFβ antibody therapy trials are underway (Nam et al., 2008; Saunier and Akhurst, 2006).
Cucurbitacin B (CuB) is a steroidal triterpenoid present in cucurbitaceous plants. A recent study indicated that the overall anti‐cancer effect of CuB was due to reactive oxygen species dependent actin aggregation leading to depletion of G‐actin pool (Kapoor, 2013). CuB was also shown to be a potent inhibitor of STAT3 (Thoennissen et al., 2009). Our previous studies have shown inhibition of HER2 and TGFβ mediated HER2‐integrin signaling by CuB (Gupta and Srivastava, 2014). The reduced protein expression of HER2 by CuB treatment was possibly due to inhibition of YB‐1/TWIST signaling known to regulate HER2 expression. Since CuB could inhibit both HER2 and TGFβ‐mediated signaling, this agent was used to evaluate its anti‐metastatic effects in our novel in vivo metastasis model of breast cancer. Furthermore, we used a highly aggressive cell line 4T‐1 representing stage IV breast cancer, which is considered to be un‐treatable. Our results clearly showed strong anti‐metastatic effects of CuB. We observed that CuB pre‐treatment prevented brain metastasis of breast cancer cells. We also observed that CuB treatment inhibited the growth of metastasized tumors in brain, lungs and liver. Hence if TGFβ signaling is targeted along with HER2, metastatic growth of highly aggressive tumors could be suppressed (Figure 11).
Figure 11.
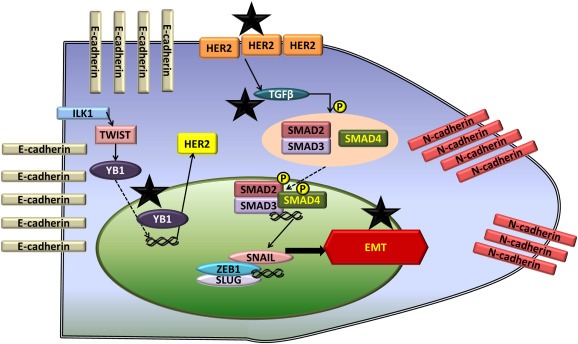
Schematic for mechanism of HER2 induced EMT and inhibition by CuB: The figure shows the molecular mechanism of EMT regulation by HER2. * symbol indicates the molecular targets of CuB leading to HER2 inhibition and suppression of EMT.
Taken together, our results showed that HER2‐mediated EMT is regulated by TGFβ leading to enhanced metastasis. EMT has also been implicated in stem cell characteristics of cancer cells as well as resistance to many anti‐cancer therapies (Mallini et al., 2014; Oliveras‐Ferraros et al., 2012). Our results thus suggested that HER2 can lead to enhanced stem‐ness through induction of EMT. However, further studies are required to confirm this possibility. Our findings are of significant importance as currently there is no therapy available for highly metastatic cancers resistant to conventional therapeutics and hence the survival of patients with metastatic breast cancer is minimal. Our study proposes a novel HER2‐regulated target, which could be crucial for designing the therapy for patients with drug resistant metastatic breast cancer.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Parul Gupta was responsible for designing the study, performing the experiments and writing the first draft of the manuscript. Sanjay K Srivastava was responsible for designing the study, analyzing the data and writing the manuscript.
Conflict of interest
Authors declare that there are no competing interests.
Acknowledgments
Kind gift of MDA‐MB‐231 (HH) cells by Dr. Patricia S. Steeg (National Cancer Institute, Maryland) and Dr. Quentin Smith (Texas Tech University Health Sciences Centre, Amarillo, Texas) and MCF‐7 (HH) cells by Dr. Huang Fei (Bristol Myers Squibb) are greatly appreciated.
Gupta Parul, Srivastava Sanjay K., (2014), HER2 mediated de novo production of TGFβ leads to SNAIL driven epithelial-to-mesenchymal transition and metastasis of breast cancer, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.06.006.
Grant support: Supported in part by R01 grant CA129038 (to S.K.S) awarded by the National Cancer Institute, NIH.
References
- Arend, R.C. , Londono-Joshi, A.I. , Straughn, J.M. , Buchsbaum, D.J. , 2013. The Wnt/Beta-catenin pathway in ovarian cancer: a review. Gynecol. Oncol. 131, 772–779. [DOI] [PubMed] [Google Scholar]
- Behrens, J. , von Kries, J.P. , Kuhl, M. , Bruhn, L. , Wedlich, D. , Grosschedl, R. , Birchmeier, W. , 1996. Functional interaction of Beta-catenin with the transcription factor Lef-1. Nature 382, 638–642. [DOI] [PubMed] [Google Scholar]
- Boreddy, S.R. , Sahu, R.P. , Srivastava, S.K. , 2011. Benzyl isothiocyanate suppresses pancreatic tumor angiogenesis and invasion by inhibiting Hif-Alpha/Vegf/Rho-Gtpases: pivotal role of Stat-3. PLoS One 6, e25799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl, M. , Seidler, B. , Haller, F. , Adamski, J. , Schmid, R.M. , Saur, D. , Schneider, G. , 2010. Ikk(Alpha) controls canonical TGF(Ss)-Smad signaling to regulate genes expressing snail and slug during Emt in Panc1 cells. J. Cell Sci. 123, 4231–4239. [DOI] [PubMed] [Google Scholar]
- Brufsky, A.M. , Mayer, M. , Rugo, H.S. , Kaufman, P.A. , Tan-Chiu, E. , Tripathy, D. , Tudor, I.C. , Wang, L.I. , Brammer, M.G. , Shing, M. , Yood, M.U. , Yardley, D.A. , 2011. Central nervous system metastases in patients with Her2-positive metastatic breast cancer: incidence, treatment, and survival in patients from registHER. Clin. Cancer Res. 17, 4834–4843. [DOI] [PubMed] [Google Scholar]
- de Herreros, A.G. , Peiro, S. , Nassour, M. , Savagner, P. , 2010. Snail family regulation and epithelial mesenchymal transitions in breast Cancer progression. J. Mammary Gland Biol. Neoplasia 15, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagotto, F. , 2013. Looking beyond the Wnt pathway for the deep nature of Beta-catenin. EMBO Rep. 14, 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheldof, A. , Berx, G. , 2013. Cadherins and epithelial-to-mesenchymal transition. Prog. Mol. Biol. Transl. Sci. 116, 317–336. [DOI] [PubMed] [Google Scholar]
- Gupta, P. , Adkins, C. , Lockman, P. , Srivastava, S. , 2013. Metastasis of breast tumor cells to brain is suppressed by phenethyl isothiocyanate in a novel in vivo metastasis model. PLoS One 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, P. , Srivastava, S.K. , 2012. Antitumor activity of phenethyl isothiocyanate in Her2-positive breast Cancer models. BMC Med. 10, 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, P. , Srivastava, S.K. , 2014. Inhibition of Her2-integrin signaling by cucurbitacin B leads to in vitro and in vivo breast tumor growth suppression. Oncotarget 5, 1812–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2011. Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Harari, D. , Yarden, Y. , 2000. Molecular mechanisms underlying Erbb2/Her2 action in breast cancer. Oncogene 19, 6102–6114. [DOI] [PubMed] [Google Scholar]
- Huang, F. , Reeves, K. , Han, X. , Fairchild, C. , Platero, S. , Wong, T.W. , Lee, F. , Shaw, P. , Clark, E. , 2007. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: rationale for patient selection. Cancer Res. 67, 2226–2238. [DOI] [PubMed] [Google Scholar]
- Irvin, W. , Muss, H.B. , Mayer, D.K. , 2011. Symptom management in metastatic breast cancer. Oncologist 16, 1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal, A. , Bray, F. , Center, M.M. , Ferlay, J. , Ward, E. , Forman, D. , 2011. Global cancer statistics. CA – A Cancer J. Clin. 61, 69–90. [DOI] [PubMed] [Google Scholar]
- Kalluri, R. , Weinberg, R.A. , 2009. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor, S. , 2013. Cucurbitacin B and its rapidly emerging role in the management of systemic malignancies besides lung carcinomas. Cancer Biother. Radiopharm 28, 359 [DOI] [PubMed] [Google Scholar]
- Katsuno, Y. , Lamouille, S. , Derynck, R. , 2013. TGF-Beta signaling and epithelial-mesenchymal transition in Cancer progression. Curr. Opin. Oncol. 25, 76–84. [DOI] [PubMed] [Google Scholar]
- Kennecke, H. , Yerushalmi, R. , Woods, R. , Cheang, M.C. , Voduc, D. , Speers, C.H. , Nielsen, T.O. , Gelmon, K. , 2010. Metastatic behavior of breast Cancer subtypes. J. Clin. Oncol. 28, 3271–3277. [DOI] [PubMed] [Google Scholar]
- Li, P. , Jing, J. , Hu, J. , Li, T. , Sun, Y. , Guan, H. , 2013. Rna Interference targeting snail inhibits the transforming growth factor Beta 2-induced epithelial-mesenchymal transition in human lens epithelial cells. J. Ophthalmol. 2013, 869101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallini, P. , Lennard, T. , Kirby, J. , Meeson, A. , 2014. Epithelial-to-mesenchymal transition: what is the impact on breast cancer stem cells and drug resistance. Cancer Treat. Rev. 40, 341–348. [DOI] [PubMed] [Google Scholar]
- Nahta, R. , 2012. Pharmacological strategies to overcome Her2 cross-talk and trastuzumab resistance. Curr. Med. Chem. 19, 1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam, J.S. , Terabe, M. , Mamura, M. , Kang, M.J. , Chae, H. , Stuelten, C. , Kohn, E. , Tang, B. , Sabzevari, H. , Anver, M.R. , Lawrence, S. , Danielpour, D. , Lonning, S. , Berzofsky, J.A. , Wakefield, L.M. , 2008. An anti-transforming growth factor Beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 68, 3835–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shaughnessy, J. , 2005. Extending survival with chemotherapy in metastatic breast Cancer. Oncologist 10, (Suppl. 3) 20–29. [DOI] [PubMed] [Google Scholar]
- Oliveras-Ferraros, C. , Corominas-Faja, B. , Cufi, S. , Vazquez-Martin, A. , Martin-Castillo, B. , Iglesias, J.M. , Lopez-Bonet, E. , Martin, A.G. , Menendez, J.A. , 2012. Epithelial-to-mesenchymal transition (Emt) confers primary resistance to trastuzumab (Herceptin). Cell Cycle 11, 4020–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padua, D. , Massague, J. , 2009. Roles of TGF-Beta in metastasis. Cell Res. 19, 89–102. [DOI] [PubMed] [Google Scholar]
- Palmieri, D. , Bronder, J.L. , Herring, J.M. , Yoneda, T. , Weil, R.J. , Stark, A.M. , Kurek, R. , Vega-Valle, E. , Feigenbaum, L. , Halverson, D. , Vortmeyer, A.O. , Steinberg, S.M. , Aldape, K. , Steeg, P.S. , 2007. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 67, 4190–4198. [DOI] [PubMed] [Google Scholar]
- Sahu, R.P. , Batra, S. , Srivastava, S.K. , 2009. Activation of Atm/Chk1 by curcumin causes cell cycle arrest and apoptosis in human pancreatic cancer cells. Br. J. Cancer 100, 1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu, R.P. , Srivastava, S.K. , 2009. The role of Stat-3 in the induction of apoptosis in pancreatic cancer cells by benzyl isothiocyanate. J. Natl. Cancer Inst. 101, 176–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Tillo, E. , de Barrios, O. , Siles, L. , Cuatrecasas, M. , Castells, A. , Postigo, A. , 2011. Beta-Catenin/Tcf4 complex induces the epithelial-to-mesenchymal transition (Emt)-Activator Zeb1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. U. S. A. 108, 19204–19209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunier, E.F. , Akhurst, R.J. , 2006. TGF Beta inhibition for Cancer therapy. Curr. Cancer Drug Targets 6, 565–578. [DOI] [PubMed] [Google Scholar]
- Schade, B. , Lesurf, R. , Sanguin-Gendreau, V. , Bui, T. , Deblois, G. , O'Toole, S.A. , Millar, E.K. , Zardawi, S.J. , Lopez-Knowles, E. , Sutherland, R.L. , Giguere, V. , Kahn, M. , Hallett, M. , Muller, W.J. , 2013. Beta-catenin signaling is a critical event in Erbb2-mediated mammary tumor progression. Cancer Res. 73, 4474–4487. [DOI] [PubMed] [Google Scholar]
- Slamon, D.J. , Clark, G.M. , Wong, S.G. , Levin, W.J. , Ullrich, A. , McGuire, W.L. , 1987. Human breast cancer: correlation of relapse and survival with amplification of the Her-2/Neu oncogene. Science 235, 177–182. [DOI] [PubMed] [Google Scholar]
- Taylor, M.A. , Parvani, J.G. , Schiemann, W.P. , 2010. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J. Mammary Gland Biol. Neoplasia 15, 169–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoennissen, N.H. , Iwanski, G.B. , Doan, N.B. , Okamoto, R. , Lin, P. , Abbassi, S. , Song, J.H. , Yin, D. , Toh, M. , Xie, W.D. , Said, J.W. , Koeffler, H.P. , 2009. Cucurbitacin B induces apoptosis by inhibition of the Jak/Stat pathway and potentiates antiproliferative effects of gemcitabine on pancreatic Cancer cells. Cancer Res. 69, 5876–5884. [DOI] [PubMed] [Google Scholar]
- Tsai, J.H. , Yang, J. , 2013. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 27, 2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, C.L. , Cobleigh, M.A. , Tripathy, D. , Gutheil, J.C. , Harris, L.N. , Fehrenbacher, L. , Slamon, D.J. , Murphy, M. , Novotny, W.F. , Burchmore, M. , Shak, S. , Stewart, S.J. , Press, M. , 2002. Efficacy and safety of trastuzumab as a single agent in first-line treatment of Her2-overexpressing metastatic breast cancer. J. Clin. Oncol. 20, 719–726. [DOI] [PubMed] [Google Scholar]
- Wang, S.E. , 2011. The functional crosstalk between Her2 tyrosine kinase and TGF-Beta signaling in breast cancer malignancy. J. Signal Transduction 2011, 804236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z.C. , Yi, M.J. , Ran, N. , Wang, C. , Fu, P. , Feng, X.Y. , Xu, L. , Qu, Z.H. , 2013. Transforming growth factor-beta1 induces bronchial epithelial cells to mesenchymal transition by activating the snail pathway and promotes airway remodeling in asthma. Mol. Med. Rep. 8, 1663–1668. [DOI] [PubMed] [Google Scholar]