

Abstract
Neurons use glucose to fuel glycolysis and provide substrates for mitochondrial respiration, but neurons can also use alternative fuels that bypass glycolysis and feed directly into mitochondria. To determine whether neuronal pacemaking depends on active glucose metabolism, we switched the metabolic fuel from glucose to alternative fuels, lactate or β-hydroxybutyrate, while monitoring the spontaneous firing of GABAergic neurons in mouse substantia nigra pars reticulata (SNr) brain slices. We found that alternative fuels, in the absence of glucose, sustained SNr spontaneous firing at basal rates, but glycolysis may still be supported by glycogen in the absence of glucose. To prevent any glycogen-fueled glycolysis, we directly inhibited glycolysis using either 2-deoxyglucose or iodoacetic acid. Inhibiting glycolysis in the presence of alternative fuels lowered SNr firing to a slower sustained firing rate. Surprisingly, we found that the decrease in SNr firing was not mediated by ATP-sensitive potassium (KATP) channel activity, but if we lowered the perfusion flow rate or omitted the alternative fuel, KATP channels were activated and could silence SNr firing. The KATP-independent slowing of SNr firing that occurred with glycolytic inhibition in the presence of alternative fuels was consistent with a decrease in a nonselective cationic conductance. Although mitochondrial metabolism alone can prevent severe energy deprivation and KATP channel activation in SNr neurons, active glucose metabolism appears important for keeping open a class of ion channels that is crucial for the high spontaneous firing rate of SNr neurons.
Keywords: excitability, glycolysis, KATP, Trp channel
Introduction
Neuronal firing is energetically expensive (Howarth et al., 2012), and therefore metabolism constrains firing and may influence neuronal processing (Kann et al., 2014). Neuronal metabolism is thought to be primarily fueled by glucose, but neurons can also use alternative fuels, such as β-hydroxybutyrate (βHB), a ketone body (DeVivo et al., 1978), during starvation states or when certain diets are consumed. These alternative fuels bypass glycolysis and directly fuel mitochondria. However, although neurons are capable of using alternative fuels, glycolysis is still crucial to drive certain cellular processes in neurons (Dhar-Chowdhury et al., 2007; Ivannikov et al., 2010; Zala et al., 2013).
Whether neuronal firing is also coupled to local energy production from glycolysis is not known. In other cell types, glycolytic enzymes are associated with the plasma membrane where they can provide rapid, local production of ATP for ion pumps (Proverbio and Hoffman, 1977; Mercer and Dunham, 1981; Paul et al., 1989; Hoffman et al., 2009). We wanted to better understand whether and how changes in glycolysis can lead to changes in neuronal excitability. We used the spontaneous firing rate of substantia nigra pars reticulata (SNr) neurons (Zhou and Lee, 2011) as a readout of metabolic influences on neuronal excitability because their high firing rates likely generate a heavy metabolic burden, and local production of ATP at sites of high energy consumption may be important to sustain their spontaneous firing.
To test whether SNr spontaneous firing requires active glycolysis, we switched between glucose metabolism and metabolism of alternative, mitochondrial fuels, which bypass glycolysis. We used several different interventions to eliminate the contribution of the glucose-specific glycolytic pathway, while maintaining cell health by supplementing with mitochondrial fuels and sufficient oxygenation conditions. We found that mitochondrial fuels, in the absence of glucose-fueled metabolism, could sustain the spontaneous firing of SNr neurons and prevent energy deprivation. The spontaneous firing was sustained at a slower rate when we used glycolytic inhibitors iodoacetate (IAA) or 2-deoxyglucose (2-DG) to eliminate any contribution of glycogen-fueled glycolysis. We determined that the lower SNr firing rate was likely produced by a reduction in a nonselective cation conductance. Our findings suggest that mitochondrial metabolism contributes a major portion of energy in SNr neurons, whereas glucose metabolism may modulate SNr firing by augmenting the activity of a nonselective cation channel.
Materials and Methods
Brain slice preparation.
All procedures involving animals were approved by the Harvard Medical Area Standing Committee on Animals. Experiments were performed using brains of male and female 13- to 20-d-old wild-type (WT) mice (C57BL/6; Charles River Laboratories), Kir6.2 knock-out (KO) mice (Miki et al., 1998) that we have backcrossed into C57/BL6 background, TRPC3 KO mice (Hartmann et al., 2008) in 129/Sv background, and KO mice lacking all seven of the canonical transient receptor potential (TRPC) channels also in 129/Sv background. The sevenfold TRPC KO line (−/− genotype for TRPC1 through TRPC7) was created by crossing the individual TRPC KO alleles (Freichel et al., 2001; Stowers et al., 2002; Dietrich et al., 2005, 2007; Hartmann et al., 2008; Perez-Leighton et al., 2011; Phelan et al., 2013) to obtain KO animals lacking all seven TRPC channels, and these mice are viable, fertile, and healthy in appearance (L. Birnbaumer, unpublished observation).
Mice were first anesthetized via isoflurane inhalation and decapitated. Using a vibrating tissue slicer (Vibratome 3000 or Campden 7000smz-2), we made acute coronal midbrain slices (275 μm) containing the substantia nigra region. Three coronal slices per animal were generally obtained and were hemisectioned to obtain six total slices containing the SNr region. All slicing procedures were performed in ice-cold slicing solution. Slices were immediately incubated in ACSF at 37°C for 35 min and afterward were kept at room temperature in ACSF for 25 min to 3 h before being used for recording. Slicing solution and ACSF were continuously oxygenated with 95% O2 and 5% CO2.
Solutions.
Slicing solution consisted of the following (in mm): 215 sucrose, 2.5 KCl, 24 NaHCO3, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgSO4, and 10 d-glucose, pH 7.4 (∼310 mOsm). ACSF consisted of the following (in mm): 125 NaCl, 2.5 or 4 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1.5 CaCl2, 1 MgSO4 (MgCl2 in barium experiments), and 10 d-glucose, pH 7.4 (∼300 mOsm). For low sodium ACSF, 125 mm NaCl was replaced with 125 mm N-methyl-d-glucamine (NMDG) chloride. Other fuel sources were provided in the ACSF solution in addition to glucose or replacing glucose as described. The pH of the ACSF was unchanged by the addition of alternate fuels. For βHB (2–3 mm), we used sodium (R)-3-hydroxybutyrate, which is the specific enantiomer of βHB that can be metabolized. This concentration of βHB is similar to circulating plasma levels observed in children consuming a ketogenic diet (Huttenlocher, 1976). For lactate addition, sodium l-lactate (5 mm) was used.
For loose patch cell-attached recordings, the pipette solution consisted of the following (in mm): 125 NaCl, 2.5 KCl, 10 HEPES, 2 CaCl2, and 1 MgCl2, pH 7.3 (∼300 mOsm). For whole-cell recordings, the pipette solution consisted of the following (in mm): 140 K-gluconate, 10 NaCl, 10 HEPES, 1 MgCl2, and 0.1 EGTA, pH 7.3 (∼300–310 mOsm). For perforated-patch recordings, amphotercin B (200 μg/ml) and Alexa Fluor 488 (10 μm; Invitrogen) were added to the whole-cell recording solution, and this solution was vortex mixed immediately before each neuron was patched.
Electrophysiology.
We recorded from GABAergic neurons of the SNr, which can be identified by several characteristics, including anatomical location, high firing rates, narrow action potentials, and minimal contribution of Ih current (Zhou and Lee, 2011). By recording at 34°C and using extracellular concentrations of calcium (1.5 mm), magnesium (1 mm), and potassium (4 mm) adjusted to near physiological values (Hansen, 1985), we found that SNr neurons fired ∼30–40 spikes/s, which is similar to firing rates observed in rodents in vivo (Sanderson et al., 1986; Gulley et al., 1999; Deransart et al., 2003; Maurice et al., 2003).
Recordings were performed in a “dual-perfusion” chamber in which the slice received a continuous supply of oxygenated ACSF from above and below (Hájos et al., 2009). The slice was placed on a metal grid (Supertech Instruments) and held down with a slice anchor (Warner Instruments; slice anchor kit for RC-22C). The chamber was produced in-house using a 3D printer (Objet 30; Stratasys) and consisted of two pieces that, when assembled, sandwiched the metal grid in between. The chamber contained two solution inputs: (1) one to flow solution above the slice; and (2) one below the slice. A total flow rate of 5 ml/min (2.5 ml/min per line) was typically used. In some experiments, the total flow rate was reduced to 1 ml/min. Bath temperature was maintained at 34°C using inline heaters (Warner Instruments) for each perfusion line. During experiments, solutions were preheated (∼36°C) by maintaining solution bottles in a water bath (VWR) to prevent out-gassing. Neurons were visualized using an upright microscope (BX51WI; Olympus) equipped with infrared differential interference contrast and controlled using TILL Vision (TILL Photonics).
Spontaneous action potentials were recorded in a loose-patch cell-attached configuration with seal resistance of ∼10–50 MΩ. Borosilicate pipettes (Warner Instruments) were used with tip resistances of ∼2 MΩ. Long-term cell-attached recordings can alter neuronal properties (Alcami et al., 2012), which we found to occur with recordings >20 min. We avoided performing very long cell-attached recordings, but, if needed, cells were patched for only short durations to obtain baseline conditions and then repatched later to record the firing rate after drug application. Only one neuron per brain slice was recorded when application of any pharmacology was tested.
Data were collected with an Axopatch 200B or Multiclamp 700B (Molecular Devices). Loose-patch recordings were low-pass filtered at either 1 or 4 kHz and sampled at 5 or 10 kHz, respectively. Current-clamp recordings were low-pass filtered at 8 kHz and sampled at 20 kHz. Voltage-clamp experiments were low-pass filtered at 4 kHz and sampled at 10 kHz. Signals were digitized using a Digidata 1321A (Molecular Devices) and acquired using pClamp 10 (Molecular Devices).
Changes in steady-state current with application of IAA and βHB were tested in voltage-clamp, perforated-patch configuration at a holding potential of −70 mV. Whole-cell current-clamp recordings of action potentials and membrane potential were performed with zero current injection. For the characterization of current–voltage (I–V) relationships, recordings were performed in whole-cell voltage-clamp configuration immediately after breaking into the neuron to avoid changes associated with dialysis of the intracellular conditions. Neurons were held at −30 mV to inactivate large voltage-gated potassium currents. Voltage steps of 150 ms duration were made in 10 mV decrements from −0 to −110 mV, and steady-state current was calculated from the average of a 10 ms window at the end of each 150 ms voltage step. These experiments were completed within 1 min of breaking into the neuron. All voltage-clamp and current-clamp experiments were corrected for liquid junction potentials, 15 mV for normal ACSF and 22 mV for NMDG-based low sodium ACSF. For whole-cell experiments, pipettes had tip resistances of 1.5–3.5 MΩ and were not fire polished.
For perforated-patch recordings, pipettes were tip filled with amphotericin-free whole-cell recording solution and then backfilled with perforated-patch solution. Neurons were sealed before the perforated-patch solution arrived at the tip of the pipette, which we monitored by imaging the Alexa Fluor 488 dye. Access resistance gradually decreased over 10 min after establishing a multi-gigaohm seal, and recordings were initiated when access resistance was <100 MΩ. The integrity of the perforated-patch recording was monitored by imaging the Alexa Fluor 488 dye, and recordings were stopped if dye entered the neuron, which usually coincided with a sharp decrease in the access resistance indicating a rupture in the membrane at the pipette tip. All perforated-patch recordings were performed at room temperature (∼23°C) to reduce the occurrence of spontaneous membrane rupture during recordings.
Pharmacology.
All chemicals used were obtained from Sigma-Aldrich. Most cell-attached recordings and all whole-cell experiments were performed in the presence of synaptic blockers of ionotropic glutamate and GABA receptors to eliminate spontaneous synaptic events. Kynurenic acid (1 mm) was used to block glutamate receptors, and picrotoxin (100 μm) was used to block GABAA receptors. Synaptic blockers were dissolved directly into the bath ACSF solution. No difference was observed in basal firing rates between experiments performed in the presence or absence of synaptic blockers, consistent with previous findings (Atherton and Bevan, 2005). Additional experiments performed in the presence of blockers of the metabotropic glutamate receptor mGluR1a (LY 367385 [(S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid]; 50 μm; Tocris Bioscience), GABAB receptors (CGP 55845 [(2S)-3-[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl)(phenylmethyl)phosphinic acid]; 2 μm; Tocris Bioscience), or adenosine A1 receptors (8-cyclopentyl-1,3-dipropylxanthine; 5 μm; Tocris Bioscience) did not alter our findings, and these experiments were not included in this analysis. All hydrophobic drugs were dissolved in DMSO to obtain stock solutions. Final DMSO concentrations in ACSF were <0.1%, and this concentration of DMSO had no effect on SNr spontaneous firing. The concentration of IAA (1 mm) was chosen to maximally inhibit GAPDH activity in brain slices and was 10-fold higher than the concentration shown to specifically block GAPDH in cultured astrocytes (Schmidt and Dringen, 2009). Slices were exposed to IAA only for short periods of time to prevent the reaction of IAA with other enzymes that can occur with long incubations.
Data analysis.
Analysis was performed using Clampfit 10 (Molecular Devices) and Origin 9.1 (Origin Lab). Recordings were digitally high-pass filtered at 1 Hz, and a threshold was used to detect individual action potentials. The number of action potentials within 10 s bins was used to determine firing frequency. For population averages, 30 s in each condition (e.g., control or IAA) were used as a measure of firing frequency. For normalized firing rate plots, individual experiments were normalized to a 30 s baseline average. Descriptive statistics are reported as mean ± SEM. Sample size reported indicates the number of neurons, and typically only one neuron was used per slice. For comparisons between two populations, paired or unpaired two-tailed Student's t test was used, and p values are reported. For multiple comparisons, one-way ANOVA with Bonferroni's post hoc test with α = 0.05 was used. An asterisk indicates significance at the p < 0.05 level.
Results
To ask how spontaneous SNr firing is affected by a metabolic fuel switch from glucose to alternative fuels, we used three manipulations that disrupt glucose-fueled metabolism while leaving mitochondrial metabolism uninhibited. In the first manipulation, we removed glucose from the bath solution while simultaneously providing the ketone body βHB, which bypasses glycolysis and directly fuels mitochondrial metabolism. In the second approach, we again replaced glucose with βHB but also added a glucose analog, 2-DG, that can inhibit glycolysis (Wick et al., 1957). In the third intervention, instead of 2-DG, we used IAA, an inhibitor of the glycolytic enzyme GAPDH (Sabri and Ochs, 1971; Schmidt and Dringen, 2009).
βHB sustains SNr firing in the absence of glucose
We initially tried the most conservative manipulation by simply replacing extracellular glucose (10 mm) with βHB (2.5 mm). We had found that, if we removed glucose without any addition of βHB, SNr firing was almost completely abolished by 20 min (Fig. 1A, red symbols). In contrast, we found the spontaneous firing rate was fully sustained for at least 20 min in the absence of glucose if we supplied the alternative fuel βHB (Fig. 1A, blue symbols). These data suggest that mitochondrial respiration of βHB can sustain, at least for tens of minutes, the high spontaneous firing rates of SNr neurons in the absence of glucose.
Figure 1.

The ketone body βHB sustains spontaneous SNr firing in the absence of glucose, but glycolytic inhibition slows the firing rate. A, SNr spontaneous firing rate in the presence of glucose (G; 10 mm; 31.4 ± 3.2 spikes/s; black symbols, n = 10). Spontaneous firing rate (29.3 ± 4.5 spikes/s; blue symbols; n = 8) recorded from neurons in the absence of glucose but in the presence of βHB (2.5 mm) for at least 20 min was not significantly different from the firing rate in glucose (p > 0.05, one-way ANOVA with Bonferroni's test). In the absence of external fuel (0 mm glucose; 0 mm βHB), SNr spontaneous firing was almost completely silent (0.9 ± 0.4 spikes/s; red symbols; n = 10). B, When glycolysis was inhibited using 2-DG (5 mm) in the absence of glucose and βHB (black line), the spontaneous firing of an SNr neuron decreased and then sharply dropped until firing was silenced. Return of glucose in the external solution rapidly restored the spontaneous firing. In the presence of βHB (2.5 mm; blue line), glycolytic inhibition with 2-DG (5 mm) in the absence of glucose decreased the spontaneous firing of a SNr neuron but did not silence it. C, When glycolysis was inhibited with IAA (1 mm) in glucose solution (10 mm; black trace), the firing rate of a representative SNr neuron decreased and then sharply stoped firing action potentials. However, in the presence of the ketone body βHB (2.5 mm; blue trace) and absence of glucose, the firing of a representative SNr neuron was sustained after glycolytic inhibition with IAA at a lower firing rate. D, Average firing rate of SNr neurons in control (Ctrl) and test conditions. The initial firing rate of SNr neurons in glucose (10 mm) was 32.7 ± 2.9 spikes/s (black squares and line; n = 12), but after removal of glucose and addition of 2-DG (5 mm), the firing rate was almost completely silenced within 15 min (0.8 ± 0.3 spikes/s). The firing rate of SNr neurons in the absence of glucose but in the presence of βHB (2.5 mm) was 33.1 ± 3.7 spikes/s (blue square and line). After addition of 2-DG (5 mm or 10 mm), the firing rate of those SNr neurons was significantly reduced but not silenced (22.2 ± 2.6 spikes/s; n = 13; p = 0.0001, Student's paired t test). Addition of IAA (1 mm) to spontaneously firing SNr neurons (37.7 ± 9.7 spikes/s; n = 6; black circles and line) in glucose (10 mm) completely silenced SNr firing. When exogenous βHB (2.5 mm; blue circles and line) replaced glucose, the spontaneous firing of SNr neurons (37.2 ± 2.7 spikes/s) was significantly decreased after addition of IAA (1 mm) but was not silenced (17.8 ± 1.4 spikes/s; n = 20; p = 4.8 × 10−7, Student's paired t test). Similarly, when lactate (5 mm; n = 5; red circles and line) replaced glucose, SNr spontaneous firing (39.8 ± 4.5 spikes/s) was decreased after addition of IAA (21.6 ± 3.4 spikes/s; p = 0.002, Student's paired t test). The mitochondrial poisons rotenone (Rot; 1 μm) and oligomycin (Oligo; 1 μm) silenced SNr firing in glucose solution (33.7 ± 3.0 vs 0.8 ± 0.5 spikes/s; n = 6; black diamonds and line). In the presence of rotenone (1 μm) and oligomycin (1 μm), βHB (2.5 mm) did not sustain SNr firing after treatment with IAA (40.2 ± 7.4 vs 0.2 ± 0.2 spikes/s; n = 4; blue diamonds and line). E, Replacement of glucose with 2-DG and βHB decreased SNr firing by 32.3 ± 5.0% (n = 13), which was significantly less than the decrease observed when using IAA to inhibit glycolysis (p < 0.05, one-way ANOVA with Bonferroni's test). Inhibition of glycolysis with IAA was performed with βHB either in the absence of glucose (blue symbols) or with 10 mm glucose (red symbols). The percentage decrease in firing rate was not significantly different (p > 0.05, one-way ANOVA with Bonferroni's test) between experiments without glucose (49.5 ± 3.4%; n = 20) or while maintaining glucose (64.2 ± 5.8%; n = 12). In the presence of the antioxidant Tempol (2 mm), the percentage decrease in firing rate (64.2 ± 5.2%; n = 8) after inhibition of glycolysis with IAA in the presence of βHB and glucose was not significantly different from control experiments without Tempol. All error bars indicate SEM; *p < 0.05.
However, neurons can contain glycogen stores that, through glycogenolysis, can provide a substrate for glycolysis in the absence of glucose (Saez et al., 2014). Although we removed the main substrate of glycolysis, glucose, we did not directly inhibit glycolysis and, therefore, our intention to shift metabolism only to mitochondria may not have been fully achieved. To address this, we used two methods to directly block glycolysis and prevent the possibility for glycogen mobilization to preserve glycolysis. The first method used the glucose analog 2-DG, which is taken up by cells and phosphorylated by hexokinase but is unable to proceed further through glycolysis. In the second method, we used IAA, an irreversible cysteine modifier that disrupts the function of the glycolytic enzyme GAPDH by alkylating its reactive cysteine residue. Each of these two methods may have nonspecific effects in addition to their inhibition of glycolysis, but if we observe similar results using both methods, we would expect these consistencies to be a result of their similar effect on glycolysis.
In the presence of mitochondrial fuels, inhibition of glycolysis with 2-DG or IAA lowers SNr firing
We repeated our experiments using glucose removal with the addition of βHB, but now we also blocked glycolysis using 2-DG (5 or 10 mm). We found that SNr firing decreased to a lower firing rate within 10 min of the switch to 2-DG and βHB (Fig. 1B, blue line), and this lower firing rate was ∼10 spikes/s (∼32%) slower than the basal firing rate (Fig. 1D, blue square). In contrast, if βHB was withheld, replacement of glucose with 2-DG (5 mm) almost completely silenced SNr firing (Fig. 1B,D, black line), indicating that βHB was used in the absence of functional glycolysis to sustain SNr firing but at a slower firing rate.
We observed a similar outcome using IAA (1 mm) instead of 2-DG to block glycolysis (Fig. 1C,D); however, the decrease in firing rate was significantly greater with IAA (Fig. 1E).
Because IAA may affect glutathione (Schmidt and Dringen, 2009) and possibly increase the amount of reactive oxygen species (ROS), we performed two manipulations that would reduce ROS. We inhibited glycolysis with IAA in the presence of βHB and glucose, which allows glucose to proceed through the pentose phosphate pathway (PPP) and increase NADPH production. In the presence of glucose, the effect of IAA was not significantly different from the effect observed in the absence of glucose (Fig. 1E). In addition, we performed experiments in the presence of an antioxidant, 4-hydroxy-TEMPO [4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (Tempol)], which has been demonstrated to be a potent scavenger of ROS (Wilcox, 2010), and we found that the decrease in firing rate after addition of IAA was not attenuated by the ROS scavenger (Fig. 1E). These results suggest that the effect of glycolytic inhibition in the presence of βHB is independent of a change in ROS or the PPP.
To determine whether the change in firing rate produced by glycolytic inhibition was specific to the mitochondrial fuel used, we inhibited glycolysis with IAA in the presence of lactate (5 mm). With lactate, inhibition of glycolysis with IAA produced a similar decrease in firing rate as with βHB, indicating that the reduction in SNr firing was not just specific to βHB (Fig. 1D, red circle). To further confirm that mitochondrial respiration was crucial to sustain SNr firing in the absence of glucose, we poisoned mitochondria with rotenone (1 μm) and oligomycin (1 μm) and found that βHB was no longer able to sustain firing in the absence of glycolysis (Fig. 1D, blue diamond). Interestingly, rotenone and oligomycin silenced SNr firing in the presence of glucose (10 mm) as well, indicating that glycolysis alone is incapable of sustaining SNr firing (Fig. 1D, black diamond). Together, these data demonstrate that, in the absence of glycolysis, mitochondrial respiration can use alternative fuels to sustain SNr firing, but the firing rate is slower.
The observed decrease in SNr firing is not mediated by KATP channel activation
The slower firing rate in the presence of mitochondrial fuels and absence of functional glycolysis may reflect the disruption of processes that are preferentially influenced by glycolysis. The ATP-sensitive potassium (KATP) channel can hyperpolarize cells when intracellular ATP levels are reduced (Ashcroft and Gribble, 1998; Nichols, 2006) and may be sensitive to changes in the ATP pool generated by glycolysis (Dhar-Chowdhury et al., 2005). SNr neurons express KATP channels (Karschin et al., 1997; Richards et al., 1997) and have been shown previously to generate changes in SNr firing during changes in metabolism (Yamada et al., 2001; Ma et al., 2007).
We tested whether an increase in KATP channel activity was responsible for the slower firing rate we observed when we inhibited glycolysis in the presence of mitochondrial fuels. When we blocked KATP channels using glibenclamide (Glib; 200 nm) and we inhibited glycolysis with 2-DG (5 mm) in the presence of βHB (2.5 mm), SNr firing was still significantly reduced (Fig. 2A), and the percentage decrease in firing rate (31.5 ± 2.9%, n = 8) was not different from the decrease observed in the absence of Glib (32.3 ± 5.0%, n = 13, p = 0.9, Student's unpaired t test). In addition, Glib (10 μm) was unable to reverse the decrease in firing rate produced by glycolytic inhibition with IAA in the presence of βHB (Fig. 2A). We also used mice lacking Kir6.2, a pore-forming subunit of the KATP channel that eliminates functional KATP channels (Miki et al., 1998). In these KATP KO animals, glycolytic inhibition with IAA in the presence of βHB still decreased SNr firing (Fig. 2A), and the percentage decrease in firing rate (51.6 ± 3.4%, n = 8) was not significantly different from the decrease observed in WT SNr neurons (49.5 ± 3.4%, n = 20, p = 0.7, Student's unpaired t test). Together, these data indicate that activation of KATP channels is not required for slower SNr firing during inhibition of glycolysis in the presence of βHB.
Figure 2.

KATP channel activation after glycolytic inhibition is conditional on the perfusion flow rate and the presence of mitochondrial fuels. A, The spontaneous firing rate of SNr neurons, recorded in the continuous presence of the KATP channel blocker Glib (200 nm), was significantly decreased (circle symbols; 28.8 ± 4.2 vs 19.3 ± 2.5 spikes/s; n = 8; p = 0.001, Student's paired t test) after inhibition of glycolysis with 2-DG (5 mm) in the absence of glucose but in the presence of βHB (2.5 mm). The decreased firing rate after inhibition of glycolysis with IAA (1 mm) in the presence of βHB (2.5 mm) was not reversed after addition of Glib (10 μm; square symbols; 18.2 ± 1.7 vs 17.0 ± 1.8 spikes/s; n = 6; p > 0.05, one-way ANOVA with Bonferroni's test). IAA (1 mm) significantly decreased the firing rate of Kir6.2 KO SNr neurons (triangle symbols; 33.3 ± 3.6 vs 15.7 ± 1.6 spikes/s; n = 8; p = 0.0003, Student's paired t test). B, With a flow rate of 5 ml/min, βHB (2.5 mm) sustained the spontaneous firing of an SNr neuron after inhibition of glycolysis with IAA (1 mm). Further addition of the KATP channel blocker Glib (10 μm) did not reverse the decrease in firing rate. a–c, Traces depict cell-attached recordings of spontaneous firing at the indicated times (calibration: 50 pA, 200 ms). C, With a lower flow rate of 1 ml/min, βHB (2.5 mm) was unable to sustain the firing rate of an SNr neuron. Addition of Glib (200 nm) could partially restore the firing rate. a–c, Traces depict cell-attached recordings of spontaneous firing at the indicated times (calibration: 20 pA, 200 ms). D, Cell-attached recordings of spontaneous firing rates with inhibition of glycolysis by IAA (1 mm) in the presence of glucose (10 mm). IAA completely silenced SNr firing of control neurons (n = 6; black trace). When KATP channels were inhibited using Glib (200 nm; 10 min preincubation; n = 4; blue trace) or eliminated in Kir6.2 KO mice (n = 6; red trace), SNr firing displayed a transient increase, followed by a complete silencing. E, Representative whole-cell recordings showing the time course of the effect of IAA (1 mm) application on the normalized firing rate of SNr neurons in the presence of glucose. In a control neuron, application of IAA promptly decreased the spontaneous firing rate without any increase in firing rate (black line). In a neuron preincubated in Glib (200 nm; blue line) or in a neuron from a Kir6.2 KO mouse (red line), the firing rate increased after application of IAA and then stopped firing. F, Summarized data from all whole-cell experiments with application of IAA in the presence of glucose. After application of IAA, control neurons had a hyperpolarized resting potential (−74.6 ± 3.5 mV; n = 9; black symbols). After IAA, neurons preincubated in Glib (200 nm; blue symbols) had more depolarized resting potentials (−60.5 ± 2.7 mV; n = 6; p < 0.05, one-way ANOVA with Bonferroni's test), and neurons from Kir6.2 KO animals (red symbols) also rested more depolarized (−53.2 ± 3.8 mV; n = 6; p < 0.05, one-way ANOVA with Bonferroni's test). All error bars indicate SEM; *p < 0.05. Ctrl, Control.
Glycolytic inhibition can activate KATP channels under slower flow rate conditions or in the absence of alternative fuels
We wondered whether our elevated flow rate conditions might contribute to the lack of KATP channel involvement by providing a highly oxygenated condition that favored robust mitochondrial metabolism. We tested this by decreasing the perfusion flow rate from 5 to 1 ml/min, a lower flow rate often used in brain slice studies. In contrast to the KATP-independent slowing of firing observed at 5 ml/min (Fig. 2B), with the slower flow rate of 1 ml/min (Fig. 2C), we found that βHB (2.5 mm) was unable to prevent the loss of SNr firing with application of IAA in six of eight neurons tested. Addition of Glib (200 nm) could partially restore firing in silenced neurons, indicating that KATP channels contributed to the loss of SNr firing and that involvement of KATP channels was dependent on the perfusion flow rate used.
To further characterize the role of KATP channel activation, we inhibited glycolysis with IAA in the presence of Glib (200 nm) and only supplied glucose (10 mm) as a fuel source. We had observed previously that with functional KATP channels, IAA could rapidly silence SNr firing when no mitochondrial fuels were provided (Fig. 2D, black line). In contrast, if we blocked KATP channels with Glib (Fig. 2D, blue line) or eliminated them genetically (Fig. 2D, red line), we observed a transient decrease in firing rate, followed by a large, transient increase in firing rate after inhibition of glycolysis with IAA. To further examine these changes in SNr firing rate, we performed whole-cell current-clamp recordings in which we again inhibited glycolysis without providing a mitochondrial fuel, in control (Fig. 2E, black line), in the presence of Glib (Fig. 2E, blue line), or from Kir6.2 KO neurons (Fig. 2E, red line). We found that WT neurons rested at hyperpolarized potentials after inhibition of glycolysis with IAA (Fig. 2F, black symbols). However, in the presence of Glib (Fig. 2F, blue symbols) or in brain slices from Kir6.2 KO mice (Fig. 2F, red symbols), SNr neurons had final resting potentials that were significantly more depolarized than in control experiments. Together, these data indicate that KATP channels can be activated by inhibition of glycolysis but only under slower flow rate conditions or when no mitochondrial fuels are provided. The activation of KATP channels under these conditions silences SNr firing and maintains SNr neurons at a hyperpolarized resting potential.
Inhibition of glycolysis with IAA in the presence of βHB decreases a nonselective cation conductance
To determine the mechanism for the KATP-independent slowing of SNr firing that we had observed with βHB-fueled metabolism during well oxygenated conditions, we examined the changes in membrane voltage and currents during inhibition of glycolysis. We recorded action potentials immediately after establishing whole-cell recordings to avoid washing out intracellular metabolites. Consistent with our cell-attached recordings, we found that basal firing rates were lower in neurons preincubated in IAA and βHB (Fig. 3A, blue trace) compared with control neurons in glucose (Fig. 3A, black trace). There was no noticeable difference in the action potential waveform between the control neurons (Fig. 3B, black line) and neurons in the presence of IAA and βHB (Fig. 3B, blue line); however, the afterhyperpolarization appeared more pronounced in the presence of IAA and βHB.
Figure 3.

Glycolytic inhibition hyperpolarizes the membrane potential by decreasing a constitutively active conductance. A, Action potentials were recorded immediately after establishing a whole-cell recording from control (Ctrl) neurons or neurons preincubated in IAA (1 mm) and βHB (2.5 mm). Representative traces from two separate neurons, one in the control condition (black line) and one preincubated in IAA and βHB (blue line), show that IAA increases interspike intervals. B, The action potential waveform of neurons incubated in IAA in the presence of βHB (n = 7; blue trace) was similar to action potentials from control neurons (n = 5; black trace) but did have a more prominent afterhyperpolarization (calibration: 20 mV, 5 ms). C, With action potentials blocked using lidocaine (1 mm) and with the KATP channel blocker Glib (200 nm) present, application of IAA (1 mm) in the continued presence of βHB (2.5 mm) decreased the membrane potential of SNr neurons recorded in perforated-patch configuration (−53.8 ± 2.0 to −67.5 ± 1.3 mV; n = 8; p = 3.0 × 10−5, Student's paired t test). D, Perforated-patch voltage-clamp recordings (holding potential of −70 mV) of SNr neurons in the presence of lidocaine (1 mm) exhibited a decrease in inward current after application of IAA (1 mm) with βHB (2.5 mm; −41.9 ± 7.4 to −6.1 ± 3.7 pA; n = 4; p = 0.0009, Student's paired t test). All error bars indicate SEM; *p < 0.05.
To monitor changes in voltage and current over time with application of IAA, we turned to a perforated-patch technique to prevent disruption of intracellular metabolites during longitudinal recordings. Because we had not observed a change in the action potential shape, we performed current-clamp recordings in the presence of the sodium channel blocker lidocaine (1 mm) to eliminate action potentials and to better observe any changes in steady-state membrane potential. We found that inhibition of glycolysis with IAA in the presence of βHB hyperpolarized the resting membrane potential by 13.7 ± 1.3 mV (Fig. 3C). These experiments were performed in the continuous presence of Glib (200 nm) to confirm that the changes in membrane potential we observed with glycolytic inhibition were independent of KATP channel activation.
Next, we used voltage clamp to monitor changes in ionic current, again during inhibition of glycolysis with IAA in the presence of βHB. In perforated-patch recordings, IAA decreased the steady-state inward current at −70 mV by 35.8 ± 4.0 pA (Fig. 3D). To better characterize the identity of this inward current, we performed an I–V analysis by stepping the voltage in 10 mV decrements. For these experiments, we used whole-cell recordings, which allowed for lower series resistance and therefore better voltage control, and measurements were made immediately after breaking into the neurons to avoid effects attributable to washout of intracellular metabolites. We also performed these recordings in the presence of lidocaine (1 mm) and tetraethylammonium (TEA; 1 mm) to eliminate large currents and improve the voltage control. When we compared neurons that had been exposed to IAA in the presence of βHB (Fig. 4A, blue squares) with control neurons in glucose (Fig. 4A, black squares), we observed a change in steady-state current over the voltage range between −60 and −30 mV. We analyzed this change at the single voltage of −50 mV, which is approximately the resting potential we observed in these neurons in the absence of action potentials. We found that the current at −50 mV was significantly different between control neurons and neurons preincubated in IAA and βHB (Fig. 4E). This change in steady-state current was also observed when experiments were performed in the continuous presence of barium (1 mm BaCl2; Fig. 4B) to block inward-rectifier potassium channels (Hibino et al., 2010), as well as members of the tandem-pore potassium channel family (Ma et al., 2011). In the presence of barium, the inward current at −50 mV was significantly reduced in the neurons preincubated in IAA and βHB (Fig. 4F), and the magnitude of the change in current was similar to that observed in experiments without barium (Fig. 4E).
Figure 4.
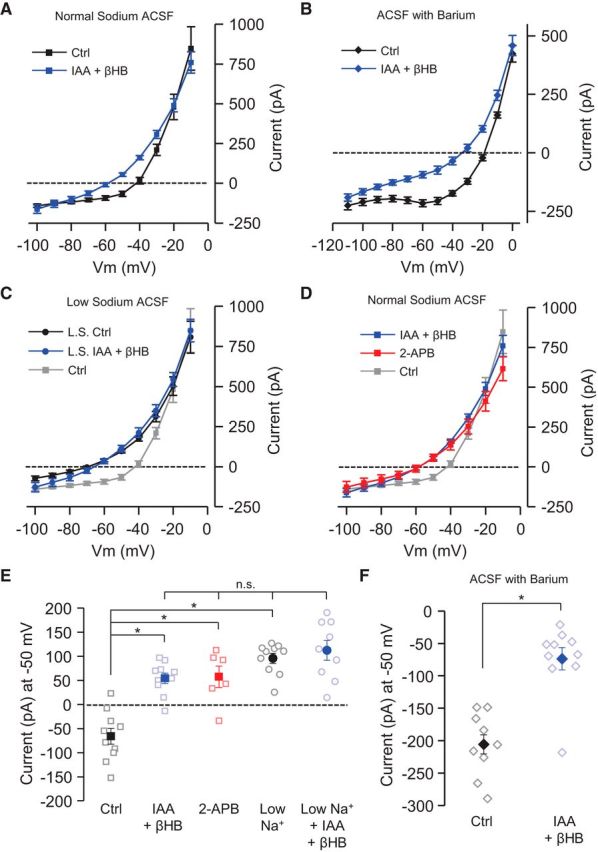
Glycolytic inhibition decreases a nonselective cationic current. A, Steady-state I–V relationship of control (Ctrl) neurons (n = 10; black filled squares) versus neurons preincubated (>10 min) in IAA (1 mm) and βHB (2.5 mm; n = 10; blue squares) over the voltage range from −100 to −10 mV. B, Steady-state currents were measured in the presence of barium chloride (1 mm) in the control condition (10 mm glucose; black diamonds; n = 10) and in neurons preincubated with IAA and βHB (blue diamonds; n = 10). Neurons in IAA and βHB had decreased steady-state inward current. C, With lowered external sodium (L.S.), the steady-state I–V relationship of control neurons (n = 10; black circles) was similar to that of neurons preincubated in IAA and βHB in lowered sodium (n = 9; blue circles). For comparison, the I–V plot of control neurons in standard sodium condition is shown (gray squares). D, Steady-state I–V relationship was similar for neurons in IAA and βHB (blue squares) and neurons preincubated (>10 min) in the nonspecific TRP channel blocker 2-APB (200 μm; n = 6; red squares). Both I–V plots from neurons in IAA and βHB and neurons in 2-APB were different than the control neurons (gray squares). E, The steady-state current (pA) at −50 mV of control neurons (−66.0 ± 16.7; n = 10) was significantly different from neurons preincubated in IAA and βHB (54.9 ± 10.7; n = 10; p < 0.05). The steady-state current at −50 mV from neurons preincubated in 2-APB (200 μm) was also significantly different from control neurons (57.9 ± 22.3; n = 6; p < 0.05) but not significantly different from neurons in IAA and βHB. Neurons in low external sodium (27 mm NaCl) had an average steady-state current at −50 mV that was also significantly different from control neurons (95.9 ± 10.3; n = 10; p < 0.05). In the low sodium condition, IAA in the presence of βHB did not significantly alter steady-state current at −50 mV compared with control neurons in low sodium (112.3 ± 20.4; n = 9; p > 0.05). Significance of pairwise comparisons at the p < 0.05 level was determined by one-way ANOVA with Bonferroni's test. F, The steady-state current (pA) at −50 mV in the presence of barium from control neurons (−205.7 ± 14.8; n = 10) was significantly different from neurons preincubated in IAA and βHB (−73.7 ± 17.4; n = 10; p = 1.7 × 10−5, Student's unpaired t test). The decrease in inward current at −50 mV generated by IAA and βHB in the presence of barium (132.0 ± 22.8 pA) was similar in magnitude as the decreased observed without barium (120.5 ± 19.8 pA). *p < 0.05. Comparisons that are not significantly different (p > 0.05) are also indicated (n.s.). All error bars indicate SEM. All experiments were performed in the presence of lidocaine (1 mm) and TEA (1 mm).
Based on the characteristics of the I–V plot, we hypothesized that the change in steady-state current could be produced by a decrease in a nonselective cation conductance. To determine whether our metabolic fuel switch altered the conductance of nonselective cation channels, we reduced the external sodium concentration from ∼152 to ∼27 mm by replacing sodium chloride with NMDG chloride. Lowering external sodium has been shown previously to eliminate a large proportion of the tonic nonselective cation conductance in SNr neurons (Atherton and Bevan, 2005; Zhou et al., 2008). In the low sodium condition, inward current through a nonselective cation channel should be reduced. Indeed, the inward steady-state current at −50 mV was significantly reduced in the low sodium condition compared with control neurons in the normal sodium condition (Fig. 4E). In addition, if inhibition of glycolysis with IAA is decreasing a nonselective cation conductance, we expected to see a smaller change in current during application of IAA in the low sodium condition. We found that, in the low external sodium condition, the steady-state current after inhibition of glycolysis with IAA in the presence of βHB (Fig. 4C,E) was not significantly different from control neurons in low sodium and also not different from neurons preincubated in IAA in the presence of βHB in the normal sodium condition (Fig. 4E). These data show that the current modulated by glycolytic inhibition in the presence of βHB is sodium dependent and consistent with a nonselective cation current.
We suspected that the nonselective cation current could be carried by a TRP channel with outward rectification (Clapham, 2003). In SNr neurons, a steady-state current with similar characteristics has been attributed previously to a TRP channel (Zhou et al., 2008). To test this, we blocked TRP channels using the nonspecific TRP channel inhibitor 2-aminoethoxydiphenyl borate (2-APB; 200 μm) and found that steady-state current in the presence of 2-APB (Fig. 4D, red squares) was altered from the control conditions (Fig. 4D, gray squares), in a manner similar to the effect of IAA with βHB (Fig. 4D, blue squares). Comparing the steady-state current at the single voltage of −50 mV, we found that both IAA with βHB and 2-APB significantly reduced the steady-state inward current, compared with controls, and by similar magnitudes (Fig. 4E). These data suggested that the change in steady-state inward current produced by IAA in the presence of βHB was consistent with a decrease in a TRP channel.
The nonselective cation conductance reduced by inhibition of glycolysis is not carried by TRPC channels
A previous study reported that murine SNr neurons express a single type of TRP channel, TRPC3, which is involved in maintaining the more depolarized potential of SNr neurons that allows them to fire spontaneously at high rates (Zhou et al., 2008). We reasoned that the constitutively active nonselective cationic current affected by IAA could be carried by TRPC3. We tested this by recording from TRPC3 KO animals and found, surprisingly, that SNr neurons were spontaneously active with firing rates comparable with WT animals (28.6 ± 4.3 spikes/s; n = 10). When glycolysis was inhibited with IAA in the presence of βHB (Fig. 5A), SNr firing was reduced in TRPC3 KO neurons, indicating that TRPC3 is not required for the decrease in firing rate produced by inhibition of glycolysis (Fig. 5B).
Figure 5.

The reduction in firing rate produced by glycolytic inhibition does not require TRPC channels. A, Loose-patch cell-attached recording of a TRPC3 KO SNr neuron showed a reduction in firing rate after application of IAA (1 mm) in the presence of βHB (3 mm). a, b, Traces depict cell-attached recordings of spontaneous firing before and after application of IAA in the presence of βHB (calibration: 50 pA, 200 ms). B, Glycolytic inhibition in the presence of βHB (2.5 or 3 mm) decreased the spontaneous firing rate of TRPC3 KO SNr neurons (28.6 ± 4.3 to 11.2 ± 2.8 spikes/s; n = 10; p = 6 × 10−5, Student's paired t test). C, SNr neurons lacking all seven TRPC channels are spontaneously active. Inhibition of glycolysis with IAA in the presence of βHB (2.5 mm) significantly reduces the firing rate of these neurons (28.7 ± 3.4 to 12.6 ± 1.8 spikes/s; n = 7; p = 0.001, Student's paired t test). Ctrl, Control.
We considered the possibility that, in the absence of TRPC3, other members of the TRPC family of TRP channels could compensate. To test this, we recorded from SNr neurons from mice lacking all seven members of the TRPC family. We found that SNr neurons lacking all TRPC channels were still spontaneously active (Fig. 5C). Furthermore, the firing rate of these neurons was still decreased after inhibition of glycolysis with IAA in the presence of βHB (Fig. 5C). These data indicate that none of the TRPC channels are required for the spontaneous firing of SNr neurons and that the decrease in firing rate produced by glycolytic inhibition in the presence of βHB is not mediated by a decrease in the activity of a TRPC channel.
Discussion
To ask whether neuronal excitability is linked to glucose-fueled metabolism, we examined how a fuel switch from glucose to an alternative fuel affected the spontaneous firing of SNr neurons. With glycolysis inhibited using 2-DG or IAA, the alternative fuels, βHB or lactate, sustain SNr firing but at a slower rate, indicating that active glycolysis is important for a portion of the spontaneous firing. The effect likely does not involve changes in the PPP because we observe similar results when we block glycolysis with IAA in the presence or absence of glucose, which should have opposite consequences on the PPP. The effect is also independent of changes in ROS because the antioxidant Tempol did not alter the response to IAA. Therefore, a change in glucose metabolism via glycolysis appears to modulate the firing rate of SNr neurons.
In the hypothalamus, changes in glucose metabolism alter KATP channel activity, which can regulate neuronal firing and control feeding behavior (Sohn, 2013), but the role of KATP channels may be different in other brain regions. We considered whether the activation of KATP channels produced the decrease in SNr firing rate. In the SNr, we find that KATP channels are not required for the decrease in firing rate after inhibition of glycolysis. Instead, inhibition of glycolysis in the presence of mitochondrial fuels appears to decrease a nonselective cation conductance, and this slows SNr firing. To activate KATP channels, we needed to inhibit glycolysis either without providing mitochondrial fuels or with a lower perfusion flow rate to decrease the oxygenation of the brain slice. Therefore, glycolysis influences SNr firing through a KATP-independent mechanism when mitochondrial metabolism is robust and a KATP-dependent mechanism during conditions of reduced mitochondrial metabolism.
These data may provide relevant insight into how a metabolic fuel switch from glucose to ketone bodies can prevent seizures, a phenomenon that is used clinically to treat pediatric epilepsy (Bailey et al., 2005). Low carbohydrate dietary therapies for treating epilepsy have been around for centuries and have recently seen a renewed interest after clinical studies demonstrating their efficacy (Neal et al., 2009). During starvation or while consuming a ketogenic diet, patients experience an increase in circulating ketone body levels (DeVivo et al., 1978). The brain uses ketone bodies as a fuel source, which likely shifts cellular metabolism away from glucose. This metabolic change is thought to underlie the mechanism of seizure protection of the ketogenic diet (Lutas and Yellen, 2013); however, the exact mechanism remains unknown. Our finding that SNr neurons fire less in the absence of glucose-fueled metabolism suggests that changes in metabolic fuel can alter neuronal excitability, which may be important for seizure protection because the SNr is implicated in the gating of seizure progression (Iadarola and Gale, 1982; McNamara et al., 1984; Depaulis et al., 1994). We also implicate the closure of a nonselective cation channel in the mechanism of the reduced excitability, and we speculate that this channel may play a role in the mechanism for seizure resistance during a ketogenic diet.
In the absence of glycolysis, KATP channel activation is conditional on perfusion flow rate and mitochondrial fuels
When we set out to test the contribution of glycolysis to SNr firing, we wanted to prevent the impairment of mitochondrial metabolism. To accomplish this, we provided the mitochondrial fuel βHB or lactate, but we also wanted to ensure that mitochondria could sufficiently use the oxidative substrate. Therefore, we used a flow rate of 5 ml/min in a dual-perfusion chamber, because this was shown previously to provide adequate oxygenation of brain slices (Hájos et al., 2009). Under this higher flow rate condition, we completely sustain spontaneous firing with βHB in the absence of glucose, and we observe no activation of KATP channels. However, we tested whether lower flow rate conditions that are often used in brain slice experiments might produce different results.
When we lower the flow rate to 1 ml/min and block glycolysis in the presence of βHB, KATP channels activate, which hyperpolarizes SNr neurons and results in the loss of firing. This resembles the activation of KATP channels that occurs when we inhibit glycolysis without providing βHB, suggesting that, at the lower flow rate of 1 ml/min, we have lowered the oxygenation to the brain slice and reduced the mitochondrial utilization of βHB. Therefore, the activation of KATP channels is dependent on the oxygenation conditions of the brain slice, and flow rate should be taken into consideration when studying KATP channel activity in brain slices. Our data support a role of KATP channels in SNr neurons during more oxygen-starved conditions but minimal KATP contribution during well oxygenated brain conditions. Although we know that the higher flow rate supplies more freshly oxygenated solution to the brain slice, the large differences in oxygen delivery—between normal perfusion of brain tissue by capillary blood containing red blood cells (and thus hemoglobin) oxygenated by 20% oxygen partial pressure and brain slice superfusion with 95% oxygen (but no oxygen carrier)—makes it difficult to know which condition more accurately models the in vivo behavior.
We also find that we can activate KATP channels if we inhibit glycolysis without providing an alternative fuel source, which disrupts all neuronal energy metabolism by eliminating substrate needed for mitochondrial ATP production. This conclusion is supported by the finding that mitochondrial blockers, such as rotenone and oligomycin, produce a loss of SNr firing similar to the loss of firing produced by glycolytic inhibition when glucose is the only fuel source provided. Under this effectively complete metabolic inhibition, KATP channels activate to hyperpolarize SNr neurons and prevent depolarization-induced hyperexcitability. In the absence of KATP channels, complete metabolic inhibition depolarizes SNr neurons and transiently increases the firing rate. This increase in firing rate resembles changes in SNr firing rate observed during hypoxia in Kir6.2 KO mice (Yamada et al., 2001). Typically, hypoxia decreases SNr firing by activating KATP channels; however, in the absence of Kir6.2-containing KATP channels, hypoxia induces an increase in SNr firing rate by a yet unknown mechanism. The findings of Yamada et al. (2001) together with our data support the idea that KATP channels prevent depolarization after metabolic inhibition (Ben-Ari et al., 1990) and therefore play an important neuroprotective role when mitochondrial oxidation is impaired (Yamada and Inagaki, 2005; Sun et al., 2006).
The KATP-independent reduction in SNr firing likely involves a decrease in a nonselective cation conductance
To determine the mechanism of the KATP-independent decrease in firing rate, we characterized the change in conductance produced by the fuel switch to non-glucose-fueled metabolism. Our data from these experiments suggest that a decrease in a constitutively active nonselective cationic current mediates the change in SNr firing. This conclusion is supported by our I–V characterization of the change produced by IAA, which indicates that a steady-state, sodium-dependent, inward current is reduced after glycolytic inhibition. The change in current produced by IAA is similar in magnitude and shape to the change in current produced by the nonspecific TRP channel blocker 2-APB, and this current resembles a TRP-like nonselective cationic current described previously in these neurons (Zhou et al., 2008). In addition, the outward rectification observed at negative voltages resembles members of the TRP channel family (Clapham, 2003).
TRPC3 is not required for spontaneous firing of SNr neurons and is not necessary for the reduction in firing produced by inhibiting glycolysis
A previous study reported that murine SNr neurons express a single TRP channel, TRPC3, that is constitutively active in SNr neurons and required for the spontaneous firing of these neurons (Zhou et al., 2008). Therefore, we tested whether the current reduced by glycolytic inhibition is carried by TRPC3 channels. Surprisingly, we find that SNr neurons from TRPC3 KO animals fire spontaneously at rates similar to WT neurons. Additionally, application of IAA decreases firing rates of TRPC3 KO neurons to the same extent as WT neurons. These data indicate that TRPC3 channels are not required for the reduction in firing rate we observe with glycolytic inhibition in the presence of βHB and suggest that TRPC3 channels are not required for the baseline activity of SNr neurons.
Other types of TRP channels have been described in SNr neurons, including the TRPM subtype in guinea pig SNr neurons (Lee et al., 2013) and multiple TRPC channel subtypes in rat midbrain GABAergic neurons (Michel et al., 2005). To test the possibility that other TRPC channel subtypes were compensating for the loss of TRPC3, we recorded from SNr neurons from mice lacking all seven TRPC channel members. Surprisingly, SNr neurons are spontaneously active in the absence of all TRPC channels, and inhibition of glycolysis in the presence of βHB decreases their firing rate. Thus, the identity of the nonselective cation channels that sustain SNr spontaneous firing remains an outstanding question.
Conclusion
Our studies demonstrate that changes in cellular metabolism can influence spontaneous firing of SNr neurons. We find that SNr neurons are capable of using mitochondrial fuels in the absence of glucose but that oxygen levels play a critical role in this ability. Similarly, KATP channels, which can have strong effects on SNr firing, are activated only under conditions that favor decreased mitochondrial respiration. Finally, we find that a nonselective cation channel, which is important for SNr spontaneous firing, closes when glycolysis is inhibited in the presence of mitochondrial fuels. These findings provide insights into how changes in glucose metabolism, possibly during starvation states or while consuming a ketogenic diet, can alter neuronal excitability.
Footnotes
This work was supported by National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke Grants R01 NS055031 (G.Y.) and F31 NS077633 (A.L.) and NIH Intramural Research Program Project Z01-ES-101684 (L.B.). We thank members of the Yellen laboratory for valuable discussions and comments. We are also grateful to Drs. Bruce Bean, Michael Do, and Chinfei Chen for helpful advice. Kir6.2 knock-out mice were generously provided by Drs. Susumu Seino and Colin Nichols.
The authors declare no competing financial interests.
References
- Alcami P, Franconville R, Llano I, Marty A. Measuring the firing rate of high-resistance neurons with cell-attached recording. J Neurosci. 2012;32:3118–3130. doi: 10.1523/JNEUROSCI.5371-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Gribble FM. Correlating structure and function in ATP-sensitive K+ channels. Trends Neurosci. 1998;21:288–294. doi: 10.1016/S0166-2236(98)01225-9. [DOI] [PubMed] [Google Scholar]
- Atherton JF, Bevan MD. Ionic mechanisms underlying autonomous action potential generation in the somata and dendrites of GABAergic substantia nigra pars reticulata neurons in vitro. J Neurosci. 2005;25:8272–8281. doi: 10.1523/JNEUROSCI.1475-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey EE, Pfeifer HH, Thiele EA. The use of diet in the treatment of epilepsy. Epilepsy Behav. 2005;6:4–8. doi: 10.1016/j.yebeh.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Krnjević K, Crépel V. Activators of ATP-sensitive K+ channels reduce anoxic depolarization in CA3 hippocampal neurons. Neuroscience. 1990;37:55–60. doi: 10.1016/0306-4522(90)90191-6. [DOI] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Depaulis A, Vergnes M, Marescaux C. Endogenous control of epilepsy: the nigral inhibitory system. Prog Neurobiol. 1994;42:33–52. doi: 10.1016/0301-0082(94)90020-5. [DOI] [PubMed] [Google Scholar]
- Deransart C, Hellwig B, Heupel-Reuter M, Léger JF, Heck D, Lücking CH. Single-unit analysis of substantia nigra pars reticulata neurons in freely behaving rats with genetic absence epilepsy. Epilepsia. 2003;44:1513–1520. doi: 10.1111/j.0013-9580.2003.26603.x. [DOI] [PubMed] [Google Scholar]
- DeVivo DC, Leckie MP, Ferrendelli JS, McDougal D., Jr Chronic ketosis and cerebral metabolism. Ann Neurol. 1978;3:331–337. doi: 10.1002/ana.410030410. [DOI] [PubMed] [Google Scholar]
- Dhar-Chowdhury P, Harrell MD, Han SY, Jankowska D, Parachuru L, Morrissey A, Srivastava S, Liu W, Malester B, Yoshida H, Coetzee WA. The glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase, triose-phosphate isomerase, and pyruvate kinase are components of the KATP channel macromolecular complex and regulate its function. J Biol Chem. 2005;280:38464–38470. doi: 10.1074/jbc.M508744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar-Chowdhury P, Malester B, Rajacic P, Coetzee WA. The regulation of ion channels and transporters by glycolytically derived ATP. Cell Mol Life Sci. 2007;64:3069–3083. doi: 10.1007/s00018-007-7332-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Mederos Y, Schnitzler M, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Kalwa H, Storch U, Mederos y, Schnitzler M, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflügers Arch. 2007;455:465–477. doi: 10.1007/s00424-007-0314-3. [DOI] [PubMed] [Google Scholar]
- Freichel M, Suh SH, Pfeifer A, Schweig U, Trost C, Weissgerber P, Biel M, Philipp S, Freise D, Droogmans G, Hofmann F, Flockerzi V, Nilius B. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat Cell Biol. 2001;3:121–127. doi: 10.1038/35055019. [DOI] [PubMed] [Google Scholar]
- Gulley JM, Kuwajima M, Mayhill E, Rebec GV. Behavior-related changes in the activity of substantia nigra pars reticulata neurons in freely moving rats. Brain Res. 1999;845:68–76. doi: 10.1016/S0006-8993(99)01932-0. [DOI] [PubMed] [Google Scholar]
- Hájos N, Ellender TJ, Zemankovics R, Mann EO, Exley R, Cragg SJ, Freund TF, Paulsen O. Maintaining network activity in submerged hippocampal slices: importance of oxygen supply. Eur J Neurosci. 2009;29:319–327. doi: 10.1111/j.1460-9568.2008.06577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiol Rev. 1985;65:101–148. doi: 10.1152/physrev.1985.65.1.101. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59:392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- Hoffman JF, Dodson A, Proverbio F. On the functional use of the membrane compartmentalized pool of ATP by the Na+ and Ca++ pumps in human red blood cell ghosts. J Gen Physiol. 2009;134:351–361. doi: 10.1085/jgp.200910270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth C, Gleeson P, Attwell D. Updated energy budgets for neural computation in the neocortex and cerebellum. J Cereb Blood Flow Metab. 2012;32:1222–1232. doi: 10.1038/jcbfm.2012.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR. Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatr Res. 1976;10:536–540. doi: 10.1203/00006450-197605000-00006. [DOI] [PubMed] [Google Scholar]
- Iadarola MJ, Gale K. Substantia nigra: site of anticonvulsant activity mediated by gamma-aminobutyric acid. Science. 1982;218:1237–1240. doi: 10.1126/science.7146907. [DOI] [PubMed] [Google Scholar]
- Ivannikov MV, Sugimori M, Llinás RR. Calcium clearance and its energy requirements in cerebellar neurons. Cell Calcium. 2010;47:507–513. doi: 10.1016/j.ceca.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O, Papageorgiou IE, Draguhn A. Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J Cereb Blood Flow Metab. 2014;34:1270–1282. doi: 10.1038/jcbfm.2014.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karschin C, Ecke C, Ashcroft FM, Karschin A. Overlapping distribution of KATP channel-forming Kir6.2 subunit and the sulfonylurea receptor SUR1 in rodent brain. FEBS Lett. 1997;401:59–64. doi: 10.1016/S0014-5793(96)01438-X. [DOI] [PubMed] [Google Scholar]
- Lee CR, Machold RP, Witkovsky P, Rice ME. TRPM2 channels are required for NMDA-induced burst firing and contribute to H2O2-dependent modulation in substantia nigra pars reticulata GABAergic neurons. J Neurosci. 2013;33:1157–1168. doi: 10.1523/JNEUROSCI.2832-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013;36:32–40. doi: 10.1016/j.tins.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening KATP channels. J Neurosci. 2007;27:3618–3625. doi: 10.1523/JNEUROSCI.0132-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XY, Yu JM, Zhang SZ, Liu XY, Wu BH, Wei XL, Yan JQ, Sun HL, Yan HT, Zheng JQ. External Ba2+ block of the two-pore domain potassium channel TREK-1 defines conformational transition in its selectivity filter. J Biol Chem. 2011;286:39813–39822. doi: 10.1074/jbc.M111.264788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Thierry AM, Glowinski J, Deniau JM. Spontaneous and evoked activity of substantia nigra pars reticulata neurons during high-frequency stimulation of the subthalamic nucleus. J Neurosci. 2003;23:9929–9936. doi: 10.1523/JNEUROSCI.23-30-09929.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO, Galloway MT, Rigsbee LC, Shin C. Evidence implicating substantia nigra in regulation of kindled seizure threshold. J Neurosci. 1984;4:2410–2417. doi: 10.1523/JNEUROSCI.04-09-02410.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer RW, Dunham PB. Membrane-bound ATP fuels the Na/K pump. Studies on membrane-bound glycolytic enzymes on inside-out vesicles from human red cell membranes. J Gen Physiol. 1981;78:547–568. doi: 10.1085/jgp.78.5.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel FJ, Fortin GD, Martel P, Yeomans J, Trudeau LE. M3-like muscarinic receptors mediate Ca2+ influx in rat mesencephalic GABAergic neurones through a protein kinase C-dependent mechanism. Neuropharmacology. 2005;48:796–809. doi: 10.1016/j.neuropharm.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci U S A. 1998;95:10402–10406. doi: 10.1073/pnas.95.18.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, Whitney A, Cross JH. A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia. 2009;50:1109–1117. doi: 10.1111/j.1528-1167.2008.01870.x. [DOI] [PubMed] [Google Scholar]
- Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- Paul RJ, Hardin CD, Raeymaekers L, Wuytack F, Casteels R. Preferential support of Ca2+ uptake in smooth muscle plasma membrane vesicles by an endogenous glycolytic cascade. FASEB J. 1989;3:2298–2301. doi: 10.1096/fasebj.3.11.2528493. [DOI] [PubMed] [Google Scholar]
- Perez-Leighton CE, Schmidt TM, Abramowitz J, Birnbaumer L, Kofuji P. Intrinsic phototransduction persists in melanopsin-expressing ganglion cells lacking diacylglycerol-sensitive TRPC subunits. Eur J Neurosci. 2011;33:856–867. doi: 10.1111/j.1460-9568.2010.07583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan KD, Shwe UT, Abramowitz J, Wu H, Rhee SW, Howell MD, Gottschall PE, Freichel M, Flockerzi V, Birnbaumer L, Zheng F. Canonical transient receptor channel 5 (TRPC5) and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Mol Pharmacol. 2013;83:429–438. doi: 10.1124/mol.112.082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proverbio F, Hoffman JF. Membrane compartmentalized ATP and its preferential use by the Na, K-ATPase of human red cell ghosts. J Gen Physiol. 1977;69:605–632. doi: 10.1085/jgp.69.5.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards CD, Shiroyama T, Kitai ST. Electrophysiological and immunocytochemical characterization of GABA and dopamine neurons in the substantia nigra of the rat. Neuroscience. 1997;80:545–557. doi: 10.1016/S0306-4522(97)00093-6. [DOI] [PubMed] [Google Scholar]
- Sabri MI, Ochs S. Inhibition of glceraldehyde-3-phosphate dehydrogenase in mammalian nerve by iodoacetic acid. J Neurochem. 1971;18:1509–1514. doi: 10.1111/j.1471-4159.1971.tb00013.x. [DOI] [PubMed] [Google Scholar]
- Saez I, Duran J, Sinadinos C, Beltran A, Yanes O, Tevy MF, Martínez-Pons C, Milán M, Guinovart JJ. Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab. 2014;34:945–955. doi: 10.1038/jcbfm.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson P, Mavoungou R, Albe-Fessard D. Changes in substantia nigra pars reticulata activity following lesions of the substantia nigra pars compacta. Neurosci Lett. 1986;67:25–30. doi: 10.1016/0304-3940(86)90202-8. [DOI] [PubMed] [Google Scholar]
- Schmidt MM, Dringen R. Differential effects of iodoacetamide and iodoacetate on glycolysis and glutathione metabolism of cultured astrocytes. Front Neuroenergetics. 2009;1:1. doi: 10.3389/neuro.14.001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JW. Ion channels in the central regulation of energy and glucose homeostasis. Front Neurosci. 2013;7:85. doi: 10.3389/fnins.2013.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers L, Holy TE, Meister M, Dulac C, Koentges G. Loss of sex discrimination and male-male aggression in mice deficient for TRP2. Science. 2002;295:1493–1500. doi: 10.1126/science.1069259. [DOI] [PubMed] [Google Scholar]
- Sun HS, Feng ZP, Miki T, Seino S, French RJ. Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2-containing ATP-sensitive K+ channels. J Neurophysiol. 2006;95:2590–2601. doi: 10.1152/jn.00970.2005. [DOI] [PubMed] [Google Scholar]
- Wick AN, Drury DR, Nakada HI, Wolfe JB. Localization of the primary metabolic block produced by 2-deoxyglucose. J Biol Chem. 1957;224:963–969. [PubMed] [Google Scholar]
- Wilcox CS. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol Ther. 2010;126:119–145. doi: 10.1016/j.pharmthera.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Inagaki N. Neuroprotection by KATP channels. J Mol Cell Cardiol. 2005;38:945–949. doi: 10.1016/j.yjmcc.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Yamada K, Ji JJ, Yuan H, Miki T, Sato S, Horimoto N, Shimizu T, Seino S, Inagaki N. Protective role of ATP-sensitive potassium channels in hypoxia-induced generalized seizure. Science. 2001;292:1543–1546. doi: 10.1126/science.1059829. [DOI] [PubMed] [Google Scholar]
- Zala D, Hinckelmann MV, Yu H, Lyra da Cunha MM, Liot G, Cordelières FP, Marco S, Saudou F. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 2013;152:479–491. doi: 10.1016/j.cell.2012.12.029. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Lee CR. Intrinsic and integrative properties of substantia nigra pars reticulata neurons. Neuroscience. 2011;198:69–94. doi: 10.1016/j.neuroscience.2011.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FW, Matta SG, Zhou FM. Constitutively active TRPC3 channels regulate basal ganglia output neurons. J Neurosci. 2008;28:473–482. doi: 10.1523/JNEUROSCI.3978-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]