

Abstract
Cells respond to mechanical forces by activating specific genes and signaling pathways that allow the cells to adapt to their physical environment. Examples include muscle growth in response to exercise, bone remodeling based on their mechanical load, or endothelial cells aligning under fluid shear stress. While the involved downstream signaling pathways and mechanoresponsive genes are generally well characterized, many of the molecular mechanisms of the initiating ‘mechanosensing’ remain still elusive. In this review, we discuss recent findings and accumulating evidence suggesting that the cell nucleus plays a crucial role in cellular mechanotransduction, including processing incoming mechanoresponsive signals and even directly responding to mechanical forces. Consequently, mutations in the involved proteins or changes in nuclear envelope composition can directly impact mechanotransduction signaling and contribute to the development and progression of a variety of human diseases, including muscular dystrophy, cancer, and the focus of this review, dilated cardiomyopathy. Improved insights into the molecular mechanisms underlying nuclear mechanotransduction, brought in part by the emergence of new technologies to study intracellular mechanics at high spatial and temporal resolution, will not only result in a better understanding of cellular mechanosensing in normal cells but may also lead to the development of novel therapies in the many diseases linked to defects in nuclear envelope proteins.
Keywords: Mechanotransduction, lamins, nesprins, nuclear envelope, force, mechanics, cell signaling
1. Introduction
Mechanotransduction describes the cellular and molecular processes of converting mechanical stimuli into biochemical signals. Since the discovery of stretch sensitive ion channels, the field has rapidly expanded, leading to the discovery of numerous force sensitive proteins within the cytoplasm and plasma membrane, such as titin, talin, vinculin, and p130Cas (Seifert and Grater, 2013). Disturbed cellular mechanotransduction causes numerous defects on the cell, tissue, and organ level (Jaalouk and Lammerding, 2009); thus, it only seems logical that the human heart, which beats on average 2.5 billion times over the course of a lifetime, requires tightly regulated and robust mechanoregulation. Beyond this, the importance of mechanoelectric feedback adds another layer of complexity to cardiac mechanotransduction. For instance, ion channels such as Cav1.2 and the TRP family of ion channels have specific locations in the heart and enable organ level responses to pressure and volume fluctuations by helping regulate action potentials (Takahashi et al., 2013). Connexins, transmembrane proteins important for gap junction formation, may act as effectors to coordinate excitation-contraction coupling (Meens et al., 2013). Beyond this, connexin-43 is upregulated in response to mechanical stimulation and precedes other cell-cell junction formations. While mechanosensing at the plasma membrane and the cytoskeleton has been well studied, our knowledge quickly diminishes as we probe deeper into (cardiac) cells. It is clear that cardiac myocyte nuclei undergo substantial deformations during contraction (Fig. 1); however, one remaining central question in cardiac mechanobiology and mechanobiology in general, revolves around the extent to which the nucleus itself can act as a mechanosensor.
Figure 1. Nuclear deformation during cardiac myocytes contraction.
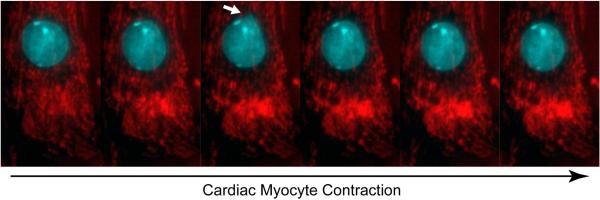
Time-lapse sequence of mouse neonatal cardiac myocytes spontaneous contracting in culture; the cytoskeleton exerts substantial forces on the cell nucleus (blue), resulting in reversible large nuclear deformations (white arrow). Mitochondria are shown in red.
A mechanosensor, as we will define it, is a protein (or, more generally, a cellular structure) that translates a mechanical input into a biochemical output, thereby initiating mechanoresponsive signaling pathways. Typically, molecular mechanosensing involves force induced conformational changes, resulting in altered interaction with binding partners or modulation of (protein) activity. It is well established that the nucleus plays an important role in mechanotransduction signaling, i.e., the process of biochemical signal propagation and processing, as most signaling pathways eventually culminate with nuclear proteins binding to specific genomic elements to modulate transcription. It is also known that the nucleus is mechanically connected to the rest of the cell via LINC (Linker of Nucleoskeleton and Cytoskeleton) complex structures in the nuclear envelope. These complexes are akin to focal adhesions at the plasma membrane, allowing for cytoskeletal and external forces to result in nuclear deformations (Lombardi and Lammerding, 2011). But, the question remains: can mechanically induced nuclear deformations directly control gene expression in a predictable, biologically-meaningful way? If so, what are the molecular mechanisms that enable the nucleus to sense and respond in this way? What are the implications for cardiac function in health and disease? Are there cardiac specific mechanisms for nuclear mechanotransduction?
The emergence of new technologies—ranging from advanced imaging and bioengineering approaches for cell-based assays, to molecular probes that can detect nanoscale forces and deformations—has enabled the scientific community to finally start addressing this important question. In the following sections, we will briefly outline current models of nuclear mechanotransduction before discussing how recent findings, ranging from cellular and subcellular studies to human diseases, and corresponding disease models are beginning to shape a clearer portrait of the nucleus as both a mechanosensor and a central processing hub for mechanoresponsive signaling pathways. Lastly, we will highlight how recent and ongoing advances in technology development will help to further elucidate the fascinating biology of nuclear mechanotransduction and its role in human health and disease.
2. The nucleus at the center of the cell
A basic overview of the mechanical connectivity linking the nucleus to the rest of the cell and its ECM surroundings (Fig. 2) provides a helpful roadmap for understanding how the nucleus may carry out mechanotransduction processes. The nucleus is encapsulated by a double lipid membrane system, composed of the inner and outer nuclear membrane (INM, ONM), that is studded with nuclear pores. Driven by recent advances in protein isolation, mass spectroscopy, and super-resolution imaging, it is now emerging that cells contain hundreds or even thousands of nuclear envelope transmembrane proteins (NETs), many of them specific to the nuclear membranes, and their expression varying widely between cell-types and tissues (de las Heras et al., 2013; Korfali et al., 2012; Schirmer et al., 2003). Underlying the INM resides the nuclear lamina, a dense 25-50 nm thick intermediate filament meshwork composed of A- and B-type lamins, which provides structural support to the nucleus and helps space nuclear pore complexes (Crisp et al., 2006a; Ho and Lammerding, 2012; Lammerding et al., 2006; Versaevel et al., 2013). Importantly, lamins bind to a large number of other nuclear (envelope) proteins, including members of the LEM protein family, comprised of LAP2, emerin, and MAN1, as well as heterochromatin. The nuclear envelope proteins interact with a variety of transcription factors in the Retinoblastoma (Rb), Wnt, TGF-β, and MAPK pathways, as well as more general transcription factors such as BAF and SREBP (Haraguchi et al., 2001; Lloyd et al., 2002; Margalit et al., 2007; Margalit et al., 2005; Moiseeva et al., 2011; Rodriguez et al., 2010; Wilson and Foisner, 2010). As such, nuclear envelope proteins are ideally situated to control critical signaling pathways involved in mechanotransduction signaling.
Figure 2. A ‘direct connection’ from the extracellular matric (ECM) to the genome.

Schematic illustration highlighting the (protein) elements that maintain the structural integrity of the cell, as well as some of the important signaling molecules and transcriptional regulators (ERK 1/2, β-catenin, nuclear, pRb) and intranuclear domains (nucleolus, Cajal bodies). Integrins at the plasma membrane connect the ECM to cytoskeletal filaments, which bind the nucleus through LINC complex proteins (nesprins and SUN proteins) and the nuclear lamina. The outer nuclear membrane (ONM), along with the 30-50 nm wide luminal space enclosed between the inner nuclear membrane (INM) and ONM, is continuous with the endoplasmic reticulum. Bridging the nuclear membranes are nuclear pores that allow for transport of large molecules, such as transcription factors or RNA, between the nucleus and the cytoplasm. Composed of both A- and B-type lamins, the lamina helps to tether heterochromatin, transcription factors, and nuclear membrane proteins to the nuclear periphery (Ho and Lammerding, 2012; Lin et al., 2000; Wilson and Foisner, 2010; Zuleger et al., 2012). This physical connectivity between nesprins, SUN-proteins, and the nuclear interior allows a direct route for mechanical signals to reach the nucleus and also impacts how biochemical signals traverse the cytoplasm and the nuclear envelope or interact with nuclear proteins once inside the nucleus. It is important to note that emerin and some nesprin isoforms can be found on both the INM and ONM (Salpingidou et al., 2007; Zhang et al., 2002); however, it remains unclear whether they fulfill distinct functions depending on their localization. Within the nesprin families, there are wide variations in size due to alternative splicing and transcriptional initiation, with the largest isoforms, referred to as nesprin-1/2 giant, respectively, reaching ~800 – 1,000 kD in size, which may enable them to reach as far as 1 μm into the cytoplasm (Rajgor et al., 2012).
In addition to stabilizing and organizing the nucleus, lamins help tether the nucleus to the surrounding cytoskeleton via a set of INM and ONM proteins comprising the LINC complex that bridges the luminal space (Crisp et al., 2006b). The LINC complex is made up of SUN protein trimers that span the INM, and at least one of five KASH-domain containing protein family members, primarily called nesprins, on the ONM (Rothballer et al., 2013a; Rothballer et al., 2013b; Roux et al., 2009; Sosa et al., 2013; Wilhelmsen et al., 2005; Zhang et al., 2002; Zhang et al., 2001). We refer to excellent recent reviews (Gundersen and Worman, 2013; Rothballer and Kutay, 2013) for a detailed discussion of LINC complex structure and nucleo-cytoskeletal coupling. The cytoplasmic domains of the various nesprins can connect to the actin, microtubule, and intermediate filament cytoskeletal systems, either by direct interaction or via molecular linkers such as plectin or the microtubule-associated motors dynein and kinesin (Rajgor and Shanahan, 2013). The cytoskeletal filaments then connect through the plasma membrane and focal adhesions to the extracellular matrix and cell-cell junctions, completing the physical connection from genome to extracellular environment. Consequently, mechanical stimuli from the cell exterior or the cytoplasm are directly transmitted to the nucleus, where they can result in substantial deformations (Maniotis et al., 1997). From this, it becomes evident that the nucleus and nuclear envelope proteins sit at the crossroads between extracellular (mechanical) signals and transcriptional response(s).
3. The theory – potential mechanisms of nuclear mechanosensing
As we gain a better understanding of the structure and function of the nuclear envelope and interior, we move ever closer to comprehending how the nucleus perceives and responds to force. Based on our current knowledge, a number of (non-mutually exclusive) molecular mechanisms have been proposed that could directly modulate nuclear structure and transcriptional regulation, thereby enabling the nucleus to transduce mechanical forces into biochemical signals (Fig. 3).
Figure 3. Potential mechanisms of nuclear mechanosensing.

The nucleus receives a vast amount of mechanical and biochemical signals from the surrounding cytoplasm. Incoming signals may trigger various responses at the nuclear envelope or within the interior that can result in changes in gene expression. (1) Nuclear deformations may alter the interactions (via change in proximity or other means) between chromatin and the nuclear lamina, causing transcriptional activation or repression. Force-induced changes in chromatin organization may also alter accessibility of chromatin to transcriptional regulators. (2) Mechanically-induced damage to the nuclear membranes, or changes in the permeability of nuclear pores or stretch-sensitive ion channels can results in altered nuclear import/export. (3) Ca2+ or other ions may enter the nucleus or be release from perinuclear stores in response to osmotic stress or other mechanical stimuli. (4) Forces transmitted across the LINC complex may lead to force-induced conformational changes in nuclear envelope proteins, resulting in their phosphorylation or altered interaction with binding partners. (5) Nuclear actin serves as both a mechanical scaffold and a signaling moiety; nuclear actin polymerization can modulate activity and import/export of MKL1/SRF. (6) Changes in membrane fluidity or curvature may alter the conformational state of bound proteins and cause assembly/disassembly of multi-protein structures at the nuclear envelope. (7) Nuclear envelope proteins can sequester transcriptional regulators at the nuclear periphery or form active/inactive complexes, thereby regulating their activity. (8) Nuclear actin (along with other structural elements such as spectrin IIα may also serve as a scaffold for transcriptional regulation by positioning DNA near transcriptional machinery or specific DNA-regulatory elements. In addition to the depicted mechanisms, additional mechanisms, such as osmotic changes in the nucleus caused by volume changes, may contribute to nuclear mechanosensing.
3.1. Conformational changes of nuclear (envelope) proteins and chromatin
Force-induced unfolding of nuclear proteins, either at the nuclear envelope or the nuclear interior, may reveal cryptic binding sites, promoting altered protein-protein interactions or phosphorylation of specific residues via nuclear kinases. Phosphorylation of specific nuclear envelope components may further alter interaction with their binding partners (Guilluy et al., 2014) or, in the case of lamins, trigger (partial) depolymerization of the nuclear lamina, similar to the events occurring during mitosis (Kochin et al., 2014; Swift et al., 2013). Even forces too low to result in partial protein unfolding (e.g., in the low picoNewton range) may still be sufficient to dissociate nuclear proteins from each other, as demonstrated for nucleosomal protein complexes (Poh et al., 2012), or to affect chromatin structure (Iyer et al., 2012; Li et al., 2011), which could alter accessibility to transcriptional regulators. Further biophysical and biochemical assays will be required to define the relevant force thresholds, identify the corresponding protein conformational changes, and evaluate the impact on protein-protein interactions and chromatin structure and organization.
3.2. Changes in gene location
In addition to changes in chromatin conformation, forces transmitted to the nuclear interior may also impact gene location. We now know that chromosomes reside with high probability in specific “territories” and that the spatial arrangement of specific DNA segments can vary widely between cell types and even change position over time within a given cell (Stancheva and Schirmer, 2014). For the most part, transcriptionally active euchromatin resides in the nuclear interior or close to nuclear pores, whereas heterochromatic and transcriptionally repressed regions associate with lamins and lamin-associated proteins at the nuclear periphery. Force-induced detachment of specific gene loci from the nuclear periphery may thus result in activation of the associated genes, whereas recruitment of genes to the nuclear periphery (for example, due to altered protein-protein or DNA-protein interactions), may result in gene silencing.
While changes in the position of specific gene loci are well documented, the underlying molecular mechanisms remain elusive, along with the potential involvement of nuclear molecular motors such as myosin. A further examination of nuclear actin and nucleosome scaffolding will be necessary to understand if/how forces may directly modulate gene localization and transcription. Interestingly, nuclear actin has the potential to integrate both biophysical and biochemical cues, making it a prime candidate for nuclear mechanosensing. The recent development of fluorescent reporters to visualize monomeric and filamentous nuclear actin (Belin et al., 2013) and other nucleoskeletal components will enable imaging dynamic changes in nuclear organization in real time.
3.3 Other potential nuclear mechanosensing mechanisms
It is intriguing to speculate on additional molecular mechanisms that may contribute to nuclear mechanotransduction. Several studies have shown that cellular and nuclear volume can change dramatically in response to mechanical force application (Guilak, 1995; Rowat et al., 2006; Versaevel et al., 2012). As the volume reduction is primarily caused by loss of water (Rowat et al., 2006), the osmolarity of the cellular compartments and concentration of nuclear and cytoplasmic proteins may change significantly, affecting biochemical signaling and intracellular mechanics. Osmotic, tensile, and compression forces may also influence nuclear membrane permeability by opening stretch-sensitive channels or by inducing transient ruptures in the membrane. Taken together, calcium entry through nuclear ion channels or nuclear volume fluctuations may be sufficient to trigger a nuclear mechanoresponse (Finan et al., 2011).
The nuclear pore complex (NPC) is another potential mechanotransduction gateway into the nucleus, yet it remains much of a black box despite advancements in theoretical modeling techniques. NPCs interact with both the cytoskeleton and the genome (directly and through lamins) and are generally considered sites of active transcription (Akhtar and Gasser, 2007) with their constituent nucleoporin proteins having a role in both mRNA export (Ball et al., 2007) and gene expression (Griffis et al., 2004; Kalverda et al., 2010). It remains to be proven whether cell stretching forces can expand the pore, thus increasing the size discrimination threshold and allowing passive diffusivity of molecules at or near the size cutoff (e.g. monomeric actin ~40 kDa). Similarly, other stretch-activated ion channels on the nuclear membranes could mediate mechanically induced influx of calcium or other ions from the cytoplasm or endoplasmic reticulum into the nuclear interior, similar to the processes observed at the plasma membrane, or sarcoplasmic reticulum. Lastly, mechanically induced changes in nuclear membrane composition or conformation may provide an additional mechanosensing mechanism. For example, free energy fluctuations in lipid bilayers may aid in force sensing by causing conformational changes in transmembrane proteins or by recruiting proteins or other factors which are sensitive to membrane curvature.
4. The case for nuclear mechanotransduction – getting to the nucleus of it
Cells subjected to mechanical stimuli respond with rapid (<30 min) activation of characteristic ‘mechanosensitive’ genes such as Egr-1, Iex-1, or Pai-1 that often represent immediate early transcription factors that turn on additional genes. Many of these responses appear quite ubiquitous and can be observed in a number of different cell lines, including fibroblasts, myotubes, and neonatal cardiac myocytes (Banerjee et al., 2014; Ho et al., 2013b; Lammerding et al., 2004). Intriguingly, cells lacking the nuclear envelope proteins lamin A/C (Lammerding et al., 2004), emerin (Lammerding et al., 2005b), or nesprins-1 and -2 (Banerjee et al., 2014) have significantly attenuated expression of Egr-1 and Iex-1 when subjected to cyclic strain, despite normal or even increased activation of cytoplasmic MAPK signaling. These findings provided the first experimental clues depicting the role of the nucleus in cellular mechanotransduction. The question whether (or to what extent) the nucleus serves as a cellular mechanosensor has occupied researchers for at least two decades (Davies, 1995; Wang et al., 2009); only in recent years have novel technologies and improved insights into nuclear architecture and nucleocytoskeletal coupling enabled more direct observations of potential nuclear mechanotransduction processes.
4.1. Mechanically induced changes in nuclear structure and organization
It is now well established that forces exerted at the cell surface or cytoskeleton can induce rapid, subnuclear deformations by propagating through the cytoskeleton and the LINC complex (Lombardi et al., 2011; Maniotis et al., 1997). However, many questions remain: What types of forces (magnitude, duration, location) are sufficient to trigger direct mechanotransduction responses from the nucleus? Which part(s) of the nucleus constitutes the mechanosensitive element(s)? Does the location of specific genes within the chromatin architecture influence the mechanoresponse? What are the timescales involved? Combining old and new technologies to both apply and record the effect of forces on the nucleus are beginning to answer these questions.
Mechanical forces can be transmitted through the cytoplasm much faster than diffusion based biochemical signals (30 m/s for mechanical stress propagation versus 1-2 μm/s for small chemical diffusion or motor-based transport) (Wang et al., 2009); therefore, rapidly induced changes in nuclear organization likely signify direct mechanosensing events (Na et al., 2008; Wang et al., 2009). For example, applying low (0.8 to 1.7 nN) forces to the surface of HeLa cells using magnetic tweezers results in a rapid (< 5 second) decrease in the fluorescence anisotropy of GFP-labelled histones, indicative of chromatin decondensation (Iyer et al., 2012). The effect is reversible upon cessation of the force; however, sustained force application (> 225 seconds) promotes irreversible changes to nuclear morphology. One caveat is that the observed intranuclear changes may also result from altered cell morphology in response to mechanical stress (Guilak, 1995; Knight et al., 2002; Legant et al., 2010), placing the nucleus downstream of these cytoplasmic changes. Nonetheless, additional evidence for molecular level changes inside the nucleus in response to external force application was provided by Poh and colleagues, who used fluorescence resonance energy transfer (FRET) assays to demonstrate rapid (<1 second) dissociation of two Cajal body (CB) proteins (SMN and coilin) in response to mechanical stress at the HeLa cell membrane. This phenomenon was irreversible and dependent on both substrate stiffness and an intact nuclear envelope.
To minimize confounding effects from upstream biochemical signals emanating from the cytoplasm, several groups have begun investigating nuclear mechanotransduction mechanisms in isolated nuclei. Using this strategy, Swift et al. (Swift et al., 2013) uncovered a cryptic site (Cys522) in the Ig-domain of lamin A that partially unfolds in response to shear stress on the order of 1 Pa. The shear-sensitive residue was identified using ‘cysteine shotgun mass spectrometry’ (CS-MS), a technique that utilizes the biochemical properties of cysteine (i.e., a hydrophobic amino acid often found buried at the core of proteins and possessing a reactive thiol-group) to covalently link the exposed amino acid to a fluorescent molecule. To what extent the observed force-induced unfolding occurs under physiological conditions in intact cells remains to be seen; nonetheless, the study presents an exciting advance in examining the ability of the nucleus to directly “sense” forces.
Perhaps some of the strongest evidence for direct nuclear mechanosensing to date comes from a recent study showing that isolated nuclei adjust their stiffness in response to force application through nesprin1 by recruiting lamin A/C to the LINC complex and thereby strengthening the nuclear envelope and nucleo-cytoskeletal coupling (Guilluy et al., 2014). The response is mediated by Src-dependent phosphorylation of emerin at two specific tyrosine residues (Tyr 74 and Tyr 95). While the study nicely illustrates that nuclear players are sufficient to dynamically regulate emerin phosphorylation and nuclear envelope composition, many important questions remain: is nuclear Src activated by mechanical stress (via nesprin1), or do the mechanical forces induce changes in emerin accessibility or localization that then result in the phosphorylation of emerin by already active Src? How does emerin phosphorylation mediate lamin A/C recruitment to the LINC complex? Does emerin phosphorylation regulate nuclear mechanics (Guilluy et al., 2014) and transcription (Ho et al., 2013b; Lammerding et al., 2005b) through distinct or overlapping pathways? Further experiments will be needed to elucidate the exact role of nesprins, emerin, and other nuclear envelope proteins in nuclear mechanotransduction signaling, as well as their dynamic posttranslational modifications and diverse interactions with signaling and structural proteins.
The potential for dynamic lamin recruitment to the mechanically perturbed-LINC complex (Guilluy et al., 2014) and, more generally, the emergence of dynamic interaction and localization of nuclear envelope proteins has interesting implications. First, it requires updating the simple model of lamins in the interphase nucleus as a static scaffold for neighboring proteins to bind. Rather, it suggests that many of the known interactions, for example, at the LINC complex, may be highly dynamic and regulated by phosphorylation or binding to other proteins such as Samp1 (Borrego-Pinto et al., 2012). Furthermore, SUN proteins, emerin, and nesprins may conversely help anchor A-type lamins at the nuclear periphery and form an interdependent network there (Banerjee et al., 2014); with the loss of any of these proteins, due to mutations or posttranslational modifications, the network and its connection to the nuclear membranes are re-arranged, modulating nuclear stability, nucleo-cytoskeletal coupling, and other nuclear envelope functions. There is also evidence that nuclear envelope components such as BAF1 may become mobile or immobile in response to stress, leading to further reorganization (Bar et al., 2014). Protein mobility studies and pull-down assay can be used to assess the dynamic behavior of the nuclear membrane, while knock down studies can help assess the impressive (partial) redundancy of NE proteins, such as SUN1/SUN2, A-Type/B-Type lamins (Lee et al., 2014), LEM-domain proteins, and nesprin1/2 (Banerjee et al., 2014). Effects of mutations or knockdown of nuclear envelope proteins go far beyond the nucleus itself. Studies in lamin A/C-, nesprin-, and emerin-deficient cells and in cells expressing dominant negative nesprin and SUN protein mutants have shown that disruption of the LINC complex and underlying nuclear lamina results in disorganized perinuclear actin and vimentin networks and impaired cell polarization and migration (Anno et al., 2012; Chambliss et al., 2013; Chancellor et al., 2010; Hale et al., 2008; Lombardi et al., 2011; Luxton et al., 2010; Morgan et al., 2011).
4.2 Mechanotransduction signaling: all roads lead to the nucleus
In addition to direct nuclear mechanosensing, several nuclear envelope proteins, including lamins and emerin, interact with a large number of transcriptional regulators (Ho and Lammerding, 2012), and therefore have the potential to modulate signaling pathways involved in mechanotransduction signaling. Many mechanotransduction signals are initiated at cytoplasmic and plasma membrane locations and are then biochemically transmitted to the cell nucleus (Wang et al., 2009). Consequently, mechanoresponsive genes can be activated even in the absence of nuclear deformations, as demonstrated in cells in which LINC complex proteins had been disrupted (Lombardi et al., 2011). Following transduction of extracellular signals at focal adhesions and cadherin complexes, intracellular signaling cascades reach the nucleus through various second-messengers and via activation of protein signaling molecules such as talin, zyxin and src family kinases that respond to mechanical stress (Schwartz, 2010). Importantly, it is now emerging that the nucleus plays a central role in modulating such cytoplasm-originating mechanotransduction signals, with a variety of direct biochemical and functional interactions between nuclear envelope proteins and transcriptional regulators contributing to this processing (Table 1). In the following, we briefly discuss some of the key mechanotransduction signaling pathways that interact with nuclear envelope proteins.
Table 1.
Transcriptional regulators and their interactions with nuclear envelope proteins.
| Transcriptional regulator |
Organism | Function | NE Interaction partner(s) |
Consequence of misregulation |
Citation |
|---|---|---|---|---|---|
| Oct-1/OCT1 | mouse/human | Regulates stress response genes | Lamin B | Increase of reactive oxygen species | (Malhas et al., 2009) |
| Tbx1 | mouse | H3K4 monomethylation of the Wnt5a gene; targets BAF chromatin remodeling complex to Wnt5a gene | BAF | Abnormal cardiac development | (Chen et al., 2012) |
| BAF/Banf1 (barrier to autointegraton factor 1) | mouse | Nuclear assembly, chromatin organization (during mitosis) | Emerin, LAP2β, MAN1, Lamin A | Altered levels of silencing and active histone marks; DNA damage repair defect | (Demmerle et al., 2012) |
| Heterochromatin protein 1 (HP1)/Chromobox protein homolog 5 (CBX5) | drosophila (yeast two-hybrid study)/human | Heterochromatin protein that contributes to repressive chromatin structures | LBR | Sister chromatid cohesion, ER stress response, VPR (C. elegans) | (Inoue et al., 2008; Ye and Worman, 1996) |
| hALP | human | Membrane-associated histone acetyltransfease (HAT) | SUN1 | Mitotic chromosome decondensationdefect; abnormal histone modifications for repression and activation | (Chi et al., 2007; Schirmer et al., 2003) |
| mGCL /germ cell less (GCL) | mouse/drosophila/human | Transcriptional repressor of E2F-dependent genes | LAP2β, emerin, lamin A* | Deacetylation of histone H4; cell cycle misregulation | (Holaska et al., 2003; Nili et al., 2001) |
| Btf (Bclafl) | human (yeast two-hybrid study) | Transcriptional repressor, apoptotic signaling | Emerin, MAN1 | Overexpression triggers apoptosis; impaired skeletal muscle function* | (Haraguchi et al., 2004; Mansharamani and Wilson, 2005) |
| Rb | human | Chromatin remodeling; tumor suppressor protein | Lamin A, LAP2α, LAP2β | Replication of damaged DNA, Cell cycle misregulation | (Chow and Dean, 1996; Harbour and Dean, 2000) |
| c-Fos | mouse/human | AP-1 family transcription factor; MAPK effector; cell growth | Lamin A | Defective growth, proliferation, differentiation | (Gonzalez et al., 2008) |
| Lmo7 | human | Emerin gene expression; relays mechanotransduction signals from cell surface to nucleus | Emerin | Deficiency leads to reduced emerin mRNA levels; muscular dystrophy phenotype | (Holaska et al., 2006) |
| Smads | human | Intracellular signaling effectors for the TGF-β family of secreted polypeptides; co-modulators of transcription | MAN1 | MAPK impairment; altered TGFβ gene expression | (Attisano and Wrana, 2000; Pan et al., 2005; Zhang et al., 1998) |
| β-catenin | human | WNT signaling transducer | Emerin, NPC proteins | Cancer; impaired WNT signaling | (Fagotto et al., 1998; Neumann et al., 2010; Rashmi et al., 2012) |
| YT521-B | human (yeast two-hybrid study) | mRNA splicing | Emerin | Myotonia and insulin resistance | (Wilkinson et al., 2003) |
| MRFs (MyoD, myogenin, Myf5, and Myf6/Mrf4) | mouse/human | Myogenic regulatory factor, essential for myogenesis, cell commitment and permanent growth arrest; myotubes formation | pRb | Failed myoblast differentiation; defective muscle regeneration; cell cycle abnormalities | (Andres and Gonzalez, 2009) (Gonzalez et al., 2008) |
| SREBP1 | human | Regulation of adipocyte differentiation | Prelamin A | Sequestration into (*toxic) nuclear aggregates | (Andres and Gonzalez, 2009; Hübner et al., 2006) |
| MOK2 | human | DNA-binding transcription repressor | A-type lamins | Sequestration into nuclear aggregates; *impaired expression of downstream target gens | (Andres and Gonzalez, 2009; Dreuillet et al., 2008) |
| TonEBP (NFAT5)* | human | Transcriptional activator of hypertonicity-induced gene transcription in kidney cells | Lamin A/C | Osmotic stress damage | (Woo et al., 2002) |
denotes speculated interaction.
4.2.1 MAPK pathway
The Mitogen Activated Protein Kinase (MAPK) pathway is responsible for growth, differentiation and cellular response to mechanical strain, heat shock, and osmotic stress (Jaalouk and Lammerding, 2009; Peti and Page, 2013) and consists of three branches: c-Jun NH2-terminal kinase (JNK), extracellular signal-related kinase 1/2 (ERK1/2), and p38. Misregulated MAPK signaling is often found in cancer and neurodegenerative disease, as well as in cardiomyopathies and muscular dystrophies caused by mutations in lamins and emerin (laminopathies)(Emerson et al., 2009; Lammerding et al., 2005b; Muchir et al., 2007a; Muchir et al., 2007b; Muchir et al., 2012a; Muchir et al., 2012b). In particular, hyperactive MAPK signaling has been demonstrated for various mouse models of muscular laminopathies and cells from laminopathy patients. Increased MAPK activity precedes the onset of severe cardiomyopathy in Lmna H222P mutant mice, suggesting that it may be a cause rather than a consequence of the disease (Muchir et al., 2007b), and inhibition of the various MAPK pathways has proven effective in ameliorating the cardiac phenotypes (Muchir et al., 2012a; Muchir et al., 2009). Functionally, lamin A mediates the interaction between ERK1/2 and the transcription factor c-Fos at the nuclear envelope, leading to the phosphorylation and release of c-Fos in response to stress (Gonzalez et al., 2008). Consequently, loss of lamins A/C can result in altered levels and activity of c-Fos. This may be part of a larger paradigm, whereby nuclear envelope proteins can sequester transcriptional regulators and control their activity on downstream target genes.
4.2.2 Wnt signaling
The Wnt pathway controls signals required for proper tissue development, a process governed extensively by mechanical forces. Disturbed Wnt signaling has been also reported in some progeroid laminopathies (Hernandez et al., 2010). β-catenin, the most well-studied Wnt regulator, transmits mechanotransduction signals from cell-cell interfaces to the nuclear interior, where it enhances cell growth and proliferation (Markiewicz et al., 2006). Since β-catenin lacks a nuclear localization signal (NLS), it is thought to be imported and exported by nuclear envelope proteins such as nesprin2 and emerin (which form complexes together with α- and β-catenin), as well as nuclear pore components (Fagotto et al., 1998; Neumann et al., 2010). Studies employing human cell knockouts indicate a model whereby nesprin2 and emerin facilitate β-catenin import and export, respectively, thereby regulating the activation of downstream target genes (Markiewicz et al., 2006; Neumann et al., 2010). In addition, β-catenin works cooperatively with chromatin remodeling proteins to orchestrate epigenetic modification and transcriptional activation at Wnt-responsive genes (Willert and Jones, 2006).
4.2.3. MKL1/SRF activation
The MKL1/SRF pathway is another important mechanoresponsive signaling pathway. MKL1 is normally sequestered by G-actin in the cytoplasm; increased actin polymerization in response to mechanical or serum stimulation results in translocation of MKL1 into the nucleus, where it, together with serum-response factor (SRF), can activate numerous cytoskeletal genes and SRF itself. At the same time, nuclear actin is required for the nuclear export of MKL1 (Olson and Nordheim, 2010). Attesting to the intricacy of mechanosingaling, MKL1 also interacts with β-catenin (Wnt pathway) and Smads (TGF-β pathway) which may have implications in cell differentiation, migration and cell cycle regulation (Charbonney et al., 2011; Scharenberg et al., 2014). Misregulation of the MKL1/SRF pathway is implicated in many diseases affecting mechanically-stressed tissue. For example, SRF-null mice develop severe cardiac defects (Parlakian et al., 2004); cells and tissues from lamin A/C-deficient and mutant mice, as well as skin fibroblasts isolated from patients with dilated cardiomyopathy caused by LMNA mutations exhibit reduced MKL1 translocation in response to serum stimulation (Ho et al., 2013a). In the latter cases, loss of emerin from the nuclear envelope disrupts both nuclear and cytoplasmic actin dynamics, causing defective nuclear import and export of MKL1 (Ho et al., 2013b). These findings, along with subsequent work by the Grosse group (Baarlink et al., 2013) suggest that emerin and the formins mDia1 and mDia2 can drive nuclear actin polymerization and modulate MKL1/SRF activity. Consequently, nuclear actin may serve as an important mechanotransduction signaling element, rapidly integrating and responding to numerous biochemical and biomechanical signals. Nuclear actin and actin-binding proteins may further contribute to the recruitment of protein kinases and histone remodelers to transcription sites and induce rapid changes in gene expression (Blessing et al., 2004).
4.2.4 Other pathways
Transcriptional regulators that shuttle from focal adhesion to the nuclear envelope have recently gained increasing attention and may provide an additional means for the nucleus to control mechanotransduction signaling. In particular, zyxin and Lmo7 are two intriguing candidates that may contribute to nuclear mechanotransduction processing. Downstream of ERK activation, the LIM-domain phosphoprotein zyxin relays signals from focal adhesions to the nucleus, where it acts as a transcription factor for genes related to adhesion and inflammation (Moon et al., 2006; Yoshigi et al., 2005). Zyxin interacts with sarcomeric elements in muscle and may directly communicate the presence of (intracellular) mechanical damage to the nucleus to activate either repair genes or a death signal, as seen in a C. elegans model for Duchenne's muscular dystrophy (Hoffman et al., 2012; Lecroisey et al., 2013). Lmo7 is a transcription factor with major roles in cardiac and skeletal muscle (Ott et al., 2008). Like zyxin, Lmo7 shuttles between focal adhesions, where it associates with the mechanosensitive protein p130Cas, and the nuclear envelope, where emerin binding modulates its activity (Sawada et al., 2006; Wozniak et al., 2013). Conversely, Lmo7 controls emerin expression, indicating a mutual co-regulatory mechanism that warrants future investigation. Lmo7 also co-localizes with F-actin and can modulate the G-actin/F-actin ratio (Hu et al., 2011), which may further affect MKL1 nuclear translocation.
While several of the molecular details of various signaling pathways are slowly coming to light, we still lack a fundamental knowledge of the basic biochemistry, structure, function, and post-translational modification of many nuclear envelope components, which may collectively constitute a nuclear control board. For example, the function and dynamic interaction of many of the nuclear membrane proteins and their binding partners remain incompletely understood. In addition to sequestering transcriptional regulators, nuclear envelope proteins can also directly affect chromatin structure and organization, thereby further modulating transcriptional activity (Stancheva and Schirmer, 2014). New insights into the role of nuclear envelope proteins into cellular mechanotransduction signaling will not only provide a better understanding of physiological processes, but also hold promise for pharmacological intervention in the diverse laminopathies (Azibani et al., 2014).
5. The consequences of impaired mechanotransduction – the nucleus and human disease
5.1 Mutations in nuclear envelope proteins cause dilated cardiomyopathy
Achieving an understanding of the mechanisms underlying human disease is one of the major drivers in biomedical sciences. A powerful illustration of the importance of mechanotransduction comes from the large number of human diseases linked to defects in cellular structure and mechanosensing, including cardiomyopathies, vascular disease, muscular dystrophy, cancer, reversal of the inner organs, and hearing defects (Jaalouk and Lammerding, 2009). Muscle diseases, such as cardiomyopathies and muscular dystrophies, are often caused by mutations or deletions of proteins that make up the force-generating sarcomeres or that connect the sarcomeres to the extracellular matrix (Harvey and Leinwand, 2011). In the 1990s, the finding that many of the same phenotypes can also result from mutations in nuclear envelope proteins, drew attention to the importance of nuclear proteins in mechanotransduction (Worman et al., 2010). Beyond this, different mutations in similar or even identical amino acid positions can cause distinct diseases. For instance, the T528M and T528K mutations in the LMNA gene result in lipodystrophy and muscular dystrophy, respectively (Bonne et al., 1999; Savage et al., 2004). The fact that mutations in very different proteins can lead to similar diseases while different mutations in the same gene (e.g. LMNA) can cause very distinct phenotypes highlights a particularly important question: how is the specific disease mechanism related to nuclear mechanics and mechanotransduction?
Cardiomyopathies are the most prevalent phenotype clinically linked to defects in the LMNA gene (Narula et al., 2012). In fact, up to 48% of all cases of dilated cardiomyopathy (DCM) are caused by genetic mutations and 5-10% of these mutations are found to be related to lamin A/C (Fatkin et al., 1999; Narula et al., 2012; Sylvius et al., 2005). Thus far, 128 LMNA mutations have been linked to DCM (http://www.umd.be/LMNA/). Other causes of DCM involving nuclear envelope defects arise from mutant forms of emerin, nesprin1/2, Lap2α and LUMA, an inner nuclear membrane protein that associates with emerin (Bengtsson and Otto, 2008; Bione et al., 1994; Taylor et al., 2005). Beyond nuclear proteins, DCM is often caused by mutations in cytoskeletal or sarcomeric proteins such as myosin, troponin, or desmin involved in cellular force generation and transmission (Kamisago et al., 2000). DCM presents with a phenotype characterized by progressive weakening of the heart muscles, thinning of the left ventricle wall, and insufficient pumping that typically leads to heart failure. LMNA mutations are particularly associated with conduction defects (30-45% of familial and 80% of non-familial cases) and arrhythmia leading to higher incidence of ventricular tachyarrhythmia and subsequent sudden cardiac arrest (Narula et al., 2012; Siu et al., 2012). In mice, lamin A/C haploinsufficiency is sufficient to produce a phenotype; in humans most disease cases are the result of heterozygous lamin mutations. Recently, it has been shown that some DCM laminopathy patients exhibit a decrease in lamin A/C expression in their myocytes and in their peripheral blood compared to healthy individuals, which could serve as a biomarker with clinical diagnostic potential (Narula et al., 2012). However, other reports in mice and laminopathy patients found that many lamin A/C mutants are expressed at similar levels as wild-type lamin A/C (Zhang et al., 2013).
On a more mechanistic level, biochemical and mechanical analyses of the structure-function consequences of specific DCM lamin mutations will lead to better mechanistic understanding not only of cardiomyopathies but also other diseases related to lamins. A recent paper demonstrated how three DCM causing mutations led to altered secondary/tertiary protein structure, association constants, and higher order assembly in lamins (Banerjee et al., 2013). Combing these types of studies with the ones done by the Neumann and Lammerding groups on the effect of specific mutations on other nuclear envelope binding and mechanics (Yang et al., 2013; Zwerger et al., 2013) will be an important step forward in determining disease mechanism. The addition of molecular modeling to this suite of experiments will only make the findings more robust for characterizing various mechanotransduction mechanisms.
Going beyond stand-alone DCM, muscular dystrophies also often present with cardiac conduction defects. Emery-Dreifuss muscular dystrophy (EDMD) can be caused by mutations in emerin (X-linked EDMD), lamins A/C (autosomal dominant EDMD), nesprins1/2 and LUMA (Bengtsson and Otto, 2008; Bione et al., 1994; Bonne et al., 1999; Isermann and Lammerding, 2013; Zhang et al., 2007). These nuclear envelope mutations produce similar, but less severe phenotypes, than the more common Duchenne's muscular dystrophy, which is caused by mutations in the dystrophin complex linking the cytoskeleton to the plasma membrane. Additionally, mutations in lamins and other nuclear envelope proteins can cause a variety of other diseases (laminopathies), some of which also have cardiac phenotypes. One such disease is Hutchinson-Gilford progeria syndrome, a premature aging disease that causes increased nuclear stiffness due to a mutant form of lamin (progerin), which alters the cellular response to mechanical stress (Dahl et al., 2006; Verstraeten et al., 2008). This change in nuclear mechanics provides a potential explanation for the lethal cardiovascular phenotypes in this disease. Please see (Isermann and Lammerding, 2013) for a detailed review of other laminopathies such as liopdystrophy and Charcot-Marie-Tooth disorder. As lamins have a broad range of functions (Ho and Lammerding, 2012), it remains unclear to what extent defective mechanotransduction contributes to the various laminopathies.
For most of these diseases, and certainly DCM, the underlying nuclear molecular details remain incompletely understood, even in cases where mutations in specific genes have been identified as the primary cause. Is the role of the nucleus primarily structural, with a decrease in nuclear stability and impaired intracellular force transmission promoting cell death and dysfunction in mechanically stressed tissues? Is it primarily biochemical, with changes in specific protein-protein interactions causing aberrant signaling and function? Or is it a combination of both, with structural and biochemical changes affecting nuclear mechanosensing and mechanotransduction processing? Furthermore, are the often tissue-specific phenotypes caused by defects in stem cell differentiation or maintenance, or do they result from some cell types being particularly sensitive to lamin mutations? Novel experimental approaches using mouse embryonic stem cells lacking specific lamins and studies on human iPSC-derived cardiac myocytes and other cell types will help address these questions.
5.2. The effect of lamin mutations on nuclear structure and mechanics
Generally, nuclear mechanics can be broken down into two main components: (1) the viscoelastic chromatin, which resists compaction higher than 60% (Guilak and Mow, 2000; Rowat et al., 2013; Rowat et al., 2006; Versaevel et al., 2013); and (2) the elastic lamina, which has an elastic modulus approximately 50-times higher than the plasma membrane in endothelial cells (Caille et al., 2002). When cells or isolated nuclei are subjected to mechanical stress, nuclei undergo rapid, mostly elastic (i.e., reversible) deformations with viscoelastic contributions on the time-scale of seconds; continued force application can result in additional viscous, plastic deformations (Rowat et al., 2005). For example, in cells exposed to shear stress, the nucleus deforms plastically with nuclear elongation and stiffening persisting for at least 24 hours after release from the substrate (Deguchi et al., 2005; Sato et al., 1987; Sato et al., 1996), suggesting long-term reorganization of nuclear structure in response to mechanical force.
Analysis of cells lacking specific nuclear envelope proteins revealed that nuclei from lamin A/C-deficient cells have a much lower elastic modulus and increased nuclear fragility compared to control cells (Broers et al., 2004; Guilluy et al., 2014; Lammerding et al., 2006; Lammerding et al., 2004; Lee et al., 2007), with nuclear stiffness increasing as a function of lamin A/C expression (Lammerding et al., 2006; Swift et al., 2013; Zwerger and Medalia, 2013). Interestingly, changes in the levels of lamin B1/B2 or emerin had only relatively minor or no effects on nuclear deformability (Lammerding et al., 2005a; Rowat et al., 2005; Rowat et al., 2006). Of note, different disease-causing lamin A/C mutations can have very distinct effects on nuclear mechanics. While mutations responsible for familial partial lipodystrophy do not disturb the structural function of lamins, many, but not all, mutations causing muscular dystrophy and dilated cardiomyopathy result in a loss of structural function, reflected in increased nuclear deformability (Zwerger et al., 2013). On the other hand, expression of progerin, the mutant lamin A responsible for Hutchinson-Gilford progeria syndrome, results in increased nuclear stiffness (Dahl et al., 2006; Verstraeten et al., 2008).
6. The road ahead - Technology leading the way
The ability to identify specific changes to gene location, chromatin structure, chromatin organization, and transcriptional activity in response to mechanical force would bring much clarity to identify specific nuclear mechanotransduction mechanisms. Emerging methods and technologies that can relate intranuclear forces with biochemical events such as conformational changes and transcriptional processes, particularly on the single cell level, have the potential to transform the way we study the nucleus (Fig. 4) and will help determine whether the nucleus itself can transduce mechanical forces into biochemical signals or whether it merely processes and relays the aftereffects of mechanotransduction events happening elsewhere in the cell.
Figure 4. Avenues for new technologies to investigate nuclear mechanosensing.

Schematic cartoon of the typical experimental workflow. Most experimental approaches begin with cells or tissues derived from animal models or human patients. Animal models can provide cells and tissues with a specific genetic background or mutation of interest. Mice with several deletions (DKO – double knock-out) can help assess synergies and redundancies between different genes. The development of advanced in vitro models, including cells cultured in 3-D environments and decellularized tissues, can provide improved accessibility of cells for experimental purposes while mimicking physiological conditions. Tissue samples, cells, or isolated nuclei can then be manipulated with an array of experimental techniques to apply either controlled mechanical forces or deformations. The biomechanical properties can be inferred from the measured force-displacement relationships. Advanced imaging modalities and emerging novel technologies that enable measuring the intracellular forces at the molecular level provide improved insights into force-induced changes of nuclear structures at higher and higher temporal and spatial resolution. Examples include genetically encoded FRET-based tension sensors or other methods such as microfabricated pressure sensors. Alternatively (or in addition), researchers may wish to employ a method of detecting molecular events or structural changes (e.g. chromatin/protein unfolding, gene translocation, etc.). Ultimately, observed mechanically induced changes in nuclear structure must be linked to readouts of cellular function to ensure their biological relevance. Such readouts can include changes in transcriptional activity (gene expression), epigenetic modifications, or kinase activation. The quantitative experimental data can provide input for computational models, ranging from molecular dynamics to systems biology analysis. These models are then validated by experimentally testing generated predictions, or refined to reflect new experimental insights.
6.1 Measuring forces at the molecular level
Experiments on primary cardiac myocytes and cultured cells have yielded valuable insights into the forces generated by the cells and transmitted to their microenvironment. In contrast, there is a tremendous need to measure the forces acting on intracellular structures, particularly the nucleus and nuclear interior, in resting cells and under physiological load, e.g. during cardiac myocyte contraction and dilation. Genetically encoded FRET-based biosensors offer a particularly attractive tool to study mechanically induced conformational changes inside living cells at high tempo-spatial resolution, as indicated by their successful use in studying the physical forces acting on vinculin at focal adhesions (Grashoff et al., 2010) and spectrin structures in the cytoskeleton (Meng and Sachs, 2011). Similar force sensors could be used to measure the forces transmitted from the cytoskeleton to the nucleus or the forces acting on specific DNA domains, which would provide valuable input for single molecule experiments and computational molecular simulations (see below). FRET-based approaches, albeit to study protein-protein interactions rather than to measure forces, have already contributed new insights into nuclear mechanosensing (Poh et al., 2012) and may be further strengthened by recent advancements in FRET technology (Rahim, Kamm, 2013; others). In addition to FRET-based probes, other new technologies have been recently developed which may be adapted to measure forces acting on or within the nucleus. Tension gauge tether (TGT) technology uses ligand-conjugated DNA tethers covalently attached to a surface to determine single molecule forces required for mechanical signaling in cells. Applied to isolated nuclei, TGT could help to characterize forces on nuclear envelope proteins (Wang and Ha, 2013). Silicon pressure sensor chips are small enough to be taken up by cells and may be used in or near the nucleus to study response to osmotic stress or other forces (Gomez-Martinez et al., 2013). Ligand-conjugated fluorocarbon oil droplets are currently too large to fit in cells, but can already help quantifying the mechanical forces in living tissue (Campas et al., 2014). Ultimately, these approaches might be further optimized to be introduced into living cells, assuming the sensors can be stably incorporated without altering cellular or nuclear integrity.
6.2 Superresolution imaging
Superresolution imaging techniques such as photoactivatable localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM) hold the promise of an improved understanding of force-induced changes in chromatin folding and localization. Currently, many changes in nuclear structure and organization may go undetected without the sufficient spatial resolving power offered by advanced microscopy methods. Stimulated emission depletion (STED) microscopy has already been used to image the remodeling of T-tubule membrane structures in cardiomyocytes following infarction (Wagner et al., 2012). One limitation of many current superresolution approaches is the long time required for complete image acquisition. As mentioned previously, mechanical forces propagate faster than chemical forces; thus, to accurately distinguish between biochemical and mechanical effects will require extremely high temporal resolution, requiring additional experimental approaches (e.g., particle tracking, fluorescence correlation spectroscopy, etc.) and further technology development. In the end, combining superresolution technologies with previously established techniques such as FRET, fluorescence lifetime imaging (FLIM), fluorescence recovery after photobleaching (FRAP), and similar variations will enhance our understanding of nuclear protein dynamics, binding and diffusion. One recent development in DNA-protein interaction studies combined binding activatable localization microscopy (BALM) with a method for preparing chromatin spreads from isolated nuclei, confirming that active RNA pol II colocalizes with decondensed regions of chromatin (Wang et al., 2014). An important next step will be to adapt this technology to image transcriptionally active sites in living cells. For a more detailed discussion of these imaging technologies, particularly in cardiac biology, we refer the reader to some excellent recent reviews (Esposito et al., 2009; Kohl et al., 2013).
By combining novel imaging platforms with some of the DNA technologies discussed in the following section (6.3), the response of specific genes of interest to mechanical stimulation can be monitored. For example, epigenetic changes such as DNA methylation and histone modification can be visualized using superresolution microscopy in conjunction with novel microfluidic technologies that allow rapid detection of multiple epigenetic markers in single DNA strands (Benitez et al., 2012). The finding that lamins and emerin are powerful modulators of epigenetic modification highlights the importance of epigenetics research. For example, mesenchymal stem cells transfected with lamin A/C siRNA do not exhibit the same reduction in histone deacetylation following compression compared to control cells (Li et al., 2011), and emerin activates histone deacetylase 3 as part of a process to control myogenic gene expression (Demmerle et al., 2012). Advanced imaging technologies may also help to identify local changes in nuclear structure in response to mechanical force, particularly on the chromatin level, as discussed below.
6.3 Detecting the effect of force on DNA, RNA and proteins
To fully characterize the effect of forces on chromatin structure and transcriptional activity, additional biomolecular techniques are needed. Providing an alternative to DNA fluorescence in situ hybridization (FISH), a GFP-labeled CRISPR-dCas9 system can be used to fluorescently tag specific genetic loci of interest in living cells to monitor changes in nuclear localization under mechanical forces. Other techniques such as such as Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE)-seq or DNase-seq can identify regions in chromatin that unfold/open up/become active in response to mechanical stress. In addition, imaging nucleosome dynamics can be used to monitor chromatin accessibility, as demonstrated by a clever imaging technique involving fluorescence correlation spectroscopy and Monte-Carlo computer simulations (Hihara et al., 2012). These techniques are essential to identify if/how global force application to the nucleus can activate specific genes, rather than cause global changes in chromatin structure and transcriptional activity.
At the protein level, questions remain regarding the composition of the nuclear envelope and the structure of its constituents, such as lamins. There has also been a report of a highly dynamic perinuclear region of unknown composition and function which may modulate forces propagating to the nucleus (Shaiken and Opekun, 2014), but further investigation is necessary. Continued advances in mass spectrometry will help to explore the role of posttranslational modifications (e.g. phosphorylation, other chemical modifications, and local opening/unfolding) in nuclear mechanotransduction. The CS-MS technology developed by Discher and colleagues to identify force-induced unfolding of the lamin Ig-fold under force (Swift et al., 2013) is a prime example of how advances in technology can drive the field of mechanobiology. Such techniques, when combined with controlled force application to cells or isolated nuclei, will be pivotal in determining how observed structural changes can affect the biological activity of the affected molecules. In addition to improving our understanding of normal nuclear mechanosensing, insight from these studies may also reveal how specific mutations in nuclear envelope proteins contribute to impaired mechanotransduction and disease.
6.4 Molecular dynamics modeling/simulations
Modeling the intracellular events, both from a mechanical/structural and chemical point, will be crucial to further understand the molecular mechanisms of nuclear mechanotransduction. Importantly, models must be capable of generating predictions that can be experimentally tested to validate the model or to guide further refinement. Given the size of nucleus and many of the involved protein complexes, computational modeling will likely require multi-scale approaches (from the nanometer range to tens of micrometers) and coarse-graining, as molecular dynamics simulation are still limited to relatively few large molecules and extremely short time frames. Furthermore, for many important nuclear envelope proteins such as lamins, their full structural details and assembly mechanisms remain incompletely understood, stimulating more experiments.
6.5 Localized force application
Studying force-induced molecular events requires controlled and localized force application to cells, isolated nuclei, or individual molecules. Single molecule force spectroscopy (SMFS) techniques such as atomic force microscopy (AFM), optical and magnetic tweezers, are powerful means of applying and quantifying picoNewton-sized forces. These methods excel at probing cell surfaces and biomolecules in vitro, but are limited in their probing depth and in their in vivo capabilities. Exposing cells seeded on flexible silicone membranes to cyclic strain enables a high throughput approach to studying cell and nuclear mechanics. This has yielded new insights on nuclear mechical properties and mechanosensitive gene activation (Table 2); however, it lacks the resolution desired by biophysicists to understand the underlying mechanisms within single cells. In addition to well established techniques such as atomic force microscopy and micropipette aspiration, novel experimental approaches such as microfluidic perfusion devices, microneedle manipulation, and compression chambers have recently been developed to study the role of disease-causing mutations on nuclear mechanics. Combined with molecular probes to study intracellular forces and molecular events inside the nucleus, these approaches will help address important questions relevant to nuclear mechanosensing (Isermann et al., 2012; Verstraeten and Lammerding, 2008). Microfluidic tools have the particular advantage that they can probe hundreds of cells/nuclei per minute, as opposed to the current AFM and micropipette aspiration techniques.
Table 2.
Nuclear mechanics and the role of nuclear proteins on cell organization/function.
| Category | Insight | Reference |
|---|---|---|
| Nuclear Mechanics | The nucleus has two main mechanical components: (1) the viscoelastic chromatin, and (2) the elastic lamina. | (Caille et al., 2002; Guilak and Mow, 2000; Rowat et al., 2013; Rowat et al., 2006; Versaevel et al., 2013) |
| The nucleus can undergo both elastic and plastic deformations. | (Deguchi et al., 2005; Sato et al., 1987; Sato et al., 1996) | |
| Cells deficient in A-type lamins, but not B-type, have increased deformation in response to mechanical stimuli. | (Broers et al., 2004; Guilluy et al., 2014; Lammerding et al., 2006; Lammerding et al., 2004; Lee et al., 2007) | |
| Disease causing lamin point mutations can increase, decrease, or have no effect on nuclear mechanical properties. | (Zwerger et al., 2013); (Dahl et al., 2006; Verstraeten et al., 2008) | |
| Force Propagation | LINC complexes are necessary for mediating the mechanical connection from the focal adhesions to the nucleus. | (Maniotis et al., 1997); (Crisp et al., 2006a; Lombardi et al., 2011) |
| Cellular Organization and Function | LINC complex disruption and emerin deficiency disrupt the underlying nuclear lamina and cause defects in cell polarization and migration. | (Anno et al., 2012; Chambliss et al., 2013; Chancellor et al., 2010; Hale et al., 2008; Lombardi et al., 2011; Luxton et al., 2010; Morgan et al., 2011) |
| The perinuclear actin and vimentin structures help to deform or stabilize the nucleus, respectively, and are disrupted by nuclear envelope protein depletion. | (Versaevel et al., 2012); (Lovett et al., 2013; Tamiello et al., 2013); (Li et al., 2014; Lu et al., 2012; Nagayama et al., 2013; Tamiello et al., 2013; Tremblay et al., 2013) | |
| Nuclear lamin A/C levels correlate with substrate stiffness. | (Swift et al., 2013) | |
| The nucleus must undergo substantial deformation during migration through constructions and transit efficiency is a strongly correlated with lamin A/C levels. | (Balzer et al., 2012; Fu et al., 2012; Harada et al., 2014; Wolf et al., 2013); (Rowat et al., 2013); Harada et al. 2014) |
A brief overview of the work done over the past 15 years on nuclear/cellular structure and mechanics and the effect of specific nuclear proteins.
Forward progress will depend on the development of smaller tools such as magnetic or dielectric beads that can be conjugated with natural biological or biosynthetic tethers to allow incorporation into specific molecular niches. Laser ablation can sever individual filaments in the cytoskeleton or nucleoskeleton, and can be used to study the redistribution of force within and around structural elements such as chromatin and nuclear actin. Recent application of this technique to cytoskeletal actin induced rapid changes in nuclear morphology and even DNA reorganization, and should be repeated using beating cardiomyocytes (Li et al., 2014; Lu et al., 2012; Nagayama et al., 2013; Tamiello et al., 2013). Taking advantage of the heart's innate excitation-contraction coupling to deliver electrical pulses which are transduced into mechanical forces is another strategy to apply forces to the nucleus. Patch-clamp technologies and emerging optogenetics can be used to gain experimental control over cardiac myocytes excitation modulate and may even record voltage potentials across the nuclear membrane in isolated nuclei.
6.6 More realistic in vitro models to study (cardiac) cells under physiological conditions
Micropatterned substrates to control cell shape and engineered substrates with varying stiffness are currently being used to better understand cellular responses to their mechanical environment, including changes in nuclear shape and morphology; such approaches should be further exploited to create better in vitro models and maintain consistent (and more physiological) experimental conditions (Lovett et al., 2013; Tamiello et al., 2013; Versaevel et al., 2012). For example, culturing neonatal cardiac myocytes on micropatterned substrates helps to control sarcomere organization and alignment, which is likely to be an important factor in transmitting forces to the nucleus. Culturing cells in three-dimensional (3-D) environments to better mimic their physiological environment is another important advance, but often comes at the cost of reduced imaging quality and experimental accessibility of the cell surface. One emerging technique for achieving even more physiological environments is the use of decellularized tissues, currently being explored mostly in tissue engineering. One aspect that is particularly needed in relation to cardiac systems is the incorporation of excitation contraction coupling – whether it is measurements of spontaneous electrical impulses in correlation with nuclear events or the application of a specific potential. A better understanding of the interplay between nuclear mechanics, nuclear mechanotransduction, and electro-mechanical coupling could help explain many of the conduction defects and arrhythmias seen in various laminopathies.
6.7 Model Organisms
Mouse models of laminopathies have been instrumental in not only investigating the disease mechanism, but also in providing an important tool to study the role of the nucleus in mechanotransduction. Sullivan et al. described the first laminopathy mouse model (Lmna−/−), resulting from a large deletion of the Lmna gene (Sullivan et al., 1999). Lmna−/− mice appear normal at birth, but rapidly decline as they develop dilated cardiomyopathy, muscular dystrophy, and growth defects until death from cardiac failure occurs at about 6 weeks of age (Nikolova et al., 2004; Sullivan et al., 1999). An independent Lmna-null model (LmnaGT−/−) generated using genetrap technology displays an even more severe phenotype (Kubben et al., 2011). Curiously, heterozygous LmnaGT+/− mice do not exhibit a phenotype, whereas the Lmna+/− heterozygotes develop electrophysiological abnormalities (at four weeks) and late-onset cardiomyopathy (Kubben et al., 2011; Wolf et al., 2008). This difference may be explained by recent reports of the presence of lamin A/C fragments in the Lmna−/− mice, suggesting that the fragments may retain some residual or dominant negative function (Jahn et al., 2012). On the other hand, recent biomechanical assays have shown that these lamin A/C fragments do not exert any dominant negative effects on nuclear stability (Zwerger et al., 2013), leaving the door open for their exact role in mediating disease phenotypes.
The fact that heterozygous Lmna+/− mice develop late-onset dilated cardiomyopathy has made these animals an attractive model to study the effect of increased mechanical stress on cardiac function. The Fatkin group sought to exacerbate the typically mild phenotype by subjecting Lmna+/− mice to chronic (treadmill) exercise or short-duration transverse aortic constriction (TAC), which results in left ventricular pressure overload. Based on their findings, they concluded that stress-induced apoptosis is not the driver of dilated cardiomyopathy (Chandar et al., 2010) however another study showed that when electrical stimulus was coupled with mechanical contraction apoptosis increased in induced patient specific pluripotent stem cells (Siu et al., 2012). Using a similar TAC model, but following mice over a longer period of time, Cupesi et al. found that Lmna+/− mice have a decreased response to left-ventricular pressure overload, reflected in impaired activation of the mechanosensitive gene Egr-1 and reduced cardiac hypertrophy and fibrosis (Cupesi et al., 2010). Subsequent studies of Lmna+/− mice suggested that impaired MKL1/SRF signaling contributes to the reduced hypertrophy in the heterozygous mice (Ho et al., 2013b).
Further follow-up studies, potentially using double knock-out and knock-in mice, may help to determine the primary mechanotransduction defect(s). For example, studies in nesprin1/2 cardiac-specific double knockout mice confirmed the importance of at least one functional LINC complex due to some redundancy in cardiac function and mechanotransduction signaling (Banerjee et al., 2014). Similarly, the generation of Lmna/Lap2α-double knockout mice helped to identify Lap2/pRb and Smad2/3 signaling pathways as mediators of defective muscle growth in Lmna−/− mice (Cohen et al., 2013). Crossing Sirt1-deficient mice with laminopathy models may draw connections between gene silencing and the nuclear envelope, as well as cell mechanics and metabolism, since Sirt1 is a histone deacetylase shown to be activated by lamin A. Interestingly, resveratrol treatment enhances Sirt1-lamin-A binding, rescues adult stem cell decline, and increases life span in mouse models of progeria (Liu et al., 2012). Sirt1-deficient mice develop cardiomyopathy linked to disturbed modulation of Mef2 transcription factors, which control mitochondrial-related genes. These mice have disturbed mitochondrial function and dilated hearts, potentially bridging the gap between two classes of cardiomyopathy: mitochondrial myopathies and dilated cardiomyopathies (Planavila et al., 2012).
Knock-in models offer an important experimental platform for studying diseases caused by lamin mutations, since the majority of human laminopathies are caused by autosomal dominant mutations, in which mutant lamins are expressed at similar levels as wild-type lamins (Zhang et al., 2013). These models reflect the tissue specific distribution with the LmnaN195K/N195K model exclusively affecting cardiac muscle, while the LmnaH222P/H222P model exhibits both cardiomyopathy and muscular dystrophy (Arimura et al., 2005; Mounkes et al., 2005). However, it is noteworthy, that these studies have not closed the gap in our understanding of how the nucleus is important in disease progression or mechanism. For a detailed review of the various laminopathy mouse models, including knock-in and knock-out models relevant to striated muscle and progeroid laminopathies, we refer the reader to one of the excellent recent reviews (Azibani et al., 2014; Zhang et al., 2013).
Importantly, mouse models not only motivate potential therapies by revealing molecular mechanisms underlying disease pathology, but they also provide the means of testing these therapies in preclinical trials. Muchir, Worman and colleagues have already provided strong evidence that pharmacological inhibition of the mechanosensitive MAPK pathway, which is hyperactive in a variety of laminopathies, can dramatically improve the cardiac phenotypes in a variety of lamin and emerin mutant mouse models, while studies with drugs targeting lamin processing (e.g., farnesyltransferase inhibitors) show promise in treating progeroid mouse models (King and Heyer, 2013; Muchir et al., 2012a). Moving forward, novel animal models will provide invaluable tools for understanding nuclear mechanotransduction and its effect on the tissue and organismal level.
Beyond the mouse models discussed above, invertebrate models such as Drosophila and C. elegans provide convenient and cost-effective ways to investigate a large number of different mutations using powerful-genetic techniques (Lyakhovetsky and Gruenbaum, 2014). The recent arrival of the CRISPR-Cas9 platform is expected to further accelerate the creation of complex disease models in a variety of organisms, allowing quicker and more efficient evaluation of the role of various nuclear envelope proteins in mechanotransduction (Shen et al., 2014).
7. Outlook & Conclusions
We can confidently proclaim that the nucleus plays an important role in cellular mechanotransduction and function, as the many diseases resulting from mutations in nuclear envelope proteins attest. Increasing evidence suggests that the nucleus itself can respond to mechanical forces and deformations, resulting in altered transcriptional regulation and cellular function. However, many of the mechanisms remain unclear and underexplored, including the precise role of nuclear envelope proteins such as lamin and emerin. Do these proteins primarily process biochemical signals, transmit mechanical forces, or integrate various mechanical and biochemical inputs? Recent studies indicate that mammalian cells have over 100 different nuclear envelope proteins, with a large fraction of them expressed only in specific cell types or certain conditions, for example, during myoblast differentiation. These tissue-/development-specific expression patterns are likely to play important roles in normal cellular function and the many diseases caused by mutations in nuclear envelope proteins. Nonetheless, the vast majority of the nuclear envelope proteins are understudied, their function often completely unknown. A detailed characterization of these proteins is needed, ranging from their identification, expression and localization in various cells/tissues, interaction partners, and function. Similarly, we are still lacking a fundamental knowledge of the dynamic organization of the proteins that make up the ‘nucleoskeleton’ or nuclear scaffold, including nucleoplasmic lamins, nuclear actin, and possibly spectrin, myosin and even titin. It is highly likely that the structure and function of these proteins has important effects on the three-dimensional (3-D) chromatin organization and transcriptional activity, particularly in response to mechanical stress. Shining light onto this nuclear ‘dark matter’ has tremendous potential and must go beyond a purely ‘DNA-centric’ view.
One powerful approach to further investigate processes involved in nuclear mechanosensing will be to combine techniques used for the identification of (dynamic) 3-D nuclear architecture and gene positioning with assays that enable imaging chromatin configuration and transcriptional processes in living cells, particularly during mechanical perturbation, so that the coordinated events that govern transcriptional regulation and activity in response to mechanical stress can be assessed. The use of new technologies, novel applications of old technologies, and a combination of tools spanning different length scales are beginning to “close the gap,” linking global forces experienced by the cell to precise biophysical ripples in the genomic landscape and transcriptional events, bringing us ever closer to fully understanding nuclear mechanotransduction.
Acknowledgements
The authors apologize to all authors whose work could not be cited due to space constraints. This work was supported by National Institutes of Health award R01 HL082792]; a Department of Defense Breast Cancer Idea Award [BC102152]; a National Science Foundation CAREER award to Lammerding J [CBET-1254846]; and a Pilot Project Award by the Cornell Center on the Microenvironment & Metastasis through Award Number U54CA143876 from the National Cancer Institute, as well as NSF graduate research fellowships to GF [2014163403] and AK [2013160437].The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhtar A, Gasser SM. The nuclear envelope and transcriptional control. Nature Reviews Genetics. 2007;8:507–517. doi: 10.1038/nrg2122. [DOI] [PubMed] [Google Scholar]
- Andres V, Gonzalez JM. Role of A-type lamins in signaling, transcription, and chromatin organization. Journal of Cell Biology. 2009;187:945–957. doi: 10.1083/jcb.200904124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anno T, Sakamoto N, Sato M. Role of nesprin-1 in nuclear deformation in endothelial cells under static and uniaxial stretching conditions. Biochemical and Biophysical Research Communications. 2012;424:94–99. doi: 10.1016/j.bbrc.2012.06.073. [DOI] [PubMed] [Google Scholar]
- Arimura T, Helbling-Leclerc A, Varnous S, Niel F, Lacene E, Fromes Y, Toussaint M, Mura AM, Keller DI, Amthor H, Isnard R, Malissen M, Schwartz K, Bonne G. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Human Molecular Genetics. 2005;14:155–169. doi: 10.1093/hmg/ddi017. [DOI] [PubMed] [Google Scholar]
- Attisano L, Wrana JL. Smads as transcriptional co-modulators. Current Opinion in Cell Biology. 2000;12:235–243. doi: 10.1016/s0955-0674(99)00081-2. [DOI] [PubMed] [Google Scholar]
- Azibani F, Muchir A, Vignier N, Bonne G, Bertrand AT. Book Striated muscle laminopathies. Elsevier; City: 2014. Striated muscle laminopathies. [DOI] [PubMed] [Google Scholar]
- Baarlink C, Wang H, Grosse R. Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science. 2013;340:864–7. doi: 10.1126/science.1235038. [DOI] [PubMed] [Google Scholar]
- Ball JR, Dimaano C, Bilak A, Kurchan E, Zundel MT, Ullman KS. Sequence preference in RNA recognition by the nucleoporin Nup153. Journal of biological chemistry. 2007;282:8734–8740. doi: 10.1074/jbc.M608477200. [DOI] [PubMed] [Google Scholar]
- Balzer EM, Tong Z, Paul CD, Hung WC, Stroka KM, Boggs AE, Martin SS, Konstantopoulos K. Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J. 2012;26:4045–56. doi: 10.1096/fj.12-211441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Rathee V, Krishnaswamy R, Bhattacharjee P, Ray P, Sood AK, Sengupta K. Viscoelastic Behavior of Human Lamin A Proteins in the Context of Dilated Cardiomyopathy. Plos One. 2013;8:13. doi: 10.1371/journal.pone.0083410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee I, Zhang J, Moore-Morris T, Pfeiffer E, Buchholz KS, Liu A, Ouyang K, Stroud MJ, Gerace L, Evans SM, McCulloch A, Chen J. Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet. 2014;10:e1004114. doi: 10.1371/journal.pgen.1004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar DZ, Davidovich M, Lamm AT, Zer H, Wilson KL, Gruenbaum Y. BAF-1 mobility is regulated by environmental stresses. Molecular biology of the cell. 2014;25:1127–1136. doi: 10.1091/mbc.E13-08-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin BJ, Cimini BA, Blackburn EH, Mullins RD. Visualization of actin filaments and monomers in somatic cell nuclei. Molecular Biology of the Cell. 2013;24:982–994. doi: 10.1091/mbc.E12-09-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson L, Otto H. LUMA interacts with emerin and influences its distribution at the inner nuclear membrane. Journal of Cell Science. 2008;121:536–548. doi: 10.1242/jcs.019281. [DOI] [PubMed] [Google Scholar]
- Benitez JJ, Topolancik J, Tian HC, Wallin CB, Latulippe DR, Szeto K, Murphy PJ, Cipriany BR, Levy SL, Soloway PD, Craighead HG. Microfluidic extraction, stretching and analysis of human chromosomal DNA from single cells. Lab on a Chip. 2012;12:4848–4854. doi: 10.1039/c2lc40955k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. IDENTIFICATION OF A NOVEL X-LINKED GENE RESPONSIBLE FOR EMERY-DREIFUSS MUSCULAR-DYSTROPHY. Nature Genetics. 1994;8:323–327. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- Blessing CA, Ugrinova GT, Goodson HV. Actin and ARPs: action in the nucleus. Trends in cell biology. 2004;14:435–442. doi: 10.1016/j.tcb.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nature Genetics. 1999;21:285–288. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- Borrego-Pinto J, Jegou T, Osorio DS, Aurade F, Gorjanaacz M, Koch B, Mattaj IW, Gomes ER. Samp1 is a component of TAN lines and is required for nuclear movement. Journal of Cell Science. 2012;125:1099–1105. doi: 10.1242/jcs.087049. [DOI] [PubMed] [Google Scholar]
- Broers JLV, Peeters EAG, Kuijpers HJH, Endert J, Bouten CVC, Oomens CWJ, Baaijens FPT, Ramaekers FCS. Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Human Molecular Genetics. 2004;13:2567–2580. doi: 10.1093/hmg/ddh295. [DOI] [PubMed] [Google Scholar]
- Caille N, Thoumine O, Tardy Y, Meister JJ. Contribution of the nucleus to the mechanical properties of endothelial cells. Journal of Biomechanics. 2002;35:177–187. doi: 10.1016/s0021-9290(01)00201-9. [DOI] [PubMed] [Google Scholar]
- Campas O, Mammoto T, Hasso S, Sperling RA, O'Connell D, Bischof AG, Maas R, Weitz DA, Mahadevan L, Ingber DE. Quantifying cell-generated mechanical forces within living embryonic tissues (vol 11, pg 183, 2014) Nature Methods. 2014;11:349–349. doi: 10.1038/nmeth.2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambliss AB, Khatau SB, Erdenberger N, Robinson DK, Hodzic D, Longmore GD, Wirtz D. The LINC-anchored actin cap connects the extracellular milieu to the nucleus for ultrafast mechanotransduction. Scientific Reports. 2013;3:9. doi: 10.1038/srep01087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chancellor T, Lee J, Thodeti CK, Lele T. Actomyosin tension exerted on the nucleus through nesprin-1 connections influences endothelial cell adhesion, migration, and cyclic strain-induced reorientation. Biophysical journal. 2010;99:115–123. doi: 10.1016/j.bpj.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandar S, Yeo LS, Leimena C, Tan JC, Xiao XH, Nikolova-Krstevski V, Yasuoka Y, Gardiner-Garden M, Wu JX, Kesteven S, Karlsdotter L, Natarajan S, Carlton A, Rainer S, Feneley MP, Fatkin D. Effects of Mechanical Stress and Carvedilol in Lamin A/C-Deficient Dilated Cardiomyopathy. Circulation Research. 2010;106:573–U82. doi: 10.1161/CIRCRESAHA.109.204388. [DOI] [PubMed] [Google Scholar]
- Charbonney E, Speight P, Masszi A, Nakano H, Kapus A. β-Catenin and Smad3 regulate the activity and stability of myocardin-related transcription factor during epithelial–myofibroblast transition. Molecular biology of the cell. 2011;22:4472–4485. doi: 10.1091/mbc.E11-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Fulcoli FG, Ferrentino R, Martucciello S, Illingworth EA, Baldini A. Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS genetics. 2012;8:e1002571. doi: 10.1371/journal.pgen.1002571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y-H, Haller K, Peloponese J-M, Jeang K-T. Histone acetyltransferase hALP and nuclear membrane protein hsSUN1 function in de-condensation of mitotic chromosomes. Journal of Biological Chemistry. 2007;282:27447–27458. doi: 10.1074/jbc.M703098200. [DOI] [PubMed] [Google Scholar]
- Chow K, Dean DC. Domains A and B in the Rb pocket interact to form a transcriptional repressor motif. Molecular and cellular biology. 1996;16:4862–4868. doi: 10.1128/mcb.16.9.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TV, Gnocchi VF, Cohen JE, Phadke A, Liu H, Ellis JA, Foisner R, Stewart CL, Zammit PS, Partridge TA. Defective skeletal muscle growth in lamin A/C-deficient mice is rescued by loss of Lap2α. Human molecular genetics. 2013 doi: 10.1093/hmg/ddt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. The Journal of Cell Biology. 2006a;172:41–53. doi: 10.1083/jcb.200509124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. 2006b;172:41–53. doi: 10.1083/jcb.200509124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupesi M, Yoshioka J, Gannon J, Kudinova A, Stewart CL, Lammerding J. Attenuated hypertrophic response to pressure overload in a lamin A/C haploinsufficiency mouse. J Mol Cell Cardiol. 2010;48:1290–7. doi: 10.1016/j.yjmcc.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10271–10276. doi: 10.1073/pnas.0601058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75:519–60. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de las Heras JI, Batrakou DG, Schirmer EC. Cancer biology and the nuclear envelope: A convoluted relationship. Seminars in Cancer Biology. 2013;23:125–137. doi: 10.1016/j.semcancer.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Deguchi S, Maeda K, Ohashi T, Sato M. Flow-induced hardening of endothelial nucleus as an intracellular stress-bearing organelle. Journal of Biomechanics. 2005;38:1751–1759. doi: 10.1016/j.jbiomech.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Demmerle J, Koch AJ, Holaska JM. The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. Journal of Biological Chemistry. 2012;287:22080–22088. doi: 10.1074/jbc.M111.325308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreuillet C, Harper M, Tillit J, Kress M, Ernoult-Lange M. Mislocalization of human transcription factor MOK2 in the presence of pathogenic mutations of lamin A/C. Biology of the Cell. 2008;100:51–61. doi: 10.1042/BC20070053. [DOI] [PubMed] [Google Scholar]
- Emerson LJ, Holt MR, Wheeler MA, Wehnert M, Parsons M, Ellis JA. Defects in cell spreading and ERK1/2 activation in fibroblasts with lamin A/C mutations. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2009;1792:810–821. doi: 10.1016/j.bbadis.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Esposito A, Schlachter S, Schierle GSK, Elder AD, Diaspro A, Wouters FS, Kaminski CF, Iliev AI. Quantitative Fluorescence Microscopy Techniques. In: Gavin RH, editor. Cytoskeleton Methods and Protocols. Humana Press Inc; Totowa: 2009. pp. 117–142. [DOI] [PubMed] [Google Scholar]
- Fagotto F, Gluck U, Gumbiner BM. Nuclear localization signal-independent and importin/karyopherin-independent nuclear import of beta-catenin. Current Biology. 1998;8:181–190. doi: 10.1016/s0960-9822(98)70082-x. [DOI] [PubMed] [Google Scholar]
- Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Spudich S, De Girolami U, Seidman JG, Seidman CE, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. New England Journal of Medicine. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- Finan JD, Leddy HA, Guilak F. Osmotic stress alters chromatin condensation and nucleocytoplasmic transport. Biochemical and Biophysical Research Communications. 2011;408:230–235. doi: 10.1016/j.bbrc.2011.03.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Chin LK, Bourouina T, Liu AQ, VanDongen AMJ. Nuclear deformation during breast cancer cell transmigration (vol 12, pg 3774, 2012) Lab on a Chip. 2012;12:5283–5283. doi: 10.1039/c2lc40477j. [DOI] [PubMed] [Google Scholar]
- Gomez-Martinez R, Hernandez-Pinto AM, Duch M, Vazquez P, Zinoviev K, de la Rosa EJ, Esteve J, Suarez T, Plaza JA. Silicon chips detect intracellular pressure changes in living cells. Nature Nanotechnology. 2013;8:517–521. doi: 10.1038/nnano.2013.118. [DOI] [PubMed] [Google Scholar]
- Gonzalez JM, Navarro-Puche A, Casar B, Crespo P, Andres V. Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. Journal of Cell Biology. 2008;183:653–666. doi: 10.1083/jcb.200805049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grashoff C, Hoffman BD, Brenner MD, Zhou RB, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T, Schwartz MA. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466:263–U143. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffis ER, Craige B, Dimaano C, Ullman KS, Powers MA. Distinct functional domains within nucleoporins Nup153 and Nup98 mediate transcription-dependent mobility. Molecular biology of the cell. 2004;15:1991–2002. doi: 10.1091/mbc.E03-10-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilak F. Compression-induced changes in the shape and volume of the chondrocyte nucleus. Journal of Biomechanics. 1995;28:1529–&. doi: 10.1016/0021-9290(95)00100-x. [DOI] [PubMed] [Google Scholar]
- Guilak F, Mow VC. The mechanical environment of the chondrocyte: a biphasic finite element model of cell-matrix interactions in articular cartilage. Journal of Biomechanics. 2000;33:1663–1673. [PubMed] [Google Scholar]
- Guilluy C, Osborne LD, Van Landeghem L, Sharek L, Superfine R, Garcia-Mata R, Burridge K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nature cell biology. 2014;16:376–381. doi: 10.1038/ncb2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen GG, Worman HJ. Nuclear Positioning. Cell. 2013;152:1376–1389. doi: 10.1016/j.cell.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CM, Shrestha AL, Khatau SB, Stewart-Hutchinson PJ, Hernandez L, Stewart CL, Hodzic D, Wirtz D. Dysfunctional Connections Between the Nucleus and the Actin and Microtubule Networks in Laminopathic Models. Biophysical Journal. 2008;95:5462–5475. doi: 10.1529/biophysj.108.139428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada T, Swift J, Irianto J, Shin J-W, Spinler KR, Athirasala A, Diegmiller R, Dingal PDP, Ivanovska IL, Discher DE. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. The Journal of cell biology. 2014;204:669–682. doi: 10.1083/jcb.201308029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi T, Holaska JM, Yamane M, Koujin T, Hashiguchi N, Mori C, Wilson KL, Hiraoka Y. Emerin binding to Btf, a death-promoting transcriptional repressor, is disrupted by a missense mutation that causes Emery–Dreifuss muscular dystrophy. European Journal of Biochemistry. 2004;271:1035–1045. doi: 10.1111/j.1432-1033.2004.04007.x. [DOI] [PubMed] [Google Scholar]
- Haraguchi T, Koujin T, Segura-Totten M, Lee KK, Matsuoka Y, Yoneda Y, Wilson KL, Hiraoka Y. BAF is required for emerin assembly into the reforming nuclear envelope. Journal of Cell Science. 2001;114:4575–4585. doi: 10.1242/jcs.114.24.4575. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes & development. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- Harvey PA, Leinwand LA. Cellular mechanisms of cardiomyopathy. Journal of Cell Biology. 2011;194:355–365. doi: 10.1083/jcb.201101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L, Roux KJ, Wong ESM, Mounkes LC, Mutalif R, Navasankari R, Rai B, Cool S, Jeong J-W, Wang H, Lee H-S, Kozlov S, Grunert M, Keeble T, Jones CM, Meta MD, Young SG, Daar IO, Burke B, Perantoni AO, Stewart CL. Functional Coupling between the Extracellular Matrix and Nuclear Lamina by Wnt Signaling in Progeria. Developmental Cell. 2010;19:413–425. doi: 10.1016/j.devcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hihara S, Pack CG, Kaizu K, Tani T, Hanafusa T, Nozaki T, Takemoto S, Yoshimi T, Yokota H, Imamoto N, Sako Y, Kinjo M, Takahashi K, Nagai T, Maeshima K. Local Nucleosome Dynamics Facilitate Chromatin Accessibility in Living Mammalian Cells. Cell Reports. 2012;2:1645–1656. doi: 10.1016/j.celrep.2012.11.008. [DOI] [PubMed] [Google Scholar]
- Ho CY, Jaalouk DE, Lammerding J. Novel insights into the disease etiology of laminopathies. Rare Diseases. 2013a;1:e27002. doi: 10.4161/rdis.27002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature. 2013b;497:507–11. doi: 10.1038/nature12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CY, Lammerding J. Lamins at a glance. Journal of Cell Science. 2012;125:2087–2093. doi: 10.1242/jcs.087288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, Jensen CC, Chaturvedi A, Yoshigi M, Beckerle MC. Stretch-induced actin remodeling requires targeting of zyxin to stress fibers and recruitment of actin regulators. Molecular biology of the cell. 2012;23:1846–1859. doi: 10.1091/mbc.E11-12-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holaska JM, Lee KK, Kowalski AK, Wilson KL. Transcriptional repressor germ cell-less (GCL) and barrier to autointegration factor (BAF) compete for binding to emerin in vitro. Journal of Biological Chemistry. 2003;278:6969–6975. doi: 10.1074/jbc.M208811200. [DOI] [PubMed] [Google Scholar]
- Holaska JM, Rais-Bahrami S, Wilson KL. Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Human molecular genetics. 2006;15:3459–3472. doi: 10.1093/hmg/ddl423. [DOI] [PubMed] [Google Scholar]
- Hu Q, Guo C, Li Y, Aronow BJ, Zhang J. LMO7 mediates cell-specific activation of the Rho-myocardin-related transcription factor-serum response factor pathway and plays an important role in breast cancer cell migration. Molecular and cellular biology. 2011;31:3223–3240. doi: 10.1128/MCB.01365-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner S, Eam J, Hübner A, Jans D. Laminopathy-inducing lamin A mutants can induce redistribution of lamin binding proteins into nuclear aggregates. Experimental cell research. 2006;312:171–183. doi: 10.1016/j.yexcr.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Inoue A, Hyle J, Lechner MS, Lahti JM. Perturbation of HP1 localization and chromatin binding ability causes defects in sister-chromatid cohesion. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2008;657:48–55. doi: 10.1016/j.mrgentox.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Isermann P, Lammerding J. Nuclear Mechanics and Mechanotransduction in Health and Disease. Current Biology. 2013;23:R1113–R1121. doi: 10.1016/j.cub.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer KV, Pulford S, Mogilner A, Shivashankar GV. Mechanical activation of cells induces chromatin remodeling preceding MKL nuclear transport. Biophys J. 2012;103:1416–28. doi: 10.1016/j.bpj.2012.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10:63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalverda B, Pickersgill H, Shloma VV, Fornerod M. Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell. 2010;140:360–371. doi: 10.1016/j.cell.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG, Seidman CE. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. New England Journal of Medicine. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- King AA, Heyer GL. Moving from gene discovery to clinical trials in Hutchinson-Gilford progeria syndrome. Neurology. 2013;81:408–409. doi: 10.1212/WNL.0b013e31829d87cd. [DOI] [PubMed] [Google Scholar]
- Knight M, Van de Breevaart Bravenboer J, Lee D, van Osch G, Weinans H, Bader D. Cell and nucleus deformation in compressed chondrocyte–alginate constructs: temporal changes and calculation of cell modulus. Biochimica et Biophysica Acta (BBA)-General Subjects. 2002;1570:1–8. doi: 10.1016/s0304-4165(02)00144-7. [DOI] [PubMed] [Google Scholar]
- Kochin V, Shimi T, Torvaldson E, Adam SA, Goldman A, Pack C-G, Melo-Cardenas J, Imanishi SY, Goldman RD, Eriksson JE. Interphase phosphorylation of lamin A. Journal of Cell Science. jcs. 2014:141820. doi: 10.1242/jcs.141820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl T, Westphal V, Hell SW, Lehnart SE. Superresolution microscopy in heart - Cardiac nanoscopy. Journal of Molecular and Cellular Cardiology. 2013;58:13–21. doi: 10.1016/j.yjmcc.2012.11.016. [DOI] [PubMed] [Google Scholar]
- Korfali N, Wilkie GS, Swanson SK, Srsen V, de las Heras J, Batrakou DG, Malik P, Zuleger N, Kerr ARW, Florens L, Schirmer EC. The nuclear envelope proteome differs notably between tissues. Nucleus-Austin. 2012;3:552–564. doi: 10.4161/nucl.22257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Voncken JW, Konings G, van Weeghel M, van den Hoogenhof MMG, Gijbels M, van Erk A, Schoonderwoerd K, van den Bosch B, Dahlmans V, Calis C, Houten SM, Misteli T, Pinto YM. Post-natal myogenic and adipogenic developmental Defects and metabolic impairment upon loss of A-type lamins. Nucleus-Austin. 2011;2:195–207. doi: 10.4161/nucl.2.3.15731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT. Lamins A and C but not lamin B1 regulate nuclear mechanics. Journal of Biological Chemistry. 2006;281:25768–25780. doi: 10.1074/jbc.M513511200. [DOI] [PubMed] [Google Scholar]
- Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. The Journal of Cell Biology. 2005a;170:781–791. doi: 10.1083/jcb.200502148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol. 2005b;170:781–91. doi: 10.1083/jcb.200502148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. Journal of Clinical Investigation. 2004;113:370–378. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecroisey C, Brouilly N, Qadota H, Mariol M-C, Rochette NC, Martin E, Benian GM, Ségalat L, Mounier N, Gieseler K. ZYX-1, the unique zyxin protein of Caenorhabditis elegans, is involved in dystrophin-dependent muscle degeneration. Molecular biology of the cell. 2013;24:1232–1249. doi: 10.1091/mbc.E12-09-0679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Tu Y, Tatar A, Wu D, Nobumori C, Jung H-J, Yoshinaga Y, Coffinier C, de Jong PJ, Fong LG. Reciprocal knock-in mice to investigate the functional redundancy of lamin B1 and lamin B2, Molecular biology of the cell. mbc. 2014:E14–01-0683. doi: 10.1091/mbc.E14-01-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JSH, Hale CM, Panorchan P, Khatau SB, George JP, Tseng Y, Stewart CL, Hodzic D, Wirtz D. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophysical Journal. 2007;93:2542–2552. doi: 10.1529/biophysj.106.102426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legant WR, Miller JS, Blakely BL, Cohen DM, Genin GM, Chen CS. Measurement of mechanical tractions exerted by cells in three-dimensional matrices. Nature Methods. 2010;7:969–971. doi: 10.1038/nmeth.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QS, Kumar A, Makhija E, Shivashankar GV. The regulation of dynamic mechanical coupling between actin cytoskeleton and nucleus by matrix geometry. Biomaterials. 2014;35:961–969. doi: 10.1016/j.biomaterials.2013.10.037. [DOI] [PubMed] [Google Scholar]
- Li Y, Chu JS, Kurpinski K, Li X, Bautista DM, Yang L, Sung KLP, Li S. Biophysical Regulation of Histone Acetylation in Mesenchymal Stem Cells. Biophysical Journal. 2011;100:1902–1909. doi: 10.1016/j.bpj.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Blake DL, Callebaut I, Skerjanc IS, Holmer L, McBurney MW, Paulin-Levasseur M, Worman HJ. MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and emerin. Journal of Biological Chemistry. 2000;275:4840–4847. doi: 10.1074/jbc.275.7.4840. [DOI] [PubMed] [Google Scholar]
- Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z, Jin G, Zheng B, Kennedy BK, Suh Y. Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell metabolism. 2012;16:738–750. doi: 10.1016/j.cmet.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Human Molecular Genetics. 2002;11:769–777. doi: 10.1093/hmg/11.7.769. [DOI] [PubMed] [Google Scholar]
- Lombardi ML, Jaalouk DE, Shanahan CM, Burke B, Roux KJ, Lammerding J. The Interaction between Nesprins and Sun Proteins at the Nuclear Envelope Is Critical for Force Transmission between the Nucleus and Cytoskeleton. Journal of Biological Chemistry. 2011;286:26743–26753. doi: 10.1074/jbc.M111.233700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi ML, Lammerding J. Keeping the LINC: the importance of nucleocytoskeletal coupling in intracellular force transmission and cellular function. Biochemical Society Transactions. 2011;39:1729–1734. doi: 10.1042/BST20110686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett DB, Shekhar N, Nickerson JA, Roux KJ, Lele TP. Modulation of Nuclear Shape by Substrate Rigidity. Cell Mol Bioeng. 2013;6:230–238. doi: 10.1007/s12195-013-0270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Schneider M, Neumann S, Jaeger V-M, Taranum S, Munck M, Cartwright S, Richardson C, Carthew J, Noh K. Nesprin interchain associations control nuclear size. Cellular and Molecular Life Sciences. 2012;69:3493–3509. doi: 10.1007/s00018-012-1034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxton GWG, Gomes ER, Folker ES, Vintinner E, Gundersen GG. Linear Arrays of Nuclear Envelope Proteins Harness Retrograde Actin Flow for Nuclear Movement. Science. 2010;329:956–959. doi: 10.1126/science.1189072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyakhovetsky R, Gruenbaum Y. Studying lamins in invertebrate models. Advances in experimental medicine and biology. 2014;773:245. doi: 10.1007/978-1-4899-8032-8_11. [DOI] [PubMed] [Google Scholar]
- Malhas AN, Lee CF, Vaux DJ. Lamin B1 controls oxidative stress responses via Oct-1. The Journal of cell biology. 2009;184:45–55. doi: 10.1083/jcb.200804155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniotis AJ, Chen CS, Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proceedings of the National Academy of Sciences. 1997;94:849–854. doi: 10.1073/pnas.94.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansharamani M, Wilson KL. Direct binding of nuclear membrane protein MAN1 to emerin in vitro and two modes of binding to barrier-to-autointegration factor. Journal of Biological Chemistry. 2005;280:13863–13870. doi: 10.1074/jbc.M413020200. [DOI] [PubMed] [Google Scholar]
- Margalit A, Brachner A, Gotzmann J, Foisner R, Gruenbaum Y. Barrier-to-autointegration factor - a BAFfling little protein. Trends in Cell Biology. 2007;17:202–208. doi: 10.1016/j.tcb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Margalit A, Segura-Totten M, Gruenbaum Y, Wilson KL. Barrier-to-autointegration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3290–3295. doi: 10.1073/pnas.0408364102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markiewicz E, Tilgner K, Barker N, van de Wetering M, Clevers H, Dorobek M, Hausmanowa-Petrusewicz I, Ramaekers FCS, Broers JLV, Blankesteijn WM, Salpingidou G, Wilson RG, Ellis JA, Hutchison CJ. The inner nuclear membrane protein Emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. Embo Journal. 2006;25:3275–3285. doi: 10.1038/sj.emboj.7601230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meens MJ, Pfenniger A, Kwak BR, Delmar M. Regulation of cardiovascular connexins by mechanical forces and junctions. Cardiovascular Research. 2013;99:304–314. doi: 10.1093/cvr/cvt095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Sachs F. Visualizing dynamic cytoplasmic forces with a compliance-matched FRET sensor. Journal of cell science. 2011;124:261–269. doi: 10.1242/jcs.071928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva O, Bourdeau V, Vernier M, Dabauvalle MC, Ferbeyre G. Retinoblastoma-independent regulation of cell proliferation and senescence by the p53-p21 axis in lamin A/C-depleted cells. Aging Cell. 2011;10:789–797. doi: 10.1111/j.1474-9726.2011.00719.x. [DOI] [PubMed] [Google Scholar]
- Moon H-S, Even-Ram S, Kleinman HK, Cha H-J. Zyxin is upregulated in the nucleus by thymosin β4 in SiHa cells. Experimental cell research. 2006;312:3425–3431. doi: 10.1016/j.yexcr.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Morgan JT, Pfeiffer ER, Thirkill TL, Kumar P, Peng G, Fridolfsson HN, Douglas GC, Starr DA, Barakat AI. Nesprin-3 regulates endothelial cell morphology, perinuclear cytoskeletal architecture, and flow-induced polarization. Molecular biology of the cell. 2011;22:4324–4334. doi: 10.1091/mbc.E11-04-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Human Molecular Genetics. 2005;14:2167–2180. doi: 10.1093/hmg/ddi221. [DOI] [PubMed] [Google Scholar]
- Muchir A, Pavlidis P, Bonne G, Hayashi YK, Worman HJ. Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery-Dreifuss muscular dystrophy. Human Molecular Genetics. 2007a;16:1884–1895. doi: 10.1093/hmg/ddm137. [DOI] [PubMed] [Google Scholar]
- Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. Journal of Clinical Investigation. 2007b;117:1282–1293. doi: 10.1172/JCI29042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchir A, Reilly SA, Wu W, Iwata S, Homma S, Bonne G, Worman HJ. Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovascular Research. 2012a;93:311–319. doi: 10.1093/cvr/cvr301. [DOI] [PubMed] [Google Scholar]
- Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Human Molecular Genetics. 2009;18:241–247. doi: 10.1093/hmg/ddn343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchir A, Wu W, Choi JC, Iwata S, Morrow J, Homma S, Worman HJ. Abnormal p38 mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Human Molecular Genetics. 2012b;21:4325–4333. doi: 10.1093/hmg/dds265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na S, Collin O, Chowdhury F, Tay B, Ouyang M, Wang Y, Wang N. Rapid signal transduction in living cells is a unique feature of mechanotransduction. Proceedings of the National Academy of Sciences. 2008;105:6626–6631. doi: 10.1073/pnas.0711704105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama K, Yahiro Y, Matsumoto T. Apical and Basal Stress Fibers have Different Roles in Mechanical Regulation of the Nucleus in Smooth Muscle Cells Cultured on a Substrate. Cellular and Molecular Bioengineering. 2013;6:473–481. [Google Scholar]
- Narula N, Favalli V, Tarantino P, Grasso M, Pilotto A, Bellazzi R, Serio A, Gambarin FI, Charron P, Meder B, Pinto Y, Elliott PM, Mogensen J, Bolognesi M, Bollati M, Arbustini E. Quantitative Expression of the Mutated Lamin A/C Gene in Patients With Cardiolaminopathy. Journal of the American College of Cardiology. 2012;60:1916–1920. doi: 10.1016/j.jacc.2012.05.059. [DOI] [PubMed] [Google Scholar]
- Neumann S, Schneider M, Daugherty RL, Gottardi CJ, Eming SA, Beijer A, Noegel AA, Karakesisoglou I. Nesprin-2 interacts with α-catenin and regulates Wnt signaling at the nuclear envelope. Journal of Biological Chemistry. 2010;285:34932–34938. doi: 10.1074/jbc.M110.119651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL, Martin D, Feneley MP, Fatkin D. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. Journal of Clinical Investigation. 2004;113:357–369. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nili E, Cojocaru GS, Kalma Y, Ginsberg D, Copeland NG, Gilbert DJ, Jenkins NA, Berger R, Shaklai S, Amariglio N. Nuclear membrane protein LAP2β mediates transcriptional repression alone and together with its binding partner GCL (germ-cell-less) Journal of cell science. 2001;114:3297–3307. doi: 10.1242/jcs.114.18.3297. [DOI] [PubMed] [Google Scholar]
- Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nature Reviews Molecular Cell Biology. 2010;11:353–365. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott EB, van den Akker N, Sakalis PA, Gittenberger-de Groot AC, Te Velthuis AJ, Bagowski CP. The lim domain only protein 7 is important in zebrafish heart development. Developmental Dynamics. 2008;237:3940–3952. doi: 10.1002/dvdy.21807. [DOI] [PubMed] [Google Scholar]
- Pan D, Estevez-Salmeron LD, Stroschein SL, Zhu XL, He J, Zhou SL, Luo KX. The integral inner nuclear membrane protein MAN1 physically interacts with the R-Smad proteins to repress signaling by the transforming growth factor-beta superfamily of cytokines. Journal of Biological Chemistry. 2005;280:15992–16001. doi: 10.1074/jbc.M411234200. [DOI] [PubMed] [Google Scholar]
- Parlakian A, Tuil D, Hamard G, Tavernier G, Hentzen D, Concordet JP, Paulin D, Li ZL, Daegelen D. Targeted inactivation of serum response factor in the developing heart results in myocardial defects and embryonic lethality. Molecular and Cellular Biology. 2004;24:5281–5289. doi: 10.1128/MCB.24.12.5281-5289.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peti W, Page R. Molecular basis of MAP kinase regulation. Protein Science. 2013;22:1698–1710. doi: 10.1002/pro.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planavila A, Dominguez E, Navarro M, Vinciguerra M, Iglesias R, Giralt M, Lope-Piedrafita S, Ruberte J, Villarroya F. Dilated cardiomyopathy and mitochondrial dysfunction in Sirt1-deficient mice: A role for Sirt1-Mef2 in adult heart. Journal of molecular and cellular cardiology. 2012;53:521–531. doi: 10.1016/j.yjmcc.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Poh Y-C, Shevtsov SP, Chowdhury F, Wu DC, Na S, Dundr M, Wang N. Dynamic force-induced direct dissociation of protein complexes in a nuclear body in living cells. Nature communications. 2012;3:866. doi: 10.1038/ncomms1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajgor D, Mellad JA, Autore F, Zhang QP, Shanahan CM. Multiple Novel Nesprin-1 and Nesprin-2 Variants Act as Versatile Tissue-Specific Intracellular Scaffolds. Plos One. 2012;7:15. doi: 10.1371/journal.pone.0040098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajgor D, Shanahan CM. Nesprins: from the nuclear envelope and beyond. Expert reviews in molecular medicine. 2013;15:e5. doi: 10.1017/erm.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashmi R, Eckes B, Glöckner G, Groth M, Neumann S, Gloy J, Sellin L, Walz G, Schneider M, Karakesisoglou I. The nuclear envelope protein Nesprin-2 has roles in cell proliferation and differentiation during wound healing. Nucleus. 2012;3:172–186. doi: 10.4161/nucl.19090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Calvo F, Gonzalez JM, Casar B, Andres V, Crespo P. ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin A complexes. Journal of Cell Biology. 2010;191:967–979. doi: 10.1083/jcb.201004067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothballer A, Kutay U. The diverse functional LINCs of the nuclear envelope to the cytoskeleton and chromatin. Chromosoma. 2013:1–15. doi: 10.1007/s00412-013-0417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothballer A, Schwartz TU, Kutay U. LINCing complex functions at the nuclear envelope: what the molecular architecture of the LINC complex can reveal about its function. Nucleus. 2013a;4:29–36. doi: 10.4161/nucl.23387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothballer A, Schwartz TU, Kutay U. LINCing complex functions at the nuclear envelope: What the molecular architecture of the LINC complex can reveal about its function. Nucleus. 2013b;4 doi: 10.4161/nucl.23387. Nucleus 2013; 4:11 - 10; PMID: 23324460; http://dx.doi.org10.4161/nucl.23387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Crisp ML, Liu Q, Kim D, Kozlov S, Stewart CL, Burke B. Nesprin 4 is an outer nuclear membrane protein that can induce kinesin-mediated cell polarization. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2194–2199. doi: 10.1073/pnas.0808602106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowat AC, Foster LJ, Nielsen MM, Weiss M, Ipsen JH. Characterization of the elastic properties of the nuclear envelope. Journal of the Royal Society Interface. 2005;2:63–69. doi: 10.1098/rsif.2004.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowat AC, Jaalouk DE, Zwerger M, Ung WL, Eydelnant IA, Olins DE, Olins AL, Herrmann H, Weitz DA, Lammerding J. Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. Journal of Biological Chemistry. 2013;288:8610–8618. doi: 10.1074/jbc.M112.441535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowat AC, Lammerding J, Ipsen JH. Mechanical properties of the cell nucleus and the effect of emerin deficiency. Biophysical Journal. 2006;91:4649–4664. doi: 10.1529/biophysj.106.086454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpingidou G, Smertenko A, Hausmanowa-Petrucewicz I, Hussey PJ, Hutchison CJ. A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. Journal of Cell Biology. 2007;178:897–904. doi: 10.1083/jcb.200702026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Levesque MJ, Nerem RM. MICROPIPETTE ASPIRATION OF CULTURED BOVINE AORTIC ENDOTHELIAL-CELLS EXPOSED TO SHEAR-STRESS. Arteriosclerosis. 1987;7:276–286. doi: 10.1161/01.atv.7.3.276. [DOI] [PubMed] [Google Scholar]
- Sato M, Ohshima N, Nerem RM. Viscoelastic properties of cultured porcine aortic endothelial cells exposed to shear stress. Journal of Biomechanics. 1996;29:461–467. doi: 10.1016/0021-9290(95)00069-0. [DOI] [PubMed] [Google Scholar]
- Savage DB, Soos MA, Powlson A, O'Rahilly S, McFarlane I, Halsall DJ, Barroso I, Thomas EL, Bell JD, Scobie I, Belchetz PE, Kelly WF, Schafer AJ. Familial partial lipodystrophy associated with compound heterozygosity for novel mutations in the LMNA gene. Diabetologia. 2004;47:753–756. doi: 10.1007/s00125-004-1360-4. [DOI] [PubMed] [Google Scholar]
- Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, Tanaka S, Sheetz MP. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell. 2006;127:1015–1026. doi: 10.1016/j.cell.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharenberg MA, Pippenger BE, Sack R, Zingg D, Ferralli J, Schenk S, Martin I, Chiquet-Ehrismann R. TGF-β-induced differentiation into myofibroblasts involves specific regulation of two MKL1 isoforms. Journal of cell science. 2014 doi: 10.1242/jcs.142075. [DOI] [PubMed] [Google Scholar]
- Schirmer EC, Florens L, Guan TL, Yates JR, Gerace L. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science. 2003;301:1380–1382. doi: 10.1126/science.1088176. [DOI] [PubMed] [Google Scholar]
- Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harbor perspectives in biology. 2010;2:a005066. doi: 10.1101/cshperspect.a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert C, Grater F. Protein mechanics: How force regulates molecular function. Biochimica Et Biophysica Acta-General Subjects. 2013;1830:4762–4768. doi: 10.1016/j.bbagen.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Shaiken TE, Opekun AR. Dissecting the cell to nucleus, perinucleus and cytosol. Sci Rep. 2014;4:4923. doi: 10.1038/srep04923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nature methods. 2014;11:399–402. doi: 10.1038/nmeth.2857. [DOI] [PubMed] [Google Scholar]
- Siu CW, Lee YK, Ho JCY, Lai WH, Chan YC, Ng KM, Wong LY, Au KW, Lau YM, Zhang JQ, Lay KW, Colman A, Tse HF. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging-Us. 2012;4:803–822. doi: 10.18632/aging.100503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa BA, Kutay U, Schwartz TU. Structural insights into LINC complexes. Current Opinion in Structural Biology. 2013;23:285–291. doi: 10.1016/j.sbi.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancheva I, Schirmer EC. Nuclear envelope: connecting structural genome organization to regulation of gene expression. Advances in experimental medicine and biology. 2014;773:209. doi: 10.1007/978-1-4899-8032-8_10. [DOI] [PubMed] [Google Scholar]
- Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. Journal of Cell Biology. 1999;147:913–919. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PDP, Pinter J, Pajerowski JD, Spinler KR, Shin J-W, Tewari M. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvius N, Bilinska ZT, Veinot JP, Fidzianska A, Bolongo PM, Poon S, McKeown P, Davies RA, Chan KL, Tang ASL, Dyack S, Grzybowski J, Ruzyllo W, McBride H, Tesson F. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. Journal of Medical Genetics. 2005;42:639–647. doi: 10.1136/jmg.2004.023283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Kakimoto Y, Toda K, Naruse K. Mechanobiology in cardiac physiology and diseases. Journal of Cellular and Molecular Medicine. 2013;17:225–232. doi: 10.1111/jcmm.12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamiello C, Kamps MA, van den Wijngaard A, Verstraeten VL, Baaijens FP, Broers JL, Bouten CC. Soft substrates normalize nuclear morphology and prevent nuclear rupture in fibroblasts from a laminopathy patient with compound heterozygous LMNA mutations. Nucleus. 2013;4:61–73. doi: 10.4161/nucl.23388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MRG, Slavov D, Gajewski A, Vlcek S, Ku L, Fain PR, Carniel E, Di Lenarda A, Sinagra G, Boucek MM, Cavanaugh J, Graw SL, Ruegg P, Feiger J, Zhu X, Ferguson DA, Bristow MR, Gotzmann J, Foisner R, Mestroni L, Familial Cardiomyopathy Registry, R. Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Human Mutation. 2005;26:566–574. doi: 10.1002/humu.20250. [DOI] [PubMed] [Google Scholar]
- Tremblay D, Andrzejewski L, Leclerc A, Pelling AE. Actin and Microtubules Play Distinct Roles in Governing the Anisotropic Deformation of Cell Nuclei in Response to Substrate Strain. Cytoskeleton. 2013;70:837–848. doi: 10.1002/cm.21148. [DOI] [PubMed] [Google Scholar]
- Versaevel M, Grevesse T, Gabriele S. Spatial coordination between cell and nuclear shape within micropatterned endothelial cells. Nature communications. 2012;3:671. doi: 10.1038/ncomms1668. [DOI] [PubMed] [Google Scholar]
- Versaevel M, Riaz M, Grevesse T, Gabriele S. Cell confinement: putting the squeeze on the nucleus. Soft Matter. 2013 [Google Scholar]
- Verstraeten V, Ji JY, Cummings KS, Lee RT, Lammerding J. Increased mechanosensitivity and nuclear stiffness in Hutchinson-Gilford progeria cells: effects of farnesyltransferase inhibitors. Aging Cell. 2008;7:383–393. doi: 10.1111/j.1474-9726.2008.00382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E, Lauterbach MA, Kohl T, Westphal V, Williams GSB, Steinbrecher JH, Streich JH, Korff B, Tuan HTM, Hagen B, Luther S, Hasenfuss G, Parlitz U, Jafri MS, Hell SW, Lederer WJ, Lehnart SE. Stimulated Emission Depletion Live-Cell Super-Resolution Imaging Shows Proliferative Remodeling of T-Tubule Membrane Structures After Myocardial Infarction. Circulation Research. 2012;111:402–U109. doi: 10.1161/CIRCRESAHA.112.274530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Tytell JD, Ingber DE. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nature Reviews Molecular Cell Biology. 2009;10:75–82. doi: 10.1038/nrm2594. [DOI] [PubMed] [Google Scholar]
- Wang XF, Ha T. Defining Single Molecular Forces Required to Activate Integrin and Notch Signaling. Science. 2013;340:991–994. doi: 10.1126/science.1231041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Maharana S, Wang MD, Shivashankar GV. Super-resolution microscopy reveals decondensed chromatin structure at transcription sites. Scientific Reports. 2014;4:7. doi: 10.1038/srep04477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen K, Litjens SH, Kuikman I, Tshimbalanga N, Janssen H, van den Bout I, Raymond K, Sonnenberg A. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. The Journal of cell biology. 2005;171:799–810. doi: 10.1083/jcb.200506083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson FL, Holaska JM, Zhang Z, Sharma A, Manilal S, Holt I, Stamm S, Wilson KL, Morris GE. Emerin interacts in vitro with the splicing-associated factor, YT521-B. European Journal of Biochemistry. 2003;270:2459–2466. doi: 10.1046/j.1432-1033.2003.03617.x. [DOI] [PubMed] [Google Scholar]
- Willert K, Jones KA. Wnt signaling: is the party in the nucleus? Genes & development. 2006;20:1394–1404. doi: 10.1101/gad.1424006. [DOI] [PubMed] [Google Scholar]
- Wilson KL, Foisner R. Lamin-binding Proteins. Cold Spring Harbor Perspectives in Biology. 2010;2:17. doi: 10.1101/cshperspect.a000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, Sherwood M, Branco DM, Wakimoto H, Fishman GI, See V, Stewart CL, Conner DA, Berul CI, Seidman CE, Seidman JG. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. Journal of Molecular and Cellular Cardiology. 2008;44:293–303. doi: 10.1016/j.yjmcc.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, te Lindert M, Krause M, Alexander S, te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. Journal of Cell Biology. 2013;201:1069–1084. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SK, Do Lee S, Na KY, Park WK, Kwon HM. TonEBP/NFAT5 stimulates transcription of HSP70 in response to hypertonicity. Molecular and cellular biology. 2002;22:5753–5760. doi: 10.1128/MCB.22.16.5753-5760.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worman HJ, Ostlund C, Wang YX. Diseases of the Nuclear Envelope. Cold Spring Harbor Perspectives in Biology. 2010;2:17. doi: 10.1101/cshperspect.a000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak MA, Baker BM, Chen CS, Wilson KL. The emerin-binding transcription factor Lmo7 is regulated by association with p130Cas at focal adhesions. PeerJ. 2013;1:e134. doi: 10.7717/peerj.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Munck M, Swaminathan K, Kapinos LE, Noegel AA, Neumann S. Mutations in LMNA Modulate the Lamin A-Nesprin-2 Interaction and Cause LINC Complex Alterations. PloS one. 2013;8:e71850. doi: 10.1371/journal.pone.0071850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Worman HJ. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. Journal of Biological Chemistry. 1996;271:14653–14656. doi: 10.1074/jbc.271.25.14653. [DOI] [PubMed] [Google Scholar]
- Yoshigi M, Hoffman LM, Jensen CC, Yost HJ, Beckerle MC. Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. The Journal of cell biology. 2005;171:209–215. doi: 10.1083/jcb.200505018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HY, Kieckhaefer JE, Cao K. Mouse models of laminopathies. Aging Cell. 2013;12:2–10. doi: 10.1111/acel.12021. [DOI] [PubMed] [Google Scholar]
- Zhang QP, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, Ragnauth CD, Yi QJ, Mellad JA, Warren DT, Wheeler MA, Ellis JA, Skepper JN, Vorgerd M, Schlotter-Weigel B, Weissberg PL, Roberts RG, Wehnert M, Shanahan CM. Nesprin-1 and -2 are involved in the pathogenesis of Emery-Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Human Molecular Genetics. 2007;16:2816–2833. doi: 10.1093/hmg/ddm238. [DOI] [PubMed] [Google Scholar]
- Zhang QP, Ragnauth C, Greener MJ, Shanahan CM, Roberts RG. The nesprins are giant actin-binding proteins, orthologous to Drosophila melanogaster muscle protein MSP-300. Genomics. 2002;80:473–481. [PubMed] [Google Scholar]
- Zhang QP, Skepper JN, Yang FT, Davies JD, Hegyi L, Roberts RG, Weissberg PL, Ellis JA, Shanahan CM. Nesprins: a novel family of spectrin-repeat-containing proteins that localize to the nuclear membrane in multiple tissues. Journal of Cell Science. 2001;114:4485–4498. doi: 10.1242/jcs.114.24.4485. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Feng XH, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature. 1998;394:909–913. doi: 10.1038/29814. [DOI] [PubMed] [Google Scholar]
- Zuleger N, Kerr ARW, Schirmer EC. Many mechanisms, one entrance: membrane protein translocation into the nucleus. Cellular and Molecular Life Sciences. 2012;69:2205–2216. doi: 10.1007/s00018-012-0929-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwerger M, Jaalouk DE, Lombardi ML, Isermann P, Mauermann M, Dialynas G, Herrmann H, Wallrath LL, Lammerding J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Human Molecular Genetics. 2013;22:2335–2349. doi: 10.1093/hmg/ddt079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwerger M, Medalia O. From lamins to lamina: a structural perspective. Histochemistry and Cell Biology. 2013;140:3–12. doi: 10.1007/s00418-013-1104-y. [DOI] [PubMed] [Google Scholar]