

Abstract
The MET receptor tyrosine kinase (RTK), implicated in risk for autism spectrum disorder (ASD) and in functional and structural circuit integrity in humans, is a temporally and spatially regulated receptor enriched in dorsal pallial-derived structures during mouse forebrain development. Here we report that loss or gain of function of MET in vitro or in vivo leads to changes, opposite in nature, in dendritic complexity, spine morphogenesis, and the timing of glutamatergic synapse maturation onto hippocampus CA1 neurons. Consistent with the morphological and biochemical changes, deletion of Met in mutant mice results in precocious maturation of excitatory synapse, as indicated by a reduction of the proportion of silent synapses, a faster GluN2A subunit switch, and an enhanced acquisition of AMPA receptors at synaptic sites. Thus, MET-mediated signaling appears to serve as a mechanism for controlling the timing of neuronal growth and functional maturation. These studies suggest that mistimed maturation of glutamatergic synapses leads to the aberrant neural circuits that may be associated with ASD risk.
Keywords: autism, glutamatergic circuit, hippocampus, MET receptor tyrosine kinase, mouse model, synaptogenesis
Introduction
Human genetic studies and brain imaging data have established MET as a risk factor for autism sprectrum disorder (ASD), a highly heritable psychiatric disorder with disrupted ontogeny of neural connectivity (Campbell et al., 2006, 2007; Geschwind and Levitt, 2007; Jackson et al., 2009; Sousa et al., 2009; Thanseem et al., 2010; Abrahams et al., 2013). We have previously shown that the rs1858830 ‘C’ allele reduces MET mRNA and protein levels in the brains of subjects with autism through altered interactions with identified transcription factors, providing a potential molecular basis for increased ASD risk (Campbell et al., 2006, 2007).
How does one go from low MET expression to influencing altered cognition, social and language skills, and executive functions seen in ASD? The rs1858830 “C” risk allele predicts atypical fMRI activation and deactivation patterns of human brain to social stimuli and reduced connectivity in temporoparietal lobes, areas known to have high levels of MET expression (Rudie et al., 2012). Moreover, in typically developing humans, the risk allele correlates with differences in trajectory of gray matter growth in temporal and posterior parietal regions (Hedrick et al., 2012), neocortical areas that express MET heavily (Judson et al., 2011; Mukamel et al., 2011). These finding are consistent with established biological roles of MET in normal CNS development, suggesting that MET signaling converges on biological processes relevant to ASD pathogenesis.
Normal brain development is instructed via molecular signaling mediated by growth factors that signal through protein receptor tyrosine kinases (RTKs) (Park and Poo, 2013). These act by complex downstream signaling, often functionally pleiotropic in nature. MET RTK and its ligand hepatocyte growth factor (HGF) mediate development of multiple peripheral organs (Cooper et al., 1984; Bottaro et al., 1991). MET and HGF also are expressed in the developing nervous system of rodents (Achim et al., 1997; Maina et al., 1997; Judson et al., 2009; Wu and Levitt, 2013), monkey (Judson et al., 2011), and humans (Mukamel et al., 2011; Hamasaki et al., 2014), where they influence many neurodevelopmental events, including neural induction, cell fate, axon guidance, and neuronal morphogenesis (Streit et al., 1995; Ebens et al., 1996; Hamanoue et al., 1996; Maina et al., 2001; Helmbacher et al., 2003; Gutierrez et al., 2004; Lim and Walikonis, 2008). While both Met heterozygous or homozygous states in mice alter local cortical interlaminar excitatory connectivity (Qiu et al., 2011), the ways through which MET signaling impacts functional synapse formation during brain development have not been defined. We postulate that disrupted development of glutamatergic circuits is a candidate mechanism to translate the lower levels of MET into the wider pathology of ASD (Südhof, 2008; Penzes et al., 2011; Clement et al., 2012; Zoghbi and Bear, 2012). Taking advantage of the enrichment of MET expression by CA1 hippocampal pyramidal neurons (Judson et al., 2009), we used complementary in vitro and in vivo methods to examine how altered MET signaling impacts synaptic development in search for a potential synaptic basis for MET-induced ASD genetic risk.
Materials and Methods
Mice.
Time-pregnant C57BL/6 mice purchased from Charles Rivers or bred in house were used for hippocampal neuron cell cultures and in utero electroporation (IUEP) studies. The day of vaginal plug detection was designated as E0.5 and the day of birth as P0. The dorsal pallial-specific conditional mutant mice (Metfx/fx; Emx1cre) were generated and genotyped using previously described methods (Judson et al., 2010). Homozygous female Metfx/fx mice were mated to hemizygote Metfx/+ male mice that also contained the Emx1Cre knock-in allele (Metfx/+; Emx1Cre) to produce the experimental mice (Metfx/fx; Emx1Cre, and its Metfx/fx controls).
Both Metfx/fx (provided by Dr. Snorri Thorgeirsson, National Institutes of Health/Center for Cancer Research, Bethesda, Maryland) and Emx1Cre breeding line (provided by Dr. Kevin Jones, University of Colorado, Boulder, Colorado) were back-crossed onto the C57BL/6J background for >10 generations in our laboratory. All research using mice were performed in accordance with animal experimental protocols approved by the Institutional Animal Care and Use Committee at University of Southern California, The University of Arizona and conformed to National Institutes of Health guidelines. Efforts were made to minimize animal suffering and to reduce the number of animals used.
DNA constructs.
Mouse full-length Met cDNA (NM_008591) expressed in the form of pMEX vector (Jeffers et al., 1998) was a generous gift from Dr. G. Vande Woude (Van Andel Institute). Neurons transfected or electroporated with this construct in combination with pEGFP-C3 are designated as ‘MET’ group. To construct an RNAi vector for MET knockdown, we initially used a lentiviral vector (PLVTHM) (Wiznerowicz and Trono, 2003) and tested the RNAi efficiency in HEK293 cells. Later, we used the pSuper vector for more efficient and faster expression of the RNAi sequences in neurons (Kim et al., 2005). Three 19-nt RNAi sequences (sequence #1, 5′-GCAGTGAATTAGTTCGCTA-3′; sequence #2, 5′-GCACAAAGCAAGCCAGATT-3′; sequence #3, 5′-GCAGCCTGATTGTGCATTT-3′) were designed to target the ORF region of Met using the BLOCK-iT online algorithm (Invitrogen). The 19-nt sequence targets mRNA transcripts and is separated by a 9-nt spacer loop (TCTCTTGAA), and followed by the reverse complement sequence of the target sequence. The sequences were cloned into the BglII and HindIII sites of pSuper vector. These RNAi sequences were unique to mouse Met gene and did not target any other sequences when queried onto the mouse genome using Blastn. Further tests for RNAi efficiency found that sequence #1 was most effective in suppressing Met gene expression. This pSuper-siRNA #1 construct was designated as ‘RNAi’ group when used for neuron transfection or electroporation in combination with pEGFP-C3 vector. The scrambled control sequence for RNAi (5′-GCTAGATAGATCTGTGTCA-3′) (‘scr RNA’) also is expressed from the pSuper vector and does not target any known mouse cDNA sequence.
IEUP.
IUEP was used to deliver exogenous DNA and RNAi molecules to developing hippocampal CA1 neurons. The procedure is similar to that described previously (Navarro-Quiroga et al., 2007). Briefly, the abdominal wall of the E14.5 time-pregnant mice (anesthetized with isoflurane) was opened by a ∼2 cm incision. The uterine horn and embryos were exposed and gently pulled outside the abdomen cavity. A sharp sterile needle filled with the nucleic acid mixture (with 0.05% fast green) was used to penetrate through the uterine wall and the cortical plate of the developing brain to reach the lateral ventricle. Approximately 1 μl solution was pressure-injected into the lateral ventricles. Next, a tweezer-shaped electrode pair was positioned to clamp the embryo heads in a mediolateral orientation so that the electrical field targeted the developing hippocampal CA1 subfield. Electrical voltage pulses (50 V, 50 ms and 5 pulses, generated by an ECM 830 electroporator) were used to electroporate the nucleic acids into the developing hippocampus. The embryos were then returned to their original position inside the dam. A sterile, size #6-0 fine monofilament medical suture was used to close the abdominal wall. Bupivacaine local anesthetic (0.25% gel) was applied topically at the incision site. Offspring born to the IUEP dam was harvested for experiments at designated postnatal time points.
Primary neuronal culture, transfection, and morphometrical analysis.
Hippocampal cultures were prepared from E16.5 time-pregnant C57BL/6 mice, similar to that previously described (Qiu and Weeber, 2007). Briefly, the dams were deeply anesthetized and the embryos harvested. The hippocampi were dissected out in ice-cold Hank's balanced salt solution, digested with 0.5 mg/ml papain at 37°C for 20 min. Where necessary, tails of the embryos were collected for genotyping. The hippocampi were then mechanically dissociated into a single-cell suspension using a plastic transfer pipette with small orifice. Neurons were grown on poly-d-lysine-coated 12 mm glass coverslips residing in 24-well plates at a density of 30,000 cells/cm2 in Neurobasal medium supplemented with 2% B27 (Invitrogen). A lower plating density (10,000 cells/cm2) was adopted when neurons were used for immunocytochemistry staining to facilitate the discrete identification of individual immunopositive puncta. Every 7 d after plating, 300 μl fresh complete culture medium supplemented with 10 μm cytosine arabinoside was added to the original 600 μl plating volume to inhibit glial cells growth.
Neurons were transfected at day 3–14 with Met cDNA and RNAi constructs, in combination with pEGFP-C3, to reveal neuron morphology. We used a calcium phosphate transfection method (ProFection mammalian transfection kit, Promega), according to the manufacturer's protocol; 8 μg total DNA was used to generate 600 μl total precipitates, which was added to each well at a 50 μl volume. Wells containing transfected cells were briefly washed with warm neurobasal medium 2 h later and then continued to grow in conditioned medium collected from sister wells until the neurons were harvested for immunocytochemistry or electrophysiology recording. For morphological analysis, neurons grown on coverslips were fixed in 4% PFA in PBS for 20 min, washed with PBS, and placed in a chamber slide for confocal microscopy analysis. Confocal images were collected at different culture day in vitro (DIV) with 20× (Plan-Apochromat, NA 0.8, for neurite growth analysis) or 63× objectives (Plan-Apochromat, NA 1.4, for dendritic spine analysis). Regions of interest were identified by selection of 4 or 5 quadrants of each coverslip. The samples were coded so that analysis was performed blind to treatment. Total axonal and dendritic length was calculated following manual tracing using ImageJ. Axons were differentiated from dendrites based on much smaller diameter and no tapering. Dendritic protrusion density, spine density, and head area size were quantified on secondary dendritic branches located between 150 and 300 μm from the pyramidal shaped soma, using a maximum intensity projected image derived from confocal Z-stacks (1024 × 1024 pixels with 2× digital zoom and 0.2 μm Z resolution). Dendritic spine head area was quantified by manual tracing in the defined regions of interests along the dendrites. The area, maximum length, and head width of each spine were measured using ImageJ. Analysis was performed in at least four different culturing sessions for each time point.
For immunocytochemical labeling and protein colocalization studies, cultured neurons at 22–28 DIV were double-fixed with 4% PFA in 0.1 m PB, followed by prechilled (−20°C) methanol for 10 min. The neurons were washed in PBS, made permeable in 0.2% Triton X-100 for 30 min, and then blocked with 10% normal donkey serum and 1% BSA. Neurons were then incubated in primary antibodies (rabbit anti-GluR1 and mouse anti-NR1, both 1:250 dilutions, rabbit anti-synapsin I, and mouse anti-PSD-95, all from Millipore) for 24 h. Neurons were washed briefly for three times, followed by an overnight incubation with AlexaFluor 488-conjugated donkey anti-rabbit secondary IgG and AlexaFluor-555-conjugated donkey anti-mouse IgG (Invitrogen). Confocal images of double staining were collected on a Zeiss LSM 710 microscope with a 63× oil-immersion objective. All digital images across experiments were acquired using identical settings for laser power, photomultiplier gain, offset and a fixed pinhole size of one airy unit. Images were blindly coded and analyzed using MetaMorph software (Molecular Devices). For colocalization analysis, at least three quadrants were selected for each coverslip and immunopositive GluA1 and GluN1, or PSD-95 and synapsin I puncta were thresholded to include all visually recognizable punctate labeling. The thresholded puncta in one channel was used to create a mask and then overlaid on the second channel to obtain the overlapping/colocalizing regions. At least eight complete neuronal profiles in each of three different culture sessions were analyzed.
CA1 neuron dendritic arbor reconstruction and spine quantification.
Offspring (P22-P25) born to IUEP dams were transcardially perfused with 4% PFA. The brains were collected and postfixed in 4% PFA overnight, sequentially cryoprotected in 10%, 20%, and 30% sucrose. The brains (n = 4–6/treatment group) were cut into 60 μm sections using a sliding microtome (Leica Microsystems), washed in 0.01 m PBS, and mounted on slides using Vectashield mounting medium (Vector Laboratories). Coronal slices containing only the middle third septotemporal levels of hippocampus were used for analysis. To minimize analyzing CA1 neurons with truncated dendritic trees, only CA1 neurons whose soma is located within the middle 20 μm were selected.
Neurons that express GFP, or in combination with MET or RNAi, were traced using a Neurolucida (Microbrightfield) platform to reconstruct their 3D dendritic arbor, similar to that described previously by our group (Judson et al., 2010). Slides were coded for blind analysis. Dendritic measures included number of primary, secondary dendrite and total dendritic length in the basal and apical compartments. In addition, Sholl analyses (Sholl, 1953) were performed in these reconstructed neurons to detect regional alterations in dendritic arbor structure and to quantify cell parameters, including dendritic length, branching complexity, and branch point locations in the denomination of a series concentric circles (20 μm radius increment from soma) on the entire dendritic arbors. Between three and five cells were reconstructed per mouse, and at least three mice were reconstructed for each treatment group.
We used Imaris software (version 7.6.5, 64 bit; Bitplane) for semiautomated dendritic spine analysis in collected confocal Z-stack images, using a similar approach described by Shen et al. (2008). For better consistency, we selected secondary apical dendritic branches that are located at 100–200 μm from center of soma for analysis. Two or three dendritic segments were quantified per neuron. Image Z-stacks were acquired using a Zeiss LSM 710 confocal microscope using a 63× (NA 1.4) oil-immersion objective, with 2× digital cropping and frame size of 1024 × 1024 pixels, and 0.2 μm Z interval. This yields a voxel size of 0.069 × 0.069 × 0.2 μm3. The confocal files were imported into Imaris software and visualized in the ‘surpass’ view. The Filament module with Autopath (no loops) was used for 3D rendering of each dendritic segment. Renderings were subsequently edited manually to include or exclude spines that are misidentified. When spine classification (stubby, mushroom, long thin, and filopodia) is implemented, we used the Imaris spine classification module with default settings. This module is executed as a graphic user interface in MATLAB that can also be user-defined based on the ratio of spine head and neck to the spine length. At least 1200 spines from three mice were constructed for each treatment group.
Surface protein biotinylation, crude synaptosome preparation, and Western blot analysis.
To assess total glutamate receptor subunit protein levels and the membrane-bound fractions at synaptic sites, we treated P14 acute hippocampal slices (pooled from 4 or 5 mice per genotype) with 0.25 mg/ml sulfo-NHS-S-S-biotin for 30 min at 4°C, using a Pinpoint cell surface protein isolation kit (Pierce Biotechnology), similar to that described previously for biotinylation in hippocampal slices (Qiu and Weeber, 2007). Micro-dissected CA1 regions were homogenized in cold buffer (containing 4 mm HEPES, 320 mm sucrose, and 1:50 protease inhibitor mixture; P8340, Sigma). The homogenates were centrifuged at 1500 × g for 10 min. The supernatant was centrifuged at 16,000 × g for 20 min to yield a pellet containing the crude synaptosome fraction. This fraction was then equally divided into two aliquots; one part was used to assess total synaptosomal proteins using SDS-PAGE and Western blot analysis, the other half was lysed and resuspended in ice-cold water containing a protease inhibitor mixture, and then mixed with immobilized NeutrAvidin beads (Pierce Biotechnology) and rotated for 2 h at 4°C. The beads were washed three times in RIPA buffer, and all biotinylated synaptic membrane proteins were eluted with 2× Laemmli buffer supplemented with 50 mm DTT. The biotinylated proteins were then subjected to Western blot analysis to probe semiquantitatively different glutamate receptor subunits located in the synaptic membrane compartments.
A standard Western blot protocol was used to assess protein contents. Proteins harvested were suspended in NP40 cell lysis buffer supplemented with proteinase inhibitor mixture (1:50, Sigma P8340) and 1 mm PMSF, and quantified using a micro-BCA assay to ensure equal loading of sample. Samples were mixed with equal amount of 2× Laemmli loading buffer and boiled for 5 min, and then separated by 9% SDS-polyacrylamide gels. The proteins were transferred to PVDF membranes (Immobilon-P, Millipore), incubated with antibodies against proteins of interest (antibody diluted in 5% nonfat dry milk) overnight at 4°C. The PVDF membranes were then washed three times in 0.01 m PBS-Tween 20 and incubated with HRP-conjugated secondary antibodies (Promega) for 2 h at room temperature. Protein signals were developed using an enhanced chemiluminescence method (ECL Plus detection system, GE Healthcare; or SignalFire, Cell Signaling Technology). The following antibodies were used: from Santa Cruz Biotechnology, anti-MET (sc-8057), neuroligin 3 (sc-137052), CaMKII (sc-9035), AKT (sc-5298), PI3K-P110α (sc-7174); from Millipore Bioscience Research Reagents, rabbit anti-GluA1 (AB1504) and anti-synapsin I (AB1543), mouse anti-GluN1 (MAB363 for Western blot; MAB1586 for immunocytochemistry), rabbit anti-GluN2A (07-632), rabbit anti-GluN2B (05-920), Homer 1 (ABN37), mouse anti-PSD-95 (AB9708), SynGap (06-900), mGluR1 (07-617). Monoclonal Shank3 antibody was purchased from NeuroMab. Rabbit anti-GAPDH was from Cell Signaling Technology. The final dilution of antibodies was between 1:1000 and 1:2000. The optical density of immunoreactive bands was quantified by densitometry using FIJI/ImageJ.
Patch-clamp whole-cell recording in CA1 neurons.
Mice born to IUEP dams were killed at P22-P28 and coronal slices containing the middle septo-temporal levels of hippocampus were used for recording. Slices from Metfx/fx; emx1cre and control mice were prepared from animals at various developmental ages (P6-P9, P12-P14, and P24-P28). Slices were cut in ice-cold ACSF saturated with 95% O2 and 5% CO2, incubated at 32°C for 30 min, and kept at room temperature before switched to the recording chamber. The chamber was circulated with 95% O2- and 5% CO2-saturated with ACSF, which contains (in mm) as follows: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1.2 MgCl2, 2.0 CaCl2, and 10 glucose, pH 7.3–7.4. We identified individual CA1 neurons that are transfected with GFP, MET, or RNAi groups under epifluorescence illumination and targeted for recording under differential interference contrast microscopy (Olympus BX-51WI platform). For mEPSC recording, the electrode (4–7 mΩ electrical resistance) contained a potassium-based solution (in mm) as follows: 130 K-gluconate, 4 KCl, 2 NaCl, 10 HEPES, 4 ATP-Mg, 0.3 GTP-Na, 1 EGTA, and 14 phosphocreatine (pH 7.2, 295 mOsm). Neurons were voltage-clamped at −65 mV (close to GABAA reversal potential) and the ACSF was supplemented with 1 μm TTX. To record mIPSCs, neuron were voltage-clamped at −65 mV. The pipette solution had symmetrical chloride ions and contained the following (in mm): 135 KCl, 2 NaCl, 10 HEPES, 2 EGTA, 5 QX314-Cl, 1 MgSO4, 4 ATP-Mg, 0.3 GTP-Tris (pH 7.4, with KOH, 290–295 mOsm), and the ACSF was supplemented with 1 μm TTX, 10 μm NBQX, and 50 μm AP5.
For experiments that require voltage clamp at higher levels than resting membrane potentials (AMPA/NMDAR current [A:N] ratio, ifenprodil sensitivity and silent synapse quantification using minimum stimulation protocols), the patch electrodes were filled with a solution containing (in mm) as follows: 125 Cs gluconate, 5 tetraethylammonium-Cl, 10 HEPES, 8 NaCl, 5 QX314-Cl, 4 Mg2+-ATP, 2.5 CsCl, 0.3 Na3GTP, 0.2 EGTA, 10 phosphocreatine, and adjusted to pH 7.2, 280–290 mOsm. The ACSF was supplemented with 5 μm bicuculline for the A:N ratio experiments. An additional 10 μm NBQX was added for the ifenprodil sensitivity experiments, during which NMDAR-mediated whole-cell current was isolated at 40 mV. Evoked synaptic currents were elicited by a stimulus delivered through a bipolar tungsten electrode (FHC) that was placed at the CA1 stratum radiatum. The stimulus was 0.1 ms in duration and generated through a Digidata 1440A interface and delivered through an iso-flex stimulus isolater (A.M.P.I.). Presynaptic fibers were stimulated at 0.2 Hz to obtain a monosynaptic response. Neuronal signals were collected using a Multiclamp 700B amplifier (Molecular Devices), low-pass filtered at 2 kHz, and digitized at 10 or 20 kHz using a Digidata 1440A board under control of pClamp 10.2 program (Molecular Devices). Series resistance (<25 mΩ) was constantly monitored, and only those with <15% variation were included for analysis.
We used a minimum stimulation protocol to activate a small number of axons or release sites to estimate the proportion of silent synapses and the quantal profiles. We adopted a stimulation intensity that produces successful AMPAR EPSCs at −70 mV holding potential that are interleaved with failures. The neurons were then voltage-clamped at 40 mV to collect more trials. The percentage of AMPAR-only silent synapses was calculated from the failure rates of AMPAR EPSCs and compound EPSCs (Liao et al., 1995; Qiu and Weeber, 2007). The analysis of quantal nature of AMPAR-mediated synaptic responses was essentially as previously described (Traynelis et al., 1993) and was implemented by MATLAB scripts. Minimum stimulation-evoked successful AMPAR events were pooled, aligned by their rising phase at the half-maximal amplitude, and averaged. Mean EPSCAMPA was peak scaled to each individual trace and was subtracted from it to obtain the difference. The decay phase variance of the current difference was calculated and plotted against the binned amplitude of the averaged response. The AMPAR single-channel current and numbers were estimated by fitting the first 50% data points back-calculated from the end-of-decay baseline to the following parabolic equation:
 |
where σ2 is the variance, i is the weighted-mean single-channel current, I is the mean current, N is the number of active AMPAR channels opening at the peak of the mean current (i.e., number of physical channels × open probability), and σb2 is the variance of the background. The single-channel conductance (γ) is calculated as follows:
 |
where V denotes driving force, which is the holding potential minus reversal potential (0 mV) for AMPAR channels.
Statistical analysis.
All results are expressed as mean ± SEM. For the electrophysiological studies, sample size n represents number of cells or slices obtained from at least three mice. For Western blot quantifications between groups, a one-way ANOVA or Student's t test was used. To compare cumulative distribution of mEPSC amplitude, Kolmogorov–Smirnov (K-S) test was used. Where necessary, Dunn's multiple-comparison post hoc test was used to compare each genotype to wild-type. Pearson's χ2 test was used to compare the classification of dendritic spine types. Comparison of spine head size was made using Kruskal–Wallis test. Statistics were processed using SPSS or GraphPad Prism 5.0 software. p < 0.05 was considered statistically significant.
Results
MET expression and signaling competency in developing mouse hippocampal neurons
Developing hippocampal CA1 neurons, including axons, dendrites, and somata, are enriched for MET during late prenatal and early postnatal ages, with highest levels evident up to weaning (Achim et al., 1997; Thewke and Seeds, 1999; Judson et al., 2009), followed by a substantial decline before puberty. Dentate granule cells and interneurons do not express MET, and only a modest number of CA3 neurons express the receptor based on in situ hybridization analysis in primate and mouse at different developmental ages (Judson et al., 2009, 2011). Thus, analysis of MET protein and manipulation of MET receptor signaling reflects responses primarily in CA1 neurons. We first examined signaling competency of the MET receptor during development. In acute hippocampal slices prepared from postnatal day 3 (P3) mice and maintained in ACSF, HGF (50 ng/ml, 30 min) treatment induces MET receptor activation, measured by an antibody that specifically detects the phosphorylated Tyr1234/1235 in the kinase domain of human MET (Naldini et al., 1991) (Fig. 1A; n = 3). The same Tyr1234/1235 phosporylation was obtained by treating embryonic hippocampal neurons grown in dissociated cultures for 7 DIV (Fig. 1A; n = 4).
Figure 1.
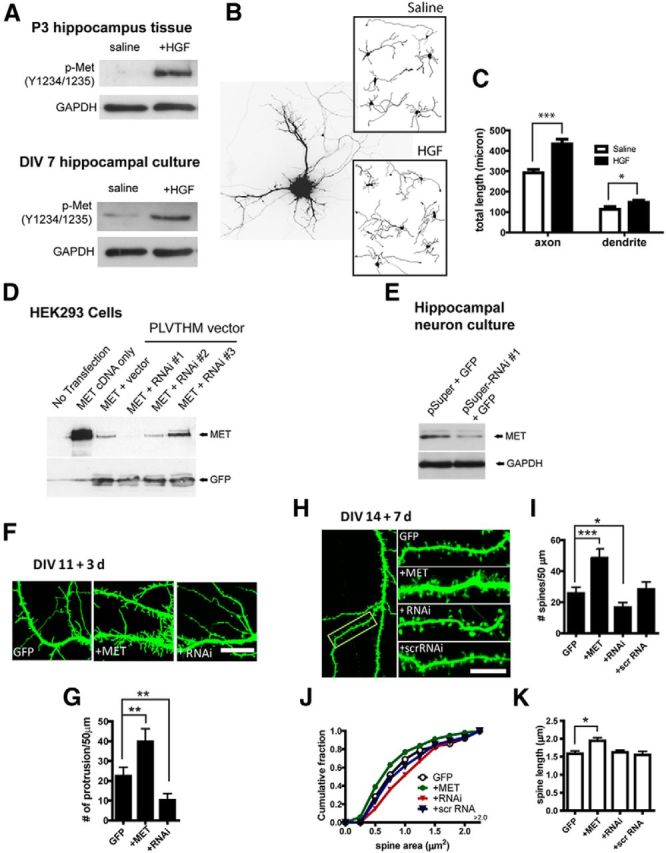
MET signaling affects growth and morphological development of hippocampal neurons in vitro. A, MET signaling competency in live P3 hippocampus slices and DIV 7 cultured hippocampal neuron. HGF stimulation leads to tyrosine (Y1234/1235) phosphorylation of MET. B, Cultured hippocampal neurons were transfected with GFP to reveal morphology. HGF treatment enhances the growth of both developing dendrites and axons. C, Quantification of neurite growth as shown in B (* p < 0.05; ***p < 0.001). D, Test of efficacy of three 19-nt RNAi sequences by cotransfection with MET cDNA in HEK293 cells. RNAi sequences were cloned into PLVTHM vector, which has bicistronic expression of GFP. RNAi #1 sequence is most efficient in reducing MET expression. E, Test of knockdown efficiency of RNAi sequence #1 on endogenous MET expression in cultured hippocampal neurons. This RNAi sequence was cloned into pSuper vector for faster expression and designated as ‘RNAi’ hereafter. F, MET OE and RNAi affect dendritic protrusions of cultured hippocampal neurons during the second week in culture. G, Quantification of F. MET OE significantly increased (**p < 0.01), while RNAi significantly decreased (**p < 0.01), the density of dendritic protrusions. H–K, MET OE or RNAi alters dendritic spine density and morphology (H). MET OE increases (***p < 0.001), while RNAi decreases (*p < 0.05), the density of dendritic spines (I). A scrambled sequence of RNAi was without effect. MET OE decreases, while RNAi increases, spine head area (J) (p < 0.01, for both effects). MET OE also significantly increases dendritic spine length (K) (*p < 0.05). Scale bars: F, H, 10 μm.
MET activation can promote neuronal growth in cultured neocortical neurons (Tyndall et al., 2007; Lim and Walikonis, 2008) and organotypic slices (Gutierrez et al., 2004). To directly examine the impact of MET signaling on CA1 neuron growth, cultures were treated with HGF (50 ng/ml) after initial plating (DIV 0), and neurite growth were determined and compared with saline-treated controls. Neurons were transfected with pEGFP-C3 (GFP) reporter plasmids at DIV 3 to reveal their morphology and their total neurite length was quantified at DIV5. HGF significantly increased both axonal and dendritic total length (Fig. 1B,C; HGF effects on axons, p < 0.001; on dendrites, p < 0.05; n = 24 neurons/3 cultures). Together with the receptor activation data, these results show that MET signaling via its ligand, HGF, is operating and capable of facilitating early neuronal growth in hippocampal CA1 neurons.
MET signaling regulates hippocampal neuron growth and dendritic morphology
We next asked whether altering MET signaling impacts neuronal morphological development. Dissociated hippocampal neurons were transfected with a Met cDNA expression construct for overexpression (OE) or an RNAi construct for loss-of-function studies. Of the three RNAi constructs tested, RNAi #1 (‘RNAi’) suppressed MET expression most efficiently in HEK293 cells (Fig. 1D; n = 2 experiments) and cultured hippocampal neurons (Fig. 1E; n = 3 experiments).
We next transfected DIV 11 neurons with GFP control vector, or in combination with MET or RNAi constructs, treated simultaneously with 50 ng/ml HGF, and neuronal morphology analyzed at 3 d after transfection. Met OE significantly increased total dendritic length (data not shown). Particularly striking was the induction of numerous dendritic protrusions by Met OE compared with neurons transfected with GFP only. In contrast, RNAi led to a significantly reduced number of dendritic protrusions (Fig. 2F,G; n = 6, p < 0.01 for both Met and RNAi effects, one-way ANOVA).
Figure 2.

Altered MET signaling affects morphological development of hippocampal neurons in vivo. A, Schematic illustration of delivering Met cDNA and RNAi to developing CA1 neurons using IUEP. Offspring were harvested at P22–P25 for morphological analysis. B, Individual CA1 neurons labeled with IUEP were subjected to morphological analysis. Dendritic arbor of CA1 neurons were traced and reconstructed with Neurolucida. High-resolution Z-stack images were then collected, and dendritic spines were reconstructed with Imaris. C, Representative neurons labeled by IUEP with control GFP, MET, RNAi, and scr RNA, and their reconstructed 3D dendritic arborization and dendritic spines. D, Sholl analysis of CA1 neurons revealed that MET OE increases, while RNAi decreases, the number of dendritic intersections at distinct compartments of the apical dendritic tree (*p < 0.05, #p < 0.05). E, F, MET OE significantly increases total apical dendritic length (E, *p < 0.05), the number of apical dendritic branches (F, *p < 0.05), whereas RNAi significantly decreases total apical dendritic length (**p < 0.01) and the number of apical dendritic branches (*p < 0.05). G, H, MET OE significantly decreases dendritic spine head volume (G, **p < 0.01) and increased spine density (H, *p < 0.05), whereas RNAi significantly enlarges dendritic spine head volume (**p < 0.01) and decreased spine density (**p < 0.01). I, Altered MET signaling changes the categorical spine classification based on the morphology. MET OE significantly decreased (p < 0.05), whereas RNAi increased (p < 0.05) the proportion of the more mature, mushroom-type spines.
Dendritic protrusions are thought to be precursors of dendritic spines (Ziv and Smith, 1996). We thus determined the effects of MET signaling specifically on dendritic spine morphogenesis in more mature neuronal cultures. Hippocampal neurons were transfected with GFP, Met, or RNAi at DIV14, treated simultaneously with 50 ng/ml HGF, and then subjected to high resolution confocal imaging 7 d later. Quantification of dendritic spine density, spine head area, and spine length was done using the maximum intensity projection image from confocal Z-stack images. Differential manipulation of MET receptor expression resulted in reciprocal effects on spine development; Met OE significantly increased, whereas RNAi significantly reduced, dendritic spine density (Fig. 1H,I; p < 0.001 for the MET, and p < 0.05 for RNAi groups, n = 16 neurons/4 cultures, one-way ANOVA). Transfection with a scramble form of RNAi (scr RNA) had no effect on spine density. Disruption of MET receptor expression also appeared to impact features of spine maturation. We found that, in Met OE neurons, dendritic spine head area was consistently smaller, whereas RNAi treatment resulted in significantly larger spine head area (Fig. 1J; n > 1100 spines/5 neurons analyzed for each group; p < 0.01 for both MET and RNAi compared with GFP, K-S test). Notably, Met OE also leads to increased averaged spine length compared with GFP controls (Fig. 1K; n = 11 neurons for all groups. p < 0.01). These data indicate that disrupting the balance of MET receptor signaling can lead to altered states of morphological spine development on CA1 neurons in vitro, indicative of potential changes of spine function.
Altered MET signaling disrupts neuronal morphology in vivo
The regulation of the morphology of spine maturation by MET may have functional implications. To determine whether this regulation occurs in the context of intact, developing hippocampal circuits, we next increased or decreased MET expression in the developing hippocampus in vivo. IUEP was used to deliver GFP, Met, and RNAi plasmids to a small population of CA1 hippocampal neurons at E14.5 (Fig. 2A). Hippocampal tissue from offspring was collected at P22-P25 for quantitative analyses of both dendrite and spine morphology. GFP-labeled neurons from different treatment groups were reconstructed with Neurolucida to analyze dendritic arborization patterns. High resolution Z-stack images of apical dendritic segments of defined locations (see Materials and Methods) were then collected, and Imaris software was used to quantify and classify dendritic spines (Fig. 2B).
As predicted, Met OE and RNAi significantly altered the dendritic arborization of CA1 neurons in an opposite direction, whereas scrRNA transfection was without effect (Fig. 2C–F). Specifically, Met OE leads to an increase in total apical dendrite length (Fig. 2E; p < 0.05, n = 15 neurons/4 mice, one-way ANOVA test) and number of apical dendrite braches (Fig. 2F; p < 0.05, n = 15). In comparison, attenuation of MET expression by RNAi decreased total apical dendrite length (p < 0.01, n = 12 neurons/3 mice) and number of branches (p < 0.05, n = 12). Sholl analysis (Fig. 2D) revealed a specific increase in dendritic intersections at middle apical arbor regions in Met OE neurons (p < 0.05, n = 7 neurons/4 mice. one-way ANOVA test), whereas a significant reduction in branching complexity in the same arbor regions was found in RNAi neurons (p < 0.05, n = 6 neurons/3 mice). Disruption of normal levels of MET expression also caused changes in dendritic spine density and spine head size. Highly consistent with the in vitro studies, Met OE increased (p < 0.05, n = 6 neurons/4 mice; one-way ANOVA test), whereas MET reduction by RNAi decreased the density of spines (p < 0.01, n = 9 neurons/3 mice) compared with control GFP neurons (n = 11 neurons/6 mice) (Fig. 2H). MET OE also led to a significant reduction (p < 0.01, nonparametric Kruskal–Wallis test), whereas RNAi resulted in a significant increase (p < 0.01, n > 1000 spines/6 neurons for each group) in spine head volume (Fig. 2G).
We further parcellated apical spines on CA1 neurons using the Imaris specification module into four distinct classes (filopodia, stubby, mushroom, and long thin) based on morphometry (Pathania et al., 2014). These morphological categories are highly correlated with glutamatergic synapse function (Matsuzaki et al., 2001; Nimchinsky et al., 2002). Met OE significantly decreased (p < 0.05, Pearson's χ2 test), whereas RNAi significantly increased (p < 0.05, χ2 test) the proportion of mushroom-type spines (Fig. 2I). This is consistent with a more mature subcategory of dendritic spines when MET expression is reduced. Collectively, the morphological analyses in vitro on dissociated neurons, and on intact CA1 neurons in vivo, show that levels of MET signaling influence the growth of CA1 neurons and, furthermore, may serve as a modulator of spine maturation state during development.
Disrupted MET signaling leads to altered CA1 glutamatergic function
Dendritic structure and spine geometry are highly correlated with functional maturity of neurons. Given the correlation of altered MET expression with human social-emotional circuit activation patterns and interlaminar excitatory drive in mouse neocortex, whole-cell patch-clamp recording on developing CA1 neurons electroporated with GFP and either Met OE or RNAi by IUEP (Fig. 3A) provided an opportunity to directly demonstrate a role for MET signaling on the maturation of specific synapses. Hippocampal slices were prepared from P22-P28 mice offspring. CA1 neuron resting membrane potentials, input resistance, and current injection-spiking relationship were measured. None of these parameters exhibited changes with either OE or RNAi, compared with the GFP-electroporated neurons (data not shown). This indicates that manipulation of MET signaling does not significantly affect membrane properties of CA1 neurons at this development age. Next, spontaneous miniature EPSCs (mEPSCs) of transfected CA1 neurons (voltage-clamped at −65 mV) were recorded as an estimate of synaptic activity (Fig. 3C). mEPSC amplitude was found significantly reduced in CA1 neurons in the context of Met OE. In contrast, CA1 neurons with RNAi showed a significant increase in the mEPSC amplitude (Fig. 3D; n > 2000 events/5 neurons for each group. p < 0.01 and p < 0.05 for both MET and RNAi group, respectively; K-S test). Furthermore, a significant reduction in the mEPSC frequency was evident in OE CA1 neurons (Fig. 3E; p < 0.05, n = 14 neurons/4 mice). The impact of altering MET signaling is specific to the excitatory glutamatergic synapse on CA1 neurons because neither Met OE nor RNAi affected the miniature IPSCs (mIPSCs, data not shown).
Figure 3.

Disrupted MET signaling in vivo alters CA1 neuron synaptic function. A, Photomicrograph illustrates labeling of neurons in hippocampal slices prepared from mice that had undergone IUEP. CA1 neurons transfected with GFP, MET, and RNAi were selected for patch-clamp recording. B, Quantification of A:N ratio. MET OE leads to reduced A:N ratio (*p < 0.05), whereas RNAi increased A:N ratio (*p < 0.05). C, Representative sample mEPSC traces recorded in CA1 neurons transfected with GFP, MET, and RNAi by IUEP. Average mEPSC from a 1 min epoch are displayed on the right. D, Plot of the cumulative fraction of mEPSC amplitude distribution. MET OE leads to significantly reduced size mEPSC amplitude (p < 0.01), whereas RNAi treatment results in significantly increased mEPSC amplitudes (p < 0.05). E, MET OE in CA1 neuron results in decreased mEPSC frequency (*p < 0.05). RNAi did not result in statistically significant changes in frequency.
Functionally, the maturation of CA1 neurons is correlated with a gradual acquisition AMPA receptors at postsynaptic sites (Isaac et al., 1995; Liao et al., 1995). Changes in the temporal pattern could account for the increased mEPSC amplitude that we observed following reduced MET. The increased AMPA receptor contribution to synaptic maturation also can be assessed by determining the A:N current ratio (Kattenstroth et al., 2004; Terashima et al., 2008). To address the possibility that MET regulates functional maturation of excitatory synapses onto CA1 neurons, the evoked EPSCs (eEPSCs) were measured in response to Schaffer collateral stimulation. Neurons were first voltage-clamped at −70 mV to record AMPA receptor-mediated whole-cell currents and then clamped at 40 mV to record the AMPA/NMDA receptor compound current response. NMDAR-mediated current was measured at 75 ms after response onset at 40 mV, during which AMPA current is decayed to negligible levels (Terashima et al., 2008). Remarkably, Met OE in CA1 neurons resulted in a significantly reduced A:N ratio compared with the nontransfected control neuron (Fig. 3B; p < 0.05, n = 13 OE neurons/3 mice; one-way ANOVA). In comparison, CA1 neurons transfected with RNAi expressed a significantly increased A:N ratio (p < 0.05, n = 7 RNAi neurons/3 mice). Compared with recordings from adjacent nontransfected (NT) CA1 neurons, transfection of GFP alone had no effect on the A:N ratio (p = 0.22, n = 5 neurons/3 mice). These data show that postsynaptic levels of MET can regulate the maturation of glutamatergic synapses, with increased expression antagonizing CA1 synapse maturation, and attenuated expression yielding accelerated maturation of glutamatergic synapses.
Genetic ablation of MET signaling facilitates developmental maturation of functional glutamatergic synapses
To gain additional evidence that the extent of MET signaling impacts glutamatergic synapse development, hippocampal neuron cultures were prepared from MET signaling-incompetent mutant mice (Met fx/fx; Emx1cre) (Judson et al., 2009, 2010) and were used to explore glutamatergic synapse development. Female Metfx/fx mice were mated to male Met fx/+; Emx1cre mice, with individual fetuses used for separate hippocampal neuron cultures. Hippocampal neurons were grown at low density, as previously described (Qiu and Weeber, 2007), and used for patch-clamp recording or immunocytochemistry labeling at DIV 22–28.
To obtain an accurate estimate of the number of glutamatergic synapses in control (Metfx/fx) and null (Met fx/fx; Emx1cre) neurons, double immunofluorescence staining was performed for the AMPAR subunit GluA1 and the obligatory NMDAR subunit GluN1. Both GluA1 and GluN1 staining resulted in punctate labeling (Fig. 4A), reflecting potential sites of glutamatergic synapses (Lissin et al., 1999; Qiu and Weeber, 2007). Colocalized immunoreactive puncta for both GluN1 and GluA1 represents a putative functional synapse. “Silent synapses” (Liao et al., 1995), which contain only NMDARs, are represented by GluN1-only positive sites. Consistent with the idea that the MET receptor impacts synapse maturation, the proportion of putative functional synapses, reflected by GluN1+, GluA1+ colabeling, was significantly increased in Metfx/fx; Emx1cre neurons (Fig. 4B; p < 0.05, n = 8 for each genotype). Yet decreased MET did not alter the density of either GluN1+ or GluA1+ individual puncta in Metfx/fx; Emx1cre neurons compared with control Met fx/fx neurons. Neurons were also double-labeled with synapsin I (presynaptic) and PSD-95 (postsynaptic) to quantitate the number of putative functional synapses. Once again, deleting Met resulted in an increase in putative functional synapses, based on the number of double-labeled, directly apposed synapsin I and PSD-95 puncta (Fig. 4A,C; p < 0.05, n = 9–11 neurons for each genotype).
Figure 4.

Ablation of MET signaling affects glutamatergic synapse development. A, Photomicrograph illustration of double labeling of GluN1 and GluA1, or PSD-95 and synapsin I. Colocalized synaptic puncta were used to represent putative functional synapses. Cultures were prepared from individual Metfx/fx; emx1cre embryos and their Metfx/fx littermate controls. B, Quantification of GluN1 and GluA1 immunolabeling. No statistical difference was found between the two genotypes for density of individual GluN1+ or GluA+ puncta. However, a significant increase in the proportion of putative functional synapse (defined by colabeling of GluN1 and GluA1 profiles) was found in Metfx/fx; emx1cre neurons (*p < 0.05). C, Quantification of PSD-95 and synapsin I immunoreactivity. No overall difference was found for density of individual PSD-95+ or synapsin I+ puncta. A significant increase in the proportion of putative functional synapse (colabeled PSD-95 and synapsin I puncta) was found in Metfx/fx; emx1cre neurons (*p < 0.05). D, Sample mEPSC recording traces obtained from Metfx/fx and Metfx/fx; emx1cre cultures. Metfx/fx; emx1cre neurons showed significantly enlarged mEPSC amplitude (p < 0.02). No change of mEPSC frequency was seen. E, Representative image on Neurolucida and Imaris reconstructions of single CA1 neuronal dendrites and spines from P21–P22 Metfx/fx mice that had undergone IUEP with CAG-GFP or CAG-GFP/cre plasmids. F–I, Quantification of single neuron knock-out of Met by IEUP. CAG-GFP/cre electroporated neurons displayed significantly reduced total dendritic length in both basal and apical compartments (F, *p < 0.05, **p < 0.01), and decreased number of apical dendrites (G, *p < 0.05). Dendritic spines from CAG-GFP/cre electroporated neurons show a significantly reduced density (H, **p < 0.01) and enlargement of head volume (I, **p < 0.01) compared with that from CAG-GFP electroporated neurons.
To complement the molecular data, functional analysis by patch-clamp recording of mEPSCs revealed that Metfx/fx; Emx1cre neurons exhibit a significant increase of the mEPSC amplitude compared with the Metfx/fx neurons, whereas there was no change in mEPSC frequency (Fig. 4D; n > 2500 events/8 neurons; p < 0.02 for mEPSC amplitude comparison, K-S test). These data indicate that elimination of Met genetically resulted in a facilitated state of glutamatergic synapse maturation, consistent with a cell autonomous mechanism in CA1 neurons in the hippocampus.
To further verify that MET loss of function leads to altered synapse development in vivo, we measured dendritic arborization and spine morphologies in CA1 neurons following genetic ablation of Met in single neurons. IUEP was used to deliver the CAG-GFP/cre or the control CAG-GFP vector (Tashiro et al., 2006) to CA1 hippocampus of the E14.5 mice expressing the conditional Met allele (Met fx/fx) (Fig. 4E). We found that, compared with CAG-GFP electroporated neurons, CAG-GFP/cre neurons had significantly reduced total dendritic length in both apical and basal dendrite compartments (Fig. 4F; p < 0.01 and p < 0.05 for apical and basal dendrites, respectively; n = 5 neurons/2 mice). In addition, CAG-GFP/cre neurons showed a significant reduction in apical dendritic branches (Fig. 4G; p < 0.05; n = 5 neurons/2 mice), a reduction of dendritic spine density (Fig. 4H; p < 0.01; n = 5 neurons/2 mice), and a significant enlargement of spine head volume (Fig. 4I; p < 0.01; n > 500 spines/6 neurons analyzed for each group, Kruskal–Wallis test). Therefore, cre-mediated single-neuron deletion of Met results in essentially the same morphological alterations as demonstrated by single-neuron RNAi (Fig. 2), with enlarged spine heads indicative of a larger proportion of functional synapses.
Genetic deletion of Met accelerates developmental maturation of intact hippocampus circuits
The morphological, molecular, and electrophysiological data together indicate that MET receptor signaling may be highly pleiotropic, engaging diverse molecular mechanisms to regulate both neuronal growth and synapse maturation in a protracted developmental window. To address this possibility, we measured the electrophysiological effects of ablation of Met in vivo on CA1 glutamatergic synapse development at different ages, using the Metfx/fx; Emx1cre and their Met fx/fx littermate controls. Spontaneous mEPSC responses in CA1 neurons prepared at an early developmental stage (P12-P14) were recorded. CA1 neurons of Metfx/fx; Emx1cre mice exhibited a significant increase in mEPSC amplitude (Fig. 5A; n > 2200 events/9 neurons analyzed for each genotype; p < 0.02, K-S test), with no change of mEPSC frequency. Next, stimulus-evoked whole-cell synaptic currents were recorded and the A:N ratio quantified. Similar to single-neuron RNAi (Fig. 3B), loss of Met during development resulted in a significantly larger A:N ratio at P14 (Fig. 5B; p < 0.05, n = 8 neurons/4 mice, t test). Remarkably, the increased A:N ratio was evident at an even earlier developing age (P6-P7; Fig. 5B; p < 0.05, n = 12 neurons/3 mice). The developmental difference in A:N ratio eventually disappeared, with comparable measures obtained in wild-type and mutant hippocampi at P24-P27 (Fig. 5B; p = 0.08, n = 7 neurons/3 mice).
Figure 5.

MET loss of function leads to accelerated glutamatergic circuit maturation. A, Analysis of mEPSC in P12–P14 acute hippocampal slices revealed an enlarged mEPSC amplitude in Metfx/fx; emx1cre neurons (p < 0.02). B, Metfx/fx; emx1cre CA1 neurons in P6–P7 and P14 slices show significantly increased A:N ratio (*p < 0.05). C, Metfx/fx; emx1cre CA1 neurons in P7–P9 and P14 slices show significantly decreased sensitivity to ifenprodil (*p < 0.05; **p < 0.01), as measured by reduction of NMDAR current after ifenprodil (3 μm). D, No change of paired pulse ratio was found at various interpulse intervals at both P7–P8 and P14. E, Illustration of isolation of whole CA1 tissue lysate (1), total synaptosome protein (2), and synaptic surface protein (3), and quantification protocol. F, Representative Western blot of different glutamate receptor subunit proteins, postsynaptic scaffold proteins, and signaling molecules in whole CA1 lysates. G, Representative blots and quantification of glutamate receptor subunits in total synaptosome fraction and synaptic membrane surface compartments. Quantitative analysis of synaptic surface proteins revealed increased GluA1, GluN2A, and decreased GluN2B levels of immunoreactive protein (*p < 0.05). No significant change of total synaptosome protein was seen (quantification not shown).
Because NMDA receptor subunit switching (i.e., the replacement of GluN2B by GluN2A) is known to mediate functional maturation of CA1 synapses during the first weeks of development (Monyer et al., 1994), we sought to quantify relative contribution of GluN2B and GluN2A to the NMDAR whole-cell current by determining the relative sensitivity of NMDAR-EPSCs to the GluN2B-selective antagonist ifenprodil (3 μm) (Qiu and Weeber, 2007; Harlow et al., 2010). In CA1 neurons obtained from P14 control Met fx/fx slices, NMDAR-EPSC total charge transfer was inhibited by 52.6 ± 8.8% (n = 10 neurons), which is significantly larger than the inhibition obtained from Metfx/fx; Emx1cre neurons (36.3 ± 5.9%, n = 7 neurons/4 mice; p < 0.05, t test) (Fig. 5C). An even larger difference in ifenprodil sensitivity was found when these two genotypes were examined at P7-P9 (Fig. 5C; p < 0.01, n = 11 neurons/4 mice, t test), with no difference was seen at P26-P28. Therefore, CA1 synapses in Metfx/fx; Emx1cre mice showed reduced GluN2B contribution and a more rapid GluN2A switch earlier in development. To investigate whether loss of MET signaling results in presynaptic changes of release probability, we measured CA1 neuron whole-cell currents in response to paired pulse stimulation across various interpulse intervals (50, 100, and 150 ms). We conducted these experiments both in P7-P8 and P14 mice, and we did not detect any statistical changes of paired pulse facilitation ratio (Fig. 5D). Together, these results show that MET uniquely regulates the pace of synapse maturation postsynaptically during an early developmental time window of hippocampal CA1 neurons.
Are there molecular correlates of the differences in synapse maturation due to MET deletion? Functional synapse maturation can be reflected by changes of synaptic protein constituents. In an additional set of experiments, we probed a number of synaptic proteins that are known to mediate excitatory synapse maturation or are downstream to MET signaling pathway. In micro-dissected whole CA1 tissue lysates from Metfx/fx; Emx1cre and its Met fx/fx controls (P14, Fig. 5E), we did not observe significant changes in expression of different glutamate receptor subunits (GluA1, GluN1, GluN2A, GluN2B, and mGluR5), postsynaptic scaffolding proteins (Shank 3, Homer 1, and PSD95), and several synaptic signaling proteins (neuroligin 3, synGap, AKT, CaMKII, and PI3 kinase) (Fig. 5F; p > 0.05 for all measurements, n = 3–5 experiments from each protein). Because functional proteins may be compartmentalized subcellularly, an alteration in glutamate receptor content at synaptic sites, in contrast to total glutamate receptors, may explain the increased A:N ratio and changed contributions of different NMDAR subunits in developing hippocampus. To address this question directly, surface proteins were biotinylated in acute hippocampus slices prepared from P14 Met fx/fx and Metfx/fx; Emx1cre mice (Qiu et al., 2006; Qiu and Weeber, 2007). The CA1 region was micro-dissected, and glutamate receptor subunit content was assessed using Western blot analysis in both surface and total synaptosome protein preparations (Fig. 5E,G). There was no difference in the expression levels of GluA1, GluN1, GluN2A, and Glu2B in the total synaptosome proteins. There was, however, a significant increase of GluA1 in the synaptic membrane preparations from Metfx/fx; Emx1cre CA1 hippocampal tissue (Fig. 5G; p < 0.05, n = 4 experiments). In addition, consistent with the electrophysiology data, there was a significant reduction of GluN2B (p < 0.05, n = 3 experiments) and a significant increase in Glu2A (p < 0.05, n = 3 experiments) in the synaptic membrane preparations from Metfx/fx; Emx1cre hippocampi. The data indicate that MET signaling regulates the rate of synapse maturation in part through a more refined mechanism of controlling synaptic protein localization rather than gross levels of expression.
To complement our findings on the functional role for MET signaling in synapse development, we used additional measures to address the developmental maturation of the CA1 glutamatergic synapse. The temporal acquisition of AMPA receptors at synaptic sites that initially contain only NMDARs (silent synapses) (Isaac et al., 1995; Liao et al., 1995) is a well-accepted measure of maturation. We thus asked whether the proportion of silent synapses was changed based on the observation of accelerated maturation in Metfx/fx; Emx1cre CA1 neurons. We adopted a minimum stimulation protocol to activate a small number of axons or release sites and probed neuronal response and failure rates at −70 mV and 40 mV (Liao et al., 1995; Qiu and Weeber, 2007). These experiments were performed at P12-P14, the peak of MET expression in the forebrain, and the time during which biochemical analysis indicated increased synaptic GluA1 and GluN2A content (Fig. 5G). CA1 neurons were voltage-clamped at −70 mV first, and minimum stimulation induced mixed events of failure and success of synaptic transmission mediated by AMPARs. Neurons were then voltage-clamped at 40 mV to obtain the synaptic events mediated by both AMPA and NMDA receptors, which has a higher success rate due to the NMDAR contribution (Fig. 6A). We observed that the difference in the rate of successful synaptic transmission at 40 mV and −70 mV is significantly smaller in Metfx/fx; Emx1cre neurons (8.5 ± 2.1%), compared with that obtained in wild-type Metfx/fx neurons (21.2 ± 2.8%) (Fig. 6B,C; p < 0.05, n = 7 or 8 neurons/4 mice for both groups). The difference in the rate of successful synaptic transmission at 40 mV and −70 mV can be eliminated in the presence of AP5 (50 μm), an NMDAR blocker (data not shown).
Figure 6.

MET loss of function reduces silent synapses and enhances AMPAR quantal transmission. A, Representative recording probes the proportion of silent synapses in P12–P14 slices of both Metfx/fx and Metfx/fx; emx1cre mice. B, C, Quantification indicates that Metfx/fx; emx1cre neurons had significantly lower proportion of silent synapses (*p < 0.05), as indicated by a smaller difference of failure rate at −70 mV and 40 mV. D, Nonstationary fluctuation analysis was applied to analyze the quantal parameters of minimum stimulation-induced EPSCAMPA. Traces from successful trails are displayed on top, with the trace averages highlighted. Two average traces from both genotypes are peak scaled to indicate no change of kinetics. Calculated variances (dotted lines) of these traces following peak scaling were displayed above. EPSC variance was plotted against binned mean EPSC amplitude, averaged AMPAR numbers that open at the peak of mean EPSC, and single-channel conductance was calculated by curve fitting from 7 or 8 experiments.
We further analyzed postsynaptic AMPAR numbers and single-channel conductance using peak-scaled nonstationary fluctuation analysis on the minimum stimulation evoked EPSCs. Figure 6D illustrates one representative experiment in P12 Metfx/fx and Metfx/fx; Emx1cre neurons, from which successful EPSC trials obtained from Metfx/fx (116 events) and Metfx/fx; Emx1cre (92 events) neurons were used for nonstationary fluctuation analysis. The mean amplitude of EPSCs was increased by 27% in the Metfx/fx; Emx1cre neuron (18.9 pA, compared with 14.8 pA in the Metfx/fx neuron), whereas the kinetics of averaged EPSCs remained unchanged. Analysis of pooled experiments (8 Metfx/fx and 7 Metfx/fx; Emx1cre neurons, P12-P14) revealed that averaged weighted mean single-channel conductance showed no statistically significance difference (Metfx/fx neurons, 15.1 ± 2.2 pS; Metfx/fx; Emx1cre neurons, 14.9 ± 2.1 pS. p = 0.23, t test). In contrast, the number of AMPARs that open at the peak of mean current exhibited a significant increase in Metfx/fx; Emx1cre neurons (15.8 ± 1.3), compared with that from Metfx/fx neurons (11.7 ± 0.9) (Fig. 6D; p < 0.05, t test). Together, these experiments indicate that, at an early developmental stage, Metfx/fx; Emx1cre neurons have a lower proportion of silent synapses and an increase in AMPAR content. These data further support our hypothesis that disruption of MET signaling leads to accelerated maturation of developing hippocampal CA1 synapses.
Discussion
The present morphological and functional studies have revealed a highly novel mechanism conferred by MET receptor signaling, which we show serves a pleiotrophic role in controlling both neuronal and spine morphology, and the time course of glutamatergic synapse maturation on CA1 hippocampal neurons. Such findings are consistent with the possibility that mistimed maturation of glutamatergic synapses underlie aberrant function of neural circuits that are enriched in MET during development. RTK signaling is well known to serve multiple roles in different cellular contexts (Park and Poo, 2013). Here, levels of MET receptor appear to impact several different aspects of neuronal maturation, shown by the use of different strategies to manipulate Met expression with morphological, electrophysiological, and biochemical measures. Specifically, higher MET expression increases, while reduced expression decreases, dendritic branching and arbor complexity. This positive effect on neuronal growth was also reflected in increased dendritic protrusions and spine density in MET OE neurons, though smaller, more densely distributed but with an immature morphology. Reduced MET signaling via RNAi decreased spine density, yet spine head size was enlarged, showing that control of levels of MET signaling can impact the morphological and functional states of excitatory neurons.
Dendritic spine volume and geometry are correlated with glutamate receptor content and functional maturity (Matsuzaki et al., 2001; Nimchinsky et al., 2002) and is implicated in neuropsychiatric conditions (Hutsler and Zhang, 2010; Penzes et al., 2011). In the present studies, manipulation of MET expression levels using several methods resulted in a bidirectional regulation of glutamate receptor content at postsynaptic sites, as well as altered maturation of hippocampal CA1 neurons. In addition, we found that the A:N ratio is higher in neurons expressing less or no MET, suggesting a more rapid acquisition of AMPA receptor in developing CA1 synapses. This result could be either a cause or an effect of increased dendritic spine size. The data are consistent with our previous findings in the neocortex demonstrating that reducing or eliminating MET signaling genetically increases interlaminar local circuit excitatory connectivity (Qiu et al., 2012).
Based on the in vivo data collected from neurons manipulated at a single-cell level, we hypothesized that one function of MET during development is to regulate the temporal profile of glutamatergic synapse maturation. This is supported by a large amount of electrophysiological data collected developmentally. CA1 neurons from Metfx/fx; Emx1cre mice displayed increased mEPSC amplitude, increased A:N ratio, faster NMDA receptor subunits developmental switch, reduced proportion of silent synapses, and increased number of postsynaptic AMPAR channels at ages significantly earlier than in normal neurons. These functional indications of early maturation are further substantiated by biochemical analysis of glutamate receptor subunit content at synaptic sites (rather than general expression levels), which reveals increased GluA1, GluN2A and decreased GluN2B synaptic targeting in Metfx/fx; Emx1cre mice at P14. Diverse mechanisms are known to recruit AMPAR to synaptic sites during patterned activity or synaptic plasticity (Hayashi et al., 2000; Hu et al., 2007; Aoto et al., 2013; Granger et al., 2013), yet the mechanisms that contribute to synaptic AMPAR acquisition in a developmental context are less well understood. Interestingly, as the hippocampus continues to mature, we found that some of these differences were eliminated. There is accumulating evidence that time-delimited disruption of signaling systems during development can have long-term consequences on function (Ansorge et al., 2004; Clement et al., 2012). Previous studies of MET are consistent with this, as there is a sustained increase in interlaminar excitatory synaptic function in young adult neocortex in which there is a genetically mediated complete or partial reduction of Met (Qiu et al., 2011). Because Met expression is enriched at synapses (Eagleson et al., 2013) and is regulated both spatially and temporally with very limited expression postpubertal, any impact of manipulating the MET receptor would be expected to have a focused impact on synaptic development that could lead to permanent functional changes.
Disruption of the molecular components that impact excitatory synapse structure and function are evident with deleterious, rare mutations of adhesion, adaptor, and signaling synaptic proteins (Durand et al., 2007; Tabuchi et al., 2007; Gilman et al., 2011; Hamdan et al., 2011). In part, the association of specific mutations with neurodevelopmental disorders has formed the basis for suggesting that synaptic development is a critical target of disruption that affects cognition, including intellectual disability, and ASD (Penzes et al., 2011; Zoghbi and Bear, 2012). As with many other synaptically enriched ASD risk genes, the specific intracellular molecular mechanisms that operate in MET-manipulated developing glutamatergic neurons that yield different phenotypes have yet to be uncovered. Nonetheless, our study presents compelling evidence that MET signaling participates in regulating dendritic arborization, timing of glutamatergic synapse formation, and functional maturation. Unlike most other synaptic proteins implicated in ASD, MET signaling may primarily engage earlier growth-associated mechanisms by controlling the pace of early neuronal growth and synaptic maturation. Although strictly correlative, the report of differences in gray matter growth in typical developing children carrying the MET promoter ‘C’ allele is consistent with this type of developmental role (Hedrick et al., 2012).
It is noteworthy that early maturation of glutamatergic synapse also has been reported with SYNGAP1 haploinsufficiency, a pathogenic mutation of the gene encoding a GTPase-activating protein that is associated with ASD and intellectual disability (Clement et al., 2012). Thus, MET and SYNGAP1 now provide two specific examples that the timing of neuronal growth and synaptogenesis may be critical to circuit function because of a projected role of these genes in laying a physical framework onto which synaptic signaling complexes, such as glutamate receptors and their downstream targets, are targeted and maintained. Notably, the lack of MET signaling during development does not seem to alter synaptic molecular constituents on a gross scale because overall levels of several prominent glutamate receptors, postsynaptic scaffolding proteins, and signaling molecules were not changed by deleting Met. One challenge, however, is that, in the absence of MET, there may be certain protein changes in the subcellular domains (e.g., synaptic structures) that cannot be resolved using whole CA1 tissue. In addition, the candidate approach of measuring certain synaptic proteins may miss changes that simply are not being assayed. Current studies are underway using discovery-based proteomic analyses to address this.
There appears to be an enrichment of ASD risk genes related to growth factor signaling (Wittkowski et al., 2014), aberrant ASD brain growth trajectories (Levitt and Campbell, 2009; Berg and Geschwind, 2012), and the coalescence of functional ASD gene networks in circuits involving superficial and deep layer glutamatergic projection neurons (Parikshak et al., 2013; Willsey et al., 2013). Furthermore, another recent ASD genetic study has reported an enrichment of genetic variations that disrupts “functional gene sets involved in cellular proliferation, projection and motility, and GTPase/Ras signaling” (Pinto et al., 2010). MET signaling is complex, capable of activating PI3 kinase, MAP kinase, and GTPase/Ras. This may explain, in part, the biologically pleiotropic effects that we observed when MET signaling was dysregulated.
Several important questions remain to be elucidated to understand mechanism of action that can be translated to our findings in humans. First, how are each of the intracellular signaling pathways engaged by MET to impact neuronal growth and synapse maturation at different times in development? Second, what controls the temporal and spatial patterns (Judson et al., 2009; Mukamel et al., 2011) of MET signaling during development? We recently demonstrated that FOXP2, which is associated with ASD-related language endophenotype, represses MET expression during human brain development (Mukamel et al., 2011). In addition, MeCP2 regulates MET transcription in an allele-specific fashion regulated by the rs1858830 ‘C’ variant (Plummer et al., 2013). Moreover, in postmortem analyses, the MET transcript is dramatically reduced in temporal neocortex of subjects with Rett syndrome compared with matched female controls. It is likely that, together with FOXP2 and MeCP2, other transcriptional regulators control the complex patterns of expression of MET. Our laboratories are in the process of identifying these factors and components of the MET protein interactome in glutamatergic circuitry. This additional work will contribute to a more in-depth understanding of the influence of MET on typical and atypical circuit development that will translate into a better understanding of ASD pathophysiology and amenability to molecular interventions.
Footnotes
This work was supported by National Institute of Mental Health Grant MH067842 and Simons Foundation Autism Research Institute Grant 137273 to P.L., and National Institute of Mental Health Grants K99MH087628 and R00MH087628, and an Institute for Mental Health Research Grant and to S.Q. We thank Dr. Daping Fan (University of South Carolina) for expert guidance in RNAi constructs, and Mariel Piechowicz for assistance with blind data analysis.
The authors declare no competing financial interests.
References
- Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, Menashe I, Wadkins T, Banerjee-Basu S, Packer A. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs) Mol Autism. 2013;4:36. doi: 10.1186/2040-2392-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achim CL, Katyal S, Wiley CA, Shiratori M, Wang G, Oshika E, Petersen BE, Li JM, Michalopoulos GK. Expression of HGF and cMet in the developing and adult brain. Brain Res Dev Brain Res. 1997;102:299–303. doi: 10.1016/S0165-3806(97)00108-9. [DOI] [PubMed] [Google Scholar]
- Ansorge MS, Zhou M, Lira A, Hen R, Gingrich JA. Early-life blockade of the 5-HT transporter alters emotional behavior in adult mice. Science. 2004;306:879–881. doi: 10.1126/science.1101678. [DOI] [PubMed] [Google Scholar]
- Aoto J, Martinelli DC, Malenka RC, Tabuchi K, Südhof TC. Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell. 2013;154:75–88. doi: 10.1016/j.cell.2013.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JM, Geschwind DH. Autism genetics: searching for specificity and convergence. Genome Biol. 2012;13:247. doi: 10.1186/gb-2012-13-7-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, Elia M, Schneider C, Melmed R, Sacco R, Persico AM, Levitt P. A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci U S A. 2006;103:16834–16839. doi: 10.1073/pnas.0605296103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DB, D'Oronzio R, Garbett K, Ebert PJ, Mirnics K, Levitt P, Persico AM. Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann Neurol. 2007;62:243–250. doi: 10.1002/ana.21180. [DOI] [PubMed] [Google Scholar]
- Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, Almonte AG, Miller BH, Wiltgen BJ, Miller CA, Xu X, Rumbaugh G. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell. 2012;151:709–723. doi: 10.1016/j.cell.2012.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM, Vande Woude GF. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311:29–33. doi: 10.1038/311029a0. [DOI] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagleson KL, Milner TA, Xie Z, Levitt P. Synaptic and extrasynaptic location of the receptor tyrosine kinase met during postnatal development in the mouse neocortex and hippocampus. J Comp Neurol. 2013;521:3241–3259. doi: 10.1002/cne.23343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebens A, Brose K, Leonardo ED, Hanson MG, Jr, Bladt F, Birchmeier C, Barres BA, Tessier-Lavigne M. Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron. 1996;17:1157–1172. doi: 10.1016/S0896-6273(00)80247-0. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger AJ, Shi Y, Lu W, Cerpas M, Nicoll RA. LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature. 2013;493:495–500. doi: 10.1038/nature11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez H, Dolcet X, Tolcos M, Davies A. HGF regulates the development of cortical pyramidal dendrites. Development. 2004;131:3717–3726. doi: 10.1242/dev.01209. [DOI] [PubMed] [Google Scholar]
- Hamanoue M, Takemoto N, Matsumoto K, Nakamura T, Nakajima K, Kohsaka S. Neurotrophic effect of hepatocyte growth factor on central nervous system neurons in vitro. J Neurosci Res. 1996;43:554–564. doi: 10.1002/(SICI)1097-4547(19960301)43:5<554::AID-JNR5>3.0.CO%3B2-H. [DOI] [PubMed] [Google Scholar]
- Hamasaki H, Honda H, Suzuki SO, Hokama M, Kiyohara Y, Nakabeppu Y, Iwaki T. Down-regulation of MET in hippocampal neurons of Alzheimer's disease brains. Neuropathology. 2014;34:284–290. doi: 10.1111/neup.12095. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Piton A, Gauthier J, Dobrzeniecka S, Krebs MO, Joober R, Lacaille JC, Nadeau A, Milunsky JM, Wang Z, Carmant L, Mottron L, Beauchamp MH, Rouleau GA, Michaud JL. De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry. 2011;69:898–901. doi: 10.1016/j.biopsych.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A. Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron. 2010;65:385–398. doi: 10.1016/j.neuron.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hedrick A, Lee Y, Wallace GL, Greenstein D, Clasen L, Giedd JN, Raznahan A. Autism risk gene MET variation and cortical thickness in typically developing children and adolescents. Autism Res. 2012;5:434–439. doi: 10.1002/aur.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmbacher F, Dessaud E, Arber S, deLapeyrière O, Henderson CE, Klein R, Maina F. Met signaling is required for recruitment of motor neurons to PEA3-positive motor pools. Neuron. 2003;39:767–777. doi: 10.1016/S0896-6273(03)00493-8. [DOI] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010;1309:83–94. doi: 10.1016/j.brainres.2009.09.120. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Jackson PB, Boccuto L, Skinner C, Collins JS, Neri G, Gurrieri F, Schwartz CE. Further evidence that the rs1858830 C variant in the promoter region of the MET gene is associated with autistic disorder. Autism Res. 2009;2:232–236. doi: 10.1002/aur.87. [DOI] [PubMed] [Google Scholar]
- Jeffers M, Fiscella M, Webb CP, Anver M, Koochekpour S, Vande Woude GF. The mutationally activated Met receptor mediates motility and metastasis. Proc Natl Acad Sci U S A. 1998;95:14417–14422. doi: 10.1073/pnas.95.24.14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Bergman MY, Campbell DB, Eagleson KL, Levitt P. Dynamic gene and protein expression patterns of the autism-associated met receptor tyrosine kinase in the developing mouse forebrain. J Comp Neurol. 2009;513:511–531. doi: 10.1002/cne.21969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Eagleson KL, Wang L, Levitt P. Evidence of cell-nonautonomous changes in dendrite and dendritic spine morphology in the met-signaling-deficient mouse forebrain. J Comp Neurol. 2010;518:4463–4478. doi: 10.1002/cne.22467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Amaral DG, Levitt P. Conserved subcortical and divergent cortical expression of proteins encoded by orthologs of the autism risk gene MET. Cereb Cortex. 2011;21:1613–1626. doi: 10.1093/cercor/bhq223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattenstroth G, Tantalaki E, Südhof TC, Gottmann K, Missler M. Postsynaptic N-methyl-d-aspartate receptor function requires alpha-neurexins. Proc Natl Acad Sci U S A. 2004;101:2607–2612. doi: 10.1073/pnas.0308626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Levitt P, Campbell DB. The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J Clin Invest. 2009;119:747–754. doi: 10.1172/JCI37934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Lim CS, Walikonis RS. Hepatocyte growth factor and c-Met promote dendritic maturation during hippocampal neuron differentiation via the Akt pathway. Cell Signal. 2008;20:825–835. doi: 10.1016/j.cellsig.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissin DV, Carroll RC, Nicoll RA, Malenka RC, von Zastrow M. Rapid, activation-induced redistribution of ionotropic glutamate receptors in cultured hippocampal neurons. J Neurosci. 1999;19:1263–1272. doi: 10.1523/JNEUROSCI.19-04-01263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maina F, Hilton MC, Ponzetto C, Davies AM, Klein R. Met receptor signaling is required for sensory nerve development and HGF promotes axonal growth and survival of sensory neurons. Genes Dev. 1997;11:3341–3350. doi: 10.1101/gad.11.24.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maina F, Panté G, Helmbacher F, Andres R, Porthin A, Davies AM, Ponzetto C, Klein R. Coupling Met to specific pathways results in distinct developmental outcomes. Mol Cell. 2001;7:1293–1306. doi: 10.1016/S1097-2765(01)00261-1. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mukamel Z, Konopka G, Wexler E, Osborn GE, Dong H, Bergman MY, Levitt P, Geschwind DH. Regulation of MET by FOXP2, genes implicated in higher cognitive dysfunction and autism risk. J Neurosci. 2011;31:11437–11442. doi: 10.1523/JNEUROSCI.0181-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Vigna E, Ferracini R, Longati P, Gandino L, Prat M, Comoglio PM. The tyrosine kinase encoded by the MET proto-oncogene is activated by autophosphorylation. Mol Cell Biol. 1991;11:1793–1803. doi: 10.1128/MCB.11.4.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Quiroga I, Chittajallu R, Gallo V, Haydar TF. Long-term, selective gene expression in developing and adult hippocampal pyramidal neurons using focal in utero electroporation. J Neurosci. 2007;27:5007–5011. doi: 10.1523/JNEUROSCI.0867-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrc3653. [DOI] [PubMed] [Google Scholar]
- Pathania M, Davenport EC, Muir J, Sheehan DF, López-Doménech G, Kittler JT. The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Transl Psychiatry. 2014;4:e374. doi: 10.1038/tp.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer JT, Evgrafov OV, Bergman MY, Friez M, Haiman CA, Levitt P, Aldinger KA. Transcriptional regulation of the MET receptor tyrosine kinase gene by MeCP2 and sex-specific expression in autism and Rett syndrome. Transl Psychiatry. 2013;3:e316. doi: 10.1038/tp.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Weeber EJ. Reelin signaling facilitates maturation of CA1 glutamatergic synapses. J Neurophysiol. 2007;97:2312–2321. doi: 10.1152/jn.00869.2006. [DOI] [PubMed] [Google Scholar]
- Qiu S, Zhao LF, Korwek KM, Weeber EJ. Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J Neurosci. 2006;26:12943–12955. doi: 10.1523/JNEUROSCI.2561-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Anderson CT, Levitt P, Shepherd GM. Circuit-specific intracortical hyperconnectivity in mice with deletion of the autism-associated Met receptor tyrosine kinase. J Neurosci. 2011;31:5855–5864. doi: 10.1523/JNEUROSCI.6569-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Aldinger KA, Levitt P. Modeling of autism genetic variations in mice: focusing on synaptic and microcircuit dysfunctions. Dev Neurosci. 2012;34:88–100. doi: 10.1159/000336644. [DOI] [PubMed] [Google Scholar]
- Rudie JD, Hernandez LM, Brown JA, Beck-Pancer D, Colich NL, Gorrindo P, Thompson PM, Geschwind DH, Bookheimer SY, Levitt P, Dapretto M. Autism-associated promoter variant in MET impacts functional and structural brain networks. Neuron. 2012;75:904–915. doi: 10.1016/j.neuron.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Sesack SR, Toda S, Kalivas PW. Automated quantification of dendritic spine density and spine head diameter in medium spiny neurons of the nucleus accumbens. Brain Struct Funct. 2008;213:149–157. doi: 10.1007/s00429-008-0184-2. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Sousa I, Clark TG, Toma C, Kobayashi K, Choma M, Holt R, Sykes NH, Lamb JA, Bailey AJ, Battaglia A, Maestrini E, Monaco AP. MET and autism susceptibility: family and case-control studies. Eur J Hum Genet. 2009;17:749–758. doi: 10.1038/ejhg.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit A, Stern CD, Théry C, Ireland GW, Aparicio S, Sharpe MJ, Gherardi E. A role for HGF/SF in neural induction and its expression in Hensen's node during gastrulation. Development. 1995;121:813–824. doi: 10.1242/dev.121.3.813. [DOI] [PubMed] [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Zhao C, Gage FH. Retrovirus-mediated single-cell gene knockout technique in adult newborn neurons in vivo. Nat Protoc. 2006;1:3049–3055. doi: 10.1038/nprot.2006.473. [DOI] [PubMed] [Google Scholar]
- Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL, McBain CJ, Isaac JT. An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron. 2008;57:872–882. doi: 10.1016/j.neuron.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanseem I, Nakamura K, Miyachi T, Toyota T, Yamada S, Tsujii M, Tsuchiya KJ, Anitha A, Iwayama Y, Yamada K, Hattori E, Matsuzaki H, Matsumoto K, Iwata Y, Suzuki K, Suda S, Kawai M, Sugihara G, Takebayashi K, Takei N, et al. Further evidence for the role of MET in autism susceptibility. Neurosci Res. 2010;68:137–141. doi: 10.1016/j.neures.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Thewke DP, Seeds NW. The expression of mRNAs for hepatocyte growth factor/scatter factor, its receptor c-met, and one of its activators tissue-type plasminogen activator show a systematic relationship in the developing and adult cerebral cortex and hippocampus. Brain Res. 1999;821:356–367. doi: 10.1016/S0006-8993(99)01115-4. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Silver RA, Cull-Candy SG. Estimated conductance of glutamate receptor channels activated during EPSCs at the cerebellar mossy fiber-granule cell synapse. Neuron. 1993;11:279–289. doi: 10.1016/0896-6273(93)90184-S. [DOI] [PubMed] [Google Scholar]
- Tyndall SJ, Patel SJ, Walikonis RS. Hepatocyte growth factor-induced enhancement of dendritic branching is blocked by inhibitors of N-methyl-d-aspartate receptors and calcium/calmodulin-dependent kinases. J Neurosci Res. 2007;85:2343–2351. doi: 10.1002/jnr.21390. [DOI] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, Murtha MT, Bichsel C, Niu W, Cotney J, Ercan-Sencicek AG, Gockley J, Gupta AR, Han W, He X, Hoffman EJ, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkowski KM, Sonakya V, Bigio B, Tonn MK, Shic F, Ascano M, Nasca C, Gold-Von Simson G. A novel computational biostatistics approach implies impaired dephosphorylation of growth factor receptors as associated with severity of autism. Transl Psychiatry. 2014;4:e354. doi: 10.1038/tp.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol. 2003;77:8957–8961. doi: 10.1128/JVI.77.16.8957-8951.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HH, Levitt P. Prenatal expression of MET receptor tyrosine kinase in the fetal mouse dorsal raphe nuclei and the visceral motor/sensory brainstem. Dev Neurosci. 2013;35:1–16. doi: 10.1159/000346367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/S0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012;4.pii:a009886. doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]