

Abstract
Traumatic brain injury (TBI) is an established risk factor for the early development of dementia, including Alzheimer's disease, and the post-traumatic brain frequently exhibits neurofibrillary tangles comprised of aggregates of the protein tau. We have recently defined a brain-wide network of paravascular channels, termed the “glymphatic” pathway, along which CSF moves into and through the brain parenchyma, facilitating the clearance of interstitial solutes, including amyloid-β, from the brain. Here we demonstrate in mice that extracellular tau is cleared from the brain along these paravascular pathways. After TBI, glymphatic pathway function was reduced by ∼60%, with this impairment persisting for at least 1 month post injury. Genetic knock-out of the gene encoding the astroglial water channel aquaporin-4, which is importantly involved in paravascular interstitial solute clearance, exacerbated glymphatic pathway dysfunction after TBI and promoted the development of neurofibrillary pathology and neurodegeneration in the post-traumatic brain. These findings suggest that chronic impairment of glymphatic pathway function after TBI may be a key factor that renders the post-traumatic brain vulnerable to tau aggregation and the onset of neurodegeneration.
Keywords: AQP4, aquaporin-4, cerebrospinal fluid, neurodegeneration, tauopathy, traumatic brain injury
Introduction
Moderate to severe traumatic brain injury (TBI) is an established risk factor for the development of neurodegeneration, including AD, with a single episode of TBI conferring an increased risk of dementia later in life (Guo et al., 2000; Plassman et al., 2000; Moretti et al., 2012; D.H. Smith et al., 2013). Prior TBI among AD patients is associated with an earlier age of disease onset than in patients without prior TBI, suggesting that TBI accelerates the development of AD pathology (Sullivan et al., 1987; Gedye et al., 1989; Nemetz et al., 1999). Indeed, postmortem histopathological examination of long-term survivors of TBI and age-matched controls revealed that among patients <60 years of age neurofibrillary tangles, intracellular aggregates composed of hyperphosphorylated tau, were present in 34% of post-traumatic brains compared with an incidence of 10% in age-matched control cases (Johnson et al., 2012). Although these findings link TBI with chronic neurodegeneration such as AD, the changes that occur in the post-traumatic brain that render it vulnerable to tau aggregation remain unknown.
Tau is an intracellular microtubule-associated protein that is released into the interstitium of the healthy young brain in response to excitatory neuronal activity (Yamada et al., 2011, 2014). Emerging evidence suggests that the movement of tau and tau aggregates through the extracellular space plays an important role in the development and spread of intracellular tau pathology (Frost and Diamond, 2010; Jucker and Walker, 2011; Walker et al., 2013). While much is known regarding the molecular pathways of tau processing in intracellular compartments (Morris et al., 2011; Chesser et al., 2013), the pathways and mechanisms of interstitial tau clearance from the brain interstitium and how these are altered in the setting of TBI are unknown.
We have defined a brain-wide paravascular pathway that facilitates the efficient clearance of interstitial proteins and peptides, including amyloid-β, from the brain parenchyma (Iliff et al., 2012, 2013a,b; Xie et al., 2013). We found that subarachnoid CSF recirculates through the brain parenchyma along paravascular spaces surrounding penetrating arteries, exchanging with the surrounding interstitial fluid (ISF) to facilitate the clearance of interstitial solutes. Paravascular CSF–ISF exchange and interstitial solute clearance is dependent upon water transport via astroglial aquaporin-4 (AQP4) water channels (Iliff et al., 2012), which are localized predominantly to perivascular astrocytic end feet. Based upon its appropriation of the lymphatic function of interstitial protein management, and its dependence upon glial water transport, we termed this paravascular pathway the “glymphatic system.” In the present study, we evaluate whether the disruption of perivascular AQP4 localization following TBI (Lu et al., 2011; Fukuda et al., 2012; Ren et al., 2013) is accompanied by post-traumatic impairment of paravascular CSF–ISF exchange and interstitial solute clearance. Finding chronic impairment of glymphatic pathway function after TBI, we define the anatomical pathway along which interstitial tau is cleared, then test whether impairment of glymphatic clearance contributes to the buildup of phosphorylated tau after TBI.
Materials and Methods
Animals.
All experiments were approved by the University Committee on Animal Resources of the University of Rochester Medical Center and the Institutional Animal Care and Use Committee of Oregon Health & Science University. Unless otherwise noted, 8- to 12-week-old male C57BL/6 mice (Charles River) were used in experiments. To define the paravascular route of tau clearance from the brain, a transgenic double reporter mouse was generated. Tg(TIE2GFP)287Sato/J (Tie2-GFP, JAX) were crossed with NG2DsRedBAC (NG2-DsRed, JAX) mice as described previously (Iliff et al., 2012). Tie2-GFP overexpress green fluorescent protein under the endothelium-specific Tie2 promoter, while NG2-DsRed mice express the red fluorescent protein DsRed under the control of the NG2 promoter. Adult NG2-DsRed animals express DsRed in brain pericytes and vascular smooth muscle cells, in addition to NG2-positive glial cells. In Tie2-GFP:NG2-DsRed mice, all blood vessels are labeled with GFP. Capillaries, venules, and veins express disperse DsRed labeling on overlaying pericytes. Arterioles and arteries exhibit a dense circumferential pattern of DsRed labeling. Aqp4−/− mice were generated as described previously (Thrane et al., 2011). These animals are global Aqp4 gene knock-outs backcrossed onto a C57BL/6 background.
“‘Hit & Run” traumatic brain injury model.
Experimental closed-skull moderate TBI was induced as described previously (Ren et al., 2013). Briefly, a pneumatic-controlled, cortical impact device (Pittsburgh Precision Instruments) was modified by mounting the metal rod horizontally. A polished stainless steel tip, which strikes the mouse's head during impact, was fitted to the end of the impactor slider. A strike depth of 10 mm and 0.1 time of impact were used with an impact velocity of 5.2 m/s. Once unresponsive to tail pinch, the mouse was anesthetized with 2.5% isoflurane for 2 min, then hung head up vertically from its incisor teeth by a mounted metal ring. The impactor was positioned normal to the skull at the loading point halfway between the ipsilateral orbit and the ear canal. Following impact, the mechanism released the injured animal onto a soft pad placed below. After anesthesia induction, it takes ∼30 s to suspend, injure, and release the animal onto the soft pad. The sham-treated mice were anesthetized, hung from the string, and allowed to fall and recover.
The benefits of the Hit & Run model against more conventional TBI models such as the controlled cortical impact (CCI) or fluid percussion injury models(FPI), including the use of a minimal period of anesthesia (<5 min), a closed skull (with no craniotomy required), and a freely moving head, have been defined in detail in our prior study characterizing this model (Ren et al., 2013). Particularly important to the present study, surgical craniectomy such as is used in both the CCI and FPI models, reduces the upregulation of AQP4 observed in the brain after closed-skull TBI (Tomura et al., 2011). Thus, to study the effect of post-traumatic changes in AQP4 expression and localization upon glymphatic pathway function, it was important to avoid this confounding influence. Additionally, an increasing number of studies suggests that exposure to clinically relevant levels of inhalation anesthetic such as isoflurane can promote tau phosphorylation and neurofibrillary pathology (Planel et al., 2009; Run et al., 2009). The minimal period of anesthesia used in the Hit & Run model reduces the likelihood that such effects influence our evaluation of tau phosphorylation at 28 d after injury induction.
Immunofluorescence.
Immunofluorescence was conducted on free-floating 100 μm PFA-fixed vibratome slices or on slide-mounted 20 μm cryostat sections. Slices were blocked for 1 h at room temperature with 3% normal donkey serum (Jackson ImmunoResearch), incubated with primary antibody overnight at 4°C, and incubated with secondary antibody for 2 h at room temperature. Primary antibodies included mouse anti-GFAP (an astrocytic marker; 1:500: Millipore), rabbit anti-AQP4 (1:500; Millipore), mouse anti-human tau (1:500; Pierce); rabbit anti-NeuN (a neuronal marker; 1:500; Millipore), mouse anti-phosphorylated tau (AT8; 1:40; Pierce), mouse anti-phosphorylated neurofilament (SMI34; 1:250; Abcam), and Iba1 (a microglia/macrophage marker; 1:250; Wako). Slices were mounted with Prolong Antifade Gold with DAPI (Life Technologies). Immunofluorescence was imaged on an inverted laser scanning confocal microscope (Zeiss LSM510), and whole-slice montages were generated using a resonance-scanning confocal microscope (Nikon A1R).
To quantify GFAP-, Iba1-, and AT8 (P-tau)-immunoreactivity, 4 20 μm slices were selected at 200 μm intervals centered around the traumatic lesion focus from each animal for imaging. In sham-treated animals, slices from equivalent rostrocaudal levels were selected. Uniform fields from the temporal ipsilateral and contralateral cortex that excluded the lesion cavity and surrounding glial scar were acquired by laser scanning confocal microscopy at 63× objective power by a blinded investigator. Confocal micrographs were analyzed in FIJI software by a blinded investigator. Fluorescence channels were separated, and the images uniformly thresholded to derive an area fraction value that represents the percentage of the imaging field. The area fractions for the four slices for each individual animal were averaged to derive a single biological replicate.
Intracisternal CSF tracer infusion.
Fluorescent CSF tracer (ovalbumin-conjugated Alexa 647, 45 kDa; Life Technologies; OA-45) was constituted in artificial CSF at a concentration of 0.5%. For intracisternal tracer infusions, anesthetized mice were fixed in a stereotaxic frame and a 30 gauge needle was inserted into the cisterna magna. Ten microliters of CSF tracer was infused at a rate of 1 μl/min for 10 min with a syringe pump (Harvard Apparatus). To visualize tracer movement from the cisternal compartments into the brain parenchyma, animals were perfusion fixed 30 min after the start of intracisternal infusion (note that the needle was left in place for the entire 30 min, even after infusion ceased). Vibratome slices (100 μm) were cut and mounted as above. Tracer influx into the brain was imaged ex vivo by whole-slice conventional fluorescence microscopy (Olympus; Stereo Investigator Software).
Tracer influx was quantified by a blinded investigator using FIJI (ImageJ) software as described previously (Iliff et al., 2012). The ipsilateral and contralateral cerebral cortex in each slice was manually outlined, and the mean fluorescence intensity within the cortical ROIs was measured. A weighted average (weighted by cortical area) of fluorescence intensity was calculated for each hemisphere between the eight slices for a single animal, resulting in a single paired (ipsilateral and contralateral) biological replicate. The effect of TBI upon CSF tracer influx into the ipsilateral or contralateral cortex was evaluated by two-way ANOVA with Sidak's post hoc test for multiple comparisons to determine differences between individual time points.
Intraparenchymal injections.
To evaluate the pathway of interstitial tau clearance, recombinant human tau (hTau; Abcam) was stereotactically microinjected into the cerebral cortex of Tie2-GFP:NG2-DsRed mice. A small burr hole was drilled and a 33 gauge stainless steel cannula (Hamilton) was inserted at the following stereotactic coordinates within the somatosensory cortex: 0.58 mm posterior, 1.90 mm lateral to bregma, 0.70 below brain surface. Thirty minutes after cannula insertion, 500 nl of 0.1 μg/μl hTau was infused over 10 min. One hour later, the cannula was removed and the animal rapidly perfusion fixed. The movement of hTau through the brain was evaluated in serial 100 μm vibratome slices labeled with a primary antibody specific for human tau.
To evaluate the rates of interstitial fluid and solute clearance from the brain, radiolabeled tracers (3H-mannitol and 14C-inulin) were injected stereotactically into the brain parenchyma. A stainless steel guide cannula was implanted into the contralateral frontal cortex of anesthetized mice 6 d after TBI with coordinates of the cannula tip at 0.7 mm anterior, 3.0 mm lateral to bregma, and 1.3 mm below the surface of the brain. Animals were allowed to recover after surgery and the experiments were performed 24 h after guide cannula implantation (7 d after TBI; Xie et al., 2013). Artificial CSF (500 nl) containing both 3H-mannitol and 14C-inulin (0.05 μCi) was infused over 5 min and 1 h after the start of infusion, the cannula was removed, and the brain dissected and prepared for radioactivity analysis. Brain samples were solubilized in 0.5 ml of tissue solubilizer overnight, followed by addition of 0.5 ml liquid scintillation cocktail. Radioactivity was analyzed in a liquid scintillation counter (Beckman-Coulter). The amount of 3H-mannitol and 14C-inulin cleared in 1 h was calculated based upon the percentage of radioactivity remaining in the brain: percentage recovery = 100 × (Nb/Ni), where Nb is the radioactivity remaining in the brain after 1 h and Ni is the radioactivity injected into the brain. The effect of TBI or Aqp4 gene deletion upon the clearance of interstitial radiotracers was evaluated by two-way ANOVA with Tukey's post hoc test for multiple comparisons to evaluate differences within individual groups.
Evaluation of traumatic lesion volume.
The effect of Aqp4 gene deletion upon traumatic lesion volume was evaluated 28 d after TBI. Eight uniformly sampled 20 μm cryostat sections were collected at 400 μm intervals from each animal and processed by conventional H&E staining. Slices were photographed with a slide scanner (Nikon) and the area of each hemisphere (excluding ventricular spaces) was manually outlined by a blinded investigator. Area measurements were integrated across eight slices to derive contralateral and ipsilateral hemispheric volume measurements for each animal. Post-traumatic changes in ipsilateral brain volume were evaluated by normalizing ipsilateral volume to the paired contralateral volumes. We have previously shown that this model of TBI does not result in loss of contralateral cortical or subcortical tissue (Ren et al., 2013). Differences in traumatic lesion volume between wild-type and Aqp4−/− mice were evaluated by unpaired Student's t test.
Evaluation of tau phosphorylation by Western blot.
Brain tissue was homogenized in solution A containing sucrose (250 mmol/L), KCl (60 mmol/L), Tris-HCl (15 mmol/L), NaCl (15 mmol/L), EDTA (5 mmol/L), EGTA (1 mmol/L), PMSF (0.5 mmol/L), and DTT (10 mmol/L), then centrifuged at 2000 g for 10 min at 4°C. Protein samples (25 μg/lane for P-tau gels, 4 μg/lane for total tau lanes) were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Blots were then blocked in 5% dry milk and incubated at 4°C overnight with a primary antibody. Primary antibodies used were rabbit anti-P-tau (pS396; Life Technologies; catalog #44–752G), mouse anti-PHF-tau (pSer202/Thr205; Thermo Scientific; catalog #MN1020), mouse anti-PHF-tau (pThr231; Thermo Scientific; catalog #MN1040); rabbit anti-P-tau (pThr205; Thermo Scientific; catalog #PA-14415); rabbit anti-P-tau (pSer404; Thermo Scientific; catalog #PA-14426); and mouse anti-tau (pan-tau; Sigma; catalog #T9450). The signal was visualized using a biotinylated secondary antibody with an ECL Plus chemiluminescence detection kit. Autoradiograms were scanned and band optical densities were quantified and expressed relative to total tau.
Assessment of post-traumatic behavioral deficits.
For open field, rotarod, novel-object recognition, and Barnes maze tests, all tests were performed on four cohorts of animals: wild-type sham-treated, wild-type TBI treated, Aqp4−/− sham treated, and Aqp4−/− TBI treated. Open field, rotarod, and novel-object recognition tests were performed 2 d before TBI, then weekly following TBI for 4 weeks. Barnes maze test was performed weekly after TBI.
In the open field test, mice were monitored by video in an activity cage to investigate spontaneous locomotor activity. Animals were evaluated for 10 min after a 1 min adaptation period and videos were analyzed using Any-Maze software (Stoelting). In the rotarod test, a mouse was placed on a rotating rod and the latency before falling from the rod was measured. The rod rotated at an initial speed of 10 rpm and accelerated 1 rpm per second. Mice were placed on the bar for a maximum of 6 min and were given three sessions per day. Mice were tested 2 d before TBI, then once per week after TBI. All post-TBI values were normalized to the individual animals' pre-TBI control performance.
In the novel-object recognition test, animals were placed in an open chamber under video surveillance and allowed to freely explore for 10 min. Three objects of different texture and design were placed in the center of the chamber and spaced equidistant from the walls. Mice were allowed to explore the objects for 10 min. On the second day, one of the objects was replaced by a novel object and animals were allowed to interact with objects for 10 min. Their interaction times were recorded with a camera and scored by individuals blinded to the treatment group of the animals. The Barnes maze test apparatus consisted of a large circular platform with 20 evenly spaced holes around the perimeter. During the test, the animal used spatial cues to locate an escape hole, which differs from other holes in that it provides an escape to a compartment that hides the animal from sight. Each day, the animal underwent four trails with a rest period of 15 min between each trial. On the first day, the animal underwent an adaptation period during which the animal was introduced to the maze and guided to the escape hole. In four test trials, the animal was allowed to explore the maze for 3 min. The trial ended if (1) the animal found the escape path or (2) after 3 min had elapsed. This process was repeated a total of four times at 1 d intervals. Performance in behavioral tests between groups was evaluated by two-way repeated-measures ANOVA with Sidak's post hoc test for multiple comparisons.
Results
Traumatic brain injury impairs paravascular clearance of interstitial solutes
Adult male mice were subjected to Hit & Run TBI injury, which we have recently described and characterized (Ren et al., 2013; Fig. 1A). In this model the animal is briefly anesthetized with isoflurane, suspended by its incisors, and a calibrated traumatic impact is delivered to the skull overlying the temporal cortex. Advantages of this model include a minimal time under anesthesia (<3 min), a freely moving head, and a closed skull. In the present experiments, impactor parameters were selected to result in a moderate-to-severe TBI grade, characterized by contusion of the underlying cortex, but not resulting in skull fracture. Sham animals were subjected to anesthesia, hanging, and falling, but no impact. We have previously provided a detailed histological characterization of this model between 3 and 28 d after TBI (Ren et al., 2013). Within 7 d of TBI, a consolidated glial scar forms around the temporal cortical lesion, and the ipsilateral cortex surrounding the glial scar is characterized by hypertrophic GFAP-positive astrocytes that are absent in control and mirror-image contralateral cortex. At 28 d post injury, the traumatic lesion is bounded by a dense GFAP-positive glial scar and surrounded by wide fields of reactive astrogliosis that extend throughout the ipsilateral cortex, underlying white matter and striatum (Fig. 1B,C). As we described previously, AQP4 localization in these regions of reactive astrogliosis in the post-traumatic cortex is markedly altered. Between 3 and 7 d post injury, the perivascular polarization of AQP4 is lost in these gliotic regions, and does not fully recover even 28 d post injury (Ren et al., 2013). This can be seen clearly when AQP4 and GFAP immunofluorescence were evaluated by confocal microscopy 28 d post, injury. In the intact cortex, AQP4 immunoreactivity is highly polarized to perivascular end foot processes (Fig. 1D,E), while regions of persistent reactive astrogliosis in the ipsilateral cortex exhibit a marked loss of perivascular AQP4 polarization (Fig. 1F), confirming that loss of perivascular AQP4 polarization is a persistent feature of the post-traumatic cortex. We confirmed the specificity of the anti-AQP4 antibody by staining tissue from sham and TBI-treated Aqp4−/− mice. Although GFAP immunoreactivity did not differ between wild-type and Aqp4−/− mice 7 d post injury, no AQP4 immunoreactivity was observed in Aqp4−/− brains (Fig. 2A–C), confirming that the loss of specific perivascular AQP4 localization is not the result of nonspecific antibody binding within the injured tissue.
Figure 1.

TBI causes loss of perivascular AQP4 polarization in astrocytic end feet. A, Schematic depicting Hit & Run model of moderate TBI (Ren et al., 2013). Mouse is briefly anesthetized with isoflurane, suspended by its incisors from a string, and a calibrated temporal impact is delivered by a pneumatic-controlled, cortical impactor device. The animal falls to an underlying pad and rapidly awakens from anesthesia. B–C, Twenty-eight days after TBI, the traumatic lesion is bounded by a dense glial scar and surrounded by a wide field of reactive astrogliosis. Changes in AQP4 expression and localization between 3 and 28 d after TBI have been extensively characterized previously (Ren et al., 2013). Within the control cortex (D), AQP4 localization is polarized to perivascular astrocytic end feet surrounding the cerebral microcirculation (arrows). Inset depicts GFAP immunoreactivity alone. Twenty-eight days after, AQP4 immunoreactivity in the contralateral cortex (E) appears normal. In regions of reactive astrogliosis (inset shows GFAP immunoreactivity alone) surrounding the traumatic lesion in the ipsilateral cortex (F), perivascular AQP4 polarization is persistently lost, as AQP4 immunoreactivity increases in nonperivascular structures (arrows identify the location of perivascular astrocytic end feet. Scale bars: 20 μm.
Figure 2.

AQP4 immunoreactivity is absent in Aqp4−/− mice. To ensure the specificity of the anti-AQP4 primary antibody used in the present study, we conducted immunolabeling of control and TBI-treated brains from Aqp4−/− mice perfused 7 d after injury. Although GFAP expression was readily detectable in control (A), contralateral TBI (B), and ipsilateral TBI (C) cortex, no AQP4 immunoreactivity was detectable. Scale bar, 20 μm.
Glymphatic clearance of interstitial solutes, including amyloid-β, is dependent upon astroglial AQP4 (Iliff et al., 2012), while perivascular polarization of AQP4 is disrupted for >1 month after experimental TBI (Fig. 1D–F). The effect of TBI upon glymphatic pathway function was first evaluated by measuring the rate of paravascular CSF–ISF exchange 1, 3, 7, and 28 d after TBI Figure 3A,B). Fluorescence tracer (ovalbumin-conjugated Alexa 555, MW 45 kDa; OA-45) was injected into the subarachnoid CSF of the cisterna magna, brains were perfusion fixed 30 min post injection, and the influx of CSF tracer into the brain parenchyma was evaluated ex vivo by whole-slice fluorescence microscopy (Iliff et al., 2012; L. Yang et al., 2013). Compared with sham controls, TBI dramatically impaired paravascular influx of CSF tracer (Fig. 3C–F). Although this effect was most pronounced in the ipsilateral cortex, glymphatic pathway impairment was additionally observed in the contralateral hemisphere and in the underlying striatum. When CSF tracer influx was quantified at t = 1, 3, 7, and 28 d after TBI, impairment of CSF influx peaked at 7 d post injury, yet remained significantly impaired even 28 d after TBI (Fig. 3G; ***p < 0.001 vs Control, ##p < 0.01 vs contralateral structure; two-way ANOVA with Tukey's post hoc test for multiple comparisons; n = 5–12 animals per group).
Figure 3.

Paravascular CSF–ISF exchange is chronically impaired after TBI. A, B, Paravascular CSF–ISF exchange was evaluated by intracisternal injection of CSF tracer (ovalbumin-conjugated Alexa 555; OA-45) 1, 3, 7, and 28 d after TBI. C–F, Ex vivo whole-slice fluorescence imaging shows that paravascular CSF influx evaluated 30 min post injection was dramatically reduced 7 d after TBI. Interestingly, reduced glymphatic influx was observed bilaterally despite the unilateral traumatic injury. Quantification of tracer influx into the cortex (G) demonstrates that the effect of TBI upon CSF influx peaks at 7 d post injury; however, a significant impairment of glymphatic function remains 28 d after injury (***p < 0.001 vs Control; ###p < 0.001 vs contralateral structure; two-way ANOVA with Tukey's post hoc test for multiple comparisons, n = 5–12 animals per group). Although impaired CSF influx is observed bilaterally in both cortex and striatum, the impairment is greatest in the ipsilateral cortex.
In prior studies (Iliff et al., 2012; Xie et al., 2013), we have demonstrated that interstitial tracers such as fluorescently labeled ovalbumin or dextrans, as well as clinically relevant solutes such as amyloid-β, are cleared from the brain along defined anatomical pathways. To evaluate whether exogenous interstitial tau is cleared along the same convective pathways, we injected recombinant human tau protein (Abcam) into the cortex of transgenic Tie2-GFP:NG2-DsRed mice (Fig. 4A). As described previously (Iliff et al., 2012), these mice express GFP in endothelial cells and the red fluorescent protein DsRed in cerebrovascular pericytes and vascular smooth muscle cells, enabling the simple identification of arteries, veins, and capillaries in fixed slices and in vivo. Sixty minutes after injection, brains were perfusion fixed, stained with an antibody specific for human tau (Abcam), and imaged by whole-slice confocal imaging. The local convective movement of interstitial tau occurred slowly through the bulk interstitium, proceeding more rapidly along the basal lamina of the nearby capillary bed. Intracortically injected hTau moved most rapidly from the injection site along the subcortical white matter of the external capsule (Fig. 4B,C). At points, most distant from the injection site, hTau appeared to accumulate along a specific subset of large-caliber draining veins, including the caudal rhinal veins that drain from the subcortical white matter tracts (Fig. 4D,E); the internal cerebral veins, which run medially within the roof of the third ventricle (Fig. 4G); and the inferior sagittal sinus at the base of the falx cerebri (Fig. 4H). Interestingly, in regions distal from the injection site, tracer accumulation occurred solely along these large-caliber draining veins, and not along penetrating arteries (Fig. 4F), which suggests that interstitial tau is cleared along the same paravascular pathways previously described for interstitial tracers and amyloid-β (Iliff et al., 2012).
Figure 4.
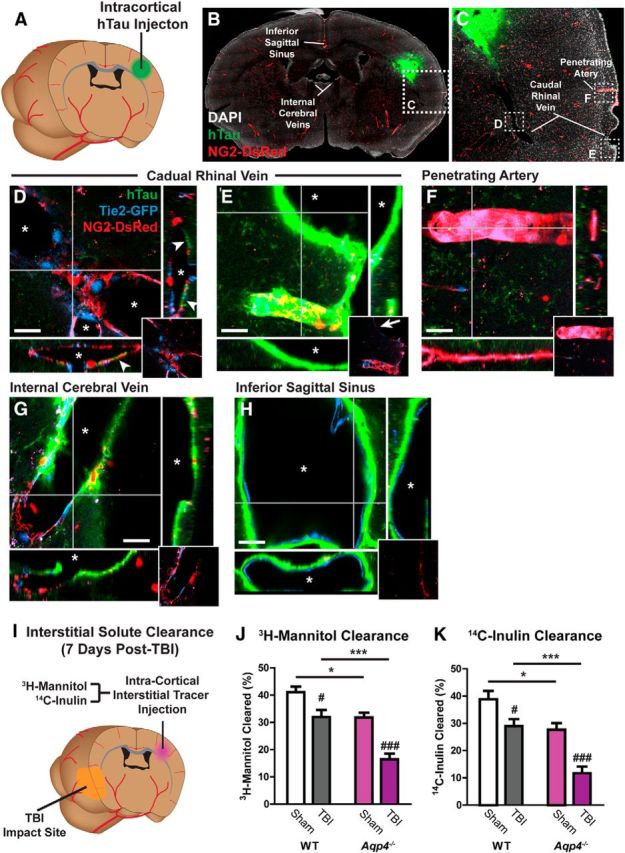
Interstitial tau is cleared along the paravascular glymphatic pathway. A, Recombinant hTau was injected into the cortex of Tie2-GFP:NG2-DsRed double-transgenic mice, which express GFP in the vascular endothelium and DsRed fluorescent protein in cerebral vascular smooth muscle and pericytes. B–H, Movement of interstitial hTau through the intact brain was evaluated 30 min post injection by immunofluorescence. hTau moved diffusely from the injection site (B, C), moving along subcortical white matter tracks and perivascular spaces, to exit the brain along large draining veins. High-power micrographs of the intraparenchymal segment (D) and the exit site from the brain (E) of the caudal rhinal vein show hTau accumulation (arrowheads) in paravascular spaces surrounding this vein. This includes the wall of the large surface venous structure present at the brain surface (arrow). Insets depict X-Z and Y-Z orthogonal views of confocal stack. Inset at bottom right depicts vascular reporter proteins without hTau fluorescence channel. Asterisk denotes vessel lumen. F, The absence of hTau along a nearby penetrating artery shows that para-arterial spaces are not long-range pathways for interstitial hTau clearance. Intense hTau labeling of the distant medial internal cerebral veins (G) and the inferior sagittal sinus (H) shows that these venous structures are major outflow routes for interstitial hTau. Scale bars: 20 μm. I, The effect of TBI upon interstitial solute clearance from the cortex was evaluated 7 d post injury. The clearance of radiolabeled 3H-mannitol (MW 182 Da; J) and 14C-inulin (MW∼5 kDa; K) was measured 60 min after infusion into contralateral frontal cortex. In wild-type mice, TBI significantly slowed the clearance of both 3H-mannitol and 14C-inulin (#p < 0.05, ###p < 0.001 TBI vs sham; two-way ANOVA with Tukey's post hoc test for multiple comparisons; n = 6 animals per group). Clearance studies conducted in Aqp4−/− mice demonstrated that impairment of solute clearance after TBI was exacerbated by Aqp4 gene deletion (*p < 0.05, ***p < 0.001 wild-type vs Aqp4−/− mice; two-way ANOVA with Tukey's post hoc test for multiple comparisons, n = 6 animals per group).
We next measured the effect of TBI upon the clearance of interstitial solutes from the cortex. Seven days after TBI, radiolabeled tracers (3H-mannitol, MW 182 Da; 14C-inulin, MW ∼5 Da) were coinjected into the contralateral frontal cortex (Fig. 4I). One hour later, the radiotracer remaining in the brain was measured by liquid scintillation counting. Mannitol and inulin were selected as interstitial tracers because neither are cleared across the blood–brain barrier, and both are cleared from the brain by convective bulk flow (Cirrito et al., 2005; Syková and Nicholson, 2008). In sham-treated mice, within 1 h of injection, ∼40% of the injected 3H-mannitol and 14C-inulin had been cleared (Fig. 4J,K). The equivalent rates of 3H-mannitol and 14C-inulin clearance, despite a ∼25-fold difference in molecular weight, is consistent with the clearance of these solutes from the brain interstitium by bulk flow (Cserr, 1971; Iliff et al., 2012). After TBI, interstitial solute clearance in wild-type mice was reduced by ∼25% (#p < 0.05, ###p < 0.001, TBI vs sham; two-way ANOVA with Tukey's post hoc test for multiple comparisons; n = 6 per group). Importantly, we conducted tracer injections contralaterally to avoid the disrupted tissue in the ipsilateral cortex. However, at 7 d post-TBI, aquaporin mislocalization (described previously in Ren et al., 2013) and glymphatic pathway impairment was more severe (Fig. 3G) in the ipsilateral cortex. Thus, we believe that this 25% reduction in glymphatic solute clearance is an underestimate of the impairment occurring after traumatic injury in the ipsilateral cortex. These data demonstrate that glymphatic pathway function, reflected by paravascular CSF–ISF exchange and interstitial solute clearance, are chronically impaired after TBI.
Impairment of glymphatic pathway function promotes post-traumatic development of tauopathy
Glymphatic pathway function is mediated by astrocytic AQP4 water channels, while genetic deletion of the Aqp4 gene in mice dramatically slows paravascular CSF–ISF exchange and impairs the clearance of intrastriate interstitial solutes such as amyloid-β (Iliff et al., 2012). These findings were confirmed in a new set of experiments where the interaction between Aqp4 gene deletion and TBI on interstitial solute clearance was evaluated. In agreement with our prior study (Iliff et al., 2012), among sham-treated animals Aqp4 gene deletion significantly reduced the amount of both intracortically injected 3H-mannitol and 14C-inulin cleared within the first hour post injection (Fig. 4J,K; *p < 0.05, wild-type sham vs Aqp4−/− sham, two-way ANOVA with Tukey's post hoc test for multiple comparisons, n = 6 per group). When Aqp4−/− mice were subjected to TBI, 7 d after injury the impairment of interstitial solute clearance was dramatically exacerbated compared with the effect observed for TBI alone (Fig. 4J,K, ***p < 0.001, wild-type TBI vs Aqp4−/− TBI, two-way ANOVA with Tukey's post hoc test for multiple comparisons, n = 6 per group). Because Aqp4−/− mice subjected to TBI exhibited a profound impairment in glymphatic pathway function, we next used this approach to evaluate whether impairment of glymphatic pathway function contributes to aberrant accumulation of phosphorylated tau after TBI.
Although studies focusing on ischemic injury demonstrated an overall protective effect of Aqp4 gene deletion on lesion development (Manley et al., 2000), more recent work in TBI models has not observed any difference in lesion volume after TBI between wild-type and Aqp4−/− mice (Lu et al., 2011). Consistent with these findings, when we assessed lesion volume by H&E staining of serial slices from wild-type and Aqp4−/− mice 28 d after TBI, we observed no differences in lesion volume (Fig. 5A–C). Thus any differences observed between wild-type and Aqp4−/− mice are likely not the result of differences in traumatic injury severity.
Figure 5.

Aqp4 gene deletion does not exacerbate chronic traumatic lesion development. The effect of Aqp4 gene deletion upon traumatic lesion volume was evaluated in brains harvested 28 d after TBI. A, B, Brains were serially sliced and brain structure was evaluated by H&E staining. Red arrow indicates site of traumatic impact and area of greatest cortical damage. Ipsilateral and contralateral cortical areas were measured for each slice, then integrated through serial slices to derive a cortical volume. Ipsilateral cortical volume was expressed as a ratio to the contralateral volume (C). No significant difference was observed in ipsilateral lesion volume between wild-type and Aqp4−/− mice (unpaired t test).
At 28 d after injury brains were evaluated by Western blot for evidence of P-tau accumulation. Using a battery of antibodies targeting the pSer202/pThr205, pThr205, pThr231, pSer396, and pSer404 P-tau epitopes, we measured P-tau accumulation in the ipsilateral and contralateral cortex of wild-type and Aqp4−/− mice. Representative blots are shown in Figure 6A, while P-tau levels normalized to total tau expression (derived from blotting with a pan-tau antibody) are presented in Figure 6B–F. Two general findings were observed across all of the data. First, TBI significantly increased P-tau levels identified with the anti-pThr205 (Fig. 6C; p < 0.05, two-way ANOVA with Tukey's post hoc test for multiple comparisons, n = 4 per group), pThr231 (Fig. 6D; p < 0.05, two-way ANOVA, n = 4 per group), and pSer396 (Fig. 6E; p < 0.01, two-way ANOVA with Tukey's post hoc test for multiple comparisons, n = 4 per group) antibodies in both the contralateral and ipsilateral hemispheres; however, the effects were greatest in the ipsilateral hemisphere. Similarly, mean P-tau levels identified by anti-pSer202/Thr205 (Fig. 6B) and pSer404 (Fig. 6F) antibodies were higher after TBI; however, these effects did not achieve statistical significance. A second finding was that after TBI, mean P-tau levels in the Aqp4−/− cortex were consistently higher than in the corresponding wild-type cortex. This was true of both contralateral and ipsilateral hemispheres; however, the effect was greatest and the levels higher in the ipsilateral cortex. Post hoc testing revealed a significant difference between wild-type and Aqp4−/− pThr231 levels in the ipsilateral cortex (Fig. 6D; *p < 0.05, wild-type TBI vs Aqp4−/− TBI; two-way ANOVA with Tukey's post hoc test for multiple comparisons, n = 4 per group).
Figure 6.

Post-traumatic P-tau accumulation is exacerbated by Aqp4 gene deletion. The effect of impairing glymphatic pathway function by Aqp4 gene deletion upon the development of tauopathy after TBI was evaluated. A, Wild-type and Aqp4−/− brains were harvested 28 d post injury and probed for the presence of P-tau epitopes. Representative blots are presented showing the effect of mouse genotype (wild-type vs Aqp4−/−), injury status (sham vs TBI), and hemisphere [contralateral (C) vs ipsilateral (I)] upon labeling by various P-tau monoclonal antibodies targeting different tau phosphorylation epitopes. Total tau was also measured (Pan-tau) and all P-tau levels were normalized to P-tau levels within each biological sample. B–F, Across all epitopes, TBI tended to increase P-tau labeling, particularly in the ipsilateral hemisphere. This effect of TBI on P-tau labeling was statistically significant when antibodies targeting the pThr205 (C), pThr231 (D), and pSer396 (E) were used (two-way ANOVA, n = 3–4 per group). Post hoc analysis revealed that Aqp4 gene deletion significantly increased pThr231 labeling after TBI (*p < 0.05, **p < 0.01, ***p < 0.001; two-way ANOVA with Tukey's post hoc test for multiple comparisons, n = 3–4 per group).
We then evaluated whether TBI or Aqp4 gene deletion exacerbated histologically evaluated tau pathology. Frozen slices were stained with the AT8 monoclonal antibody (which detects the pSer202/pThr205 P-tau epitopes) and counterlabeled with the neuronal marker NeuN 28 d after TBI. Within the control cortex, no P-tau immunoreactivity was evident (Fig. 7A). After TBI, the wild-type ipsilateral cortex exhibited elevated, but still diffuse, P-tau immunoreactivity (Fig. 7B). Within Aqp4−/− mice, P-tau immunoreactivity in the ipsilateral cortex was dramatically altered. Intense P-tau immunoreactivity was present in NeuN-positive neuronal soma and surrounding neurites (Fig. 7C–E). These findings are in general agreement with the quantitative results obtained by Western blot, demonstrating that Aqp4−/− mice, in which glymphatic pathway function and interstitial solute clearance are profoundly impaired (Iliff et al., 2012), also exhibit higher levels of tau pathology in the chronic phase after TBI.
Figure 7.

Neuronal P-tau accumulation and axonal degeneration after TBI in Aqp4−/− mice. Wild-type and Aqp4−/− mice were subjected to TBI and the presence of P-tau was evaluated by immunofluorescence 28 d post injury. Double labeling with the AT8 P-tau antibody (specific for pSer202/pThr205 epitopes) and the neuronal marker NeuN showed that in the wild-type cortex, P-tau immunoreactivity was not observed under control conditions (A). After TBI, P-tau immunoreactivity was diffusely increased, but at a low level (B). In the Aqp4−/− cortex (C), marked P-tau labeling was observed after TBI, both within neuronal soma (arrows) and in surrounding neurites (arrowheads). Quantification of P-tau immunoreactivity in cortical (D) and striatal (E) regions proximal to the traumatic lesion showed that P-tau accumulation was dramatically increased in the Aqp4−/− brain after TBI. F–K, Axonal degeneration was evaluated in the ipsilateral cortex and underlying corpus callosum 28 d after TBI by staining for phosphorylated neurofilament with the SMI34 monoclonal antibody. In the control cortex (F) and corpus callosum (I), no axonal spheroids or varicosities were evident. In the wild-type cortex after TBI (G), SMI34-immunoreactive varicosities were sparsely present (arrows). In the Aqp4−/− cortex (H) and corpus callosum (K), SMI34-positive spheroids and varicosities were readily detectable (arrows).
Aqp4 gene deletion promotes neurodegeneration and neuroinflammation
Neuroinflammation and cognitive decline are hallmarks of neurodegenerative processes, including Alzheimer's disease (Eikelenboom et al., 2002) and chronic traumatic encephalopathy (CTE; Goldstein et al., 2012). In our initial characterization of the Hit & Run model of TBI we observed that at 14 d post injury, evidence of axonal degeneration was readily apparent in the ipsilateral cortex (Ren et al., 2013). When we evaluated axonal degeneration by immunostaining for phosphorylated neurofilament with the SMI34 monoclonal antibody, we observed that SMI34-positive axonal varicosities and retraction bulbs were detectible in wild-type mice in the ipsilateral cortex 28 d after TBI (Fig. 7F,G,I,J), but were sparsely distributed. Axonal degeneration was more extensive in the ipsilateral cortex as well as within the underlying corpus callosum of Aqp4−/− mice (Fig. 7H,K).
In our prior study characterizing the development of reactive gliosis after TBI in the Hit & Run model (Ren et al., 2013), we observed that widespread reactive astrogliosis and microgliosis develops across large areas of the cortex and underlying striatum and hippocampus in the days following traumatic injury, which would slowly subside over the weeks following injury (Ren et al., 2013). Four weeks after injury, although intense astrocyte and microglial activation persisted in tissue adjacent to the traumatic lesion, GFAP immunoreactivity in regions not immediately associated with the glial scar remained only somewhat elevated in the ipsilateral cortex of wild-type mice (Fig. 8A–C,G). In Aqp4−/− mice, however, GFAP expression in these remote cortical regions was dramatically increased in the ipsilateral cortex (Fig. 8D–G; **p < 0.01, Aqp4−/− ipsilateral TBI vs wild-type ipsilateral TBI; ##p < 0.01 vs contralateral structure; two-way ANOVA with Tukey's post hoc test for multiple comparisons; n = 4 per group). A similar exacerbation of Iba1 labeling was observed in the ipsilateral cortex of Aqp4−/− mice after TBI compared with wild-type mice after TBI (Fig. 8H; #p < 0.05, Aqp4−/− ipsilateral vs Aqp4−/− contralateral; two-way ANOVA with Tukey's post hoc test for multiple comparisons; n = 4 per group).
Figure 8.

Aqp4 gene deletion promotes neuroinflammation after TBI. The effect of Aqp4 gene deletion upon the persistence of neuroinflammation after TBI was evaluated. A–F, Wild-type and Aqp4−/− mice were subjected to TBI and reactive astrogliosis (GFAP expression), and microgliosis (Iba1 expression) in the contralateral and ipsilateral cortex was evaluated by immunofluorescence 28 d post injury. Compared with wild-type mice after TBI, markedly elevated GFAP- and Iba1-immunoreactivity were observed in the ipsilateral cortex of Aqp4−/− mice after TBI. G, Quantification of GFAP labeling demonstrated significantly increased reactive astrogliosis in the ipsilateral cortex of Aqp4−/− mice (###p < 0.001, Ipsilateral vs Contralateral Aqp4−/−; two-way ANOVA with Tukey's post hoc test for multiple comparisons; n = 4 animals per group), which was not present in wild-type mice (**p < 0.01, Aqp4−/− Ipsilateral vs WT Ipsilateral). H, Quantification of Iba1 labeling showed that microglial activation was similarly increased in the ipsilateral cortex of Aqp4−/− mice (#p < 0.05, Ipsilateral vs Contralateral Aqp4−/−).
When behavioral tests were used to evaluate motor and cognitive function after TBI, we similarly found that Aqp4 gene deletion worsened functional outcome. Mice underwent behavioral evaluation 2 d before TBI, then weekly after injury (Fig. 9A). When gross motor function was evaluated by open field test, no significant difference was observed in wild-type mice after TBI while in Aqp4−/− mice, open field test performance was impaired after TBI (Fig. 9B; #p < 0.05 Aqp4−/− Control vs Aqp4−/− TBI; two-way repeated-measures ANOVA; n = 9–14 per group). When motor coordination was evaluated with the rotarod test, we found that while TBI did not significantly impair function in wild-type mice, in Aqp4−/− mice performance was significantly impaired after injury (Fig. 9C; #p < 0.05 Aqp4−/− Control vs Aqp4−/− TBI; two-way repeated-measures ANOVA with Sidak's post hoc test for multiple comparisons, n = 9–14 animals per group). Cognitive function was evaluated both with the novel-object recognition test and with the Barnes maze spatial learning test. TBI impaired cognitive function both in wild-type and Aqp4−/− mice [Fig. 9D,E; *p < 0.05, **p < 0.01 WT Control vs WT TBI; #p < 0.05, ##p < 0.05 Aqp4−/− Control vs Aqp4−/− TBI; two-way repeated-measures ANOVA with Sidak's post hoc test for multiple comparisons; n = 9–14 animals per group (Novel-Object Recognition), n = 5 per group (Barnes Maze)]. Post-traumatic cognitive deficits, however, were more pronounced in Aqp4−/− mice compared with wild-type animals (†p < 0.05 WT TBI vs Aqp4−/− TBI). In the case of both the rotarod test (Fig. 9C) and the Novel-Object Recognition Test (Fig. 9D), performance of the Aqp4−/− TBI group persistently tracked below that of the corresponding WT TBI group, through the full 4 weeks of evaluation post injury. Similarly, the deficit in Barnes Maze performance was observed when the test was administered beginning 28 d after TBI (Fig. 9E). These results demonstrate that Aqp4 gene deletion, in addition to causing glymphatic pathway dysfunction and promoting post-traumatic neuroinflammation, persistently exacerbates cognitive deficits after TBI.
Figure 9.
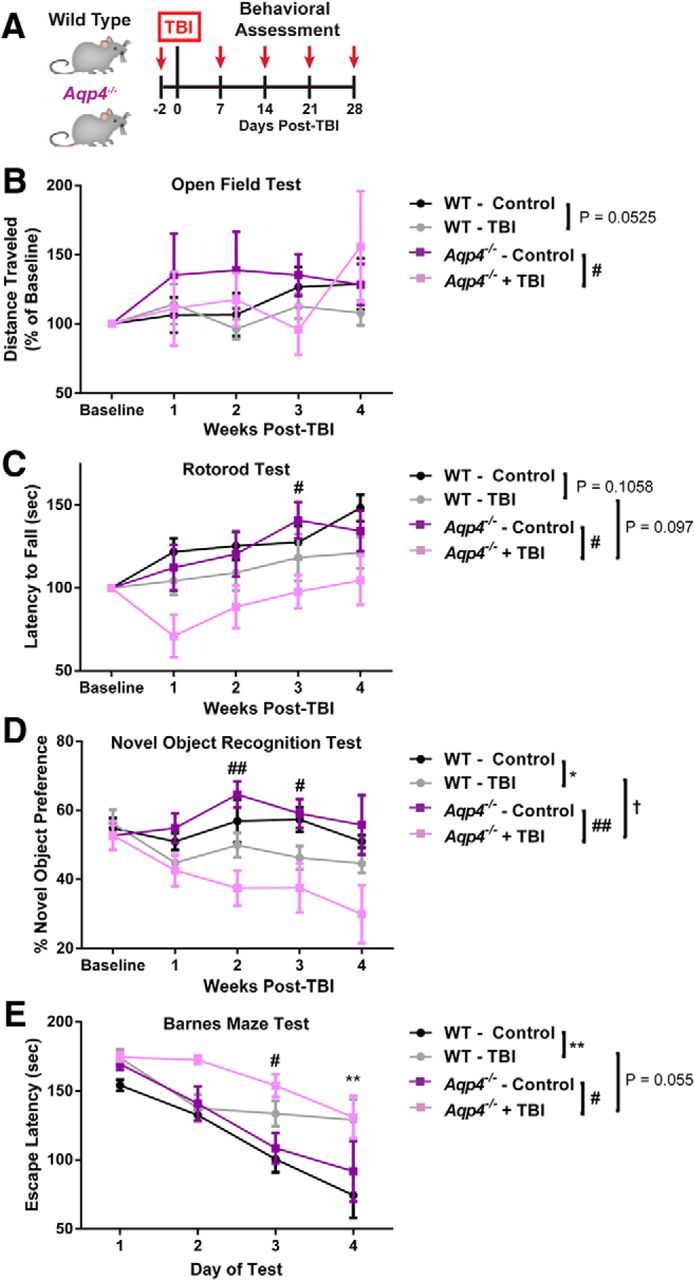
Aqp4 gene deletion exacerbates post-traumatic cognitive impairment. A, The effect of impairing the glymphatic pathway upon post-traumatic cognitive deficits was evaluated in wild-type and Aqp4−/− mice subjected to TBI. Animals underwent baseline behavioral testing 2 d before injury, then weekly after TBI. B, Gross motor behavior was assessed by the open field test. TBI did not significantly alter performance in the open field test in wild-type mice; however, performance in the open field test was significantly impaired after TBI in the Aqp4−/− mice (#p < 0.05 Aqp4−/− Control vs Aqp4−/− TBI; two-way repeated-measures ANOVA; n = 9–14 per group). C, Motor coordination was evaluated by the rotarod test. While TBI did not significantly impair rotarod performance in wild-type animals, in Aqp4−/− mice TBI significantly impaired test performance (#p < 0.05 Aqp4−/− Control vs Aqp4−/− TBI; two-way repeated-measures ANOVA with Sidak's post hoc test for multiple comparisons, n = 9–14 animals per group). Cognitive function was evaluated with the novel-object recognition test (D) and the Barnes maze test (E). In both tests, TBI impaired cognitive performance among both wild-type and Aqp4−/− mice [*p < 0.05, **p < 0.01 WT Control vs WT TBI; #p < 0.05, ##p < 0.05 Aqp4−/− Control vs Aqp4−/− TBI; two-way repeated-measures ANOVA with Sidak's post hoc test for multiple comparisons; n = 9–14 animals per group (Novel-Object Recognition), n = 5 per group (Barnes Maze)]. Post-traumatic cognitive impairment was exacerbated in Aqp4−/− mice compared with wild-type animals (†p < 0.05 WT TBI vs Aqp4−/− TBI).
Discussion
Moderate-to-severe TBI is an established risk factor for the development of neurodegenerative disease, including Alzheimer's disease (Guo et al., 2000; Lye and Shores, 2000; Plassman et al., 2000; D.H. Smith et al., 2013). The basis for this association has remained unclear; however, the presence of amyloid plaques in the brains of young patients that died from severe TBI (Roberts et al., 1991, 1994) and the presence of neurofibrillary pathology in the brains of TBI survivors (Johnson et al., 2012) has suggested that pathogenic processes initiated after TBI are related to those that occur in the aging brain in the setting of Alzheimer's disease.
Disruption of cells and cellular function within the perivascular domains that constitute the glymphatic pathway are established features of the brain in the setting of aging and Alzheimer's disease. Loss of pericyte coverage and blood–brain barrier disruption occur in the brains of Alzheimer's patients (Zlokovic, 2005; Sengillo et al., 2013), while loss of perivascular AQP4 polarization in the surrounding perivascular astrocytic end feet is observed in the aging (Kress et al., 2014) and Alzheimer's disease brain (Wilcock et al., 2009; J. Yang et al., 2011). These changes within perivascular domains are associated with the impairment of interstitial amyloid- β clearance (Zlokovic, 2005; Kress et al., 2014), suggesting that disruption of perivascular cellular function may render the aging brain vulnerable to misaggregation of amyloid-β and other proteins, and subsequent neurodegeneration. We have reported that after both mild and moderate TBI in mice, the perivascular polarization of AQP4 is chronically disrupted in reactive astrocytes after injury (Ren et al., 2013). Consistent with the dependence of brain interstitial solute clearance upon perivascular AQP4 (Iliff et al., 2012), we observed that moderate TBI impaired glymphatic pathway function for >1 month after injury, slowing the clearance of interstitial solutes from the brain parenchyma. The prolonged impairment of the glymphatic pathway after TBI, a key contributor to amyloid-β clearance from the brain interstitium22,24, may promote both early amyloid plaque deposition after severe TBI (Roberts et al., 1991, 1994) and the accelerated development of amyloid pathology in the aging brain after TBI (Guo et al., 2000; Lye and Shores, 2000; Plassman et al., 2000; D.H. Smith et al., 2013).
More recently, the development of neurodegeneration in the clinical condition CTE after exposure to repeated mild TBI has gained much attention. This condition, like Alzheimer's disease, is classified as a tauopathy, and is characterized by intracellular tau aggregates observed first in perivascular foci in the superficial frontal cortex to other cortical areas, the hippocampus, and wider brain regions (Omalu et al., 2011; McKee et al., 2013). Although tau, a microtubule-associated protein, is conventionally regarded as an intracellular protein, emerging research suggests that in the healthy young brain tau is released from active neurons into the brain interstitium (Yamada et al., 2011, 2014). After brain injury, including TBI, high levels of tau are released into the brain ISF and CSF (Morris et al., 2011; Magnoni et al., 2012; Tsitsopoulos and Marklund, 2013), presumably as a result of axonal damage. Interstitial tau is taken up into cells of the CNS, and the uptake of interstitial tau aggregates into neurons is now thought to underlie the “prionic” spread of tauopathy in progressive neurodegenerative diseases such as Alzheimer's disease (Kfoury et al., 2012; Holmes et al., 2013; Walker et al., 2013). In the present study, we report that in addition to cellular uptake, a large proportion of interstitial monomeric tau is cleared from the cortex by bulk flow along the white matter tracks and paravascular spaces of the glymphatic pathway.
After TBI, the clearance of interstitial solutes from the brain parenchyma was impaired, even when clearance was measured from the more modestly affected contralateral hemisphere. Yamada et al. (2011) and (2013) have proposed that increasing interstitial tau concentrations promote tau aggregation both in the intracellular and extracellular compartments. We tested whether Aqp4 gene deletion, which is associated with a profound global impairment of glymphatic pathway function and interstitial solute clearance, promotes the buildup of phosphorylated tau after TBI. In wild-type mice, cortical P-tau levels were mildly elevated 28 d after TBI, as shown by both Western blot using a battery of P-tau antibodies and immunofluorescence using the AT8 antibody. In Aqp4−/− mice, pronounced P-tau immunoreactivity was evident in the cortex surrounding the traumatic lesion 28 d after TBI. Western blot analysis similarly revealed elevated P-tau levels in Aqp4−/− compared with wild-type mice after TBI. Accumulation of P-tau in Aqp4−/− mice was accompanied by increasing axonal degeneration, the development of pronounced neuroinflammation, and exacerbation of post-traumatic cognitive deficits. Thus, we propose that loss of perivascular AQP4 polarization after TBI impairs the clearance of interstitial solutes, including the protein tau, along the paravascular glymphatic pathway. The failure to clear interstitial tau in turn promotes intracellular tau aggregation and neurodegeneration, exacerbating neurocognitive function after TBI (shown in schematic form in Fig. 10).
Figure 10.
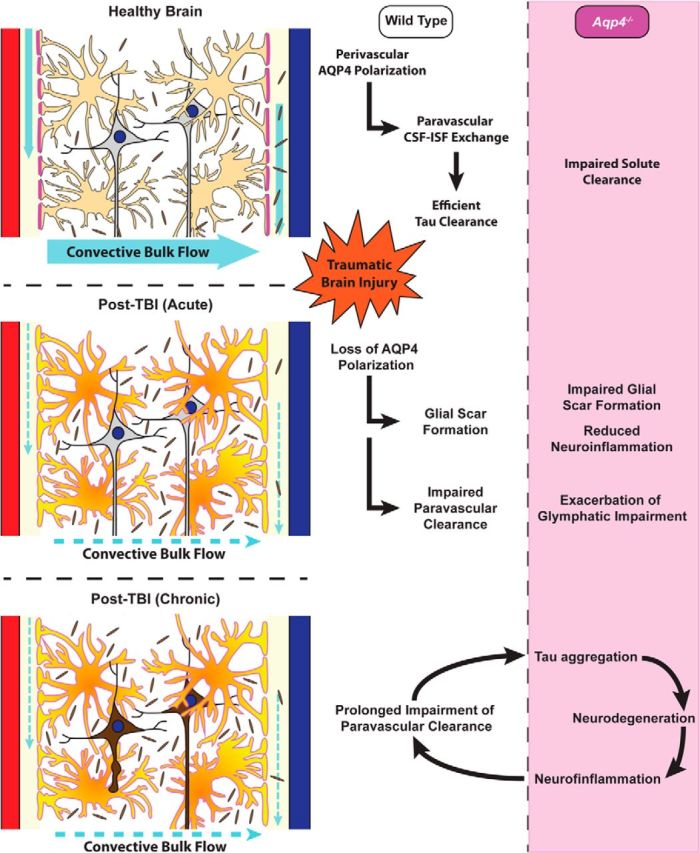
Post-traumatic impairment of glymphatic pathway function promotes tau aggregation. Schematic representation of proposed relationship between perivascular AQP4 polarization, glymphatic pathway function, and interstitial tau clearance after TBI. We propose that chronic loss of perivascular AQP4 polarization after TBI impairs paravascular clearance of interstitial tau, promoting tau aggregation, neurodegeneration, and persistent neuroinflammation in the post-traumatic brain.
In our prior study (Iliff et al., 2012), and in the present study, Aqp4 gene deletion impaired the clearance of interstitial solutes, including amyloid-β, and promoted the accumulation of phosphorylated tau after TBI. In the case of amyloid-β, a substantial fraction of interstitial clearance was insensitive to Aqp4 gene deletion (Iliff et al., 2012), suggesting that multiple processes may be responsible for the clearance of this compound from the brain interstitium. One such mechanism is trans-endothelial efflux across the blood–brain barrier via efflux transporters such as the low-density lipoprotein-related protein-1 (Shibata et al., 2000; Zlokovic, 2005). Given that these two mechanisms occur within the same perivascular domains, it is plausible that these two processes interact with one another. One intriguing possibility is that convective bulk flow along paravascular pathways serves to distribute transporter substrates along perivascular spaces, allowing more efficient clearance of solutes like amyloid-β across the blood–brain barrier. It is currently unclear whether interstitial tau is a substrate for any specific or saturable transporter system at the blood—brain barrier. Thus, whether processes other than cellular uptake (Holmes et al., 2013) or paravascular bulk flow contribute to the clearance of interstitial tau remains unknown.
An extensive prior literature has focused upon the role of AQP4 in the setting of traumatic injury. AQP4 appears to participate both in the formation of cytotoxic edema in addition to the resolution of vasogenic edema after brain injury (Badaut et al., 2011; Thrane et al., 2014). In the acute setting, AQP4 expression is increased after TBI and in a recent experimental study using an open-skull model of TBI in juvenile rats, siRNA-based knockdown of AQP4 around the time of injury reduced the formation of cerebral edema, suggesting that AQP4 participates in the development of cerebral edema (Fukuda et al., 2013). In a closed-skull model of adult TBI, we recently reported that changes in AQP4 expression and localization peaked after cerebral edema and elevated intracranial pressure normalized (Ren et al., 2013), suggesting that changes in AQP4 expression and localization may facilitate the resolution of vasogenic edema after TBI. This notion is consistent with observations in other experimental models of vasogenic edema that report exacerbation of injury with Aqp4 gene knock-out (Papadopoulos et al., 2004). The present study suggests that in addition to these effects of AQP4 in the formation and resolution of cerebral edema in the acute stage of TBI, the long-term mislocalization of AQP4 after TBI (Ren et al., 2013) additionally impairs glymphatic clearance of interstitial solutes from the brain, contributing to tau aggregation and neurodegeneration in the chronic phase after traumatic injury.
More recently, a second role for AQP4 in the development of brain injury has emerged with the recognition that this water channel supports astroglial migration. Deletion of the Aqp4 gene impaired glial scar formation after traumatic injury (Lu et al., 2011), while in primary cell culture Aqp4 gene deletion impairs astrocyte migration (Auguste et al., 2007). The role of AQP4 in supporting astroglial migration appears to be attributable to a pool of AQP4 distinct from the AQP4 that exhibits polarized perivascular localization. Within astrocytes, AQP4 is expressed in two variant forms that are the result of alternative transcription initiation sites: the longer AQP4-M23 variant that is predominant in healthy brain and exhibits highly polarized perivascular localization (Nagelhus and Ottersen, 2013) and the shorter AQP4-M1 form that does not polarize (Furman et al., 2003; Crane et al., 2009), but facilitates astrocyte migration (A.J. Smith et al., 2014). Although this has not been explicitly tested, we surmise that the loss of the AQP4-M1 variant and its facilitation of astroglial migration accounts for the effect of Aqp4 gene deletion on glial scar formation. In contrast, we propose that the effect of TBI and Aqp4 gene deletion upon glymphatic pathway function and tau aggregation result from the loss of the perivascular pools of AQP4 that facilitate the movement of fluid into and out of the paravascular CSF influx and ISF clearance pathways (Iliff et al., 2012). One intriguing possibility is that astroglial upregulation of AQP4-M1 is protective in the acute and subacute setting by promoting glial scar formation, but also reduces perivascular AQP4 polarization, setting the stage for chronic glymphatic pathway impairment, protein aggregation, and neurodegeneration (shown in schematic form in Fig. 10).
The loss of perivascular AQP4 polarization is a key feature of reactive astrocytes after both mild and moderate TBI (Ren et al., 2013) and in a mouse model of diffuse micro-infarcts (Wang et al., 2012). In the aging mouse brain it is associated with the impairment of glymphatic pathway dysfunction and impairment of interstitial amyloid-β clearance from the brain (Kress et al., 2014). These findings, with those of the present study. suggest that the association between mild-to-moderate reactive astrogliosis, loss of perivascular AQP4 polarization and impairment of glymphatic pathway function may be common features of both the aging and the injured brain that promote the misaggregation of proteins like amyloid-β or tau and the development of neurodegeneration. Lu et al. (2007) have demonstrated in a rat model of TBI that 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) are effective in improving neuronal survival, neurogenesis, and functional recovery after traumatic injury. In subsequent studies, this group suggests that the protective effects of statins in the setting of TBI stem from their ability to inhibit the development of post-traumatic gliosis (Li et al., 2009; Wu et al., 2010). It is intriguing to speculate that by inhibiting the development of reactive astrogliosis, statins preserve perivascular polarization of AQP4 and maintain normal glymphatic function after TBI, preventing post-traumatic neurodegeneration. Based upon this premise, the present results suggest that therapeutic interventions targeting post-traumatic perivascular AQP4 mislocalization, either through inhibition of the development of reactive astrogliosis or by stabilizing AQP4 localization to the perivascular end feet (perhaps by inhibiting the induction of the AQP4-M1 variant after brain injury), may offer a novel approach to preserve glymphatic pathway function, to maintain efficient clearance of interstitial solutes such as amyloid-β or tau, and to prevent the onset of neurodegeneration after TBI. A therapeutic approach such as this would conceivably be effective preventing the onset of post-traumatic neurodegeneration, by preventing the development of the earliest steps of tau pathology in the brain after TBI.
Footnotes
This study was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke (NS078167 and NS078304 to M.N. and NS073373 to J.J.I.) and the American Heart Association (12SDG11820014 to J.J.I.). We thank Dr. Marjorie Grafe, Xiao Jing Nie, and Dr. Lijunan Liu of the Oregon Health & Science University Department of Anesthesiology and Peri-Operative Medicine Histology Core for expert technical assistance performing histological studies.
The authors declare no competing financial interests.
References
- Auguste KI, Jin S, Uchida K, Yan D, Manley GT, Papadopoulos MC, Verkman AS. Greatly impaired migration of implanted aquaporin-4-deficient astroglial cells in mouse brain toward a site of injury. FASEB J. 2007;21:108–116. doi: 10.1096/fj.06-6848com. [DOI] [PubMed] [Google Scholar]
- Badaut J, Ashwal S, Obenaus A. Aquaporins in cerebrovascular disease: a target for treatment of brain edema? Cerebrovasc Dis. 2011;31:521–531. doi: 10.1159/000324328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesser AS, Pritchard SM, Johnson GV. Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer Disease. Front Neurol. 2013;4:122. doi: 10.3389/fneur.2013.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115:3285–3290. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JM, Bennett JL, Verkman AS. Live cell analysis of aquaporin-4 m1/m23 interactions and regulated orthogonal array assembly in glial cells. J Biol Chem. 2009;284:35850–35860. doi: 10.1074/jbc.M109.071670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserr HF. Physiology of the choroid plexus. Physiol Rev. 1971;51:273–311. doi: 10.1152/physrev.1971.51.2.273. [DOI] [PubMed] [Google Scholar]
- Eikelenboom P, Bate C, Van Gool WA, Hoozemans JJ, Rozemuller JM, Veerhuis R, Williams A. Neuroinflammation in Alzheimer's disease and prion disease. Glia. 2002;40:232–239. doi: 10.1002/glia.10146. [DOI] [PubMed] [Google Scholar]
- Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. 2010;11:155–159. doi: 10.1038/nrn2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda AM, Pop V, Spagnoli D, Ashwal S, Obenaus A, Badaut J. Delayed increase of astrocytic aquaporin 4 after juvenile traumatic brain injury: possible role in edema resolution? Neuroscience. 2012;222:366–378. doi: 10.1016/j.neuroscience.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda AM, Adami A, Pop V, Bellone JA, Coats JS, Hartman RE, Ashwal S, Obenaus A, Badaut J. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J Cereb Blood Flow Metab. 2013;33:1621–1632. doi: 10.1038/jcbfm.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman CS, Gorelick-Feldman DA, Davidson KG, Yasumura T, Neely JD, Agre P, Rash JE. Aquaporin-4 square array assembly: opposing actions of M1 and M23 isoforms. Proc Natl Acad Sci U S A. 2003;100:13609–13614. doi: 10.1073/pnas.2235843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedye A, Beattie BL, Tuokko H, Horton A, Korsarek E. Severe head injury hastens age of onset of Alzheimer's disease. J Am Geriatr Soc. 1989;37:970–973. doi: 10.1111/j.1532-5415.1989.tb07283.x. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra60. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316–1323. doi: 10.1212/WNL.54.6.1316. [DOI] [PubMed] [Google Scholar]
- Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–3147. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M, Benveniste H. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest. 2013a;123:1299–1309. doi: 10.1172/JCI67677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, Deane R, Nedergaard M. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci. 2013b;33:18190–18199. doi: 10.1523/JNEUROSCI.1592-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287:19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, Plog BA, Ding F, Deane R, Nedergaard M. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014 doi: 10.1002/ana.24271. Advance online publication. Retrieved Oct. 10, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Mahmood A, Lu D, Wu H, Xiong Y, Qu C, Chopp M. Simvastatin attenuates microglial cells and astrocyte activation and decreases interleukin-1beta level after traumatic brain injury. Neurosurgery. 2009;65:179–185. doi: 10.1227/01.NEU.0000346272.76537.DC. discussion 185–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Qu C, Goussev A, Jiang H, Lu C, Schallert T, Mahmood A, Chen J, Li Y, Chopp M. Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J Neurotrauma. 2007;24:1132–1146. doi: 10.1089/neu.2007.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu DC, Zador Z, Yao J, Fazlollahi F, Manley GT. Aquaporin-4 reduces post-traumatic seizure susceptibility by promoting astrocytic glial scar formation in mice. J Neurotrauma. 2011 doi: 10.1089/neu.2011.2114. Advance online publication. Retrieved Oct. 10, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye TC, Shores EA. Traumatic brain injury as a risk factor for Alzheimer's disease: a review. Neuropsychol Rev. 2000;10:115–129. doi: 10.1023/A:1009068804787. [DOI] [PubMed] [Google Scholar]
- Magnoni S, Esparza TJ, Conte V, Carbonara M, Carrabba G, Holtzman DM, Zipfel GJ, Stocchetti N, Brody DL. Tau elevations in the brain extracellular space correlate with reduced amyloid-beta levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain. 2012;135:1268–1280. doi: 10.1093/brain/awr286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti L, Cristofori I, Weaver SM, Chau A, Portelli JN, Grafman J. Cognitive decline in older adults with a history of traumatic brain injury. Lancet Neurol. 2012;11:1103–1112. doi: 10.1016/S1474-4422(12)70226-0. [DOI] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, Ottersen OP. Physiological roles of aquaporin-4 in brain. Physiol Rev. 2013;93:1543–1562. doi: 10.1152/physrev.00011.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, Kurland LT. Traumatic brain injury and time to onset of Alzheimer's disease: a population-based study. Am J Epidemiol. 1999;149:32–40. doi: 10.1093/oxfordjournals.aje.a009724. [DOI] [PubMed] [Google Scholar]
- Omalu B, Bailes J, Hamilton RL, Kamboh MI, Hammers J, Case M, Fitzsimmons R. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery. 2011;69:173–183. doi: 10.1227/NEU.0b013e318212bc7b. discussion 183. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18:1291–1293. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- Planel E, Bretteville A, Liu L, Virag L, Du AL, Yu WH, Dickson DW, Whittington RA, Duff KE. Acceleration and persistence of neurofibrillary pathology in a mouse model of tauopathy following anesthesia. FASEB J. 2009;23:2595–2604. doi: 10.1096/fj.08-122424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/WNL.55.8.1158. [DOI] [PubMed] [Google Scholar]
- Ren Z, Iliff JJ, Yang L, Yang J, Chen X, Chen MJ, Giese RN, Wang B, Shi X, Nedergaard M. ‘Hit and Run’ model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J Cereb Blood Flow Metab. 2013;33:834–845. doi: 10.1038/jcbfm.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts GW, Gentleman SM, Lynch A, Graham DI. beta A4 amyloid protein deposition in brain after head trauma. Lancet. 1991;338:1422–1423. doi: 10.1016/0140-6736(91)92724-G. [DOI] [PubMed] [Google Scholar]
- Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1994;57:419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Run X, Liang Z, Zhang L, Iqbal K, Grundke-Iqbal I, Gong CX. Anesthesia induces phosphorylation of tau. J Alzheimers Dis. 2009;16:619–626. doi: 10.3233/JAD-2009-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer's disease. Brain Pathol. 2013;23:303–310. doi: 10.1111/bpa.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Jin BJ, Ratelade J, Verkman AS. Aggregation state determines the localization and function of M1- and M23-aquaporin-4 in astrocytes. J Cell Biol. 2014;204:559–573. doi: 10.1083/jcb.201308118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol. 2013;9:211–221. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan P, Petitti D, Barbaccia J. Head trauma and age of onset of dementia of the Alzheimer type. JAMA. 1987;257:2289–2290. doi: 10.1001/jama.257.17.2289. [DOI] [PubMed] [Google Scholar]
- Syková E, Nicholson C. Diffusion in brain extracellular space. Physiol Rev. 2008;88:1277–1340. doi: 10.1152/physrev.00027.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrane AS, Rappold PM, Fujita T, Torres A, Bekar LK, Takano T, Peng W, Wang F, Rangroo Thrane V, Enger R, Haj-Yasein NN, Skare Ø, Holen T, Klungland A, Ottersen OP, Nedergaard M, Nagelhus EA. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci U S A. 2011;108:846–851. doi: 10.1073/pnas.1015217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrane AS, Rangroo Thrane V, Nedergaard M. Drowning stars: reassessing the role of astrocytes in brain edema. Trends Neurosci. 2014;S0166-2236(14) doi: 10.1016/j.tins.2014.08.010. 00153-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomura S, Nawashiro H, Otani N, Uozumi Y, Toyooka T, Ohsumi A, Shima K. Effect of decompressive craniectomy on aquaporin-4 expression after lateral fluid percussion injury in rats. J Neurotrauma. 2011;28:237–243. doi: 10.1089/neu.2010.1443. [DOI] [PubMed] [Google Scholar]
- Tsitsopoulos PP, Marklund N. Amyloid-beta peptides and tau protein as biomarkers in cerebrospinal and interstitial fluid following traumatic brain injury: a review of experimental and clinical studies. Front Neurol. 2013;4:79. doi: 10.3389/fneur.2013.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, Diamond MI, Duff KE, Hyman BT. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 2013;70:304–310. doi: 10.1001/jamaneurol.2013.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Iliff JJ, Liao Y, Chen MJ, Shinseki MS, Venkataraman A, Cheung J, Wang W, Nedergaard M. Cognitive deficits and delayed neuronal loss in a mouse model of multiple microinfarcts. J Neurosci. 2012;32:17948–17960. doi: 10.1523/JNEUROSCI.1860-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Vitek MP, Colton CA. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer's disease. Neuroscience. 2009;159:1055–1069. doi: 10.1016/j.neuroscience.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Mahmood A, Lu D, Jiang H, Xiong Y, Zhou D, Chopp M. Attenuation of astrogliosis and modulation of endothelial growth factor receptor in lipid rafts by simvastatin after traumatic brain injury. J Neurosurg. 2010;113:591–597. doi: 10.3171/2009.9.JNS09859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O'Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, Binder LI, Mandelkow EM, Diamond MI, Lee VM, Holtzman DM. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31:13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Liao F, Stewart FR, Mahan TE, Cirrito JR, Patel TK, Hochgrafe K, Mandelkow EM, Holtzman DM. Excitatory neuronal activity regulates extracellular tau in vivo. Soc Neurosci Abstr. 2013;39:598.01. [Google Scholar]
- Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, Cirrito JR, Patel TK, Hochgräfe K, Mandelkow EM, Holtzman DM. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 2014;211:387–393. doi: 10.1084/jem.20131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lunde LK, Nuntagij P, Oguchi T, Camassa LM, Nilsson LN, Lannfelt L, Xu Y, Amiry-Moghaddam M, Ottersen OP, Torp R. Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer's disease. J Alzheimers Dis. 2011;27:711–722. doi: 10.3233/JAD-2011-110725. [DOI] [PubMed] [Google Scholar]
- Yang L, Kress BT, Weber HJ, Thiyagarajan M, Wang B, Deane R, Benveniste H, Iliff JJ, Nedergaard M. Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J Transl Med. 2013;11:107. doi: 10.1186/1479-5876-11-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]