

Abstract
The sleep-promoting ventrolateral preoptic nucleus (VLPO) shares reciprocal inhibitory inputs with wake–active neuronal nuclei, including the locus ceruleus. Electrophysiologically, sleep-promoting neurons in the VLPO are directly depolarized by the general anesthetic isoflurane and hyperpolarized by norepinephrine, a wake-promoting neurotransmitter. However, the integration of these competing influences on the VLPO, a sleep- and anesthetic-active structure, has yet to be evaluated in either brain slices in vitro or the intact organism. Single-cell multiplex RT-PCR conducted on both isoflurane-activated, putative sleep-promoting VLPO neurons and neighboring, state-indifferent VLPO neurons in mouse brain slices revealed widespread expression of α2A-, α2B- and α2C-adrenergic receptors in both populations. Indeed, both norepinephrine and the highly selective α2 agonist dexmedetomidine each reversed the VLPO depolarization induced by isoflurane in slices in vitro. When microinjected directly into the VLPO of a mouse lightly anesthetized with isoflurane, dexmedetomidine increased behavioral arousal and reduced the depressant effects of isoflurane on barrel cortex somatosensory-evoked potentials but failed to elicit spectral changes in spontaneous EEG. Based on these observations, we conclude that local modulation of α-adrenergic activity in the VLPO destabilizes, but does not fully antagonize, the anesthetic state, thus priming the brain for anesthetic emergence.
Keywords: adrenergic, anesthesia, dexmedetomidine, emergence, sleep, VLPO
Introduction
When anesthetics were discovered in the mid-19th century, their ability to rapidly and reversibly eliminate consciousness was recognized as a pivotal advance in surgical medicine. Many theories have been proposed to describe how anesthetics exert their hypnotic effects. The “wet blanket” theory suggests that anesthetics envelope the entire brain and extinguish consciousness through widespread disruption of both protein and lipid interactions (Sukhotinsky et al., 2007). Although anesthetics do bind promiscuously throughout the brain (Eckenhoff and Eckenhoff, 1998), focal anesthetic interactions targeting critical CNS sites might be sufficient to disrupt the essential neuronal activity required for conscious perception. Neuronal targets for anesthetics include regions well established for modulating endogenous sleep and arousal (Lydic and Baghdoyan, 2005; Franks, 2008; Kottler et al., 2013). Notably, sleep-promoting neurons in the ventrolateral preoptic nucleus (VLPO) are activated by anesthetic drugs and directly depolarized by the general anesthetic isoflurane (Nelson et al., 2002, 2003; Lu et al., 2008; Li et al., 2009; Moore et al., 2012).
The VLPO shares reciprocal connections with various arousal-promoting nuclei, including the tuberomammillary nucleus, dorsal raphe, ventral periaqueductal gray, and locus ceruleus (Sherin et al., 1996, 1998; Chou et al., 2002; Hsieh et al., 2011), thereby providing the VLPO with rich monoaminergic innervation. Electrophysiological evidence reveals that norepinephrine exerts inhibitory effects on sleep-promoting VLPO neurons (Gallopin et al., 2000, 2005; Matsuo et al., 2003). A variety of adrenergic receptors have been localized to neurons within the preoptic anterior hypothalamus (Kolaj and Renaud, 2001, 2007; Kumar et al., 2006, 2007; Jha and Mallick, 2009; Frontini and Giordano, 2010; Berridge et al., 2012), but α2 adrenoceptors are hypothesized to mediate the inhibition of sleep–active neurons in vitro by norepinephrine (Osaka and Matsumura, 1995; Bai and Renaud, 1998). Neurons in the preoptic anterior hypothalamus that are known to be sleep-active based on c-Fos expression have also been found to be GABAergic and contain α2 adrenoceptors (Modirrousta et al., 2004). These GABAergic VLPO neurons also express c-Fos after exposure to hypnotic doses of general anesthetics (Lu et al., 2008; Moore et al., 2012; Han et al., 2014).
Herein we evaluate the effects of acute administration of adrenergic agonists on anesthetic-activated VLPO neurons in both slices and anesthetized mice. After electrophysiological recordings in the VLPO, neurons were evaluated for the presence of various α adrenoceptors using multiplex RT-PCR. Subsequently, adrenergic drugs were microinjected into the VLPO of anesthetized, chronically instrumented mice. Behavioral responses were measured by both a qualitative arousal assessment and a change in time spent moving. Additional evidence of arousal was evaluated using changes in spontaneous electroencephalogram and barrel cortex somatosensory evoked local field potentials (LFPs) of mice receiving acute microinjection of dexmedetomidine into the VLPO. Together, these experiments address the extent to which adrenergic stimulation opposes isoflurane-induced activation of the VLPO.
Materials and Methods
Animals.
All studies were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania. Wild-type male C57BL/6J mice (The Jackson Laboratory) aged 2–4 months were used for all experiments. For 2 weeks before experiments, animals were given access to food and water ad libitum and acclimatized to a reverse 12 h light/dark cycle with zeitgeber time 0 (lights on) at 7:00 P.M.
In vitro slice preparation and electrophysiology.
Hypothalamic slice preparation and electrophysiological recordings were performed as described previously (Moore et al., 2012). Briefly, anesthetic-naive mice were killed by cervical dislocation and then decapitated, followed by immediate dissection of the brain. Brains were kept in a 4°C solution containing 248 mm sucrose, 2.5 mm KCl, 1.25 mm NaH2PO4, 2.0 mm MgSO4, 2.5 mm CaCl2, 10 mm dextrose, and 26 mm NaHCO3 while they were sliced coronally at a thickness of 200 μm using a vibratome (Leica Microsystems). VLPO-containing slices were transferred to a submerged chamber and then continuously superfused with oxygenated artificial CSF (aCSF) at 34°C while equilibrating for 60 min before recordings. The aCSF consisted of 124 mm NaCl, 2.5 KCl mm, 1.25 mm Na2HPO4, 2.0 mm MgSO4, 2.5 mm CaCl2, 10 mm dextrose, and 26 mm NaHCO3, pH 7.4, and osmolality of 300–305 mOsm. After the 60 min incubation period, slices were maintained in continuously oxygenated aCSF at room temperature until being recorded. During recordings, slices were maintained in a recording chamber continuously perfused with oxygenated 32°C aCSF at a rate of 2 ml/min. Data were obtained using whole-cell patch-clamp techniques. Micropipettes (resistance of 4–8 mΩ) were filled with an intracellular solution of 130 mm K-gluconate, 5 mm NaCl, 10 mm phosphocreatine disodium salt, 1 mm MgCl2, 10 mm HEPES, 0.02 mm EGTA, 0.5 mm Na2GTP, 2 mm MgATP, and 0.1% biocytin at pH 7.3, and osmolality of 280–290 mOsm. Biocytin was used to allow post hoc confirmation of the anatomic location of the cells and their morphology.
Electrophysiology analysis.
Input resistance was calculated from the slope of voltage–current plots produced from a series of hyperpolarizing and depolarizing current pulses. Only cells with stable membrane potentials more negative than −43 mV were selected for analysis. To be considered isoflurane activated, cells had to exhibit a change in membrane potential ≥4 mV in response to 320 μm isoflurane administration. We quantified the response to isoflurane by calculating the mean membrane potential from three 2 min epochs: (1) the baseline membrane potential immediately before isoflurane administration; (2) the peak isoflurane effect after steady-state concentration had been achieved; and (3) the peak drug combination effect after steady-state concentration of the combination drug solution had been achieved.
Electrophysiology drugs.
Isoflurane was sonicated into preoxygenated aCSF to achieve a concentration of 320 μm. This concentration was chosen to simulate a deep surgical anesthetic plane corresponding to ∼1.6% (Franks and Lieb, 1996; Moore et al., 2012). All cells were exposed to isoflurane under current-clamp conditions. Isoflurane-containing aCSF was delivered to the brain slices via a closed system using syringe pumps. After observing the response of VLPO neurons to an initial isoflurane exposure, cells were then treated with the following drugs or combinations thereof: 100 μm norepinephrine, 100 nm dexmedetomidine, 50 μm phenylephrine, and 1 μm tetrodotoxin (TTX). Combination solutions were mixed with preoxygenated isoflurane-containing aCSF before administration to achieve the final concentrations listed above. After a subset of recordings, final isoflurane drug concentrations were verified to be 320 ± 19 μm (mean ± SD, n = 8) using HPLC as described previously (Wei et al., 2005; Kelz et al., 2008).
Single-cell PCR analysis.
After completion of electrophysiology experiments, negative pressure was applied to the cytoplasm of 15 isoflurane-activated and 13 isoflurane-non-activated neurons using a 10 ml syringe connected through a three-way stopcock to the recording electrode. The contents of the recording electrodes were transferred to 200 μl PCR tubes and frozen at −80°C. Samples underwent RT and first-strand cDNA synthesis using a Superscript III kit as indicated by the protocol of the manufacturer (catalog #12574-018; Invitrogen). Single-cell cDNA amplification was divided into two steps. The first step was multiplex PCR with pairs of gene-specific outside primers for glutamic acid decarboxylase (GAD1; Lazarenko et al., 2010) and adrenoceptors (α1A, α1B, α1D, α2A, α2B, and α2C). The second step was conducted using the corresponding nested primers (Table 1). The PCR reaction conditions for multiplex PCR were as follows: 94°C for 2 min, 35 cycles of 94°C (15 s), 55°C (30 s), 68°C (1 min), and 72°C for 10 min. The conditions for the nested PCR were as follows: 94°C for 2 min, 35 cycles of 94°C (15 s), 55°C (30 s), 68°C (50 s), and 72°C for 10 min. The nested PCR products were separated on 2% agarose gels. Positive RNA controls for the RT-PCR assay were made using mouse brain tissue. Tissue RNA was extracted using an RNeasy Mini kit (catalog #74106; Qiagen). RT, cDNA synthesis, and subsequent PCR were completed simultaneously with experimental samples.
Table 1.
Primer sequences for α-adrenergic receptors and GAD
| Primer | Sequence (5′-3′) | PCR product (bp) |
|---|---|---|
| Adrenergic α1A | ||
| Outside | ||
| Forward | GAG CAA GTG ACG CTC CGT AT | 348 |
| Reverse | ACA CTG GAT TCG CAG CAC AT | |
| Nested | ||
| Forward | ATT GTG GTG GGA TGC TTC GT | 95 |
| Reverse | ACT GTT TCC GGT GGC TTG AA | |
| Adrenergic α1B | ||
| Outside | ||
| Forward | TTT CAT GAG GAC ACC CTC AGC AGT | 303 |
| Reverse | CAT GAA GGC GCG TTT GAA CTC CTT | |
| Nested | ||
| Forward | CCT TGG GCA TTG TAG TCG GAA TGT | 136 |
| Reverse | TTG AAG TAG CCC AGC CAG AAC ACT | |
| Adrenergic α1D | ||
| Outside | ||
| Forward | TCC GTA AGG CTG CTC AAG TT | 176 |
| Reverse | TAG CCC AGC CAG AAG ATG AC | |
| Nested | ||
| Forward | CAA GAC CCT AGC CAT CGT CG | 112 |
| Reverse | GAA GAC GCC CTC TGA TGG TT | |
| Adrenergic α2A | ||
| Outside | ||
| Forward | TGG CCA ACG AGG TTA TGG GTT ACT | 210 |
| Reverse | AGA TGA CCC ACA CGG TGA CAA TGA | |
| Nested | ||
| Forward | TCT ATT TGG CTC TCG ACG TGC TCT | 115 |
| Reverse | GTG CGC TTC AGG TTG TAC TCG AT | |
| Adrenergic α2B | ||
| Outside | ||
| Forward | TCT TGG CTT CCA GCA TCG GAT CTT | 457 |
| Reverse | AAG GGT GGG TTG AAT ACT GAG GCA | |
| Nested | ||
| Forward | TGT AAT TGC CAA ACG CAG CCA CTG | 162 |
| Reverse | TGT CCA TTA GCC TCT CCG ACA GAA | |
| Adrenergic α2C | ||
| Outside | ||
| Forward | AAG TGT GGT GTG GTG TAT ACC TGG | 291 |
| Reverse | AGC CTA TGC AGG AGG ACA AGA TGT | |
| Nested | ||
| Forward | ATC GTG CAC CTG TGT GCC ATT AGT | 139 |
| Reverse | GGA AGG AGA TGA CAG CGG AGA TGA | |
| GAD1 | ||
| Outside | ||
| Forward | ACA ACC TTT GGC TGC ATG TGG ATG | 331 |
| Reverse | AAT CCC ACG GTG CCC TTT GCT TTC | |
| Nested | ||
| Forward | AGT CAC CTG GAA CCC TC | 128 |
| Reverse | GCT TGT CTG GCT GGA A |
Cannula implants.
Eighteen mice were implanted with bilateral cannulae aimed at the VLPO. General anesthesia was induced with ∼1.8% isoflurane, and animals were placed in a stereotaxic frame. Hair was removed from the scalp, the area was sterilized with Betadine, and a 2 cm midline incision exposed the skull. Holes were drilled at 0.05 mm anterior to bregma and ±0.65 mm lateral to bregma, allowing insertion of a two-barrel 26 gauge guide cannula (3 mm long) with a 33 gauge dummy cannula (5.5 mm long; Plastics One) placed inside. The pedestal of the cannula was secured with dental cement. After surgery, animals were housed individually to avoid damage to the implant. Animals recovered for 2 weeks before behavioral testing.
In vivo drug delivery for behavioral experiments.
In individual chambers, each mouse was exposed to 1.2% isoflurane for 10 min to induce hypnosis. The mouse was then moved to a nose cone for maintenance of the hypnotic state at 1.2% isoflurane. The dummy cannula was removed and replaced with an injector cannula attached to prefilled plastic tubing connected to two Hamilton syringes controlled by a syringe pump (Harvard Apparatus). The mouse was rolled onto its back while remaining in the nose cone. Isoflurane concentration was reduced to 0.8%, or 1.0% for a subset of experiments, for 10 min. These doses were identified as the EC5 and EC1, respectively, for emergence in these animals using previously published methods (McCarren et al., 2013). After this 10 min preinjection period, 25 nl of drug was delivered bilaterally to the VLPO over the course of 1 min. This rate of delivery limited the potential for backflow up the cannula tract (Morrison et al., 1999). Drugs used for in vivo experiments were 5.91 mm norepinephrine, 500 μm dexmedetomdine, normal saline, and 60 mm phenylephrine. Each mouse received each drug in a randomized order with at least 24 h between drug exposures.
Behavioral and movement scoring.
The 10 min before drug injection and the 5 min after the start of drug injection were defined as the preinjection and postinjection periods, respectively. An assignment of “responsive” or “nonresponsive” was made for the postinjection periods for each drug exposure in each mouse. Responsive behavior was defined as any full-body attempt to right with concentrated movements of the tail, hips, and limbs or a successful return of all four paws to the ground. Behaviors that were not considered to be responsive included small movements of the limbs and transient slow lifting of the hips. No mouse displayed any responsive behaviors during any preinjection periods. Video recordings of each preinjection and postinjection period were analyzed post hoc for total amount of movement using ANY-maze software with a customized freezing protocol (Stoelting).
Acute dexmedetomidine microinjection with concurrent EEG recording.
A total of six mice were each anesthetized with ∼1.8% isoflurane and placed in a stereotaxic frame. Hair was removed from the scalp, and a 2 cm midline incision exposed the skull. Holes were drilled for EEG electrode placement (relative to bregma) over the frontal cortex (2.6 mm anterior, 2.25 mm bilateral), motor cortex (1.5 mm anterior, 2.0 mm left lateral), cingulate cortex (1.5 mm anterior, 0.3 mm right lateral), parietal cortex (1.8 mm posterior, 1.0 mm bilateral), and visual cortex (2.6 mm posterior, 2.5 mm bilateral). EEG leads consisted of blunted needles that were curved to hook under the skull and were secured with super glue. A ground screw was also placed over the cerebellum (5.5 mm posterior, 1.0 mm right lateral). Needle electrodes were placed through the neck muscle for EMG recordings. Two additional holes were made, and injection pipettes were lowered with one going straight down at 0.05 mm anterior, −0.65 mm left lateral, 5.65 mm ventral and the second at a 10° angle at 0.05 mm anterior, 1.6 mm right lateral, 5.7 mm ventral. A 50 μl volume of 0.25% bupivacaine was targeted at zygomaticotemporal, auriculotemporal, great auricular, and lesser occipital nerves bilaterally to alleviate potential discomfort from the ear bars at lighter anesthetic concentrations. Anesthetic concentration was lowered to 0.8% isoflurane, and collection of EEG and EMG signals began. Signals were amplified with a Telefactor TM103D amplifier (Grass Technologies) and recorded using custom in-house MATLAB-based software. Bipolar pairs of leads in the same coronal plane were referenced to the ground screw and sampled at a frequency of 200 Hz/channel. After giving the animal 45–60 min to equilibrate to the lighter anesthetic, 50 nl of 500 μm dexmedetomidine was pressure injected bilaterally over 30–60 s using a Picospritzer III (Parker Hannifin). A total of three bilateral injections were performed in each mouse with at least 10 min between each injection.
Acute dexmedetomidine microinjection with concurrent somatosensory-evoked potential recordings.
Eight mice were prepared for surgery, and holes were drilled for VLPO microinjection as described above. A craniotomy window measuring ∼2 × 2 mm was made over the left barrel cortex, centered around the coordinates 1.0 mm posterior, 3.0 mm left lateral. A silicon probe (Neuronexus) containing 16 electrodes, arranged vertically and spaced 100 μm apart, was lowered into the brain as described previously (Roy et al., 2011). A capillary tube attached to a pressure injection system was placed ∼1 cm above the right-side whisker pad so that a 50-ms-long, 30 pounds/inch2 puff of air displaced most of the larger whiskers contralateral to the recording site. After performing bupivacaine scalp blocks as above, the anesthetic concentration was lowered to 0.8% isoflurane and maintained for 45–60 min before recording started. Injection pipettes were lowered into place at this time. Before and after each drug injection, 5 min of data were recorded with the air-puff stimulus given every 3.11 s. Four mice received three bilateral injections each of 50 nl of 500 μm dexmedetomidine with 20 min between each injection. The other four mice received two injections of dexmedetomidine and two injections of saline in a randomized order (either both saline injections first or both dexmedetomidine injections first), again with 20 min between each injection. LFPs were amplified with a Lynx-8 amplifier (Neuralynx) and recorded at a rate of 10,101 Hz/channel using Cheetah acquisition software. The electrode at 900 μm deep was used as reference for all other electrodes. At the end of the experiment, anesthetic concentration was raised to 1.5% isoflurane, and a ground screw was placed over the cerebellum for subsequent marking of the injection sites by iontophoresis (see below).
Cannula and microinjection localization.
After behavioral testing was complete in cannulated animals, 50 nl of fluorescent microspheres (488 nm Fluospheres; Life Technologies) were injected bilaterally. At the end of each microinjection experiment, an electrode containing 2% pontamine sky blue dye in 0.5 m sodium acetate and a wire filament was lowered to each injection site. A square wave alternating between 0 and −25 V with a frequency of 5 Hz was applied for 10 min using a Grass SD9 Stimulator (Grass Technologies). After fluorescent microsphere injection or dye iontophoresis, mice were deeply anesthetized with isoflurane and perfused transcardially with ice-cold saline, followed by 4% paraformaldehyde. Brains were dissected and postfixed in 4% paraformaldehyde at 4°C overnight. The brains were then cryoprotected in 30% sucrose until sinking and flash frozen with liquid nitrogen. Tissue was sectioned at 40 μm, and any section showing evidence of cannula tracks or blue dye was slide mounted. Slides from cannula experiments were coverslipped with Dapi-Fluoromount G (Southern Biotech), whereas those from microinjection experiments were counterstained with Nuclear Fast Red (Vector Laboratories), dehydrated, and coverslipped with DPX mountant (Sigma-Aldrich). Cannula tips and microinjections were localized to their epicenter by visualizing the fluorescent microspheres or bright-field blue-labeled cells on an Olympus BX41 microscope (Olympus America). Stitched images of the entire brain section showing cannula tips and microinjection sites were created using Fiji (Preibisch et al., 2009) and scaled to corresponding coronal planes of the mouse brain atlas (Franklin and Paxinos, 2007). Sites found to be within 300 μm (for 25 nl injections) or 360 μm (for 50 nl injections) of any edge of the VLPO were considered to be “hits”; those outside of this range were labeled “misses.” These distances were chosen based on the estimated radius of 25 and 50 nl pressure injections into brain (Nicholson, 1985).
EEG and evoked potential data analyses.
EEG data were passed through a low-pass 35 Hz Butterworth filter. Spectral analysis was conducted on the 2 min before and after injections using MATLAB 7.9 (MathWorks) with custom code that uses the built-in “periodogram” function. Relative band powers were obtained by averaging the signal power across the frequency range of each band (delta = 0.5–4 Hz, theta = 4–8 Hz, alpha = 8–12 Hz, beta = 12–20 Hz, and gamma = 20–35 Hz) and then dividing by the total power from 0.5–80 Hz. For evoked potential experiments, data from the 50 ms preceding and 100 ms after each stimulus were averaged together for each collection period (preinjection and postinjection). Every channel was analyzed except the one containing data from the 12th electrode on the array (1100 μm deep), which proved nonfunctional. The maximum amplitude of the absolute value of the average LFP as well as the latency to the first peak after stimulus were identified using MATLAB.
Statistics.
All statistical analyses were performed using GraphPad Prism version 4.0b (GraphPad Software; Research Resource Identifier 000081) except Cox regressions, which were performed using Stata version 12.1 (StataCorp; Research Resource Identifier nlx_156918). Baseline membrane and action potential characteristics for isoflurane-activated and isoflurane-non-activated cells were compared by a t test. Repeated-measures one-way ANOVAs with post hoc Bonferroni's correction were used to analyze data from isoflurane plus norepinephrine, isoflurane plus dexmedetomidine, and TTX electrophysiology experiments. Membrane potentials and firing rates before and after phenylephrine exposure were compared using paired t tests. A χ2 test was used to compare differences in the number of isoflurane-activated and isoflurane-non-activated cells that expressed each adrenoceptor. Behavioral scores for norepinephrine, dexmedetomidine, saline, and phenylephrine injections were compared using Cochran's Q test for responsivity. Preinjection and postinjection movement scores for each drug injection were compared using a Cox regression. Both the changes in relative EEG power for each frequency band for a given channel and the average changes in LFP peak amplitude after dexmedetomidine and saline injections into the VLPO were compared with a theoretical change of 0 compared with preinjection using two-tailed one-sample t tests with a Holm's sequential Bonferroni's correction for multiple comparisons. A two-way ANOVA was used to compare the effects of drug (saline vs dexmedetomidine) and electrode location on changes in LFP amplitude. One-way ANOVAs were used to compare the evoked potential peak latency for preinjection, post-dexmedetomidine, and post-saline, as well as to compare the effects of dexmedetomidine on change in peak amplitude across channels. In all cases, a p value <0.05 was considered significant.
Results
Electrophysiological properties of VLPO neurons
Observations were based on data from 44 neurons anatomically localized to the VLPO in 44 different brain slices. VLPO neurons have been categorized previously using a variety of different schemes, including their responses to somnogenic and arousal-promoting neurotransmitters (Gallopin et al., 2000, 2004, 2005; Morairty et al., 2004). Alternatively, in the present study, VLPO neurons were classified according to their response to an initial exposure to 320 μm isoflurane, corresponding to that found in a deep surgical plane of anesthesia, and subsequently categorized into two groups: (1) isoflurane-activated (25 of 44, 57%), defined by depolarization of the resting membrane potential of a neuron of at least 4 mV; or (2) isoflurane-non-activated (19 of 44, 43%). In agreement with previous literature (Gallopin et al., 2000, 2004; Moore et al., 2012), basic membrane properties and action potential characteristics for these two classes of cells were not statistically different (Table 2). After bath application of 320 μm isoflurane, isoflurane-activated neurons were depolarized by 9.27 ± 0.98 mV, whereas isoflurane-non-activated VLPO neurons were hyperpolarized by 1.79 ± 1.50 mV.
Table 2.
Membrane and action potential characteristics of isoflurane-activated and isoflurane-non-activated cells
| Isoflurane-activated | Isoflurane-non-activated | p | |
|---|---|---|---|
| Resting membrane potential | −58.26 ± 0.98 mV | −58.73 ± 1.29 mV | 0.77 |
| Input resistance | 618.1 ± 25.3 MΩ | 602.3 ± 34.9 MΩ | 0.71 |
| Tau | 23.17 ± 1.49 ms | 24.89 ± 1.65 ms | 0.45 |
| Action potential threshold | −27.59 ± 0.68 mV | −28.34 ± 0.89 mV | 0.50 |
| Action potential amplitude | 55.60 ± 1.82 mV | 58.72 ± 1.65 mV | 0.23 |
| Action potential duration | 1.58 ± 0.11 ms | 1.53 ± 0.14 ms | 0.79 |
Adrenergic stimulation antagonizes isoflurane-induced activation of VLPO neurons
VLPO neurons that were depolarized by isoflurane were subsequently exposed to either norepinephrine or dexmedetomidine in the continued presence of isoflurane. In nine of nine neurons, relative to 320 μm isoflurane by itself, the addition of 100 μm norepinephrine resulted in a significant membrane hyperpolarization of 12.06 ± 2.18 mV (F(2,24) = 22.94, p = 0.039 overall, p < 0.001 for isoflurane vs isoflurane plus norepinephrine) along with a complete cessation of firing in all cells (isoflurane alone, 0.5 ± 0.15 Hz; F(2,24) = 11.70, p = 0.0007 overall, p < 0.01 for isoflurane vs isoflurane plus norepinephrine; Fig. 1). Similarly, in a separate cohort of eight neurons, the addition of 100 nm dexmedetomidine to 320 μm isoflurane also overcame the excitatory effect of isoflurane, resulting in a membrane hyperpolarization of 6.92 ± 1.18 mV compared with isoflurane alone (F(2,21) = 33.63, p < 0.0001 overall, p < 0.001 for isoflurane vs isoflurane plus dexmedetomidine) and a decrease in firing rate from 0.85 ± 0.33 to 0.10 ± 0.05 Hz (F(2,21) = 5.64, p = 0.016 overall, p < 0.05 for isoflurane vs isoflurane plus dexmedetomidine; Fig. 2).
Figure 1.

Norepinephrine reversed isoflurane-induced activation of VLPO neurons. Membrane potential (A) and firing rate (B) of nine neurons at baseline, during isoflurane exposure (Iso), and during concomitant isoflurane and norepinephrine exposure (Iso + NE). Connected points represent paired responses of individual neurons; colored lines represent group means. Data were analyzed with a one-way ANOVA and post hoc Bonferroni's-corrected comparisons. **p < 0.01; ***p < 0.001. C, Representative image of an electrode patched onto an isoflurane-activated VLPO neuron. D, Representative membrane potential trace from a current-clamped VLPO neuron showing depolarization and increased firing caused by isoflurane exposure (blue line) and ensuing hyperpolarization and decreased firing caused by simultaneous exposure to isoflurane and norepinephrine (red and blue lines).
Figure 2.

Dexmedetomidine reversed isoflurane-induced activation of VLPO neurons. Membrane potential (A) and firing rate (B) of eight neurons at baseline, during isoflurane exposure (Iso), and during concomitant isoflurane and dexmedetomidine exposure (Iso + Dex). Connected points represent paired responses of individual neurons; colored lines represent group means. Data were analyzed with a one-way ANOVA and post hoc Bonferroni's-corrected comparisons. *p < 0.05; ***p < 0.001. C, Representative membrane potential trace from a current-clamped VLPO neuron showing depolarization and increased firing caused by isoflurane exposure (blue line) and ensuing hyperpolarization and decreased firing caused by simultaneous administration of isoflurane and dexmedetomidine (blue and purple lines).
Single-cell α-adrenergic receptor mRNA analysis
Multiplex PCR for α adrenoceptors was performed on 15 isoflurane-activated and 13 isoflurane-non-activated neurons that were studied electrophysiologically. The summary profile of α adrenoceptors as a function of the response of the neuron to anesthetic exposure is shown in Figure 3. χ2 testing confirmed that isoflurane-activated cells were more likely to contain mRNA for α1 adrenoceptors compared with the non-activated cells (73.33 vs 30.77%, p = 0.02). The α1A and α1D adrenoceptor mRNA was present in 60.00 and 26.67% of the isoflurane-activated cells, respectively, compared with 30.77 and 7.69% of the isoflurane-non-activated cells. The α1B adrenoceptor mRNA was not detected in any VLPO neuron studied. Isoflurane-activated and isoflurane-non-activated VLPO neurons showed similar α2 adrenoceptor profiles (86.67 vs 84.62%, p = 0.88). The α2A, α2B, and α2C adrenoceptor subtypes were detected in 86.67, 60.00, and 73.33% of isoflurane-activated cells and 84.62, 69.23, and 61.53% of isoflurane-non-activated cells, without any significant difference in expression as a function of cell type.
Figure 3.

Qualitative multiplex RT-PCR showed a difference in α1 adrenergic receptor expression between isoflurane-activated and isoflurane-non-activated VLPO neurons. A, Representative gel revealing the expression of α-adrenergic receptors and GAD1 PCR products from an isoflurane-non-activated neuron, an isoflurane-activated neuron, and whole-brain library cDNA positive controls. B, Percentage of α-adrenergic receptor subtypes detected in isoflurane-non-activated neurons (gray) and isoflurane-activated neurons (black). C, Percentage of each cell type containing any subtype of α1 receptors and α2 receptors. Data were analyzed by χ2 test, *p < 0.05.
Dexmedetomidine hyperpolarizes isoflurane-activated VLPO neurons even after generation of action potentials is blocked
To determine whether direct α2-adrenoceptor stimulation without recurrent excitatory activity reverts isoflurane-induced depolarization, additional recordings were performed in the presence of 1 μm TTX. In accordance with previously published data (Moore et al., 2012), a one-way ANOVA demonstrated that, in the presence of TTX, 320 μm isoflurane retained its ability to significantly depolarize VLPO neurons by 8.23 ± 1.38 mV (F(2,14) = 17.27, p = 0.0012 overall, p < 0.01 for TTX vs TTX plus isoflurane). Moreover, the addition of 100 nm dexmedetomidine to the bath solution containing both isoflurane and TTX caused a significant 4.88 ± 1.13mV hyperpolarization (p < 0.05) and returned resting membrane potentials to a level statistically indistinguishable from that of the baseline TTX condition (Fig. 4).
Figure 4.

Reversal of isoflurane-induced depolarization in anesthetic-activated VLPO neurons by dexmedetomidine is mediated via postsynaptic mechanisms. A, Membrane potential of five neurons recorded during the continuous presence of TTX, after addition of isoflurane (TTX + Iso), and during concomitant TTX, isoflurane, and dexmedetomidine exposure (TTX + Iso + Dex). Connected points represent paired responses of individual neurons; colored lines depict group means. Data were analyzed with a one-way ANOVA and post hoc Bonferroni's-corrected comparisons. *p < 0.05, **p < 0.01. B, Representative membrane potential trace from a current-clamped anesthetic-activated VLPO neuron during exposure to TTX (gray line) showing depolarization by isoflurane (gray and blue lines) and subsequent hyperpolarization caused by simultaneous isoflurane and dexmedetomidine together with TTX (gray, blue, and purple lines).
Phenylephrine depolarizes both populations of VLPO neurons
Because of our identification of an increased probability of α1A and α1D adrenoceptors being expressed in isoflurane-activated cells, we examined how VLPO neurons responded to the α1-adrenergic agonist phenylephrine. In four of four isoflurane-activated neurons, after isoflurane washout, 50 μm phenylephrine caused significant depolarization with a change in membrane potential of 9.56 ± 2.80 mV (p = 0.04 compared with baseline) and a nonsignificant 0.74 ± 0.33 Hz increase in firing rate (p = 0.11). Similarly, in three of three isoflurane-non-activated VLPO neurons, phenylephrine exposure also resulted in a significant depolarization of 5.71 ± 1.08 mV (p = 0.034) and nonsignificant change in firing rate of −0.06 ± 0.17 Hz (p = 0.756).
Dexmedetomidine injection into the VLPO causes behavioral arousal
A representative image of bilateral cannulae that successfully targeted the VLPO, as well as markers indicating the target locations for all microinjection experiments, is shown in Figure 5. Injections of dexmedetomidine in the VLPO of supine, chronically cannulated mice increased behavioral arousal in 6 of 10 mice (Fig. 6). Responsive behavior was defined as full-body attempts to right or successful return of all paws to the ground. In comparison, when injected with saline, just 1 of 10 mice responded. Phenylephrine was similarly ineffective, with only 1 of 10 mice responding. Surprisingly, norepinephrine increased behavioral arousal in only 1 of 10, despite its hyperpolarizing actions on isoflurane-activated VLPO neurons in vitro (Fig. 1). The average latency to response in animals with cannulae targeting the VLPO after dexmedetomidine injection was 72.83 ± 40.43 s (mean ± SEM). The latency to response after any other drug in the VLPO (in the three cases in which a response occurred) was 166.7 ± 40.44 s. Cochran's Q test rejects the null hypothesis that all drugs were equally effective at causing responsiveness (Q = 8.43, p = 0.038). However, the efficacy of dexmedetomidine to partially antagonize the anesthetic state was dependent on anesthetic depth. When dexmedetomidine injections were performed at a higher dose of 1% isoflurane [corresponding to an ED1 for spontaneous return of righting reflex (RORR)] rather than 0.8% (corresponding to an ED5 for spontaneous RORR; Fig. 7), no mice responded. Notably, all drugs injected in eight mice with cannulae implanted just outside of VLPO were equally ineffective at increasing behavioral arousal (Q = 7.58, p = 0.056).
Figure 5.
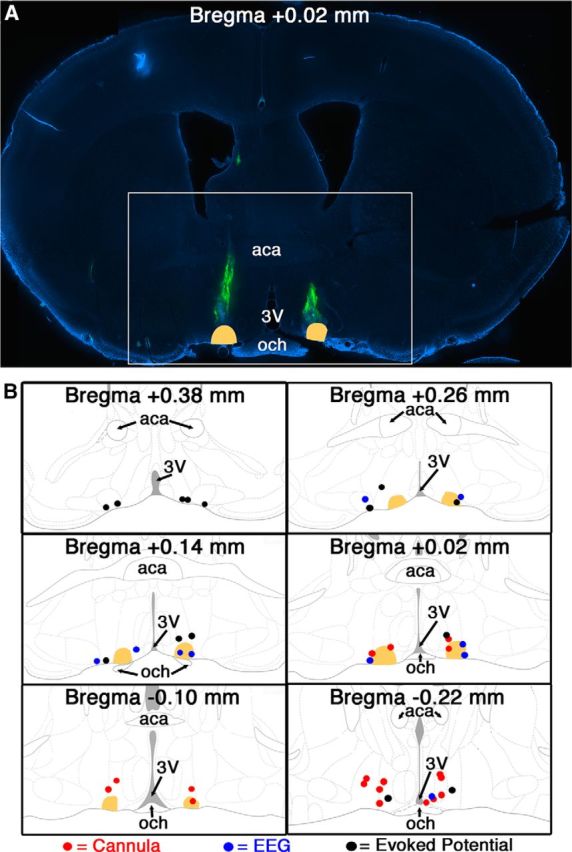
Cannula tips and microinjection sites within 300 μm (25 nl injections) or 360 μm (50 nl injections) of the VLPO were considered to be hits. A, Representative image of a VLPO-containing brain slice with 488 nm Fluospheres injected bilaterally (green) to permit localization of a chronically implanted cannula tip. The white box indicates the area shown in B. B, Summary of injection locations in all successful hits for cannula experiments (red), EEG experiments (blue), and evoked potential experiments (black). In all panels, the VLPO is colored in peach. aca, Anterior commissure; 3V, third ventricle; och, optic chiasm.
Figure 6.

Injection of dexmedetomidine into the VLPO increased behavioral arousal. Shown are the number of mice that exhibited signs of behavioral arousal when drugs were injected into the VLPO (n = 10) (A) or >300 μm from the edge of the VLPO (n = 8) (B). Drugs tested in animals anesthetized with 0.8% isoflurane include norepinephrine (NE), dexmedetomidine (Dex), phenylephrine (Phen), and saline. Dexmedetomidine was also injected in animals anesthetized with 1.0% isoflurane (A only). C and D show the fraction of mice that were immobile for a given percentage of the observation period after injection of drugs into the VLPO (C) or >300 μm from the edge of the VLPO (D) in mice anesthetized with 0.8% isoflurane. Data were analyzed by Cox regression.
Figure 7.

Induction and emergence curves displayed on a logarithmic x-axis for mice with cannulae directed at the VLPO. Least-squares best-fit line for induction is shown in solid and for emergence is shown in dashed. Shaded areas bracketing each curve depict the best-fit, 95% confidence intervals for each curve. The EC1 and EC5 for emergence are labeled in red.
Dexmedetomidine injection into the VLPO increases movement
In addition to increased behavioral responsiveness, injection of dexmedetomidine into the VLPO increased the fraction of time that mice spent moving. In contrast, saline, phenylephrine, and norepinephrine failed to affect movement. Because of the bounded nature and non-normal distribution of these measurements, Cox regression survival statistics were used to examine the likelihood that the population of mice spent at least a given fraction of time immobile (Fig. 6C,D; Jahn-Eimermacher et al., 2011). There were significant effects of both drug (p = 0.038) and preinjection versus postinjection (p = 0.014) on the fraction of time spent immobile. When injected into the VLPO, hazard ratios suggest that norepinephrine, phenylephrine, and saline were 72, 79, and 47%, respectively, less efficacious than dexmedetomidine at antagonizing anesthetic-induced immobility. Outside of the VLPO, no significant effect of drug (p = 0.26) or time (p = 0.12) was observed, indicating that behavioral arousal was specifically increased only when dexmedetomidine injections targeted the VLPO. Notably, the observation period was terminated early in mice that fully righted and removed the nose cone used to deliver anesthetic, thus minimizing the apparent efficacy of the drug injections that caused the greatest responses.
Intra-VLPO dexmedetomidine injection does not affect the spontaneous cortical EEG but reduces the depressant effects of isoflurane on barrel cortex somatosensory-evoked responses
Given signs of behavioral arousal after intra-VLPO dexmedetomidine, we next sought evidence of cortical arousal in six mice acutely implanted with electroencephalographic leads. Increased cortical arousal has been indicated by a shift in relative power from lower frequencies to higher frequencies in spontaneous EEG with arousal-promoting stimuli (Ma and Leung, 2006; Orth et al., 2006; Sukhotinsky et al., 2007; Van Dort et al., 2009; Pillay et al., 2011; Solt et al., 2011). Figure 8 surprisingly illustrates the lack of spectral changes in the EEG after intra-VLPO dexmedetomidine in a prototypical mouse stably anesthetized at 0.8% isoflurane and shows the absence of significant changes in the power of any EEG band frequency after intra-VLPO dexmedetomidine in recordings simultaneously acquired over frontal, motor/cingulate, parietal, and visual cortices. Although spontaneous cortical activity was not altered, evoked activity was significantly affected by drug injection. At steady-state 0.8% isoflurane, whisker stimuli repeatedly evoked large, stimulus-locked LFPs in the barrel cortex. In a separate cohort of eight mice, bilateral injections of dexmedetomidine into the VLPO increased the average amplitude of evoked LFPs compared with the preinjection period (Fig. 9; p = 0.000056). Conversely, bilateral injections of an identical volume of saline into the VLPO lowered evoked LFP amplitude (p = 0.000066), possibly as a result of a transient increase in intracranial pressure during injection. Intra-VLPO acute injections were slightly more rostral than cannula injections but nevertheless successfully targeted VLPO (Fig. 5). A two-way ANOVA that compared the effects of drug (saline vs dexmedetomidine) and electrode location found a significant effect of drug only (F(1, 270) = 29.99, p < 0.0001 for drug; F(13, 270) = 0.36, p = 0.981 for location) and no interaction between the two variables (F(13, 270) = 0.70, p = 0.763). A subsequent one-way ANOVA found no effect of individual channels on the response to dexmedetomidine (F(13,200) = 1.02, p = 0.44), indicating that the observed increase in LFP amplitude with dexmedetomidine injection was present throughout the primary barrel cortex. Microinjections of drugs into VLPO did not alter evoked potential latency, which occurred at 28.34 ± 0.41 ms before drug injection, 28.27 ± 0.46 ms after injection of dexmedetomidine, and 29.27 ± 0.62 ms after saline injections (F(2,230) = 1.12, p = 0.328).
Figure 8.
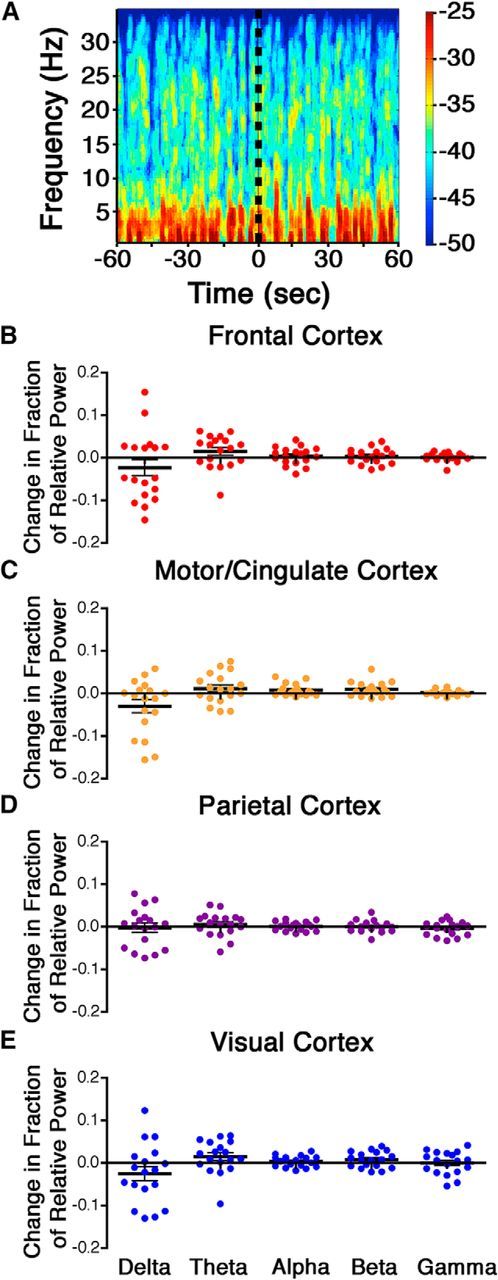
There is no change in spontaneous cortical EEG power after dexmedetomidine injection in the VLPO. A shows a representative spectrogram from the parietal cortex of a mouse before and after injection of dexmedetomidine into the VLPO (indicated by the dotted black line). The change in relative power from preinjection to postinjection was measured over the frontal cortex (B), motor/cingulate cortex (C), parietal cortex (D), and visual cortex (E) for five different frequency bands: delta = 0.5–4 Hz, theta = 4–8 Hz, alpha = 8–12 Hz, beta = 12–20 Hz, and gamma = 20–35 Hz. Black bars show the mean with SE for each dataset. Data were analyzed by two-tailed one-sample t tests with a Holm's sequential Bonferroni's correction for multiple comparisons.
Figure 9.
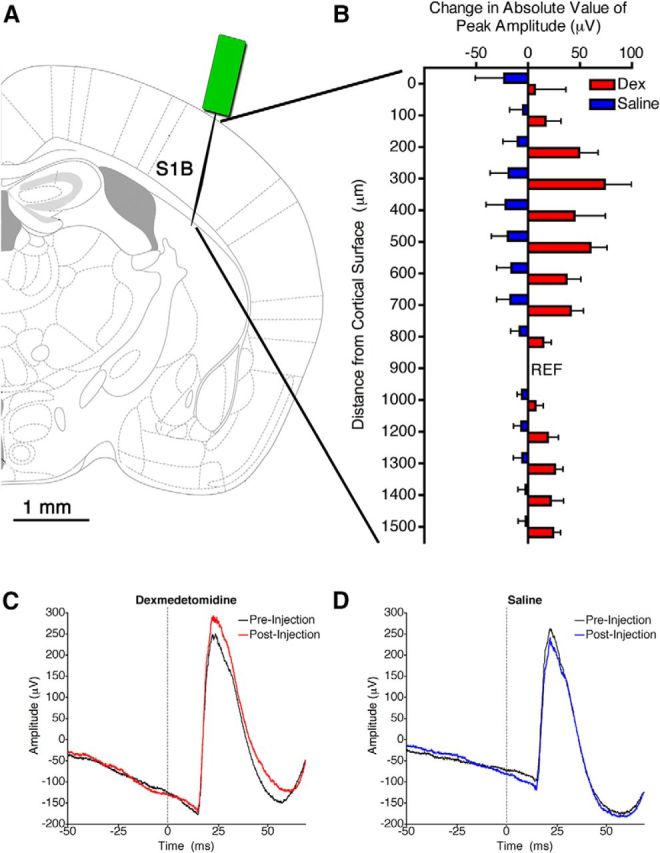
Dexmedetomidine in the VLPO increased the amplitude of somatosensory-evoked potentials. A, Graphic representation of the location in the primary barrel cortex (S1B) in which the recording electrode was placed. B, The change in the absolute value of the amplitude of evoked LFPs after injection of dexmedetomidine (red; Dex) or saline (blue) at each recording site throughout the depth of the barrel cortex. The reference electrode is indicated by REF. The electrode at 1100 μm depth was nonfunctional and thus is not shown. A two-way ANOVA indicated a significant effect of drug (p < 0.0001). C, D, Representative traces of the average LFP evoked by a whisker stimulus before and after injection of dexmedetomidine (C) or saline (D) from the electrode at 200 μm depth. Stimulus time is marked by a dotted gray line.
Discussion
We used adrenergic drugs to probe the functional significance of isoflurane-induced activation of putative sleep-promoting VLPO neurons. Numerous studies have demonstrated that norepinephrine has the capacity to inhibit activity in sleep-promoting VLPO neurons (Gallopin et al., 2000; Matsuo et al., 2003; Saint-Mleux et al., 2004; Moore et al., 2012). Sleep-promoting neurons are known to possess α2 adrenoceptors (Modirrousta et al., 2004). Hence, our finding that dexmedetomidine could be used to reverse isoflurane-induced activation in eight of the eight cells tested in vitro strongly implicates VLPO α2 adrenergic signaling in modulation of arousal state.
Attempting to distinguish adrenergic receptor expression profiles among anesthetic-activated and non-activated VLPO neurons, we performed single-cell multiplex RT-PCR analyses on 28 neurons in which we obtained electrophysiological recordings. There was an increased likelihood of detecting α1A or α1D adrenoceptors in isoflurane-activated cells but a similar incidence of detecting all three α2 adrenoceptor subtypes in both populations. Despite the increased probability of finding α1 adrenoceptor mRNA in anesthetic-activated cells, 50 μm phenylephrine depolarized both the isoflurane-activated and non-activated VLPO neurons. One study limitation is that only a single dose of phenylephrine—chosen to activate all three α1 adrenoceptors—was tested. Its lack of selectivity in depolarizing both VLPO cell populations discouraged additional α1 agonist investigation. In an effort to determine whether the ability of dexmedetomidine to antagonize isoflurane-induced activation of VLPO neurons was mediated by direct action on the postsynaptic VLPO neurons themselves or potentially presynaptically through actions on neighboring non-activated cells or more distant cells also present in the slice, such as those of the median preoptic area (Kolaj and Renaud, 2001), we suppressed synaptic transmission by blocking action potential generation with TTX. Isoflurane still depolarized a subset of VLPO neurons, consistent with previous findings (Moore et al., 2012). In the presence of TTX, isoflurane-induced depolarization was reversed by dexmedetomidine, consistent with direct hyperpolarizing actions occurring through α2 adrenoceptors expressed on the postsynaptic VLPO neurons. Conversely, systemic rather than local administration of dexmedetomidine appears to indirectly depolarize VLPO neurons (Nelson et al., 2003), likely as a net consequence of silencing inhibitory input to the VLPO. Although the hypnotic mechanism underlying systemic delivery of dexmedetomidine remains unknown, previous studies prove that its essential α2-adrenergic effects occur on non-adrenergic neurons (Lakhlani et al., 1997; Gilsbach et al., 2009; Hu et al., 2012).
The robust antagonistic effects of local application of α2-adrenergic agents on isoflurane-induced VLPO depolarization in vitro led to in vivo investigations. Both qualitative and quantitative measures of behavior were used to determine whether injection of adrenergic drugs into the VLPO of anesthetized mice would increase arousal. The qualitative test relied on the return of the righting reflex, a common surrogate readout for restoration of consciousness in rodents. Although the definition of this test was expanded to consider mice that made a clear effort to right as responsive, the readout was still binary and failed to address the significance of smaller behaviors that may be associated with arousal, such as facial grooming and tail swinging (Pillay et al., 2011). For this reason, the percentage of time spent moving before and after drug injections was also examined. Such measures provide a more quantitative and continuous assessment of arousal in settings in which interventions fail to fully antagonize the anesthetic state.
In accordance with in vitro slice experiments, we demonstrated both subjective and objective increases in arousal when dexmedetomidine was injected into the VLPO of lightly anesthetized mice. Microinjection of histamine into the rat VLPO, which reduces the firing of norepinephrine-hyperpolarized, putative sleep- and anesthetic-activated neurons, has also been shown to increase behavioral activity (Liu et al., 2010). The 0.8% isoflurane dose used in this study corresponded to a calculated EC5 for spontaneous RORR in this cohort of mice. When dexmedetomidine was administered during the deeper plane of 1.0% isoflurane, corresponding to an EC1 for spontaneous return of righting, no arousing response was seen despite the ability of dexmedetomidine to reverse isoflurane-induced activation of VLPO neurons in vitro at an even higher anesthetic concentration. This suggests that the VLPO may contribute to the transition between arousal states but that recruitment of other brain regions, neurotransmitter systems, and protein targets are likely necessary to enact a full return to arousal (Alkire et al., 2007, 2009; Kelz et al., 2008; Pillay et al., 2011; Solt et al., 2011; Taylor et al., 2013; van Swinderen and Kottler, 2014). Although global modulation of adrenergic signaling affects both anesthesia induction and emergence and local activation of the locus ceruleus powerfully facilitates emergence, local modulation of adrenergic signaling within the basal forebrain could only elicit transient microarousals (Mason et al., 1983; Kushikata et al., 2011; Pillay et al., 2011; Hu et al., 2012; Vazey and Aston-Jones, 2014). Hence, a major unanswered question remains regarding the adrenoceptive neuronal targets capable of eliciting a complete response.
Despite the observed signs of behavioral arousal, EEG recordings during acute dexmedetomidine injection into the VLPO showed no evidence of spectral changes suggestive of cortical activation. It is possible that application of other processed EEG measures may be more sensitive to subtle changes in spontaneous cortical activity. Together, the results of our in vivo experiments suggest an apparent dissociation between behavioral activation and EEG arousal, a phenomenon that has been observed previously (Feldman and Waller, 1962; Vanderwolf, 1992). However, to substantiate this hypothesis that intra-VLPO dexmedetomidine destabilizes the anesthetic state, we recorded somatosensory-evoked potentials. Evoked responses have been shown previously to be sensitive to changes in anesthetic-related arousal state, with LFP amplitudes increasing as anesthetic concentration decreases (Erchova et al., 2002; Antunes et al., 2003; Banoub et al., 2003; Rojas et al., 2006). At a fixed anesthetic concentration, increased evoked potential amplitudes would thus indicate a higher fidelity sensory gating to the cortex, consistent with an apparent decrease in the depth of the anesthetic state. We observed an increase in the amplitude of evoked responses after injection of dexmedetomidine into the VLPO, further supporting the theory that hyperpolarization of anesthetic-activated cells in the VLPO primes the anesthetized brain for arousal. Whisker stimuli thus provide another tool suggesting that VLPO hyperpolarization may cause subcortical network adaptations that ultimately affect thalamocortical transmission.
In contrast to in vitro observations, mice did not show increased arousal with local delivery of norepinephrine into the VLPO. Multiplex PCR data indicated that a variety of adrenoceptors were present in anesthetic-activated and neighboring VLPO neurons. It is possible that norepinephrine itself failed to induce arousal in mice as a result of opposing actions within the VLPO microenvironment, which was the intended target of cannula drug delivery. Such an effect could potentially be mediated by the isoflurane-non-activated subset of VLPO cells or by off-target actions of the single, high concentration of norepinephrine chosen based on the literature (Pillay et al., 2011). A complete neurotransmitter receptor profile would be required to determine all of the VLPO targets on which norepinephrine may be working along with a full dose–response curve, but it is beyond the scope of this study.
Based on the exquisite selectivity of medetomidine, the racemic mixture of dexmedetomidine and its stereoisomer, which display a 1600:1 preference for α2 adrenoceptors over α1 adrenoceptors (Virtanen et al., 1988), the most congruent interpretation of our pharmacologic data is that stimulation of α2 adrenergic receptors on VLPO neurons antagonizes the activating effects of isoflurane on these cells in vitro. Dexmedetomidine injected directly into the VLPO of anesthetized mice destabilized the anesthetic state, favoring behavioral arousal, but only at lighter anesthetic doses. At 0.8% isoflurane, the system was already primed for emergence, and subsequent adrenergic hyperpolarization of the VLPO further destabilized the hypnotic state, making the transition to arousal even more probable. This “paradoxical” antagonism of the anesthetic state by dexmedetomidine—an anesthetic drug—is reminiscent of the functional recovery observed in patients with disorders of consciousness after administration of zolpidem (Clauss et al., 2000; Cohen et al., 2004; Brefel-Courbon et al., 2007; Shames and Ring, 2008; Whyte and Myers, 2009; Williams et al., 2013). Such non-intuitive drug effects highlight the critical requirement to understand the circuits on which general anesthetics act for mechanistic insight. Although manipulating the activity of VLPO neurons is not a guaranteed means to precipitate an arousal state transition, predicted prolonged depolarization of these anesthetic-active neurons with a glutamatergic agonist markedly delayed emergence from isoflurane (McCarren et al., 2013). Hence, anesthetic-activated neurons in the VLPO may form an intrinsic switch that prepares the brain for state transitions, but future in vivo unit recordings are needed to confirm this prediction. Given the response to noxious stimuli, it is not surprising that a surgical plane of anesthesia cannot be antagonized by a surge in central adrenergic tone, although activation of central adrenergic signaling clearly prepares the brain for emergence (Vazey and Aston-Jones, 2014). Cumulatively, our findings indicate that VLPO activity contributes to the stability of the anesthetic state and reinforce the notion that the VLPO may be a potential bellwether capable of heralding state changes before their actual occurrence. Anesthetic drugs, as a class, are likely working simultaneously on numerous and varied components of brain circuitry to produce their many effects. Identification of the VLPO as an effector of changes in anesthetic sensitivity will help to guide future experiments that look at synergistic effects of multiple arousal-related nuclei.
Footnotes
This work was supported by National Institutes of Health Grants R01 GM088156 and T32 HL007713-18 and the Department of Anesthesiology and Critical Care at the University of Pennsylvania. We acknowledge Matthew Fleisher for his experimental assistance in localizing VLPO neurons, and we also thank Rebecca Hoffman and Rachel Kelz for their assistance with Cox regression analysis. Finally, we thank Alex Proekt for his suggestions on spectral analysis.
The authors declare no competing financial interests.
References
- Alkire MT, McReynolds JR, Hahn EL, Trivedi AN. Thalamic microinjection of nicotine reverses sevoflurane-induced loss of righting reflex in the rat. Anesthesiology. 2007;107:264–272. doi: 10.1097/01.anes.0000270741.33766.24. [DOI] [PubMed] [Google Scholar]
- Alkire MT, Asher CD, Franciscus AM, Hahn EL. Thalamic microinfusion of antibody to a voltage-gated potassium channel restores consciousness during anesthesia. Anesthesiology. 2009;110:766–773. doi: 10.1097/ALN.0b013e31819c461c. [DOI] [PubMed] [Google Scholar]
- Antunes LM, Golledge HDR, Roughan JV, Flecknell PA. Comparison of electroencephalogram activity and auditory evoked responses during isoflurane and halothane anaesthesia in the rat. Vet Anaesth Analg. 2003;30:15–23. doi: 10.1046/j.1467-2995.2003.00085.x. [DOI] [PubMed] [Google Scholar]
- Bai D, Renaud LP. Median preoptic nucleus neurons: an in vitro patch-clamp analysis of their intrinsic properties and noradrenergic receptors in the rat. Neuroscience. 1998;83:905–916. doi: 10.1016/S0306-4522(97)00435-1. [DOI] [PubMed] [Google Scholar]
- Banoub M, Tetzlaff JE, Schubert A. Pharmacologic and physiologic influences affecting sensory evoked potentials: implications for perioperative monitoring. Anesthesiology. 2003;99:716–737. doi: 10.1097/00000542-200309000-00029. [DOI] [PubMed] [Google Scholar]
- Berridge CW, Schmeichel BE, España RA. Noradrenergic modulation of wakefulness/arousal. Sleep Med Rev. 2012;16:187–197. doi: 10.1016/j.smrv.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brefel-Courbon C, Payoux P, Ory F, Sommet A, Slaoui T, Raboyeau G, Lemesle B, Puel M, Montastruc JL, Demonet JF, Cardebat D. Clinical and imaging evidence of zolpidem effect in hypoxic encephalopathy. Ann Neurol. 2007;62:102–105. doi: 10.1002/ana.21110. [DOI] [PubMed] [Google Scholar]
- Chou TC, Bjorkum AA, Gaus SE, Lu J, Scammell TE, Saper CB. Afferents to the ventrolateral preoptic nucleus. J Neurosci. 2002;22:977–990. doi: 10.1523/JNEUROSCI.22-03-00977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauss RP, Güldenpfennig WM, Nel HW, Sathekge MM, Venkannagari RR. Extraordinary arousal from semi-comatose state on zolpidem. A case report. South Afr Med J Suid-Afr Tydskr Vir Geneeskd. 2000;90:68–72. [PubMed] [Google Scholar]
- Cohen L, Chaaban B, Habert MO. Transient improvement of aphasia with zolpidem. N Engl J Med. 2004;350:949–950. doi: 10.1056/NEJM200402263500922. [DOI] [PubMed] [Google Scholar]
- Eckenhoff MF, Eckenhoff RG. Quantitative autoradiography of halothane binding in rat brain. J Pharmacol Exp Ther. 1998;285:371–376. [PubMed] [Google Scholar]
- Erchova IA, Lebedev MA, Diamond ME. Somatosensory cortical neuronal population activity across states of anaesthesia. Eur J Neurosci. 2002;15:744–752. doi: 10.1046/j.0953-816x.2002.01898.x. [DOI] [PubMed] [Google Scholar]
- Feldman SM, Waller HJ. Dissociation of electrocortical activation and behavioural arousal. Nature. 1962;196:1320–1322. doi: 10.1038/1961320a0. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Ed 3. San Diego: Elsevier; 2007. [Google Scholar]
- Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–386. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- Franks NP, Lieb WR. Temperature dependence of the potency of volatile general anesthetics: implications for in vitro experiments. Anesthesiology. 1996;84:716–720. doi: 10.1097/00000542-199603000-00027. [DOI] [PubMed] [Google Scholar]
- Frontini A, Giordano A. Leptin-sensitive neurons in mouse preoptic area express α1A- and α2A-adrenergic receptor isoforms. Neurosci Lett. 2010;471:83–88. doi: 10.1016/j.neulet.2010.01.016. [DOI] [PubMed] [Google Scholar]
- Gallopin T, Fort P, Eggermann E, Cauli B, Luppi PH, Rossier J, Audinat E, Mühlethaler M, Serafin M. Identification of sleep-promoting neurons in vitro. Nature. 2000;404:992–995. doi: 10.1038/35010109. [DOI] [PubMed] [Google Scholar]
- Gallopin T, Luppi PH, Rambert FA, Frydman A, Fort P. Effect of the wake-promoting agent modafinil on sleep-promoting neurons from the ventrolateral preoptic nucleus: an in vitro pharmacologic study. Sleep. 2004;27:19–25. [PubMed] [Google Scholar]
- Gallopin T, Luppi PH, Cauli B, Urade Y, Rossier J, Hayaishi O, Lambolez B, Fort P. The endogenous somnogen adenosine excites a subset of sleep-promoting neurons via A2A receptors in the ventrolateral preoptic nucleus. Neuroscience. 2005;134:1377–1390. doi: 10.1016/j.neuroscience.2005.05.045. [DOI] [PubMed] [Google Scholar]
- Gilsbach R, Röser C, Beetz N, Brede M, Hadamek K, Haubold M, Leemhuis J, Philipp M, Schneider J, Urbanski M, Szabo B, Weinshenker D, Hein L. Genetic dissection of α2-adrenoceptor functions in adrenergic versus nonadrenergic cells. Mol Pharmacol. 2009;75:1160–1170. doi: 10.1124/mol.109.054544. [DOI] [PubMed] [Google Scholar]
- Han B, McCarren HS, O'Neill D, Kelz MB. Distinctive recruitment of endogenous sleep-promoting neurons by volatile anesthetics and a nonimmobilizer. Anesthesiology. 2014;121:999–1009. doi: 10.1097/ALN.0000000000000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh KC, Gvilia I, Kumar S, Uschakov A, McGinty D, Alam MN, Szymusiak R. c-Fos expression in neurons projecting from the preoptic and lateral hypothalamic areas to the ventrolateral periaqueductal gray in relation to sleep states. Neuroscience. 2011;188:55–67. doi: 10.1016/j.neuroscience.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu FY, Hanna GM, Han W, Mardini F, Thomas SA, Wyner AJ, Kelz MB. Hypnotic hypersensitivity to volatile anesthetics and dexmedetomidine in dopamine β-hydroxylase knockout mice. Anesthesiology. 2012;117:1006–1017. doi: 10.1097/ALN.0b013e3182700ab9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn-Eimermacher A, Lasarzik I, Raber J. Statistical analysis of latency outcomes in behavioral experiments. Behav Brain Res. 2011;221:271–275. doi: 10.1016/j.bbr.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha SK, Mallick BN. Presence of α-1 norepinephrinergic and GABA-A receptors on medial preoptic hypothalamus thermosensitive neurons and their role in integrating brainstem ascending reticular activating system inputs in thermoregulation in rats. Neuroscience. 2009;158:833–844. doi: 10.1016/j.neuroscience.2008.10.038. [DOI] [PubMed] [Google Scholar]
- Kelz MB, Sun Y, Chen J, Cheng Meng Q, Moore JT, Veasey SC, Dixon S, Thornton M, Funato H, Yanagisawa M. An essential role for orexins in emergence from general anesthesia. Proc Natl Acad Sci U S A. 2008;105:1309–1314. doi: 10.1073/pnas.0707146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaj M, Renaud LP. Norepinephrine acts via α2 adrenergic receptors to suppress N-type calcium channels in dissociated rat median preoptic nucleus neurons. Neuropharmacology. 2001;41:472–479. doi: 10.1016/S0028-3908(01)00090-9. [DOI] [PubMed] [Google Scholar]
- Kolaj M, Renaud LP. Presynaptic {alpha}-adrenoceptors in median preoptic nucleus modulate inhibitory neurotransmission from subfornical organ and organum vasculosum lamina terminalis. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1907–R1915. doi: 10.1152/ajpregu.00763.2006. [DOI] [PubMed] [Google Scholar]
- Kottler B, Bao H, Zalucki O, Imlach W, Troup M, van Alphen B, Paulk A, Zhang B, van Swinderen B. A sleep/wake circuit controls isoflurane sensitivity in Drosophila. Curr Biol. 2013;23:594–598. doi: 10.1016/j.cub.2013.02.021. [DOI] [PubMed] [Google Scholar]
- Kumar VM, Vetrivelan R, Mallick HN. Alpha-1 adrenergic receptors in the medial preoptic area are involved in the induction of sleep. Neurochem Res. 2006;31:1095–1102. doi: 10.1007/s11064-006-9109-8. [DOI] [PubMed] [Google Scholar]
- Kumar VM, Vetrivelan R, Mallick HN. Noradrenergic afferents and receptors in the medial preoptic area: neuroanatomical and neurochemical links between the regulation of sleep and body temperature. Neurochem Int. 2007;50:783–790. doi: 10.1016/j.neuint.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Kushikata T, Yoshida H, Kudo M, Kudo T, Kudo T, Hirota K. Role of coerulean noradrenergic neurones in general anaesthesia in rats. Br J Anaesth. 2011;107:924–929. doi: 10.1093/bja/aer303. [DOI] [PubMed] [Google Scholar]
- Lakhlani PP, MacMillan LB, Guo TZ, McCool BA, Lovinger DM, Maze M, Limbird LE. Substitution of a mutant α2a-adrenergic receptor via “hit and run” gene targeting reveals the role of this subtype in sedative, analgesic, and anesthetic-sparing responses in vivo. Proc Natl Acad Sci U S A. 1997;94:9950–9955. doi: 10.1073/pnas.94.18.9950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarenko RM, Fortuna MG, Shi Y, Mulkey DK, Takakura AC, Moreira TS, Guyenet PG, Bayliss DA. Anesthetic activation of central respiratory chemoreceptor neurons involves inhibition of a THIK-1-like background K+ current. J Neurosci. 2010;30:9324–9334. doi: 10.1523/JNEUROSCI.1956-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li KY, Guan YZ, Krnjević K, Ye JH. Propofol facilitates glutamatergic transmission to neurons of the ventrolateral preoptic nucleus. Anesthesiology. 2009;111:1271–1278. doi: 10.1097/ALN.0b013e3181bf1d79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YW, Li J, Ye JH. Histamine regulates activities of neurons in the ventrolateral preoptic nucleus. J Physiol. 2010;588:4103–4116. doi: 10.1113/jphysiol.2010.193904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Nelson LE, Franks N, Maze M, Chamberlin NL, Saper CB. Role of endogenous sleep-wake and analgesic systems in anesthesia. J Comp Neurol. 2008;508:648–662. doi: 10.1002/cne.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydic R, Baghdoyan HA. Sleep, anesthesiology, and the neurobiology of arousal state control. Anesthesiology. 2005;103:1268–1295. doi: 10.1097/00000542-200512000-00024. [DOI] [PubMed] [Google Scholar]
- Ma J, Leung LS. Limbic system participates in mediating the effects of general anesthetics. Neuropsychopharmacology. 2006;31:1177–1192. doi: 10.1038/sj.npp.1300909. [DOI] [PubMed] [Google Scholar]
- Mason ST, King RAJ, Banks P, Angel A. Brain noradrenaline and anaesthesia: behavioural and electrophysiological evidence. Neuroscience. 1983;10:177–185. doi: 10.1016/0306-4522(83)90091-X. [DOI] [PubMed] [Google Scholar]
- Matsuo S, Jang IS, Nabekura J, Akaike N. alpha 2-Adrenoceptor-mediated presynaptic modulation of GABAergic transmission in mechanically dissociated rat ventrolateral preoptic neurons. J Neurophysiol. 2003;89:1640–1648. doi: 10.1152/jn.00491.2002. [DOI] [PubMed] [Google Scholar]
- McCarren HS, Moore JT, Kelz MB. Assessing changes in volatile general anesthetic sensitivity of mice after local or systemic pharmacological intervention. J Vis Exp. 2013;(80):e51079. doi: 10.3791/51079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modirrousta M, Mainville L, Jones BE. Gabaergic neurons with [alpha]2-adrenergic receptors in basal forebrain and preoptic area express c-Fos during sleep. Neuroscience. 2004;129:803–810. doi: 10.1016/j.neuroscience.2004.07.028. [DOI] [PubMed] [Google Scholar]
- Moore JT, Chen J, Han B, Meng QC, Veasey SC, Beck SG, Kelz MB. Direct activation of sleep-promoting VLPO neurons by volatile anesthetics contributes to anesthetic hypnosis. Curr Biol. 2012;22:2008–2016. doi: 10.1016/j.cub.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morairty S, Rainnie D, McCarley R, Greene R. Disinhibition of ventrolateral preoptic area sleep-active neurons by adenosine: a new mechanism for sleep promotion. Neuroscience. 2004;123:451–457. doi: 10.1016/j.neuroscience.2003.08.066. [DOI] [PubMed] [Google Scholar]
- Morrison PF, Chen MY, Chadwick RS, Lonser RR, Oldfield EH. Focal delivery during direct infusion to brain: role of flow rate, catheter diameter, and tissue mechanics. Am J Physiol Regul Integr Comp Physiol. 1999;277:R1218–R1229. doi: 10.1152/ajpregu.1999.277.4.R1218. [DOI] [PubMed] [Google Scholar]
- Nelson LE, Guo TZ, Lu J, Saper CB, Franks NP, Maze M. The sedative component of anesthesia is mediated by GABAA receptors in an endogenous sleep pathway. Nat Neurosci. 2002;5:979–984. doi: 10.1038/nn913. [DOI] [PubMed] [Google Scholar]
- Nelson LE, Lu J, Guo T, Saper CB, Franks NP, Maze M. The alpha2-adrenoceptor agonist dexmedetomidine converges on an endogenous sleep-promoting pathway to exert its sedative effects. Anesthesiology. 2003;98:428–436. doi: 10.1097/00000542-200302000-00024. [DOI] [PubMed] [Google Scholar]
- Nicholson C. Diffusion from an injected volume of a substance in brain tissue with arbitrary volume fraction and tortuosity. Brain Res. 1985;333:325–329. doi: 10.1016/0006-8993(85)91586-0. [DOI] [PubMed] [Google Scholar]
- Orth M, Bravo E, Barter L, Carstens E, Antognini JF. The differential effects of halothane and isoflurane on electroencephalographic responses to electrical microstimulation of the reticular formation. Anesth Analg. 2006;102:1709–1714. doi: 10.1213/01.ane.0000205752.00303.94. [DOI] [PubMed] [Google Scholar]
- Osaka T, Matsumura H. Noradrenaline inhibits preoptic sleep-active neurons through [alpha]2-receptors in the rat. Neurosci Res. 1995;21:323–330. doi: 10.1016/0168-0102(94)00871-C. [DOI] [PubMed] [Google Scholar]
- Pillay S, Vizuete JA, McCallum JB, Hudetz AG. Norepinephrine infusion into nucleus basalis elicits micro-arousal in desflurane-anesthetized rats. Anesthesiology. 2011;115:733–742. doi: 10.1097/ALN.0b013e31822c5ee1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinforma. 2009;25:1463–1465. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas MJ, Navas JA, Rector DM. Evoked response potential markers for anesthetic and behavioral states. Am J Physiol Regul Integr Comp Physiol. 2006;291:R189–R196. doi: 10.1152/ajpregu.00409.2005. [DOI] [PubMed] [Google Scholar]
- Roy NC, Bessaih T, Contreras D. Comprehensive mapping of whisker-evoked responses reveals broad, sharply tuned thalamocortical input to layer 4 of barrel cortex. J Neurophysiol. 2011;105:2421–2437. doi: 10.1152/jn.00939.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Mleux B, Eggermann E, Bisetti A, Bayer L, Machard D, Jones BE, Mühlethaler M, Serafin M. Nicotinic enhancement of the noradrenergic inhibition of sleep-promoting neurons in the ventrolateral preoptic area. J Neurosci. 2004;24:63–67. doi: 10.1523/JNEUROSCI.0232-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shames JL, Ring H. Transient reversal of anoxic brain injury-related minimally conscious state after zolpidem administration: a case report. Arch Phys Med Rehabil. 2008;89:386–388. doi: 10.1016/j.apmr.2007.08.137. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998;18:4705–4721. doi: 10.1523/JNEUROSCI.18-12-04705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt K, Cotten JF, Cimenser A, Wong KFK, Chemali JJ, Brown EN. Methylphenidate actively induces emergence from general anesthesia. Anesthesiology. 2011;115:791–803. doi: 10.1097/ALN.0b013e31822e92e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhotinsky I, Zalkind V, Lu J, Hopkins DA, Saper CB, Devor M. Neural pathways associated with loss of consciousness caused by intracerebral microinjection of GABAA-active anesthetics. Eur J Neurosci. 2007;25:1417–1436. doi: 10.1111/j.1460-9568.2007.05399.x. [DOI] [PubMed] [Google Scholar]
- Taylor NE, Chemali JJ, Brown EN, Solt K. Activation of D1 dopamine receptors induces emergence from isoflurane general anesthesia. Anesthesiology. 2013;118:30–39. doi: 10.1097/ALN.0b013e318278c896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dort CJ, Baghdoyan HA, Lydic R. Adenosine A1 and A2A receptors in mouse prefrontal cortex modulate acetylcholine release and behavioral arousal. J Neurosci. 2009;29:871–881. doi: 10.1523/JNEUROSCI.4111-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Swinderen B, Kottler B. Explaining general anesthesia: a two-step hypothesis linking sleep circuits and the synaptic release machinery. BioEssays. 2014;36:372–381. doi: 10.1002/bies.201300154. [DOI] [PubMed] [Google Scholar]
- Vanderwolf CH. The electrocorticogram in relation to physiology and behavior: a new analysis. Electroencephalogr Clin Neurophysiol. 1992;82:165–175. doi: 10.1016/0013-4694(92)90164-D. [DOI] [PubMed] [Google Scholar]
- Vazey EM, Aston-Jones G. Designer receptor manipulations reveal a role of the locus coeruleus noradrenergic system in isoflurane general anesthesia. Proc Natl Acad Sci U S A. 2014;111:3859–3864. doi: 10.1073/pnas.1310025111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen R, Savola JM, Saano V, Nyman L. Characterization of the selectivity, specificity and potency of medetomidine as an α2-adrenoceptor agonist. Eur J Pharmacol. 1988;150:9–14. doi: 10.1016/0014-2999(88)90744-3. [DOI] [PubMed] [Google Scholar]
- Wei H, Kang B, Wei W, Liang G, Meng QC, Li Y, Eckenhoff RG. Isoflurane and sevoflurane affect cell survival and BCL-2/BAX ratio differently. Brain Res. 2005;1037:139–147. doi: 10.1016/j.brainres.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Whyte J, Myers R. Incidence of clinically significant responses to zolpidem among patients with disorders of consciousness: a preliminary placebo controlled trial. Am J Phys Med Rehabil. 2009;88:410–418. doi: 10.1097/PHM.0b013e3181a0e3a0. [DOI] [PubMed] [Google Scholar]
- Williams ST, Conte MM, Goldfine AM, Noirhomme Q, Gosseries O, Thonnard M, Beattie B, Hersh J, Katz DI, Victor JD, Laureys S, Schiff ND. Common resting brain dynamics indicate a possible mechanism underlying zolpidem response in severe brain injury. eLife. 2013;2:e01157. doi: 10.7554/eLife.01157. [DOI] [PMC free article] [PubMed] [Google Scholar]