

Abstract
Long-term depression (LTD) of the efficacy of synaptic transmission is now recognized as an important mechanism for regulation of information storage and control of actions, as well as synapse, neuron, and circuit development. Studies of LTD mechanisms have focused mainly on postsynaptic AMPA receptor trafficking. However, the focus has now expanded to include presynaptically expressed plasticity; the predominant form being initiated by presynaptically expressed Gi/o-coupled metabotropic receptor (Gi/o-GPCR) activation. Several forms of LTD involving activation of different presynaptic Gi/o-GPCRs as a “common pathway” are described. Here, we review the literature on presynaptic Gi/o-GPCR-mediated LTD, discuss known mechanisms, gaps in our knowledge, and evaluate if all Gi/o-GPCR are capable of inducing presynaptic LTD.
Keywords: Long-term synaptic plasticity, Gi/o-GPCR, Synaptic inhibition, Presynaptic plasticity, Neurotransmitter release, Vesicle release machinery, Plasticity mechanisms
Defining LTD
It is increasingly clear that long-term depression of synaptic transmission (LTD) is critical to shaping lasting changes in circuit function, learning, memory, and behavior. LTD is often thought of as a mechanism that weakens synaptic strength (Glossary) [1]. However it is a common misconception that LTD is “forgetting” and long-term potentiation (LTP) is “memory.” Several studies now indicate that LTD supports various forms of learning [2–4]. LTD may function as a signal/noise filter, as the strongest afferent inputs would have the most influence on postsynaptic neuronal output. LTD may thus increase the amount of convergent input required to generate output from a particular circuit element. It is conceivable that if both presynaptically-expressed LTD and postsynaptically-expressed LTP exist at the same synapse, the earliest response to a burst of afferent inputs would be augmented relative to subsequent responses. Therefore, LTD contributes to frequency filtering at synapses that are critical for encoding novel behaviors or memories.
In contrast to short-term depression (STD; Fig. 1a), which by definition lasts for only seconds to minutes, LTD persists for tens of minutes or more (Box 1, Fig. 1b). Operational definitions of what constitutes LTD vary, however. The more liberal definition of LTD is any suppression that persists over time and may or may not be reversed by a receptor antagonist. Here, we refer to LTD that is reversible by application of receptor antagonists after LTD is established as “labile” LTD (Fig. 1c). There are a few reasons suppression can endure yet be reversible, including persistent neurotransmitter release, persistent receptor binding, or poor agonist clearance. Indeed, extracellular levels of neuromodulators can reach levels that tonically activate receptors [5, 6]. Receptor conformational states may also play a role in the reversibility of LTD [7]. A more stringent definition of LTD is a persistent depression that is not reversible by application of receptor antagonists after LTD is established, which we refer to as “static” LTD (Fig. 1d).
Figure 1. Operational definitions for distinct forms of synaptic depression.
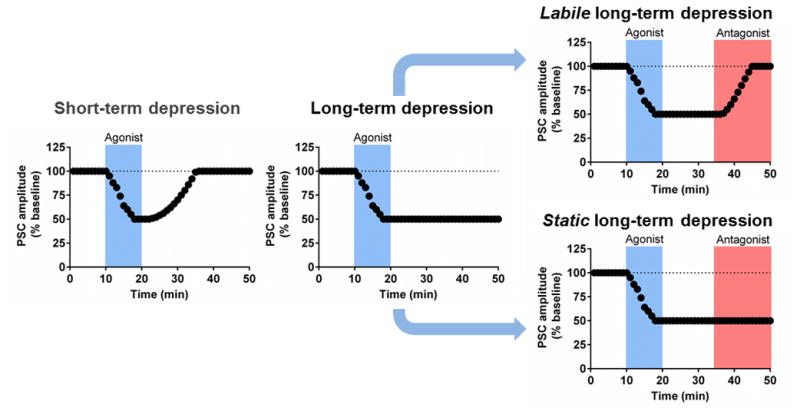
(a) Short-term synaptic depression (STD) persists for only as long as the neurotransmitter or agonist is present in the preparation (usually seconds to minutes) and therefore reverses upon washout of receptor agonist or termination of a stimulation induction protocol. (b) Depression that lasts for tens of minutes or longer following termination of receptor agonist application or an induction protocol is defined as long-term depression (LTD). LTD may be further subdivided into two operational definitions; “labile” and “static” LTD. (c) Evidence for labile LTD may be obtained by a chase with a receptor antagonist subsequent to LTD induction, as persistent receptor activation will therefore be blocked and LTD reversed. (d) We refer to the second definition of LTD as “static” LTD. Static LTD does not reverse when a receptor agonist application (or LTD-induction protocol) is followed by a receptor antagonist chase.
Box 1. Presynaptic Gi/o-GPCR STD versus LTD: binary states or two ends of a continuum?
Many factors may determine if a particular presynaptic Gi/o-GPCR mediates LTD. It is not as simple as a Gi/o-GPCR belonging to a specific structural class. For example, class A/rhodopsin GPCRs, such as CB1R, 5-HT1bR, and the opioid receptors and the class C/glutamate GPCRs, the group II/III mGluRs [10], can induce LTD following activation. Even within classes there appears to be divergence; activation of certain presynaptic Gi/o-GPCRs, such as CB1R, is known to induce either STD or LTD, depending on the induction protocol that is delivered [100].
The level of activity in the presynaptic terminal appears to be important for the transition from STD to LTD, at least at some synapses. At GABAergic synapses in hippocampus and striatum and at corticostriatal synapses in dorsal striatum, decreasing presynaptic activity with TTX determines whether CB1R activation induces STD or LTD [24, 43, 101]. However, for 5-HT1bR, application of an agonist in the presence of TTX does result in corticostriatal LTD [87]. Furthermore, the level of presynaptic activity at inhibitory synapses in the hilar region of the dentate gyrus does not affect eCB-LTD expression [102]. These functional differences also occur across synapses. In contrast to the effect of CB1R agonist on excitatory synapses on to striatal MSNs in TTX, a CB1R agonist depresses inhibitory synapses on to MSNs in TTX, but this depression is reversed by application of a CB1R antagonist [43]. This apparent difference across synapse types then suggests what dictates the expression of STD versus LTD is a combination of the particular presynaptic Gi/o-GPCR and the complement of downstream effectors that are expressed in a particular presynaptic terminal. Are the mechanisms underlying STD and LTD dissociable? Possibly at some synapses STD is solely mediated by presynaptic VDCC blockade, whereas LTD results from extended agonist binding or activation of a high number of receptors, along with presynaptic terminal activation, leading to long-term maintenance of synaptic depression involving AC inhibition, de novo local protein synthesis, and other intra-terminal Ca2+ signaling mechanisms. It is also possible that presynaptic VDCC blockade, downstream of AC inhibition, may also contribute to protein synthesis necessary for LTD maintenance. As we have discussed, there are multiple examples of presynaptic Gi/o-GPCRs that can induce LTD, and some that induce STD. Experimental evidence suggests that synaptic depression is more complicated than this binary (either LTD or STD) picture would suggest. Perhaps it could be better described as a continuum, from STD to LTD, encompassing reversible labile and irreversible, likely protein synthesis-dependent static LTD. Is a synapse that undergoes LTD forever depressed? It would not be unreasonable to think that even these forms of LTD could be reversed via engagement of mechanisms that are as yet undiscovered.
Mechanistically, LTD can be placed into two categories: those with presynaptic and those with postsynaptic expression mechanisms. Postsynaptically-expressed LTD generally involves the removal of receptor proteins, such as AMPA receptors, from the postsynaptic membrane, resulting in reduced transmission. These mechanisms are reviewed elsewhere [8]. Presynaptic LTD is expressed through various mechanisms including presynaptic NMDA receptor (NMDAR) activation [9] or activation of select presynaptically-localized metabotropic receptors that couple to Gi/o G proteins (Gi/o-GPCRs). This review focuses on this latter, presynaptic Gi/o-GPCR-dependent form of LTD. Furthermore, we discuss a wide range of receptors that mediate presynaptic LTD and explore the general principle that activation of any presynaptic Gi/o-GPCR is capable of inducing LTD.
GPCRs and presynaptic LTD
GPCRs are a diverse family of metabotropic receptors that signal through associated heterotrimeric G proteins and scaffolding proteins. G proteins exist in complexes consisting of a Gα subunit and a Gβγ dimer pair of subunits. Receptor activation induces these G protein subunits to couple to numerous effector proteins, allowing a single receptor to modulate diverse cellular processes.
Gα subunits are divided into four classes. Gαs stimulates adenylyl cyclase (AC), Gαi inhibits AC, Gαq/11 activates phospholipases and subsequently intracellular Ca2+ levels, whereas Gα12/13 operates through more complex pathways [10]. Specific GPCRs are referred to as coupling to a specific Gα class, but this preferred coupling is mutable [10]. Gβγ protein subunits couple to ion channels such as voltage dependent Ca2+ channels (Cav2, VDCCs) and the G protein-coupled Kir3 class of potassium channels (GIRKs) [11]. Gβγ also directly interacts with vesicular fusion machinery (consisting of SNARE, Ca2+ sensing and regulatory proteins) [11] as well as kinases, phospholipases, and even AC [12]. Scaffolding proteins such as beta-arrestin unite GPCRs with other signaling proteins such as kinases that provide additional routes for GPCRs to influence cellular function [10].
Activation of presynaptic Gi/o-GPCRs produces a decrease in synaptic efficacy through inhibition of neurotransmitter release. Table 1 lists the effectors through which Gi/o-GPCRs exert their control of neurotransmission. Fig. 2 details how some of these effectors may induce LTD subsequent to receptor activation.
Table 1.
Coupling of G protein subunits with effectors and the cellular consequences of such interactions.
| G protein subunit | Target | Cellular Effect | Effect on Neurotransmission | References |
|---|---|---|---|---|
| Gαi/o | Adenylyl cyclase | Inhibition, reduction in cAMP | Inhibition of neurotransmitter release | [26, 108] |
| Gβγ | VDCC inhibition | Reduced ICa | Reduced neurotransmitter release | [109, 110] |
| Gβγ | K channel activation | Increased IK, reduced cellular excitability | Reduced neurotransmitter release | [111–113] |
| Gβγ | Vesicular release machinery | Inhibition of vesicular release | Inhibition of neurotransmitter release | [114–116] |
| Gβγ | PLCβ | PKC activation, increase intracellular Ca2+ | Inhibition of neurotransmitter release | [117] |
| Gβγ | Adenylyl cyclase | Activation or inhibition, increase or decrease in cAMP | Inhibition or enhancement of neurotransmitter release1 | [26, 108] |
| Gβγ | Kinases: MAPK, βARK, PI3K | Phosphorylation of target proteins | Inhibition or enhancement of neurotransmitter release1 | [118–120] |
Gβγ-mediated effects on presynaptic-modulation neurotransmission for the indicated effectors remain to be directly demonstrated.
Figure 2. A schematic diagram of the possible mechanisms of presynaptic Gi/o coupled GPCR activation-induced LTD.
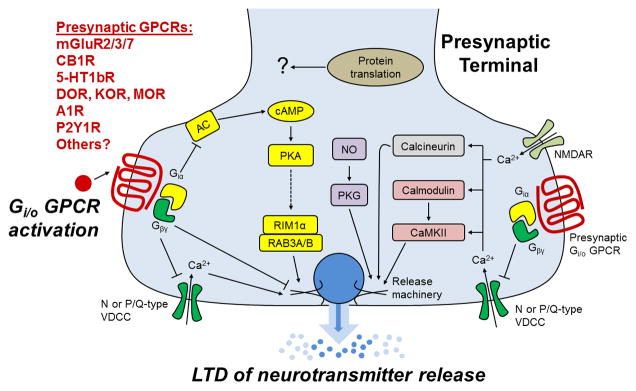
Activation of presynaptic Gi/o-coupled GPCRs results in the dissociation of the Gαi and Gβγ subunits from the receptor complex. Gβγ subunits directly and negatively couple to voltage-dependent Ca2+ channels (VDCCs) resulting in reduced Ca2+ entry into the presynaptic terminal. Reduced Ca2+ influx decreases vesicle fusion and neurotransmitter release probability. In addition, Gβγ may directly inhibit components of the vesicular release machinery (e.g. SNAP25). The Gαi subunits negatively couple to adenylyl cyclase (AC) resulting in decreased cAMP levels. A decrease in cAMP levels results in dampened PKA activity, which is associated with reduced functionality of RIM1α. RIM1α complexes with the vesicular proteins RAB3A and RAB3B to enhance neurotransmitter release. Therefore, reduced PKA activity directly or indirectly inhibits this complex’s ability to promote neurotransmission. In addition, Ca2+ entry through VDCCs or NMDARs activates the presynaptic kinases calmodulin and CaMKII and the protein phosphatase calcineurin. GPCR modulation of presynaptic Ca2+ levels likely influences the activity of these proteins, which subsequently alters neurotransmitter release. Although not directly modulated by presynaptic Gi/o GPCRs, nitric oxide (NO) signaling intersects with GPCR-mediated signaling to promote LTD. NO signaling results in PKG activation that influences release machinery function. Finally, at some synapses protein translation appears to be a necessary component of LTD maintenance. The mechanisms that engage the protein translation apparatus as well as the proteins that are expressed are currently unknown.
Ca2+ influx from VDCCs or release from internal stores influences vesicular release as well as Ca2+-sensitive signal transduction pathways that influence synaptic plasticity. The sensitivity of the release machinery in a particular synapse to Ca2+ and/or the degree to which receptors couple to VDCCs confers synapse-specific effects of receptor mediated, Ca2+-dependent regulation of neurotransmitter release [13, 14]. Gi/o-GPCRs regulate presynaptic Ca2+ levels by inhibiting VDCCs. Blocking VDCCs often disrupts LTD [15–21], but this may be due to reduced Ca2+-sensitive protein function rather than direct involvement of VDCC inhibition in LTD expression. Indeed, Ca2+ activates proteins, such as calmodulin and Ca2+/calmodulin-dependent protein kinase II (CaMKII), which may play important roles in presynaptic LTD expression [22, 23]. The Ca2+ sensitive phosphatase calcineurin can contribute to LTD, but this appears to be synapse-specific [24, 25].
Gi/o-GPCR-induced LTD with mechanisms downstream of VDCC activation is likely due to an effect on vesicular release machinery. Many signaling pathways engaged by Gi/o-GPCRs converge on these proteins. Presynaptic Gi/o-GPCRs negatively couple to AC, attenuating cAMP-protein kinase A (PKA) signaling [26], which influences release machinery function and indirectly modulates VDCCs [21, 27–30]. Kinases, such as protein kinase C (PKC) [31–34], diacylglycerol kinase (DGK) [35], and mitogen-activated protein kinase (MAPK) might have a role in presynaptic Gi/o-GPCR-mediated LTD [36]. Presynaptic NMDARs may also play a role in GPCR-mediated presynaptic LTD, albeit in a supporting role [31, 37–39]. Nitric oxide (NO) signaling can intersect with GPCR-mediated LTD through NO-signaling pathway-mediated Ca2+ release from presynaptic internal stores and subsequent activation of Ca2+-sensitive LTD mediators. Protein kinase G (PKG)-mediatedphosphorylation of release machinery may also contribute to these forms of LTD [40–42]. While these mechanisms can be recruited for presynaptic LTD induction, the maintenance of this form of LTD requires long-lasting changes at axon terminals possibly accomplished by alterations in protein translation or posttranslational modification. Presynaptic protein synthesis plays a role in presynaptic LTD maintenance at some synapses [43–45], but not at others [16, 46].
Presynaptic Gi/o-GPCRs implicated in LTD
Group II and III mGluRs
LTD mediated by the group II mGluRs, mGluR2 and mGluR3, was first identified at mossy fiber (MF)-area CA3 hippocampal synapses [47] and has since been found in many brain regions. For example, mGluR2/3 agonists induce LTD (mGluR2/3-LTD) in nucleus accumbens (NAc), prefrontal cortex, and substantia nigra pars reticulata [21, 48, 49], among others. mGluR2/3-LTD may be achieved via modulation of cAMP-PKA signaling. Maintaining high cAMP levels prevents mGluR2/3-LTD induction at MF-CA3 synapses and PKA inhibition is occlusive of mGluR2/3-LTD [47]. PKA inhibition similarly occludes mGluR2/3-LTD in basolateral amygdala [50]. mGluR2/3-LTD at excitatory synapses onto medium spiny neurons (MSNs) in the NAc is also dependent upon cAMP-PKA signaling [21]. Interestingly, this LTD can be occluded by blockade of P/Q-type presynaptic VDCCs, suggesting an interaction between suppression of PKA activity and VDCC inhibition, as previously reported [21, 51].
NMDARs may serve a supporting role for mGluR2/3-LTD. In dentate gyrus mGluR2-LTD is partially sensitive to NMDAR antagonists and is completely blocked by both PKA and PKC inhibitors [31]. The fact that this LTD is not completely ablated in the presence of NMDAR antagonists suggests that NMDARs are not of critical importance. However, it is possible that NMDARs are downregulated along with VDCCs to ultimately result in some forms of Gi/o-GPCR initiated LTD.
mGluR2/3 can target SNAP25, a SNARE protein. Botulinum toxin A treatment cleaves SNAP25 and disrupts mGluR2/3-LTD at hippocampal CA3-CA1 synapses [52]. Saturation of LTD occludes this toxin’s effect, which is not prevented by increasing Ca2+ levels. Sequestration of Gβγ with C-terminal SNAP25 peptide fragment infused presynaptically into CA3 neurons prevents LTD induction. This blocking peptide only slightly reduced Ca2+ transients, thus this LTD pathway occurs independently of Ca2+ and is likely due to a direct action of G proteins on SNAP25 or other components of the interacting release machinery [52].
Activation of the group III mGluR7 receptor, another presynaptically-localized Gi/o-GPCR, by L-AP4 or high frequency stimulation (HFS) also induces LTD (mGluR7-LTD). Interestingly, mGluR7-LTD at excitatory synapses onto CA3 hippocampal interneurons requires postsynaptic Ca2+ entry through AMPA receptors. Thus, a retrograde signaling molecule cannot be ruled out that may act in concert with mGluR7 activation to induce LTD [53]. HFS of MF synapses onto CA3 stratum lucidum interneurons induces presynaptic mGluR7-LTD, but only in slices not previously exposed to mGluR7 agonist [33]. This mechanism functions through inhibition of P/Q-type VDCC inhibition, as P/Q-channel inhibition occludes mGluR7-LTD [54]. Consistent with these findings, Ca2+ transients in the filopodial extensions of mGluR7-LTD-sensitive MF boutons are persistently depressed following LTD induction by HFS [54]. This indicates that a lasting inhibition of Ca2+ influx through presynaptic P/Q-channels, rather than a transient inhibition of influx triggering a downstream signaling event, may be a necessary feature of presynaptic Gi/o-GPCR mediated LTD.
The cAMP pathway is recruited upstream of P/Q channel inhibition [55, 56]. Following activation, mGluR7 is internalized, which induces a switch in cAMP production. mGluR7 internalization allows HFS to activate AC and PKA, resulting in LTP of neurotransmitter release. Thus, mGluR7 acts as a state-dependent switch of bidirectional plasticity. mGluR7 acts similarly at glutamatergic synapses on magnocellular neurosecretory cells (MNCs) in the paraventricular nucleus of the hypothalamus (PVN), although its activation only induces STD at these synapses [3]. Further investigation is necessary to determine if other presynaptic Gi/o-GPCRs are capable of this function.
The kinase activation profile during presynaptic mGluR-dependent LTD is not limited to PKA. At MF-stratum lucidum interneuron synapses (in CA3), presynaptic mGluR7 activation results in PKC- but not PKA-mediated LTD [33]. Priming synapses with PKC activation using the phorbol ester PMA lowers the threshold for LTD induction at Schaffer collateral (SC)-CA1 hippocampal synapses [34]. In addition to mGluR activation in CA3, CaMKII activation is also required for presynaptic LTD of MF synapses [22, 57]. In neonatal mice, presynaptic DGKi plays a role in mGluR2/3-LTD at SC-CA1 synapses and PKC inhibition restores mGluR2/3-LTD that was lost in DGKi KO mice [35]. mGluR2/3-LTD in the striatum is unaffected by cAMP-PKA pathway modulation but is partially blocked by MAPK kinase1/2 inhibitors suggesting the importance of the MAPK signaling pathway [36]. However, it is unclear whether this MAPK activity dependence is localized to the presynaptic terminal.
CB1 receptor
The first hint that activation of the presynaptically-expressed Gi/o-coupled type 1 cannabinoid receptor (CB1R) might induce LTD came from a study of glutamatergic transmission from parallel fibers onto cerebellar Purkinje cells. A CB1R agonist induced a persistent, but reversible depression of transmission (CB1R-LTD) [58]. Following the discovery of pharmacologically-induced CB1R-LTD, HFS-induced-LTD of glutamatergic transmission on to MSNs in the dorsal striatum (corticostriatal synapses) was demonstrated to occur via presynaptic CB1R activation, providing the first evidence that this form of LTD is inducible by an endocannabinoid (eCB) [59]. Shortly thereafter moderate-frequency stimulation-induced eCB-LTD was discovered at cortical inputs to MSNs in NAc [60]. eCB-dependent LTD is now known to be inducible at striatal GABAergic synapses and in many other brain regions [3, 61–64]. In contrast to mGluR2/3-LTD, where the putative source of the endogenous agonist resulting in LTD is the presynaptic terminal itself, the source of eCBs is most likely the postsynaptic neuron [65].
CB1R-LTD of glutamatergic transmission in midbrain dopamine neurons involves presynaptic inhibition of PKA [66]. Experiments in NAc and hippocampus indicate that manipulating AC function and cAMP levels prevent induction of CB1R-LTD [20, 28]. However, postsynaptic AC5 appears to have important roles in eCB-LTD induction at glutamatergic synapses onto striatal MSNs [67]. Thus, it will be important to separate pre- and postsynaptic AC roles, at least at these synapses.
As with other Gi/o-GPCRs, activation of CB1R directly inhibits VDCCs, but according to studies in single neurons this inhibition does not persist following removal of a CB1R agonist [68, 69]. Indirect CB1R-VDCC interactions through cAMP-PKA may also be involved in LTD. For example, the irreversible phase of eCB-LTD in NAc depends on the cAMP-PKA cascade and ultimately suppression of P/Q-type VDCCs [20], much like what was found with amphetamine-induced eCB-LTD in the amygdala [19]. In the lateral amygdala, CB1R mediates LTD through PKA modulation of N-type VDCCs, but this may be an indirect effect [27].
CB1R coupling to potassium channels may play some role in LTD. Glutamatergic transmission onto MSNs of the NAc is depressed by application of a CB1R agonist, and this depression is reversible by a CB1R antagonist [70]. This reversible form of CB1R-dependent synaptic depression is not mediated by the cAMP-PKA cascade, but instead by potassium channel activation. Potassium channel blockers also prevent CB1R-mediated LTD at hippocampal SC-CA1 synapses [71], in the lateral amygdala [72] and synaptic depression (not-necessarily LTD) at cerebellar parallel fiber-Purkinje cell synapses [73]. Moreover, activation of GIRKs produces STD that can prevent the induction of CB1R-dependent LTD at some synapses [27] and may play a role in CB1R-dependent LTD induction at others [72]. In addition to presynaptic VDCC and potassium channels, some evidence exists to support a role for presynaptic NMDARs as downstream targets of CB1R activation [37, 74], as well as NO at striatal synapses [40, 75, 76].
PKA inhibition appears to be critical for CB1R-LTD. But what is downstream of this? A potential PKA target is the release machinery-associated protein RIM1α [30, 77]. CB1R-LTD is absent in RIM1α null mice and PKA inhibition fails to induce LTD in these mice as well [28]. Interestingly, CB1R agonist application induces STD at these synapses rather than LTD. RIM1α deletion also prevents a CB1R-mediated component of LTD at glutamatergic synapses in NAc [78]. RIM1α interacts with RAB3, and both are predominantly presynaptic proteins associated with release machinery at inhibitory terminals [30]. The RAB3B null mouse has normal basal neurotransmission but has greatly reduced inhibitory LTD in CA1and lacks LTD at MF-CA3 synapses [43, 70, 79]. Phosphorylation of RIM1α induces LTP at cerebellar parallel fiber-Purkinje cell synapses [80], thus it is possible that dephosphorylation allows for LTD to occur. The protein phosphatase calcineurin has been implicated in CB1R-LTD at hippocampal GABAergic synapses, and this protein could also contribute to presynaptic dephosphorylation needed for LTD expression [24].
Opioid and neuropeptide receptors
Opioid receptor mediated-LTD (OP-LTD) was first identified in the PVN [81]. Kappa opioid receptor (KOR) activation induces LTD (KOP-LTD) of excitatory inputs to MNCs. Dynorphin is somatodendritically released from vasopressin MNCs to act on presynaptic KORs. Here, KOP-LTD is gated by the presynaptic modulation of glutamate release by eCB signaling [82]. GABAergic synapses onto paraventricular neuroendocrine cells also undergo LTD when moderate frequency stimulation is paired with postsynaptic depolarization in slices treated with cortisol or in slices made from previously stressed animals [83]. This LTD, which is induced by retrograde opioid signaling on to presynaptic mu opioid receptors (MORs) (MOP-LTD), is reversed by an antagonist chase.
In hippocampus area CA2, HFS of the SC pathway produces presynaptically expressed LTD of parvalbumin positive interneuron-pyramidal cell synapses that is not blocked by MOR or CB1R antagonists, but is blocked by delta opioid receptor (DOR) antagonists [84]. This DOR-mediated LTD (DOP-LTD) is also induced by moderate frequency stimulation and by DOR receptor agonists. DOR agonist effects are mutually occlusive with HFS-induced LTD. DOP-LTD is not present in CA1 as DOR activation there only produces STD.
OP-LTD also occurs in the dorsal striatum, mediated by DOR, KOR, and MOR [85]. Both exogenously applied and endogenously released opioid peptides produce LTD of excitatory inputs on to MSNs. Opioid receptor antagonists fail to reverse depression elicited by activation of each receptor. The OP-LTD mediated by activation of each receptor is dissociable from the others. Whereas MOP-LTD and DOP-LTD are clearly presynaptically expressed, KOP-LTD appears to have a presynaptic component, but is not unequivocally presynaptic. MOP-LTD is mutually occlusive with CB1R-LTD, whereas DOP-LTD is additive with both MOP- and CB1R-LTD, suggesting synapse-specificity or parallel routes of LTD induction.
Neuropeptide Y (NPY) application causes a persistent decrease in excitatory transmission in the suprachiasmatic nucleus [86]. However, NPY also causes a lasting depression of postsynaptic Ca2+ levels. Therefore, the mechanisms underlying NPY-LTD require clarification. NPY-LTD is not dependent on NMDARs, L- or N-type VDCCs, but is dependent on Gi/o proteins. Since NPY1 and NPY2 receptors were ruled out, the exact identity of the NPY receptor that produces the NPY-LTD remains a mystery. However, the other NPY receptors NPY4 and NPY5 are also Gi/o-coupled.
5-HT1b receptor
Activation of the presynaptic serotonin 5-HT1bR with exogenous 5-HT, endogenous 5-HT, (secondary to application of a selective 5-HT reuptake inhibitor) or the 5-HT1bR-specific agonist CP93129, induces LTD of corticostriatal neurotransmission in dorsal striatum [87]. This 5-HT-LTD is AC-cAMP-dependent and is not reversible. 5-HT-LTD is mutually occlusive with eCB-LTD, indicating that both 5-HT1bR and CB1R co-express on corticostriatal terminals. In addition, 5-HT-LTD mediated by 5-HT1bR is also present at dorsal striatal GABAergic terminals and in the NAc [2, 29, 87]. 5-HT-LTD in NAc is sensitive to protein kinase inhibitors [29]. Interestingly, in this same study the authors found evidence that was suggestive of increased phosphorylation of release machinery following in vivo cocaine exposure that prevented the induction of 5-HT-LTD [29]. 5-HT-LTD mediated by 5-HT1bR in NAc is also induced by presynaptic oxytocin receptor activation on inputs from the dorsal raphe nucleus, and appears to be a critical component of socially conditioned place preference [2].
A1 adenosine and purinergic receptors
Data from in vivo studies and reduced preparations indicate that synapses in many brain regions are subjected to tonic, low-level presynaptic A1 adenosine receptor (A1R)-mediated synaptic depression due to ambient levels of adenosine [88, 89]. A1R activation often results in STD, however, a more recent report investigating hippocampal CA3-CA1 synapses of morphine-dependent rats showed that theta (5 Hz)-burst stimulation results in LTD only in morphine dependent animals. This LTD is blocked by an A1R-specific antagonist or an NMDAR antagonist, but not both [90]. In addition, in cerebellar slices delivery of a train of 5 electrical pulses at 100 Hz, paired with postsynaptic depolarization, results in LTD of parallel fiber-Purkinje cell transmission that is largely blocked by an A1R antagonist [91].
Activation of P2Y1 receptors produces presynaptic NMDAR-independent LTD in cultured hippocampal neuron pairs and in hippocampal area CA1 [15]. Receptor activation via endogenous release of ATP from optogenetically stimulated astrocytes, or by exogenous application of ATP or a P2Y agonist, induces this form of plasticity. These effects are blocked by a P2Y1 antagonist and the astrocytic-stimulated LTD is occluded by electrical stimulation-induced LTD.
Manifold control
Given the number of known presynaptic Gi/o-GPCRs involved in LTD and the potential for more (Table 2), it is quite possible that a given presynaptic terminal may be under the control of multiple Gi/o-GPCRs. At the corticostriatal synapse alone, GABABR, 5-HT1bR, CB1R, D2R, A1R, mGluR2, H3 histamine receptor (H3R), M4 muscarinic receptor (M4R), DOR, and MOR are at least known to be expressed. Fig. 3 conceptualizes three possible interactions of different presynaptic Gi/o-GPCRs expressed on the same terminal. First, it is clear that at some synapses, LTD induced by different receptors is mutually occlusive [85, 87, 92]. It is therefore possible that presynaptic LTD operates via a common pathway with a “winner-take-all” process such that the modulator that manages to induce LTD prevents others from doing so (Fig. 3a). A possible mechanistic example comes from heterologous CB1R expression in superior cervical ganglion neurons. CB1R sequesters Gi/o G proteins such that it prevents other endogenously expressed Gi/o-coupled receptors from inhibiting VGCCs, but mGluR2 expression had no effect on these receptors [93]. However, evidence that this sort of sequestration occurs in native presynaptic terminals is currently lacking. Second, it is conceivable that full activation of two Gi/o-GPCRs on the same terminal could operate in parallel through distinct pathways to produce an additive effect on LTD induction (Fig. 3b), although this has not been demonstrated experimentally to our knowledge. Third, despite full activation, some Gi/o-GPCRs do not induce LTD on their own, but facilitate the ability of other receptors to induce LTD possibly through enhancing inhibition of the cAMP-PKA pathway [41, 94, 95] (Fig. 3c). A variant of this possibility may be that simultaneous, but insufficient activation of two Gi/o-GPCRs on the same terminal is capable of summating to sufficiently induce LTD.
Table 2.
A list of inhibitory Gi/o-coupled GPCRs that may be presynaptically-expressed and potentially mediate LTD at various synapses throughout the CNS.
| Receptor Family | Receptor |
|---|---|
| Serotonin | 5-HT1a |
| 5-HT1b | |
| 5-HT1d | |
| 5-HT1e | |
| 5-HT1f | |
| 5-HT5a | |
| Acetylcholine | M2 |
| M4 | |
| Adenosine | A1 |
| A3 | |
| Adrenergic | A2a |
| A2b | |
| A2c | |
| Anaphylatoxin | C5a1/CD88 |
| Angiotensin | AT1 |
| AT2 | |
| Apelin | APJ |
| Cannabinoid | CB1 |
| Chemokine | CCR2 |
| CCR10 | |
| CXCR4 | |
| CX3CR1 | |
| Dopamine | D2 |
| D3 | |
| D4 | |
| Galanin | GAL1 |
| GAL2 | |
| GAL3 | |
| Histamine | H3R |
| Leukotriene | BLT1 |
| BLT2 | |
| OXE | |
| FPR2/AXE | |
| Lysophospholipid | LPA3 |
| S1P1 | |
| S1P3 | |
| S1P5 | |
| MCH | MCH1 |
| Melatonin | MT1 |
| MT2 | |
| MT3 | |
| Neuropeptides | DOR |
| KOR | |
| MOR | |
| NOR/ORL-1 | |
| NPFF1 | |
| NPFF2 | |
| NPBW1 | |
| NPBW2 | |
| Orexin | OX2 |
| Purinergic | P2Y1 |
| P2Y12 | |
| P2Y13 | |
| Prokineticin | PKR2 |
| Prostanoid | EP3 |
| DP2 | |
| Relaxin | RXFP2 |
| RXFP3 | |
| RXFP4 | |
| Somatostatin | SSTR1 |
| SSTR2 | |
| SSTR3 | |
| SSTR4 | |
| SSTR5 | |
| Ca2+ Sensing | CaS |
| GABA | GABAB |
| mGluR | mGluR2 |
| mGluR3 | |
| mGluR4 | |
| mGluR6 | |
| mGluR7 | |
| mGluR8 |
Figure 3. Possible interactions between two Gi/o-coupled receptors co-expressed on the same presynaptic terminal.
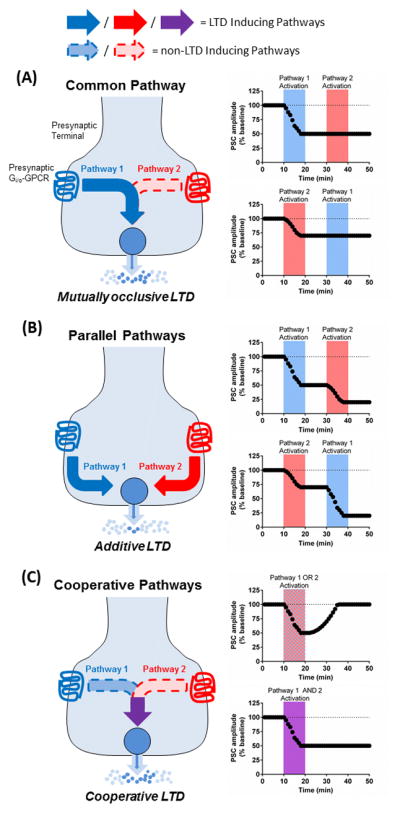
(a) Two presynaptic Gi/o-GPCRs may signal through pathways that converge upon the same downstream effectors to induce LTD. Alternatively the two receptors may signal through pathways that not only converge, but are nearly identical. In either case, the two receptors will compete at some level for access to signaling proteins. The end result of this interaction will be mutually occlusive: LTD expression mediated by one receptor will prevent LTD by the second receptor. (b) A second possible interactive scenario could occur if two receptors operate through independent, non-converging signaling pathways that each achieved the same end result of inducing LTD. In this case LTD induction by one GPCR would be added to during induction by the second GPCR to increase the degree to which the terminal is depressed resulting in greater, additive LTD. (c) A final possible interaction between two presynaptic receptors is cooperation. In this relationship neither receptor is able to induce LTD on its own, but the ability of one receptor to do so is bolstered by signaling from the other. As such, one receptor would be required for LTD induction and the other would serve a facilitating role. In each of these scenarios, the timing of receptor activation would be critical. Coincident receptor activation could be needed for the hypothesized outcomes to occur, or possibly there may be a time window in which the two pathways could still interact.
Slice electrophysiology only provides one piece of evidence for interactions between given presynaptic Gi/o-GPCRs, and is not necessarily an accurate reflection of how the system is actually functioning in vivo. To advance our understanding of how coordination and competition between multiple Gi/o-GPCRs contributes to learning and memory, it will be important to understand the context specific patterning of neurotransmission amongst differentially excitable dendritic arbor subregions on a given neuron.
It is evident that much more work is needed to clarify the role of known Gi/o-GPCR contributors to LTD in vitro and in vivo, and to investigate the potential for other receptors in this class to modulate synaptic transmission at different central synapses (Box 2). Given the wide ranging and powerful effect of presynaptic Gi/o-GPCR activation on LTD, it is not surprising that many of these receptors are involved in pathophysiology and are current therapeutic targets for a host of neuropsychiatric disorders ranging from anxiety and depression, to addiction, to Parkinson’s disease [96–99]. Induction of LTD may well be one desirable consequence of such therapeutic receptor targeting. Tapping into this powerful neurotransmitter release control system using circuit-specific tools could represent a fruitful strategy for the development of future neurotherapeutics. Furthermore, clarifying the role of individual receptors and the interactions between receptors in presynaptic LTD will further our understanding of how these receptors shape learning, memory, and behavior.
Box 2. Open questions.
Are there more forms of presynaptic Gi/o-GPCR-mediated LTD to be discovered?
While it is established that certain presynaptic Gi/o-GPCRs induce LTD, there are many other Gi/o-GPCRs that may be localized presynaptically and positioned to exert LTD (see Table 2). Evidence for involvement of novel forms of LTD already exists. In rats, H3R activation by histamine produces a persistent decrease in glutamate release onto striatal MSNs [103], and from medial perforant pathway synapses of the dentate gyrus [104]. M2 muscarinic receptor activation in somatosensory cortex results in a lasting depression of glutamate-evoked discharges [105]. Presynaptic M4Rs at corticostriatal terminals might also induce LTD [106]. Finally, in the amygdala, alpha2-adrenoreceptor activation inhibits excitatory transmission on to principal neurons and prevents LTD induction by low frequency stimulation [17]. More work is needed to clearly define the role of these proteins in inducing presynaptic static or labile LTD. Given the number of Gi/o-GPCRs remaining to be studied, the potential is high for the discovery of more forms of presynaptic Gi/o-GPCR-mediated LTD.
What determines whether a Gi/o-GPCR induces labile or static LTD?
Some Gi/o-GPCRs induce labile LTD at one synapse and static LTD at another. For example MOR activation in hypothalamus produces labile LTD [83] whereas in dorsal striatum it produces static LTD [85]. One hypothesis to explain this diversity is that the efficiency with which the receptor at a particular synapse couples to downstream effectors dictates the persistence of synaptic depression. For instance, eCB-LTD in dorsal striatum is only reversed by a CB1R antagonist applied 1 min, but not 10 min, after induction [107]. There may be a requirement for the receptor to have sufficient time to engage signaling pathways and the efficiency of its coupling may determine the window in which it needs to remain active. Also, a recent report suggests that during LTD induction receptors may become constitutively activated but are still in equilibrium with inactive conformations [7]. Thus an antagonist can “reverse” LTD if it stabilizes LTD-inducing receptors in an inactive conformation. It is possible that upon removal of antagonist the receptors can return to a constitutively active conformation and restore LTD. The presynaptic proteome may also play a role. CB1R-LTD of inhibitory synapses in hippocampus and basolateral amygdala is changed to STD when RIM1α is absent [28]. Perhaps an unknown set of presynaptic proteins performs similar roles in determining whether labile or static LTD is the end result of receptor activation. Much more work is needed to determine why different Gi/o-GPCRs or the same Gi/o-GPCRs at different synapses induce differing forms of LTD.
What is the nature and significance of the interaction between multiple Gi/o-GPCRs at a given presynaptic terminal?
Multiple receptors exist at a given synaptic terminal, so what is the significance of their inevitable interaction? Is there a common pathway for presynaptic Gi/o-GPCR mediated LTD induction or are there multiple routes to achieve LTD? It is apparent that there are many effectors between receptor activation and manifestation of LTD. Are kinase activation, ion channel inhibition, and release machinery interactions all interconnected mechanisms or are there parallel pathways that yield the same end result of LTD? Unraveling this mystery requires a greater understanding of how different presynaptic Gi/o-GPCRs expressed at the same terminals interact over time to affect synaptic depression and, ultimately, behavior.
Glossary Box
- Gi/o G protein-coupled receptor (Gi/o-GPCR)
An inhibitory metabotropic neurotransmitter receptor characterized by its seven transmembrane helical structure and its ability to activate associated heterotrimeric G proteins upon agonist binding. These receptors couple to the Gi/o class of G proteins that, when localized presynaptically, allow them to inhibit neurotransmitter release through modulation of adenylyl cyclase and ion channels
- Presynaptic long-term depression
Long-term depression (LTD) of synaptic efficacy that results from a reduction in the probability of neurotransmitter release. Presynaptic LTD is distinguished from postsynaptic LTD, which results from reduced responsiveness to neurotransmitter due to a reduction in postsynaptic receptor levels
- Short-term depression
Short-term depression (STD) of synaptic efficacy is a form of synaptic plasticity that reduces neurotransmission on a time scale of seconds to minutes
Acknowledgments
This work was supported by the National Institute on Alcohol Abuse and Alcoholism (NIAAA), US National Institutes of Health (NIH) K22 AA021414 for B.N.M. and the Division of Intramural Clinical and Biological Research of the NIAAA/NIH for B.K.A. and D.M.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yoon BJ, et al. Essential role for a long-term depression mechanism in ocular dominance plasticity. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9860–9865. doi: 10.1073/pnas.0901305106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dolen G, et al. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature. 2013;501:179–184. doi: 10.1038/nature12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuzmiski JB, et al. Metaplasticity of hypothalamic synapses following in vivo challenge. Neuron. 2009;62:839–849. doi: 10.1016/j.neuron.2009.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yin HH, et al. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nature neuroscience. 2009;12:333–341. doi: 10.1038/nn.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang CC, et al. Repeated cocaine administration impairs group II metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:2958–2968. doi: 10.1523/JNEUROSCI.4247-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neu A, et al. Postsynaptic origin of CB1-dependent tonic inhibition of GABA release at cholecystokinin-positive basket cell to pyramidal cell synapses in the CA1 region of the rat hippocampus. The Journal of physiology. 2007;578:233–247. doi: 10.1113/jphysiol.2006.115691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lodge D, et al. Antagonists reversibly reverse chemical LTD induced by group I, group II and group III metabotropic glutamate receptors. Neuropharmacology. 2013;74:135–146. doi: 10.1016/j.neuropharm.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 8.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez-Moreno A, et al. Presynaptic NMDA Receptors and Spike Timing-Dependent Depression at Cortical Synapses. Frontiers in synaptic neuroscience. 2010;2:18. doi: 10.3389/fnsyn.2010.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Latek D, et al. G protein-coupled receptors--recent advances. Acta biochimica Polonica. 2012;59:515–529. [PMC free article] [PubMed] [Google Scholar]
- 11.Betke KM, et al. GPCR mediated regulation of synaptic transmission. Progress in neurobiology. 2012;96:304–321. doi: 10.1016/j.pneurobio.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oldham WM, Hamm HE. Structural basis of function in heterotrimeric G proteins. Quarterly reviews of biophysics. 2006;39:117–166. doi: 10.1017/S0033583506004306. [DOI] [PubMed] [Google Scholar]
- 13.Dittman JS, Regehr WG. Contributions of Ca2+-dependent and Ca2+-independent mechanisms to presynaptic inhibition at a cerebellar synapse. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1996;16:1623–1633. doi: 10.1523/JNEUROSCI.16-05-01623.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamasaki M, et al. Miniature synaptic events elicited by presynaptic Ca2+ rise are selectively suppressed by cannabinoid receptor activation in cerebellar Purkinje cells. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:86–95. doi: 10.1523/JNEUROSCI.2258-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, et al. Heterosynaptic long-term depression mediated by ATP released from astrocytes. Glia. 2013;61:178–191. doi: 10.1002/glia.22425. [DOI] [PubMed] [Google Scholar]
- 16.Connelly T, et al. Distinct mechanisms contribute to agonist and synaptically induced metabotropic glutamate receptor long-term depression. European journal of pharmacology. 2011;667:160–168. doi: 10.1016/j.ejphar.2011.04.063. [DOI] [PubMed] [Google Scholar]
- 17.DeBock F, et al. Alpha2-adrenoreceptor activation inhibits LTP and LTD in the basolateral amygdala: involvement of Gi/o-protein-mediated modulation of Ca2+-channels and inwardly rectifying K+-channels in LTD. The European journal of neuroscience. 2003;17:1411–1424. doi: 10.1046/j.1460-9568.2003.02544.x. [DOI] [PubMed] [Google Scholar]
- 18.Huang CC, et al. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. The Journal of physiology. 2001;532:731–748. doi: 10.1111/j.1469-7793.2001.0731e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang YC, et al. Mediation of amphetamine-induced long-term depression of synaptic transmission by CB1 cannabinoid receptors in the rat amygdala. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:10311–10320. doi: 10.1523/JNEUROSCI.23-32-10311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mato S, et al. Role of the cyclic-AMP/PKA cascade and of P/Q-type Ca++ channels in endocannabinoid-mediated long-term depression in the nucleus accumbens. Neuropharmacology. 2008;54:87–94. doi: 10.1016/j.neuropharm.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 21.Robbe D, et al. Role of p/q-Ca2+ channels in metabotropic glutamate receptor 2/3-dependent presynaptic long-term depression at nucleus accumbens synapses. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:4346–4356. doi: 10.1523/JNEUROSCI.22-11-04346.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi K, et al. Ca2+-dependent mechanisms involved in presynaptic long-term depression at the hippocampal mossy fibre-CA3 synapse. The European journal of neuroscience. 1999;11:1633–1638. doi: 10.1046/j.1460-9568.1999.00578.x. [DOI] [PubMed] [Google Scholar]
- 23.Stanton PK, Gage AT. Distinct synaptic loci of Ca2+/calmodulin-dependent protein kinase II necessary for long-term potentiation and depression. Journal of neurophysiology. 1996;76:2097–2101. doi: 10.1152/jn.1996.76.3.2097. [DOI] [PubMed] [Google Scholar]
- 24.Heifets BD, et al. Interneuron activity controls endocannabinoid-mediated presynaptic plasticity through calcineurin. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10250–10255. doi: 10.1073/pnas.0711880105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li ST, et al. Calcineurin plays different roles in group II metabotropic glutamate receptor- and NMDA receptor-dependent long-term depression. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:5034–5041. doi: 10.1523/JNEUROSCI.22-12-05034.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiological reviews. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- 27.Azad SC, et al. Activation of CB1 specifically located on GABAergic interneurons inhibits LTD in the lateral amygdala. Learning & memory. 2008;15:143–152. doi: 10.1101/lm.741908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chevaleyre V, et al. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang CC, et al. A single in vivo cocaine administration impairs 5-HT(1B) receptor-induced long-term depression in the nucleus accumbens. Journal of neurochemistry. 2013;125:809–821. doi: 10.1111/jnc.12227. [DOI] [PubMed] [Google Scholar]
- 30.Kaeser PS, Sudhof TC. RIM function in short- and long-term synaptic plasticity. Biochemical Society transactions. 2005;33:1345–1349. doi: 10.1042/BST0331345. [DOI] [PubMed] [Google Scholar]
- 31.Huang L, et al. Activation of mGluRII induces LTD via activation of protein kinase A and protein kinase C in the dentate gyrus of the hippocampus in vitro. Neuropharmacology. 1999;38:73–83. doi: 10.1016/s0028-3908(98)00168-3. [DOI] [PubMed] [Google Scholar]
- 32.Kamsler A, et al. Presynaptic m1 muscarinic receptors are necessary for mGluR long-term depression in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1618–1623. doi: 10.1073/pnas.0912540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pelkey KA, et al. mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron. 2005;46:89–102. doi: 10.1016/j.neuron.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 34.Stanton PK. Transient protein kinase C activation primes long-term depression and suppresses long-term potentiation of synaptic transmission in hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:1724–1728. doi: 10.1073/pnas.92.5.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J, et al. DGKiota regulates presynaptic release during mGluR-dependent LTD. The EMBO journal. 2011;30:165–180. doi: 10.1038/emboj.2010.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kahn L, et al. Group 2 metabotropic glutamate receptors induced long term depression in mouse striatal slices. Neuroscience letters. 2001;316:178–182. doi: 10.1016/s0304-3940(01)02397-7. [DOI] [PubMed] [Google Scholar]
- 37.Bender VA, et al. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crozier RA, et al. Deprivation-induced synaptic depression by distinct mechanisms in different layers of mouse visual cortex. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1383–1388. doi: 10.1073/pnas.0609596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sjostrom PJ, et al. Endocannabinoid-dependent neocortical layer-5 LTD in the absence of postsynaptic spiking. Journal of neurophysiology. 2004;92:3338–3343. doi: 10.1152/jn.00376.2004. [DOI] [PubMed] [Google Scholar]
- 40.Chepkova AN, et al. Developmental alterations of DHPG-induced long-term depression of corticostriatal synaptic transmission: switch from NMDA receptor-dependent towards CB1 receptor-dependent plasticity. Pflugers Archiv: European journal of physiology. 2009;459:131–141. doi: 10.1007/s00424-009-0714-7. [DOI] [PubMed] [Google Scholar]
- 41.Santschi LA, et al. Activation of receptors negatively coupled to adenylate cyclase is required for induction of long-term synaptic depression at Schaffer collateral-CA1 synapses. Journal of neurobiology. 2006;66:205–219. doi: 10.1002/neu.20213. [DOI] [PubMed] [Google Scholar]
- 42.Upreti C, et al. Role of presynaptic metabotropic glutamate receptors in the induction of long-term synaptic plasticity of vesicular release. Neuropharmacology. 2013;66:31–39. doi: 10.1016/j.neuropharm.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adermark L, et al. Endocannabinoid-dependent plasticity at GABAergic and glutamatergic synapses in the striatum is regulated by synaptic activity. The European journal of neuroscience. 2009;29:32–41. doi: 10.1111/j.1460-9568.2008.06551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yin HH, et al. The role of protein synthesis in striatal long-term depression. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:11811–11820. doi: 10.1523/JNEUROSCI.3196-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zakharenko SS, et al. Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron. 2002;35:1099–1110. doi: 10.1016/s0896-6273(02)00898-x. [DOI] [PubMed] [Google Scholar]
- 46.Jung KM, et al. Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nature communications. 2012;3:1080. doi: 10.1038/ncomms2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tzounopoulos T, et al. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- 48.Johnson KA, et al. Activation of group II metabotropic glutamate receptors induces long-term depression of excitatory synaptic transmission in the substantia nigra pars reticulata. Neuroscience letters. 2011;504:102–106. doi: 10.1016/j.neulet.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kasanetz F, et al. Prefrontal synaptic markers of cocaine addiction-like behavior in rats. Molecular psychiatry. 2013;18:729–737. doi: 10.1038/mp.2012.59. [DOI] [PubMed] [Google Scholar]
- 50.Lin HC, et al. Activation of group II metabotropic glutamate receptors induces long-term depression of synaptic transmission in the rat amygdala. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20:9017–9024. doi: 10.1523/JNEUROSCI.20-24-09017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Surmeier DJ, et al. Modulation of Ca2+ currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- 52.Zhang XL, et al. Gbetagamma and the C terminus of SNAP-25 are necessary for long-term depression of transmitter release. PloS one. 2011;6:e20500. doi: 10.1371/journal.pone.0020500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laezza F, et al. Long-term depression in hippocampal interneurons: joint requirement for pre- and postsynaptic events. Science. 1999;285:1411–1414. doi: 10.1126/science.285.5432.1411. [DOI] [PubMed] [Google Scholar]
- 54.Pelkey KA, et al. Compartmentalized Ca(2+) channel regulation at divergent mossy-fiber release sites underlies target cell-dependent plasticity. Neuron. 2006;52:497–510. doi: 10.1016/j.neuron.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 55.Pelkey KA, et al. State-dependent cAMP sensitivity of presynaptic function underlies metaplasticity in a hippocampal feedforward inhibitory circuit. Neuron. 2008;60:980–987. doi: 10.1016/j.neuron.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pelkey KA, McBain CJ. Target-cell-dependent plasticity within the mossy fibre-CA3 circuit reveals compartmentalized regulation of presynaptic function at divergent release sites. The Journal of physiology. 2008;586:1495–1502. doi: 10.1113/jphysiol.2007.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi K, et al. Presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapse. Science. 1996;273:648–650. doi: 10.1126/science.273.5275.648. [DOI] [PubMed] [Google Scholar]
- 58.Levenes C, et al. Cannabinoids decrease excitatory synaptic transmission and impair long-term depression in rat cerebellar Purkinje cells. The Journal of physiology. 1998;510 (Pt 3):867–879. doi: 10.1111/j.1469-7793.1998.867bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gerdeman GL, et al. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nature neuroscience. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- 60.Robbe D, et al. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heifets BD, Castillo PE. Endocannabinoid signaling and long-term synaptic plasticity. Annual review of physiology. 2009;71:283–306. doi: 10.1146/annurev.physiol.010908.163149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lovinger DM, Mathur BN. Endocannabinoids in striatal plasticity. Parkinsonism & related disorders. 2012;18(Suppl 1):S132–134. doi: 10.1016/S1353-8020(11)70041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mathur BN, et al. Voltage drives diverse endocannabinoid signals to mediate striatal microcircuit-specific plasticity. Nature neuroscience. 2013;16:1275–1283. doi: 10.1038/nn.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Younts TJ, Castillo PE. Endogenous cannabinoid signaling at inhibitory interneurons. Current opinion in neurobiology. 2013;26C:42–50. doi: 10.1016/j.conb.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mathur BN, Lovinger DM. Endocannabinoid-dopamine interactions in striatal synaptic plasticity. Frontiers in pharmacology. 2012;3:66. doi: 10.3389/fphar.2012.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haj-Dahmane S, Shen RY. Regulation of plasticity of glutamate synapses by endocannabinoids and the cyclic-AMP/protein kinase A pathway in midbrain dopamine neurons. The Journal of physiology. 2010;588:2589–2604. doi: 10.1113/jphysiol.2010.190066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kheirbek MA, et al. Adenylyl cyclase type 5 contributes to corticostriatal plasticity and striatum-dependent learning. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:12115–12124. doi: 10.1523/JNEUROSCI.3343-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo J, Ikeda SR. Endocannabinoids modulate N-type Ca2+ channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Molecular pharmacology. 2004;65:665–674. doi: 10.1124/mol.65.3.665. [DOI] [PubMed] [Google Scholar]
- 69.Twitchell W, et al. Cannabinoids inhibit N- and P/Q-type Ca2+ channels in cultured rat hippocampal neurons. Journal of neurophysiology. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- 70.Robbe D, et al. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2001;21:109–116. doi: 10.1523/JNEUROSCI.21-01-00109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yasuda H, et al. Regulation of excitability and plasticity by endocannabinoids and PKA in developing hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3106–3111. doi: 10.1073/pnas.0708349105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Azad SC, et al. Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learning & memory. 2003;10:116–128. doi: 10.1101/lm.53303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Daniel H, et al. Mechanisms underlying cannabinoid inhibition of presynaptic Ca2+ influx at parallel fibre synapses of the rat cerebellum. The Journal of physiology. 2004;557:159–174. doi: 10.1113/jphysiol.2004.063263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sjostrom PJ, et al. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- 75.Centonze D, et al. Dopamine, acetylcholine and nitric oxide systems interact to induce corticostriatal synaptic plasticity. Reviews in the neurosciences. 2003;14:207–216. doi: 10.1515/revneuro.2003.14.3.207. [DOI] [PubMed] [Google Scholar]
- 76.Fino E, et al. Asymmetric spike-timing dependent plasticity of striatal nitric oxide-synthase interneurons. Neuroscience. 2009;160:744–754. doi: 10.1016/j.neuroscience.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 77.Wang Y, et al. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature. 1997;388:593–598. doi: 10.1038/41580. [DOI] [PubMed] [Google Scholar]
- 78.Grueter BA, et al. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nature neuroscience. 2010;13:1519–1525. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsetsenis T, et al. Rab3B protein is required for long-term depression of hippocampal inhibitory synapses and for normal reversal learning. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:14300–14305. doi: 10.1073/pnas.1112237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lonart G, et al. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- 81.Iremonger KJ, Bains JS. Retrograde opioid signaling regulates glutamatergic transmission in the hypothalamus. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:7349–7358. doi: 10.1523/JNEUROSCI.0381-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Iremonger KJ, et al. Dual regulation of anterograde and retrograde transmission by endocannabinoids. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:12011–12020. doi: 10.1523/JNEUROSCI.2925-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wamsteeker Cusulin JI, et al. Glucocorticoid feedback uncovers retrograde opioid signaling at hypothalamic synapses. Nature neuroscience. 2013;16:596–604. doi: 10.1038/nn.3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Piskorowski RA, Chevaleyre V. Delta-opioid receptors mediate unique plasticity onto parvalbumin-expressing interneurons in area CA2 of the hippocampus. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:14567–14578. doi: 10.1523/JNEUROSCI.0649-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Atwood BK, et al. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nature neuroscience. 2014 doi: 10.1038/nn.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van den Pol AN, et al. Neuropeptide Y-mediated long-term depression of excitatory activity in suprachiasmatic nucleus neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1996;16:5883–5895. doi: 10.1523/JNEUROSCI.16-18-05883.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mathur BN, et al. Serotonin induces long-term depression at corticostriatal synapses. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:7402–7411. doi: 10.1523/JNEUROSCI.6250-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Burnstock G, et al. Adenosine and ATP receptors in the brain. Current topics in medicinal chemistry. 2011;11:973–1011. doi: 10.2174/156802611795347627. [DOI] [PubMed] [Google Scholar]
- 89.Dunwiddie TV, Diao L. Regulation of extracellular adenosine in rat hippocampal slices is temperature dependent: role of adenosine transporters. Neuroscience. 2000;95:81–88. doi: 10.1016/s0306-4522(99)00404-2. [DOI] [PubMed] [Google Scholar]
- 90.Hosseinmardi N, et al. Theta pulse stimulation: a natural stimulus pattern can trigger long-term depression but fails to reverse long-term potentiation in morphine withdrawn hippocampus area CA1. Brain research. 2009;1296:1–14. doi: 10.1016/j.brainres.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 91.Sun CL, et al. Cerebellar long-term depression is deficient in Niemann-Pick type C disease mice. Cerebellum. 2011;10:88–95. doi: 10.1007/s12311-010-0233-2. [DOI] [PubMed] [Google Scholar]
- 92.Mato S, et al. Presynaptic homeostatic plasticity rescues long-term depression after chronic Delta 9-tetrahydrocannabinol exposure. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:11619–11627. doi: 10.1523/JNEUROSCI.2294-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vasquez C, Lewis DL. The CB1 cannabinoid receptor can sequester G-proteins, making them unavailable to couple to other receptors. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1999;19:9271–9280. doi: 10.1523/JNEUROSCI.19-21-09271.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chiu CQ, et al. Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:7236–7248. doi: 10.1523/JNEUROSCI.0736-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pan B, et al. D2 dopamine receptor activation facilitates endocannabinoid-mediated long-term synaptic depression of GABAergic synaptic transmission in midbrain dopamine neurons via cAMP-protein kinase A signaling. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:14018–14030. doi: 10.1523/JNEUROSCI.4035-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kheder A, Nair KP. Spasticity: pathophysiology, evaluation and management. Practical neurology. 2012;12:289–298. doi: 10.1136/practneurol-2011-000155. [DOI] [PubMed] [Google Scholar]
- 97.Munoz A, et al. Combined 5-HT1A and 5-HT1B receptor agonists for the treatment of L-DOPA-induced dyskinesia. Brain: a journal of neurology. 2008;131:3380–3394. doi: 10.1093/brain/awn235. [DOI] [PubMed] [Google Scholar]
- 98.Olive MF. Metabotropic glutamate receptor ligands as potential therapeutics for addiction. Current drug abuse reviews. 2009;2:83–98. doi: 10.2174/1874473710902010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pacher P, Kunos G. Modulating the endocannabinoid system in human health and disease--successes and failures. The FEBS journal. 2013;280:1918–1943. doi: 10.1111/febs.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chevaleyre V, et al. Endocannabinoid-mediated synaptic plasticity in the CNS. Annual review of neuroscience. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- 101.Singla S, et al. Mechanisms for synapse specificity during striatal long-term depression. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:5260–5264. doi: 10.1523/JNEUROSCI.0018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nahir B, et al. mGluR-mediated and endocannabinoid-dependent long-term depression in the hilar region of the rat dentate gyrus. Neuropharmacology. 2010;58:712–721. doi: 10.1016/j.neuropharm.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Doreulee N, et al. Histamine H(3) receptors depress synaptic transmission in the corticostriatal pathway. Neuropharmacology. 2001;40:106–113. doi: 10.1016/s0028-3908(00)00101-5. [DOI] [PubMed] [Google Scholar]
- 104.Brown RE, Haas HL. On the mechanism of histaminergic inhibition of glutamate release in the rat dentate gyrus. The Journal of physiology. 1999;515 (Pt 3):777–786. doi: 10.1111/j.1469-7793.1999.777ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin Y, Phillis JW. Muscarinic agonist oxotremorine-M-induced long-term depression in rat cerebral cortex. Brain research bulletin. 1991;27:115–117. doi: 10.1016/0361-9230(91)90291-q. [DOI] [PubMed] [Google Scholar]
- 106.Pancani T, et al. M4 mAChR-Mediated Modulation of Glutamatergic Transmission at Corticostriatal Synapses. ACS chemical neuroscience. 2014 doi: 10.1021/cn500003z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ronesi J, et al. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24:1673–1679. doi: 10.1523/JNEUROSCI.5214-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Taussig R, et al. Distinct patterns of bidirectional regulation of mammalian adenylyl cyclases. The Journal of biological chemistry. 1994;269:6093–6100. [PubMed] [Google Scholar]
- 109.Herlitze S, et al. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 110.Ikeda SR. Voltage-dependent modulation of N-type Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 111.Dascal N, et al. Atrial G protein-activated K+ channel: expression cloning and molecular properties. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:10235–10239. doi: 10.1073/pnas.90.21.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kretz R, et al. Presynaptic inhibition produced by an identified presynaptic inhibitory neuron. II. Presynaptic conductance changes caused by histamine. Journal of neurophysiology. 1986;55:131–146. doi: 10.1152/jn.1986.55.1.131. [DOI] [PubMed] [Google Scholar]
- 113.Logothetis DE, et al. The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- 114.Blackmer T, et al. G protein betagamma directly regulates SNARE protein fusion machinery for secretory granule exocytosis. Nature neuroscience. 2005;8:421–425. doi: 10.1038/nn1423. [DOI] [PubMed] [Google Scholar]
- 115.Blackmer T, et al. G protein betagamma subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science. 2001;292:293–297. doi: 10.1126/science.1058803. [DOI] [PubMed] [Google Scholar]
- 116.Yoon EJ, et al. Gbetagamma interferes with Ca2+-dependent binding of synaptotagmin to the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. Molecular pharmacology. 2007;72:1210–1219. doi: 10.1124/mol.107.039446. [DOI] [PubMed] [Google Scholar]
- 117.Diverse-Pierluissi M, et al. Transmitter-mediated inhibition of N-type Ca2+ channels in sensory neurons involves multiple GTP-binding proteins and subunits. Neuron. 1995;14:191–200. doi: 10.1016/0896-6273(95)90254-6. [DOI] [PubMed] [Google Scholar]
- 118.Stephens L, et al. A novel phosphoinositide 3 kinase activity in myeloid-derived cells is activated by G protein beta gamma subunits. Cell. 1994;77:83–93. doi: 10.1016/0092-8674(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 119.Pitcher JA, et al. Role of beta gamma subunits of G proteins in targeting the beta-adrenergic receptor kinase to membrane-bound receptors. Science. 1992;257:1264–1267. doi: 10.1126/science.1325672. [DOI] [PubMed] [Google Scholar]
- 120.Crespo P, et al. Ras-dependent activation of MAP kinase pathway mediated by G-protein beta gamma subunits. Nature. 1994;369:418–420. doi: 10.1038/369418a0. [DOI] [PubMed] [Google Scholar]