

Abstract
Frontotemporal dementia (FTD) is a neurodegenerative behavioral disorder that selectively affects the salience network, including the ventral striatum and insula. Tau mutations cause FTD, but how mutant tau impairs the salience network is unknown. Here, we address this question using a mouse model expressing the entire human tau gene with an FTD-associated mutation (V337M). Mutant, but not wild-type, human tau transgenic mice had aging-dependent repetitive and disinhibited behaviors, with synaptic deficits selectively in the ventral striatum and insula. There, mutant tau depleted PSD-95, resulting in smaller postsynaptic densities and impaired synaptic localization of NMDA receptors (NMDARs). In the ventral striatum, decreased NMDAR-mediated transmission reduced striatal neuron firing. Pharmacologically enhancing NMDAR function with the NMDAR co-agonist cycloserine reversed electrophysiological and behavioral deficits. These results indicate that NMDAR hypofunction critically contributes to FTD-associated behavioral and electrophysiological alterations and that this process can be therapeutically targeted by a Food and Drug Administration–approved drug.
Keywords: cycloserine, frontotemporal dementia, NMDA receptor, postsynaptic density, salience network, tau
Introduction
Frontotemporal dementia (FTD) is a devastating disease that causes a variety of behavioral symptoms with brain region–specific atrophy and dysfunction. Common presenting symptoms include repetitive behavior, personality changes, social withdrawal, apathy, and disinhibition (Mendez et al., 1997; Neary et al., 1998; Mendez and Perryman, 2002; Mendez and Shapira, 2008; Rascovsky et al., 2011). These symptoms are associated with region-specific atrophy and reduced connectivity in the salience network (Rabinovici et al., 2007; Seeley et al., 2009; Seeley, 2010; Zhou et al., 2010). The salience network includes the anatomically connected insular cortex and ventral striatum. Insular cortex is among the first regions to atrophy in FTD (Seeley, 2010), and its dysfunction correlates with clinical progression (Zhou et al., 2010). Ventral striatum dysfunction correlates with severity of repetitive behaviors (Josephs et al., 2008; Halabi et al., 2013).
The neurobiology underlying region-specific dysfunction and behavioral symptoms in FTD is poorly understood. As a result, disease-modifying treatments are lacking. Mutations in the MAPT gene (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998), which encodes tau protein, are a known cause of FTD. Here we used a mouse model that expresses the entire human tau gene with the FTD-associated V337M mutation (hT-337M mice; McMillan et al., 2008) and found several aging-dependent behavioral abnormalities, including repetitive behavior that, like in FTD patients, was linked to ventral striatum dysfunction. Therefore, this mouse model provides a unique opportunity to determine how tau causes salience network dysfunction and related behavioral abnormalities and to identify potential treatment targets.
To address these questions, we studied behavior, physiology, biochemistry, and neuropathology in several cohorts of hT-337M mice at different ages. Our findings illuminate the molecular mechanisms of mutant tau-mediated behavioral abnormalities and point to a potential and feasible treatment target for FTD.
Materials and Methods
Transgenic mice
All experimental procedures were performed under a protocol approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham. Two transgenic mouse lines expressing the human tau gene (MAPT) were studied (McMillan et al., 2008): hT-PAC-337M, expressing human tau with the FTD-associated V337M mutation (referred to here as hT-337M), and hT-PAC-N, a control line expressing wild-type human tau (referred to here as hT-WT). These lines express the 61D6 P1 artificial chromosome (PAC) with the entire human tau gene, including 5′ and 3′ regulatory regions, so expression is driven by the tau promoter. The mouse tau gene was not disrupted. Hemizygous transgenic mice were compared with their nontransgenic (Ntg) siblings. Both transgenic mouse lines were on a congenic C57BL/6J background. Male and female mice were used for all experiments except when specified. Mice were kept on a 12 h light/dark cycle with ad libitum access to food (NIH-31 Open Formula Diet, catalog #7917; Harlan) and water.
Behavior
All behavior experiments were conducted during the light phase of the diurnal cycle and analyzed with the researchers blind to genotype.
Grooming.
Each mouse was placed in a clean, empty cage (15.25 × 7.8 × 9.5 inches) to habituate for 10 min. Next, each mouse was moved into a StereoScan behavior monitoring chamber (Med Associates) and was video-recorded for 10 min. Grooming behavior was defined as any rubbing, scratching, or licking of any part of the face or body and was scored by an observer blind to genotype.
Nest building.
Mice were moved from group housing to single housing at the start and for the duration of each 72 h nesting experiment. A new nestlet (2 × 2 inches) was weighed and placed in the center of each cage. Then, each mouse's cage was observed after 24, 48, and 72 h. For each observation, the cage was photographed, and the remaining, unused nestlet was weighed. Each nest was scored based on a modified six-point scale (Deacon, 2012): 0, the nestlet is almost entirely untouched (>99% intact); 1, the nestlet is mostly intact (>90% intact); 2, the nestlet is partially torn up (50–90% intact); 3, the nestlet is mostly torn up (50–10% intact); 4, the nestlet is almost entirely torn up (<10% intact) with nest walls higher than the mouse body on <50% of its circumference; and 5, the nestlet is almost entirely torn up (<10% intact) with nest walls higher than the mouse body on >50% of its circumference. For scores 1–3, 0.5 was added if there was an identifiable nest site.
Elevated plus maze.
Mice were habituated to the testing room with lights and white noise on for at least 1 h. The elevated plus maze (EPM; Med Associates) was 1 m high with 2-inch-wide arms. Each mouse was placed in the center of the maze to begin the 10-min trial. The mouse's activity throughout the maze was measured by video-tracking software (CleverSys).
Cycloserine treatment.
To measure the effect on grooming, mice were intraperitoneally injected with saline on one day and then with 20 mg/kg cycloserine (Blundell et al., 2010; C6880; Sigma-Aldrich) on the next day. After either saline or cycloserine injection, mice were returned to their home cage for 20 min. Then they were put in a clean, empty cage for 10 min to habituate. Finally, they were video recorded for 10 min. Grooming behavior was scored by an observer blind to genotype and treatment.
For the effect on the EPM and nesting behaviors, mice were semirandomly assigned to receive 7017 NIH-31 Open Formula diet (Harlan Laboratories) either with or without cycloserine (20 mg · kg−1 · d−1). Groups were balanced for mouse age and weight. Mice were weighed and food consumption was monitored for several weeks. We determined that the average mouse weighed 26 g and consumed 2.7 g/d food. A formulation with 192.6 mg of cycloserine per kg of diet thus yielded an effective dose of 20 mg · kg−1 · d−1. Mice were monitored daily for changes in weight and food consumption that might indicate an aversion to the diet, and none were noted. After 2 weeks on the diet, the mice were tested in the EPM and then for nesting behaviors.
Biochemistry
Brain region subdissection.
For subregion-specific biochemistry, we dissected several brain regions from mouse hemibrains. To get hemibrains, mice were killed with 100 mg/kg Fatal-Plus (Vortech Pharmaceuticals) and decapitated, and hemibrains were flash frozen on dry ice. Frozen hemibrains were chopped into 450 μm sections with a tissue chopper, and the sections were transferred to a Petri dish filled with ice-cold PBS plus protease and phosphatase inhibitors. Using a dissecting microscope, the sections were subdissected into various brain regions based on anatomical landmarks (Franklin and Paxinos, 2008). Each brain region was then stored at −80°C until use.
Synaptosomal preparations.
Synaptosomes were prepared from a modified and scaled-down biochemical fractionation protocol (Hallett et al., 2008). Frozen, subdissected brain regions were defrosted in ice-cold TEVP buffer (pH 7.4, in mM: 10 Tris base, 5 NaF, 1 Na3VO4, 1 EDTA, and 1 EGTA) with 320 mm sucrose and protease and phosphatase inhibitors. Tissue was homogenized for 15 s, a small volume of whole homogenate (WH) was saved, and then the remaining homogenate was centrifuged at 800 × g for 10 min at 4°C. The supernatant (S1) was removed, and the pellet (P1, nuclei and large debris) was resuspended in TEVP buffer with protease and phosphatase inhibitors and saved. A small volume of S1 was saved, and then the remaining S1 was centrifuged at 9200 × g for 15 min at 4°C. The supernatant (S2) was removed and saved. The pellet (P2, crude synaptosomal membranes) was resuspended in TEVP buffer with 35.6 mm sucrose and protease and phosphatase inhibitors. A small volume of P2 was saved, and then the remaining P2 was centrifuged at 25,000 × g for 20 min at 4°C. The supernatant (LS1) was removed and saved. The pellet (LP1, synaptosomal fraction) was resuspended in TEVP buffer with protease and phosphatase inhibitors and saved. All pellet fractions were sonicated with a probe sonicator for 10 s. A protein assay was performed on all fractions, which were then stored at −80°C. Only the WH and LP1 fractions were used for protein level comparison by Western blot to determine the synaptic localization of glutamate receptors.
Western blotting.
Samples were run on NuPage 4–12% gels with NuPage MOPS running buffer for 2 h at 110 V. For WHs, 12 μg of protein was loaded into each well. For synaptosomes (LP1), 1.5 μg of protein was loaded into each well. Gels were transferred to PVDF membranes in NuPage transfer buffer for 1.5 h at 30 V. Membrane blots were blocked in Odyssey blocking buffer for 1 h at room temperature (RT). Primary antibodies were incubated overnight at 4°C or, in some cases, for 1 h at RT. Primary antibodies were from Millipore and used at 1:1000 unless specified otherwise. Primary antibodies used were as follows: GluR1 (catalog #AB1504), GluR2 (L21/32 clone, 1:500; Antibodies Inc.), NR1 (N308/48 clone, 1:500; Antibodies Inc.), NR2A (catalog #AB10531), NR2B (N59/36 clone; NeuroMabs), postsynaptic density-95 (PSD-95; 7E3-1B8 clone), tau (catalog #A0024; Dako), tau5 (catalog #MAB361), ptau–12E8 (Peter Seubert, Elan Pharmaceuticals, South San Francisco, CA), and ptau–CP9 (Peter Davies, Feinstein Institute for Medical Research, Manhasset, NY). IRDye secondary antibodies (1:20,000 + 0.5% SDS; LI-COR) were incubated for 2 h at RT. Blots were visualized with Odyssey Infrared Imager (LI-COR).
Histology
Immunohistochemistry.
Mice were killed with 100 mg/kg Fatal-Plus and decapitated, and then hemibrains were drop fixed in 4% paraformaldehyde. Hemibrains were serially sectioned at 30 μm with a sliding microtome to produce free-floating sections. Sections to be stained were washed, quenched, and blocked as in the protocol of Palop et al. (2011). Sections were incubated overnight at 4°C in primary antibody [12E8, 1:500; CP-13, 1:100; neuronal nuclei (NeuN), 1:1000], then for 2 h at RT in biotinylated secondary antibody (1:500), and finally for 5 min at RT in DAB reaction mixture. Stained sections were mounted onto gelatin-coated slides and then coverslipped using Cytoseal mounting media. Microscopy was performed on a MicroBrightField system (MBF Bioscience).
Stereological neuron counts.
Brain sections were stained for neurons with anti-NeuN monoclonal antibody (MAB377; Millipore Bioscience Research Reagents) and counted by unbiased stereological methods using Stereo Investigator (MBF Bioscience). The optical fractionator probe was used to count three sections (each 30 μm thick) with a section interval of 10 between +1.70 and +0.74 mm from bregma. For estimates of neuron populations in the ventral striatum, a minimum of 30 sampling sites per section were sampled on a grid size of 233 × 206 μm and a counting frame size of 30 × 30 μm. For the insular cortex, at least 60 sites were sampled on a grid size of 200 × 150 μm and a counting frame size of 30 × 30 μm. For all tissue, the mounted thickness was measured at each sampling site, and a dissector height of 10 μm was used. The estimate of the neuron population was finally calculated using the weighted section thickness.
Morphological analysis from Golgi-stained tissue.
Male mouse brains were processed for morphological analysis using the FD Rapid GolgiStain Kit (FD NeuroTechnologies) according to the protocol of the manufacturer. Mice were killed with 100 mg/kg Fatal-Plus and decapitated, and then the brains were removed. Hemibrains were submerged in impregnation solution for 9 d and then transferred to a cryoprotectant for an additional 7 d. Next, brains were cut into 200 μm sections with a cryostat (Leica Microsystems) and mounted on gelatin-coated slides. After mounting, the slides were rinsed in ddH2O and incubated in developing solution. After development, the sections were rehydrated with increasing concentrations of ethanol and cleared with xylene. Last, slides were coverslipped using EUKITT mounting medium (Electron Microscopy Sciences).
Microscopy was performed on a MicroBrightField system (MBF Bioscience). For dendritic spines, image stacks were taken of 10–40 μm segments of second-order dendrites on medium spiny neurons at 100× (oil-immersion objective). All image stacks were manually traced using Neurolucida and analyzed using Neurolucida Explorer.
Electron microscopy.
Male mice were anesthetized with 120 mg/kg Nembutal intraperitoneally and transcardially perfused with ice-cold saline, followed by a 4% paraformaldehyde and 0.1% glutaraldehyde mixture in 0.1 m phosphate buffer. The brains were removed and fixed overnight at 4°C. Brains were coronally sliced (50 μm thick) on a vibratome and stored in phosphate buffer, pH 7.4. The sections were put in 1% osmium tetroxide for 1 h and then dehydrated in increasing concentrations of ethanol. Sections were placed in 1% uranyl acetate in 70% ethanol for 1 h, further dehydrated with ethanol, cleared with propylene oxide, and then infiltrated with Epon resin. The ventral striatum (nucleus accumbens) was blocked and cut into ultrathin sections (90 nm thick) on a Leica EM UC6 ultramicrotome (Leica Microsystems). Ultrathin sections were mounted on a copper slot grid and imaged on a Hitachi TEM model H-7650-II equipped with a Hamamatsu Orca HR digital camera.
For each mouse, a photographer blind to genotype took 30 random, non-overlapping images at 25,000×, providing a total field of view of 1228.26 μm2 (40.942 μm2/picture) per mouse. Greater than 130 synapses were identified per mouse. Synapses were classified into categories of either asymmetric axospinous or asymmetric axodendritic. Images were imported into NIH ImageJ, in which PSD areas were traced and measured.
Electrophysiology
Field and whole-cell patch-clamp recordings were performed on acute striatal slices from 14- to 18-month-old mice. Mice were anesthetized in isoflurane and decapitated. The brains were rapidly removed and placed in ice-cold cutting solution (in mm: 75 sucrose, 85.0 NaCl, 2.5 KCl, 4.0 MgSO4, 0.5 CaCl2, 1.25 NaH2PO4, 25.0 NaHCO3, and 25.0 glucose) saturated with 95% O2 and 5% CO2. Coronal slices (400 μm thick) were prepared on a Vibratome, and acute striatal slices were stored in RT aCSF solution (in mm: 119.0 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1.0 NaH2PO4, 26.0 NaHCO3, and 11.0 glucose) saturated with 95% O2 and 5% CO2. Slices were allowed to equilibrate for at least 1 h before being transferred to the recording chambers.
Field recordings.
Field recordings were performed in an interface chamber (Fine Science Tools) with constant perfusion of 27–30°C aCSF. Pictrotoxin at 100 μm (Sigma-Aldrich) was included to block inhibitory transmission. Extracellular stimuli were applied with bipolar tungsten electrodes dorsal to recording electrodes in the ventral striatum (nucleus accumbens core) and dorsomedial to recording electrodes in dorsal striatum. Glass recording electrodes (1–3 MΩ) were filled with aCSF. Input–output curves were generated by applying a range of stimuli from 0.5 to 25 V in 0.5 V increments with an ISO-STIM 01D stimulus isolator (npi electronic).
NMDA receptor (NMDAR)-dependent field recordings were performed in a custom submersion chamber (made at the University of Alabama at Birmingham machine shop) with constant perfusion of 27–30°C aCSF. Picrotoxin at 100 μm was included in all recording solutions to block inhibitory transmission. A bipolar nichrome electrode was used for extracellular stimulation. Electrode placement was as described above, and extracellular stimuli were applied in 10 s intervals. After establishing a 10 min baseline, slices were perfused with 0 mm Mg2+ aCSF for 40 min to remove Mg2+ from solution and then with 0 mm Mg2+ aCSF with 100 μm GYKI 52466 [4-(8-methyl-9H-1,3-dioxolo [4,5-h][2,3]benzodiazepin-5-yl)-benzenamine hydrochloride; Abcam] for 10 min to block AMPA receptors (AMPARs). Input–output curves were generated by applying a range of stimuli from 40 to 320 μA in 40 μA increments with a constant-current isolated stimulator (Digitimer). The area of NMDAR field EPSPs (fEPSPs) was measured in a 20 ms timeframe starting at ∼6–8 ms after stimulation. Five traces per stimulus intensity were averaged for each slice. NMDAR fEPSPs were completely blocked by adding 100 μm APV (Tocris Bioscience).
Whole-cell recordings.
Blind whole-cell patch-clamp recordings were performed in a custom submersion chamber with constant perfusion of 27–30°C aCSF. Patch pipettes (2.5–5.0 MΩ) were pulled from filamented borosilicate glass (Sutter Instruments) and filled with one of two types of pipette solution containing 0.4% biocytin (Sigma-Aldrich) for later identification of neuron type. For voltage-clamp recordings, the pipette solution contained the following (in mm): 120.0 Cs-methansulfonate, 0.6 EGTA, 2.8 NaCl, 5.0 MgCl2, 2.0 ATP, 0.3 GTP, 20 HEPES, and 5.0 QX-314. For current-clamp recordings, the pipette solution contained the following (in mm): 120.0 K-gluconate, 0.6 EGTA, 5.0 MgCl2, 2.0 ATP, 0.3 GTP, and 20 HEPES. Whole-cell recordings were collected with a Digidata-1440A digitizer and pClamp 10.2 software (Molecular Devices), low-pass filtered at 2 kHz with an Axopatch-200B amplifier, and digitally sampled at 100 Hz.
EPSCs were recorded in the presence of 100 μm picrotoxin to block inhibitory transmission. Spontaneous (sEPSC) and miniature (mEPSC) EPSCs were recorded at a holding voltage of −70 mV. For mEPSCs, action potentials were blocked with 1 μm tetrodotoxin (TTX; Abcam). sEPSCs and mEPSCs were analyzed using Clampfit 10.3 (Molecular Devices). Synaptic events >4 pA were detected after creating a unique template for each neuron. The first 200 events for each cell were analyzed.
Evoked EPSCs were generated in 10 s intervals with a bipolar nichrome electrode dorsal to the patch pipette in the ventral striatum. To determine NMDAR/AMPAR ratios, neurons were recorded at a holding voltage of −20 mV to relieve the Mg2+ plug of NMDARs. After a 5 min baseline, 100 μm APV (Sigma-Aldrich) was bath applied to block NMDARs. The remaining current was AMPAR mediated and could be completely blocked with 10 μm NBQX. Using Clampfit 10.3, the NMDAR-mediated component was generated by subtracting the AMPAR-mediated components from the baseline currents. The peak amplitude of the NMDAR-mediated component was divided by the peak amplitude of the AMPAR-mediated component to produce the NMDAR/AMPAR ratio. Series and input resistance were monitored throughout each experiment, and neurons were excluded if either of these changed by >15%.
To record action potentials, neurons were injected with current until they rested at −70 mV. This provided a common starting point for the current injection steps. The neurons were injected with current steps from −100 to 300 pA in 25 pA increments to elicit action potentials. The first action potential elicited from these injection steps was analyzed for threshold, amplitude, maximum rate of rise, and half-width (Kaphzan et al., 2013). Threshold potential was calculated as the voltage at which the first derivative of the action potential trace reached 30 mV/ms. Action potential amplitude, maximum rate of rise, and half-width were analyzed using Clampfit 10.3. Neurons were excluded if no action potentials were fired by at least +150 pA of injected current. For subthreshold recordings, neurons were injected with enough current to depolarize the cell to −40 mV, and action potentials were recorded for 5 min. For drug effects, slices were incubated in 100 μm APV (Tocris Bioscience) for at least 10 min or in 20 μm cycloserine for at least 30 min, but typically for 120 min, before recordings.
Data analysis and statistical methods
Based on pilot studies in our laboratory and similar studies in the literature, we powered each study based on the magnitude of the predicted effect and its variability. Sample sizes of ∼8–12 mice/slices/samples provided at least a power of 80% to find such differences. Sample size for each experiment is indicated in the figure legends.
The data were normally distributed unless otherwise stated. Normally distributed data are represented in bar graphs and x–y plots with error bars indicating SEM. For normally distributed data, we used either a two- or one-tailed t test for direct comparisons or two-way ANOVA for multiple comparisons. A one-tailed t test was used for analyzing glutamate receptor levels because of the predicted decrease in receptor levels given the large effect on excitatory transmission already determined with electrophysiology. For non-normal data, we used nonparametric tests such as the Kruskal–Wallis test with Dunn's multiple comparisons test. We used the Kolmogorov–Smirnov test for cumulative probability plots.
Results
hT-337M mice have aging-dependent behavioral abnormalities
The transgenic mice studied here express the entire human tau gene. Wild-type (hT-WT) and mutant (hT-337M) human tau transgenes are on a P1-derived artificial chromosome and express all six isoforms of tau under control by the tau promoter (McMillan et al., 2008). The tau promoter avoids problems with heterologous promoters, and the transgenes drive only moderate overexpression of tau (2.5- to 3-fold compared with Ntg mice; Fig. 1).
Figure 1.

Similar tau overexpression in hT-337M and hT-WT mice. A, B, Representative Western blots and quantification of tau levels (tau5 antibody) in brain tissue from Ntg, hT-WT, and hT-337M mice. The magnitude of tau overexpression was similar in hT-WT and hT-337M mice. ***p < 0.001 by ANOVA with Bonferroni's multiple comparisons test.
Although hT-337M mice did not have obvious abnormalities through midlife (∼12–14 months), in older mice we noticed a striking pattern of repetitive grooming that increased as the mice aged. We quantified this behavior, first in a cross-sectional study of cohorts at different ages. With aging, hT-337M mice spent abnormally high amounts of time grooming, unlike their Ntg littermates, in which grooming time did not increase with age (Fig. 2A). We then followed a cohort longitudinally; grooming increased in hT-337M mice by 15 months (Fig. 2B). Repetitive grooming was severe enough to cause age-dependent facial lesions in hT-337M mice by ∼15–18 months, with full penetrance by 28 months (Fig. 2C,D). These lesions were self-induced, because they occurred in single-housed hT-337M mice and only in transgenic mice when transgenic and Ntg mice were group housed. Repetitive grooming and lesions did not develop in hT-WT mice (Fig. 2D,E), suggesting that the effect is driven by the FTD-associated tau mutation and not simply by tau overexpression.
Figure 2.

hT-337M mice have aging-dependent behavioral abnormalities. A, Percentage of time spent grooming during a 10 min observation in Ntg and hT-337M mice across various ages. hT-337M mice had an age-dependent increase in grooming. n = 94 Ntg, 72 hT-337M. ***p < 0.0001 comparison of best-fit curves. B, hT-337M mice followed longitudinally had an age-dependent increase in grooming by 15 months. n = 10 per genotype. *p < 0.05, ***p < 0.001, two-way repeated-measures ANOVA for genotype and age. C, Representative pictures of Ntg mice without and hT-337M mice with repetitive grooming-induced facial lesions. D, Aging-dependent appearance of facial lesions in hT-337M mice (***p < 0.0001 by log rank) but not in hT-WT mice. E, hT-WT mice did not spend more time grooming. n = 21 per genotype. F–I, EPM in 15- to 18-month-old mice. Percentage time spent in the open arms (F, H) and distance traveled (G, I) of the EPM. hT-337M mice spent more time in the open arms, whereas hT-WT mice did not, and neither line had abnormalities in ambulatory distance traveled. n = 17 Ntg, 15 hT-337M. **p < 0.01. J, Quantification of nest building by 3- to 5-month-old mice showed impaired nest building in hT-337M mice. n = 66 Ntg, 23 hT-WT, 33 hT-337M. **p < 0.01, two-way ANOVA with Bonferroni's multiple comparisons test.
These findings prompted testing the mice in a more thorough behavioral battery. Social behavior, motor coordination, and spatial learning and memory were intact (data not shown).
We used the EPM to assess potential anxiety-like behaviors. The amount of time spent in the open arms was similar between Ntg and hT-337M mice at 9–11 months of age (data not shown). However, by 15–18 months of age, hT-337M mice spent more time in the open arms compared with Ntg mice (Fig. 2F), suggesting an aging-dependent, anti-anxiety or disinhibited phenotype in hT-337M mice. There was no difference in distance traveled between genotypes, indicating similar activity levels during testing (Fig. 2G). In contrast to hT-337M mice, hT-WT mice had open-arm time in the EPM similar to Ntg mice (Fig. 2H), demonstrating that this behavioral abnormality is also mutation-specific.
Finally, we compared nest building behavior in Ntg and hT-337M mice. Nests from hT-337M mice were indistinguishable from Ntg mice at 1–2 months of age (data not shown). However, hT-337M mice developed nest building deficits by 3–5 months of age, the earliest abnormality we detected in these mice (Fig. 2J). Nest building was intact in hT-WT mice (Fig. 2J).
Decreased excitatory transmission and impaired network activity in the ventral striatum of hT-337M mice
Because repetitive behavior was the most striking behavioral abnormality in hT-337M mice and repetitive behavior in FTD is linked to ventral striatum dysfunction (Josephs et al., 2008; Halabi et al., 2013), we determined whether there were synaptic functional changes in the ventral striatum of hT-337M mice. Using extracellular fEPSP recordings in acute striatal slices prepared from 14-month-old mice, the age at which repetitive behaviors became apparent, we found a significant deficit in excitatory transmission in hT-337M mice compared with Ntg mice (Fig. 3A). Interestingly, no difference in excitatory transmission was observed in recordings from the dorsal striatum between hT-337M and Ntg mice (Fig. 3B), indicating selective deficits in the ventral striatum, and there was no difference in recordings from the ventral striatum between hT-WT and Ntg mice (Fig. 3C), indicating mutation-specific deficits, consistent with the behavior data.
Figure 3.
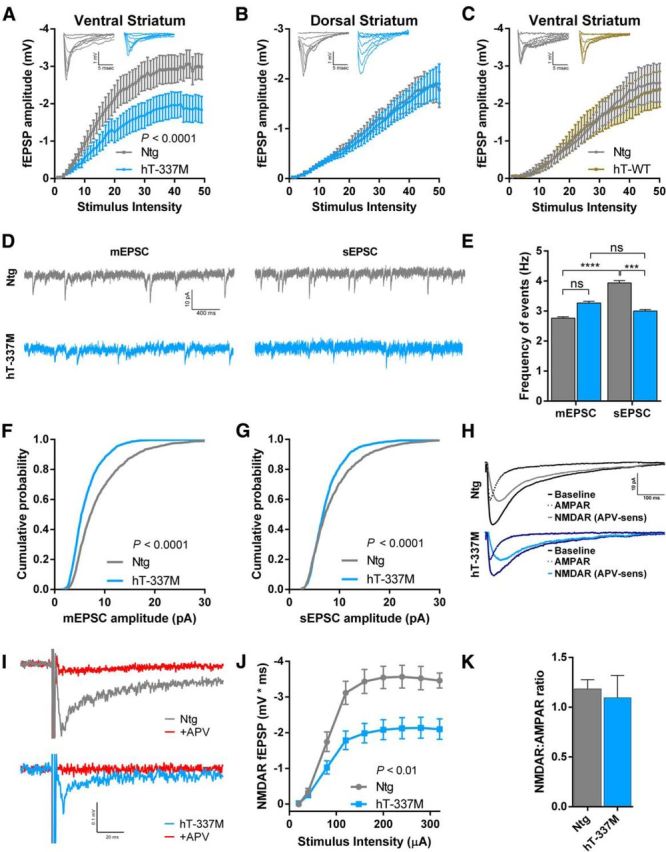
Subregion-specific depression of excitatory glutamatergic transmission in aged hT-337M mice. A, Input–output curves from the ventral striatum demonstrated depressed synaptic transmission in hT-337M mice compared with Ntg. n = 11 slices in 5 Ntg mice, 12 slices in 5 hT-337M mice. B, Input–output curves from the dorsolateral striatum were not different in hT-337M compared with Ntg mice. n = 12 slices in 5 Ntg mice, 9 slices in 5 hT-337M mice. p = 0.83. C, Input–output curves from the ventral striatum were not different in hT-WT mice compared with Ntg mice. n = 6 slices in 3 Ntg mice, 7 slices in 3 hT-WT mice. p = 0.85. Insets in A–C show representative traces. D, Representative traces of mEPSC and sEPSC events with and without TTX, respectively, from whole-cell patch-clamp recordings of ventral striatal neurons held at −70 mV. E, Frequency of spontaneous and miniature events from ventral striatal neurons. sEPSC frequency was reduced in hT-337M mice and, unlike in Ntg mice, was not significantly higher than mEPSC frequency. ***p < 0.001, ****p < 0.0001 by Kruskal–Wallis test with Dunn's multiple comparisons test. F, G, Cumulative probability plots showed decreased mEPSC (F) and sEPSC (G) amplitudes in ventral striatal neurons from aged hT-337M mice. n = 8 neurons in 3 mice per genotype; Kolmogorov–Smirnov tests. H, Representative whole-cell traces of evoked transmission onto ventral striatal neurons before and after application of APV to isolate the AMPAR and NMDAR (APV-sensitive) components. I, Representative traces of NMDAR-dependent fEPSPs in the ventral striatum at maximum stimulus intensity (320 μA) showing complete block after APV application (red). J, Input–output curves demonstrated decreased NMDAR-dependent transmission in ventral striatal neurons from hT-337M mice. n = 10 slices in 3 Ntg mice, 11 slices in 3 hT-337M mice. K, Quantification of the NMDAR/AMPAR ratio from H showed no change in the ventral striatum.
Because fEPSPs in the ventral striatum are mediated predominantly by AMPARs (Zhang and Warren, 2008), these findings suggested a possible deficit in AMPAR function. To better understand these changes, we recorded mEPSCs and sEPSCs from medium spiny neurons in the ventral striatum using whole-cell voltage clamp (Fig. 3D). sEPSCs include action potential–dependent and –independent events, whereas mEPSCs, which occur in the presence of TTX, include only action potential–independent events. As an index of excitatory synaptic input to the ventral striatum, we compared the frequency of sEPSCs and mEPSCs. mEPSC frequency was unchanged in hT-337M mice compared with Ntg mice (Fig. 3D,E), suggesting intact presynaptic function and normal numbers of synapses in hT-337M mice. In Ntg mice, sEPSC events were significantly more frequent than mEPSC events (Fig. 3D,E), consistent with occurrence of spontaneous presynaptic action potentials driving some synaptic activity in these slices. However, in hT-337M mice, there was no difference between the frequency of sEPSC and mEPSC events (Fig. 3D,E), suggesting less action potential–dependent input onto medium spiny neurons.
We also examined the amplitudes of mEPSCs and sEPSCs, which generally reflect postsynaptic function. Both mEPSC and sEPSC amplitudes were decreased in hT-337M mice (Fig. 3D,F,G). This is consistent with the reduced fEPSP amplitudes, indicating decreased postsynaptic AMPAR function.
Next we determined whether NMDAR function was also impaired, in addition to AMPAR impairments, in the ventral striatum of hT-337M mice. To address this question, we first calculated NMDAR/AMPAR ratios of evoked EPSCs. NMDAR/AMPAR ratios did not differ between Ntg and hT-337M mice (Fig. 3H,K). Given the large decrease in AMPAR function (Fig. 3A,F,G), the lack of any change in the NMDAR/AMPAR ratio suggests a parallel decrease in NMDAR function.
To directly examine NMDAR function in the ventral striatum, we measured NMDAR-dependent fEPSPs by recording from slices in aCSF with GYKI 52466 (an AMPAR antagonist) and without magnesium. NMDAR-dependent fEPSPs were decreased in hT-337M mice (Fig. 3I,J). These data demonstrate NMDAR hypofunction in the ventral striatum of hT-337M mice.
Abnormal biochemical composition of excitatory synapses in insulostriatal regions of hT-337M mice
Because hT-337M mice had impaired excitatory transmission in the ventral striatum, we determined whether there were biochemical changes at excitatory synapses in the ventral striatum of hT-337M mice. We compared the levels of AMPAR and NMDAR subunits in the ventral striatum of 14-month-old mice, because AMPARs and NMDARs are the primary receptors for excitatory transmission and the electrophysiological recordings indicated that both were decreased in the ventral striatum of hT-337M mice. Although AMPAR and NMDAR subunit levels in WHs of the ventral striatum were unchanged (Fig. 4A,B), levels in synaptosomal preparations were significantly decreased (Fig. 4C). These data suggest impaired synaptic localization of glutamate receptors in the ventral striatum. In the dorsal striatum, both WH and synaptosomal levels of AMPARs or NMDARs were unchanged (Fig. 4D–F), consistent with the fEPSP results (Fig. 3B) and again indicating selective vulnerability in the ventral striatum.
Figure 4.

Impaired synaptic localization of AMPARs and NMDARs in the ventral but not dorsal striatum of aged hT-337M mice. A, Representative Western blots from whole homogenates (left) and synaptosomes (right) from the ventral striatum of 14-month-old mice. B, Normalized quantification of whole homogenates for AMPAR (GluA1, GluA2) and NMDAR (GluN1, GluN2A, GluN2B) subunits demonstrated no differences between Ntg and hT-337M mice. n = 11 Ntg, 13 hT-337M. p > 0.05 by one-tailed t test. C, Normalized quantification of synaptosomes showed decreased AMPARs and NMDARs. n = 10 per genotype. *p < 0.05, #p = 0.0780. D, Representative Western blots from whole homogenates (left) and synaptosomes (right) from the dorsal striatum of 14-month-old mice. E, F, Normalized quantification of whole homogenates (n = 13 per genotype) (E) and synaptosomes (n = 11 Ntg, 10 hT-337M) (F) of the dorsal striatum showed no differences between genotypes.
Next we determined whether synaptic localization of glutamate receptors was also altered in the insular cortex, which is one of the primary cortical inputs to the ventral striatum and is also selectively vulnerable in FTD patients. As in the ventral striatum, AMPAR and NMDAR subunit levels in the insular cortex were unchanged in WHs (Fig. 5A) but decreased in synaptosomes in hT-337M mice (Fig. 5B). However, in the motor cortex, a nearby and cytoarchitecturally similar region to the insular cortex, synaptosomal levels of AMPAR and NMDAR subunits were unchanged (Fig. 5C). Together, these data suggest impaired synaptic localization of glutamate receptors in selectively vulnerable brain regions of hT-337M mice.
Figure 5.
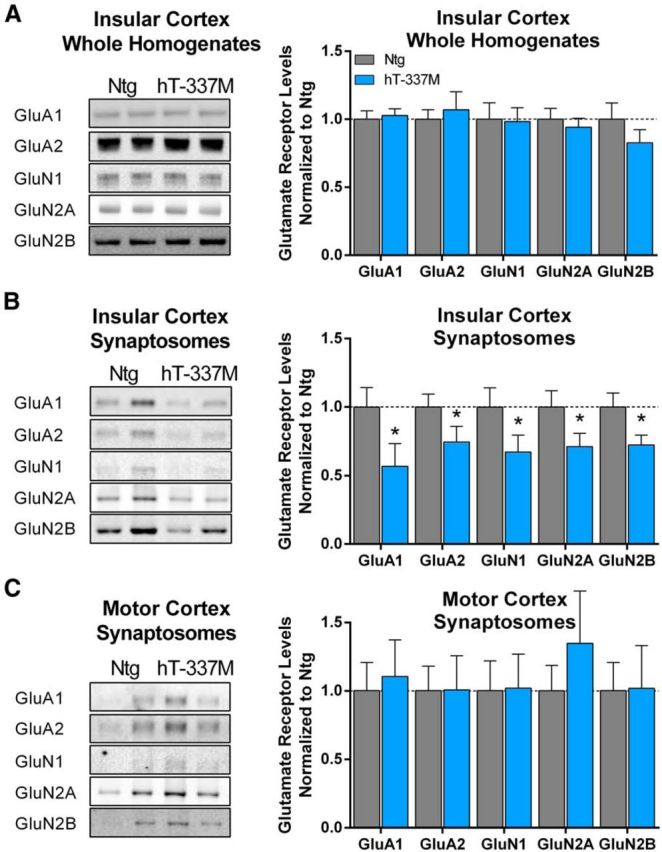
Impaired synaptic localization of AMPARs and NMDARs in the insular cortex. A, Representative Western blots and quantification of whole homogenates from insular cortex of 14-month-old mice. There were no differences in AMPAR (GluA1 or GluA2) or NMDAR (GluN1, GluN2A, GluN2B) subunits between genotypes. n = 10–13 per genotype. B, Representative blots and quantification of synaptosomes from the insular cortex of 14-month-old mice. There were significant decreases of all AMPAR and NMDAR subunits examined. n = 10–11 per genotype. *p < 0.05. C, Representative blots from synaptosomes of the motor cortex from 14-month-old mice. Normalized quantification of synaptosomes for AMPAR (GluA1, GluA2) and NMDAR (GluN1, GluN2A, GluN2B) subunits showed no differences between genotypes. n = 9–11 per genotype.
Smaller postsynaptic densities at excitatory synapses in hT-337M mice
Next we examined the neuroanatomic basis of these synaptic changes. Decreased excitatory transmission in hT-337M mice could have been attributable to fewer neurons. We determined whether there was neuron loss in the ventral striatum or the insula in hT-337M mice by comparing unbiased stereological neuronal counts in 22-month-old Ntg and hT-337M mice. There were no differences in neuron counts in either the ventral striatum or insular cortex (Fig. 6A).
Figure 6.

Dendritic spine PSDs are smaller in the ventral striatum of aged hT-337M mice. A, Estimated population of neurons using stereological counts and weighted section thickness from NeuN-stained sections showed no change in either region of interest in hT-337M mice. Three 30-μm-thick sections per mouse, n = 9–11 per genotype. B, Representative photomicrograph of Golgi-stained medium spiny neuron dendrites in ventral striatum of 24-month-old Ntg and hT-337M mice. Scale bar, 5 μm. C, Quantification of dendritic spine density showed no change in hT-337M mice. n = 17 Ntg, 19 hT-337M dendrites from 3 mice per genotype. D, Spine head diameter was quantified during tracing and reconstruction of dendrites and showed no change in hT-337M mice. E, Representative electron microscopy images showing asymmetric axospinous (AS) synapses. sp, Dendritic spine head. PSD denoted by arrow. Presynaptic vesicles denoted by arrowheads. Scale bar, 0.50 μm. F, There was no difference in the total number of AS or asymmetric axodendritic (AD) synapses per 1000 μm2 in the ventral striatum of hT-337M mice. n = 3 mice per genotype. G, Total PSD area per 1000 μm2 was decreased at asymmetric axospinous synapses in ventral striatum of hT-337M mice. n = 3 mice per genotype. **p < 0.01, two-way repeated-measures ANOVA for genotype and synapse type with Bonferroni's multiple comparisons test. H–J, Cumulative probability plots showed decreased PSD area (H), thickness (I), and length (J) in the ventral striatum of hT-337M mice (Kolmogorov–Smirnov tests). H–J, Insets show histograms with Gaussian fit curves.
Fewer or smaller dendritic spines, which are the site of excitatory synapses, could also have contributed to the synaptic deficits we observed. To examine the density, shape, and diameter of dendritic spines, we compared Golgi-stained medium spiny neurons in the ventral striatum of 24-month-old mice. Even at this late age, dendritic spine density was similar in Ntg and hT-337M mice (Fig. 6B,C). Furthermore, there was no difference in the proportions of different type/shapes of spines (data not shown), and the average spine head diameter was unchanged in hT-337M mice (Fig. 6D).
Given the normal density and size of dendritic spines and recent evidence that tau can localize to the PSD of excitatory synapses (Hoover et al., 2010; Ittner et al., 2010; Chabrier et al., 2012; Mondragón-Rodríguez et al., 2012; Kimura et al., 2014), we determined whether the PSD of dendritic spines was affected. We counted synapses and measured the area, length, and width of PSDs in ventral striatum of 14-month-old mice by electron microscopy (Fig. 6E). Consistent with the mEPSC recordings and Golgi data, there was no change in the number of excitatory synapses, either onto dendritic spines or shafts (Fig. 6F). However, the total PSD area measured at excitatory synapses onto spines was decreased in hT-337M mice (Fig. 6E,G). Comparing the distribution of individual PSD sizes at dendritic spines confirmed that hT-337M mice had decreased PSD area, length, and width (Fig. 6H–J).
The decreased PSD size in hT-337M mice predicted reduced levels of PSD-95, the major scaffolding protein for glutamate receptors at the PSD. Consistent with this hypothesis, PSD-95 levels were decreased in the ventral striatum of 22-month-old hT-337M mice (Fig. 7A,B). PSD-95 levels were also decreased in the insular cortex (Fig. 7D,E). Consistent with the regional selective vulnerability seen in the electrophysiological and biochemical studies, changes in PSD-95 levels were not observed in the dorsal striatum (Fig. 7A,B) or motor cortex (Fig. 7D,E). Thus, these changes in the biochemical composition of excitatory synapses are selective to regions within the salience network. In hT-WT mice, PSD-95 levels were not decreased even in the ventral striatum and insular cortex at 22 months (Fig. 7C,F), consistent with the behavioral and electrophysiological data and indicating that these synaptic changes are not simply attributable to tau overexpression.
Figure 7.

Region-specific depletion of PSD-95 with aging in hT-337M mice. A, B, Representative Western blots and quantification of PSD-95 levels in the ventral (VS) and dorsal (DS) striatum of hT-337M mice. B, In the ventral striatum of hT-337M mice, PSD-95 levels were decreased as early as 14 months (n = 11 Ntg, 13 hT-337M) and remained decreased at 22 months in hT-337M mice (n = 15 Ntg, 14 hT-337M). PSD-95 levels were not different in the dorsal striatum even by 22 months. n = 12 Ntg, 10 hT-337M. C, Representative blot and quantification of PSD-95 levels in the ventral striatum of 22-month-old Ntg and hT-WT mice showing no differences. n = 9 Ntg, 10 hT-WT. D, E, Representative Western blots and quantification of PSD-95 levels in the insular cortex and motor cortex of hT-337M mice. E, In the insular cortex of hT-337M mice, PSD-95 levels were decreased as early as 2 months (n = 10 Ntg, 8 hT-337M) and remained decreased at 22 months in hT-337M mice (n = 15 Ntg, 14 hT-337M). PSD-95 levels were not different in the motor cortex even by 22 months (n = 15 Ntg, 14 hT-337M). F, Representative blot and quantification of PSD-95 levels in the insular cortex of 22-month-old Ntg and hT-WT mice showing no differences (n = 9 Ntg, 10 hT-WT). *p < 0.05, **p < 0.01, ***p < 0.001, #p = 0.0938.
We next determined the age dependence of the decreased PSD-95 levels in the ventral striatum and insular cortex (Fig. 7B,E). PSD-95 levels were normal at weaning in both regions. In 2-month-old mice (before behavioral abnormalities were present), PSD-95 was decreased in the insular cortex but was normal in the ventral striatum, indicating earlier onset of abnormalities in the insula. In 14-month-old mice (the age of onset of repetitive behaviors), PSD-95 levels were decreased in ventral striatum of hT-337M mice. This age dependence and regional specificity is interesting given that the insular cortex is one of the first regions with reduced connectivity in FTD and ventral striatum dysfunction correlates with repetitive behaviors in FTD (Josephs et al., 2008; Zhou et al., 2010; Halabi et al., 2013).
Regional pattern of phosphorylated tau predicts selective vulnerability in hT-337M mice
We next examined whether regional differences in tau phosphorylation could explain the selective vulnerability of the ventral striatum and insular cortex. In vitro, tau phosphorylation is necessary and sufficient for tau to mislocalize into dendritic spines and impair the synaptic localization of glutamate receptors (Hoover et al., 2010). We looked at phosphorylation residues on tau that have been linked to somatodendritic mislocalization (Zempel et al., 2010). Levels of tau phosphorylated at serine-262/serine-356 (12E8) were much higher in ventral striatum than in dorsal striatum, in both Ntg and hT-337M mice at 14 months of age (Fig. 8A). The higher levels of phosphorylated tau in the ventral striatum of hT-337M mice was not simply attributable to more human tau expression in ventral striatum, because human tau levels there were actually lower in the ventral striatum than in the dorsal striatum (Fig. 8B). Together, this suggests a predilection for increased phosphorylated tau in the ventral striatum.
Figure 8.

Increased phosphorylated tau in salience network regions. A, Representative Western blots and quantification of phosphorylated tau using the 12E8 antibody. Levels of phosphorylated tau were much higher in the ventral striatum than in the dorsal striatum and were increased in both regions of 14-month-old hT-337M mice. n = 11–13 per genotype per region. **p < 0.01, ***p < 0.001 by two-way ANOVA. VS, Ventral striatum; DS, dorsal striatum. B, Representative blots and quantification of human tau expression in the ventral and dorsal striatum of hT-337M mice shows that the higher levels of phosphorylated tau in the ventral striatum was not attributable to higher transgene expression there, because levels of total human tau were actually somewhat higher in the dorsal striatum. n = 6 for ventral striatum, 5 for dorsal striatum. *p < 0.05 by two-tailed t test. C, Immunoreactivity of phosphorylated tau using the 12E8 antibody. There was intense staining in the insular cortex in Ntg and hT-337M mice (arrows). D, E, Immunoreactivity of phosphorylated tau using the CP-13 antibody in the insular cortex (D) and ventral striatum (E) of Ntg, hT-337M, and hT-WT mice. D, There was an age-dependent increase of phosphorylated tau in the insula in both Ntg and hT-337M mice. In hT-337M mice, phospho-tau pathology appeared in the insula as early as 2 months (D) and in the ventral striatum by 14 months (E). Scale bars, 10 μm.
To better visualize the regional pattern of phosphorylated tau, we used immunohistochemistry. We compared tissue from 2-, 14-, and 22-month-old Ntg and hT-337M mice using multiple phosphorylated tau antibodies. Most strikingly, levels of phosphorylated tau (12E8, serine-262/serine-356) were markedly higher in the insula than in other cortical areas in both Ntg and hT-337M mice (Fig. 8C). Additionally, phosphorylated tau (CP-13, serine-202) was present in the somatodendritic compartment in the insula in both Ntg and hT-337M mice, with more intense pretangle-like pathology in hT-337M mice (Fig. 8D). Interestingly, this phospho-tau pathology appeared as early as 2 months of age in the insula of hT-337M mice (Fig. 8D), the same age as the decreased PSD-95 levels in the insula (Fig. 7D,E). Phospho-tau pathology did not develop in the insula of hT-WT mice even as late as 28 months of age (Fig. 8D).
In the ventral striatum, somatodendritic mislocalization of phosphorylated tau was detectable at 2 months of age in hT-337M mice (Fig. 8E), albeit much less abundant and less intense than in the insula (Fig. 8D). In the ventral striatum of both Ntg and hT-337M mice, staining intensity increased with age. hT-337M mice developed phospho-tau pathology in the ventral striatum by 14 months of age (Fig. 8E), the same age as the decreased PSD-95 (Fig. 7A,B), with more granular staining at 22 months. Phospho-tau pathology did not develop in the ventral striatum of hT-WT mice even as late as 28 months of age (Fig. 8E).
Interestingly, phosphorylated tau staining was much less intense in the motor cortex than in the insular cortex across all ages in Ntg mice (Fig. 8C). Similarly, phosphorylated tau staining was less intense in the dorsal striatum than ventral striatum in Ntg mice, consistent with the biochemical data (Fig. 8A). This regional pattern was repeated in hT-337M mice. Together, these data suggest a natural, age-dependent mapping of phosphorylated tau staining onto salience network regions in Ntg mice, which may predispose these regions to the abnormalities seen with mutant tau in hT-337M mice.
Increasing NMDAR function restores striatal neuron firing
Next, we determined how the decreased excitatory synaptic input affected action potential output of ventral striatal neurons in hT-337M mice. NMDARs regulate the action potential firing of ventral striatal neurons during depolarized up states (Kerr and Plenz, 2002; Carter and Sabatini, 2004; Wolf et al., 2005). Given the NMDAR hypofunction in hT-337M mice, we determined whether action potential output from ventral striatal neurons was decreased using whole-cell current-clamp recordings from ventral striatal neurons. Importantly, independent of synaptic input, the intrinsic membrane properties and properties of single action potentials did not differ between Ntg and hT-337M neurons (Table 1).
Table 1.
Intrinsic membrane properties and ability to fire action potentials were unchanged in the ventral striatum of hT-337M mice
| Ntg | hT-337M | p | |
|---|---|---|---|
| Input resistance (MΩ) | 132.4 (4.7) | 132.3 (6.5) | 0.9876 |
| Resting membrane potential (mV) | −80.9 (0.7) | −80.0 (0.6) | 0.3797 |
| AP threshold (mV) | −27.4 (1.3) | −27.2 (2.2) | 0.9390 |
| AP amplitude (mV) | 67.8 (3.3) | 66.0 (5.8) | 0.7732 |
| AP maximum rate of rise (mV/ms) | 105.8 (7.6) | 100.3 (10.7) | 0.6800 |
| AP half-width (ms) | 1.24 (0.06) | 1.25 (0.06) | 0.8945 |
Whole-cell current-clamp recordings demonstrated that neurons in the ventral striatum of hT-337M mice have normal membrane excitability and are able to fire normal action potentials in response to current injections (n = 23 neurons in 9 Ntg mice, 11 neurons in 5 hT-337M mice; p value from two-tailed t test for each measurement) AP, Action potential.
To determine the effect of NMDAR hypofunction on action potential firing, we depolarized ventral striatal neurons to a subthreshold potential that acts as an artificial up state whereby excitatory inputs can further depolarize the cell to evoke action potentials. hT-337M neurons had dramatically fewer action potentials in this artificial up state (Fig. 9A,D), consistent with NMDAR hypofunction. In fact, action potential firing in hT-337M neurons was similar to that in Ntg neurons treated with APV, an NMDAR antagonist (Fig. 9B,D). These results indicate that NMDAR hypofunction in hT-337M mice severely reduces the output of ventral striatal neurons.
Figure 9.

Increasing NMDAR function in hT-337M mice restores action potential output of ventral striatal neurons. A–C, Representative traces of action potential firing in ventral striatal neurons held at a subthreshold potential (artificial up state) in untreated slices (aCSF, A) and slices treated with either APV (B) or cycloserine (C). D, Quantification of the number of action potentials fired from cells held at a subthreshold potential. hT-337M neurons fired fewer action potentials than Ntg in aCSF. Ntg neurons in APV fired fewer action potentials than Ntg neurons in aCSF.hT-337M neurons in aCSF and APV fired action potentials similarly. hT-337M neurons in cycloserine (Cyclo) fired more action potentials than hT-337M neurons in aCSF. aCSF neurons, n = 9 Ntg, 9 hT-337M; APV neurons, n = 7 Ntg, 6 hT-337M; cycloserine neurons, n = 3 Ntg, 7 hT-337M; mice for all recordings, n = 7 Ntg, 6 hT-337M, aged 14–18 months. **p < 0.01, ***p < 0.001 by two-way ANOVA with Tukey's multiple comparisons test.
We next asked whether boosting NMDAR function using the NMDAR co-agonist cycloserine (Watson et al., 1990; Dravid et al., 2010) would restore action potential firing in hT-337M mice. Cycloserine completely reversed the impairment of striatal neuron firing (Fig. 9C,D). Action potential frequency in slices from hT-337M mice treated with cycloserine did not differ from untreated Ntg slices. This finding suggests that NMDAR hypofunction in hT-337M mice reduces the calcium influx needed to generate action potentials and indicates that this deficit can be rescued by enhancing NMDAR function.
Increasing NMDAR function alleviates behavioral abnormalities in hT-337M mice
To determine whether targeting NMDAR hypofunction could also alleviate the behavioral abnormalities in hT-337M mice, we increased NMDAR function by systemic administration of cycloserine. First, we injected cycloserine acutely (20 mg/kg, i.p.) and measured its effect on grooming in 14- to 20-month-old mice. As expected, grooming time in saline-injected hT-337M mice was much higher than normal (Fig. 10A). Consistent with its beneficial effects on striatal neuron firing, cycloserine significantly decreased grooming in hT-337M mice (Fig. 10A).
Figure 10.
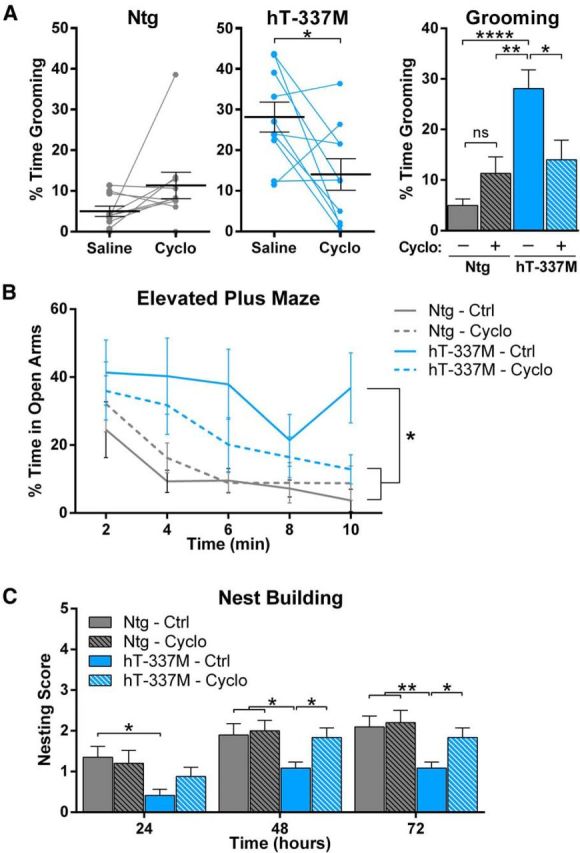
Increasing NMDAR function alleviates behavioral abnormalities in hT-337M mice. A, Percentage of time grooming in Ntg (left) and hT-337M (middle) mice after treatment with either saline or cycloserine (Cyclo). Cycloserine significantly decreased grooming in hT-337M mice (each line is one mouse). *p < 0.05 by paired t test. Right, Grouped data. n = 10 mice per genotype receiving each treatment. *p < 0.05, **p < 0.01, ****p < 0.0001 with two-way ANOVA and Tukey's multiple comparisons test. B, C, Two week treatment with either control or cycloserine diet in 18-month-old animals. n = 10 Ntg mice for each treatment, n = 12 hT-337M for each treatment. *p < 0.05, **p < 0.01 with two-way ANOVA and Newman–Keuls multiple comparisons test. B, hT-337M treated with cycloserine spent significantly less time in the open arms of EPM than untreated hT-337M mice. C, hT-337M treated with cycloserine built significantly better nests than untreated hT-337M mice.
We also examined the effect of cycloserine on the other behavioral abnormalities in hT-337M mice. Because the EPM is sensitive to the stressful nature of acute intraperitoneal injections and nesting is assessed over several days, we fed the mice cycloserine (20 mg · kg−1 · d−1 in chow) for several weeks; these studies were conducted in 18-month-old mice. In the EPM, hT-337M mice fed normal chow spent more time in the open arms, as expected, whereas hT-337M mice fed cycloserine chow spent less time in the open arms (Fig. 10B). Consistent with the nesting deficits seen in younger (5-month-old) mice (Fig. 2J), hT-337M mice fed normal chow had impaired nest building (Fig. 10C). However, even >12 months after the onset of nesting deficits, cycloserine improved nest building in hT-337M mice such that it was indistinguishable from controls (Fig. 10C). These results indicate that targeting NMDAR hypofunction with cycloserine can reverse behavioral deficits in hT-337M mice.
Discussion
In this study, we sought to understand how a tau mutation causes salience network dysfunction and behavioral abnormalities, and how these deficits might be corrected. We capitalized on the hT-337M mouse model, which we found had a relevant pattern of selective vulnerability in salience network regions (ventral striatum and insula) with related, aging-dependent behavioral abnormalities. Our results indicate that salience network regions have higher levels of phosphorylated tau. In these more susceptible regions, mutant tau depletes PSD-95 and reduces the size of the PSD, which impairs synaptic localization of glutamate receptors and results in AMPAR and NMDAR hypofunction, selectively in salience network regions in hT-337M mice. NMDAR hypofunction appears to be an attractive therapeutic target, because boosting NMDAR function with cycloserine was sufficient to reverse electrophysiological and behavioral abnormalities in hT-337M mice.
hT-337M mice have several interesting features related to selective vulnerability of specific brain regions. First, both electrophysiological (Figs. 3, 10) and biochemical (Figs. 4, 5, 7) deficits were found in the insular cortex and ventral striatum but not in nearby and cytoarchitecturally similar regions (motor cortex and dorsal striatum, respectively). The insular cortex and ventral striatum are nodes in the salience network, which is selectively vulnerable in patients with FTD (Rabinovici et al., 2007; Seeley et al., 2009; Seeley, 2010; Zhou et al., 2010), whereas motor cortex and dorsal striatum are not. Thus, abnormalities in hT-337M mice seem to target anatomically and functionally connected networks, just as in human neurodegenerative disease. Second, the insular cortex may be the most vulnerable region in FTD (Seeley, 2010) and was also the first area affected in hT-337M mice (Fig. 7E). This is intriguing because the insular cortex in mice does not contain von Economo neurons, which are an especially vulnerable population in the human insular cortex (Kim et al., 2012). One potential explanation for selective vulnerability of the insula is the natural accumulation of phosphorylated tau in both Ntg mice (Fig. 8) and in healthy aging humans (Price and Morris, 1999), which might prime the region and may contribute to its vulnerability in FTD. In future studies, it will be interesting to determine what makes these susceptible regions either more efficient at tau phosphorylation or less efficient at dephosphorylation. The insula was the first region in hT-337M mice with phospho-tau pathology, followed by appearance in the ventral striatum (Fig. 8). In future studies, it will be interesting to determine whether this network vulnerability in hT-337M mice is linked to recent findings that tau can spread trans-synaptically to anatomically linked regions (de Calignon et al., 2012; Liu et al., 2012). Third, this anatomic predilection for the insular cortex and ventral striatum most likely explains why the major behavioral abnormality in hT-337M mice is repetitive behavior, which is a core feature of FTD (Rascovsky et al., 2011) and has been linked to ventral striatum in several studies of FTD patients (Josephs et al., 2008; Halabi et al., 2013). The fact that tau expression in hT-337M mice is driven by the human tau promoter makes this pattern of selective vulnerability particularly interesting, because it is not an artifact of the heterologous promoters often used in transgenic mice. In combination, these features make this mouse model advantageous for studying pathophysiological mechanisms that are characteristic of FTD, particularly aspects of FTD related to ventral striatal dysfunction and repetitive behavior, a core feature of the disease (Roberson, 2012).
Our finding that mutant tau impedes the proper synaptic localization of glutamate receptors is consistent with a previous study showing similar effects in cultured neurons expressing a different tau mutation (P301L; Hoover et al., 2010), emphasizing the importance of impaired glutamate receptor localization as a mechanism of tauopathy. Here, we identified a mechanism for this effect and showed that it occurs selectively in salience network regions (Figs. 6–8), results in functional synaptic deficits in the ventral striatal circuit (Figs. 3, 9), and can be effectively targeted to improve behavioral symptoms (Fig. 10). Our data suggest that impaired synaptic localization of glutamate receptors in hT-337M mice may be attributable to mutant tau-mediated depletion of PSD-95 (Fig. 7), which would provide less synaptic scaffolding for glutamate receptors at each synapse. In both tau-expressing cultured neurons (Hoover et al., 2010) and hT-337M mice (Fig. 6A), abnormalities in synaptic glutamate receptor localization occur independent of neuron loss. The absence of neuron loss in this model enabled us to study early aspects of how tau produces synaptic and network dysfunction in specific brain regions, without confounds in the biochemical and electrophysiological analyses from loss of neurons. Interestingly, in both cultured neurons (Hoover et al., 2010) and hT-337M mice (Fig. 8), impaired synaptic localization of glutamate receptors was associated with increased phosphorylated tau. The region and age dependence of tau phosphorylation corresponded with the region- and age-dependent decrease of PSD-95 in the insula and ventral striatum. Identifying which factors make these regions more prone to phosphorylated tau is an important goal for future studies.
Our studies suggest that NMDAR hypofunction may be a desirable treatment target for FTD. We found biochemical and electrophysiological evidence of NMDAR hypofunction, and the NMDAR co-agonist cycloserine protected against both electrophysiological and behavioral abnormalities. Despite the decrease in synaptic glutamate receptors, treatment with cycloserine restored action potential output from ventral striatal neurons (Fig. 9) and reversed behavioral deficits in hT-337M mice (Fig. 10). The behavioral improvement included correction of nesting deficits that had been present for >1 year; thus, even long after onset, these abnormalities remained reversible.
Interestingly, although the NMDAR antagonist memantine is approved for treating patients with Alzheimer's disease, a recent study found that memantine worsened deficits in patients with FTD (Boxer et al., 2013), consistent with the idea that NMDAR hypofunction contributes to FTD pathophysiology. Cycloserine is Food and Drug Administration-approved to treat patients with tuberculosis but can have neurological side effects at the high doses needed to act as an inhibitor of bacterial cell-wall synthesis. At lower doses, cycloserine has been used clinically to treat patients with social anxiety disorder (Hofmann et al., 2006) and tobacco dependence (Santa Ana et al., 2009) and is well tolerated at these low, neuro-active doses (Posey et al., 2004; Wilhelm et al., 2008; Otto et al., 2010; Cain et al., 2014). We hypothesize that increasing NMDAR function may benefit FTD patients. With additional preclinical validation, this hypothesis could be tested in clinical trials using cycloserine, which represents a drug repurposing opportunity for FTD.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants R01NS075487, F30AG046088, P01AG017586, T32GM008361, P30NS057098, and P30NS047466, the Consortium for Frontotemporal Dementia Research, and the Stephen Bechtel Fund. We thank James Black and Miriam Roberson for colony maintenance and tissue processing, Joy Roche for processing EM tissue, and Tarek Midani for scoring grooming videos.
The authors declare no competing financial interests.
References
- Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, Bolliger MF, Südhof TC, Powell CM. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J Neurosci. 2010;30:2115–2129. doi: 10.1523/JNEUROSCI.4517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer AL, Knopman DS, Kaufer DI, Grossman M, Onyike C, Graf-Radford N, Mendez M, Kerwin D, Lerner A, Wu CK, Koestler M, Shapira J, Sullivan K, Klepac K, Lipowski K, Ullah J, Fields S, Kramer JH, Merrilees J, Neuhaus J, Mesulam MM, Miller BL. Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2013;12:149–156. doi: 10.1016/S1474-4422(12)70320-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain CK, McCue M, Bello I, Creedon T, Tang DI, Laska E, Goff DC. D-Cycloserine augmentation of cognitive remediation in schizophrenia. Schizophr Res. 2014;153:177–183. doi: 10.1016/j.schres.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Chabrier MA, Blurton-Jones M, Agazaryan AA, Nerhus JL, Martinez-Coria H, LaFerla FM. Soluble Aβ promotes wild-type tau pathology in vivo. J Neurosci. 2012;32:17345–17350. doi: 10.1523/JNEUROSCI.0172-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon R. Assessing burrowing, nest construction, and hoarding in mice. J Vis Exp. 2012;(59):e2607. doi: 10.3791/2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravid SM, Burger PB, Prakash A, Geballe MT, Yadav R, Le P, Vellano K, Snyder JP, Traynelis SF. Structural determinants of d-cycloserine efficacy at the NR1/NR2C NMDA receptors. J Neurosci. 2010;30:2741–2754. doi: 10.1523/JNEUROSCI.5390-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. 3rd Ed. San Diego: Academic; 2008. [Google Scholar]
- Halabi C, Halabi A, Dean DL, Wang PN, Boxer AL, Trojanowski JQ, Dearmond SJ, Miller BL, Kramer JH, Seeley WW. Patterns of striatal degeneration in frontotemporal dementia. Alzheimer Dis Assoc Disord. 2013;27:74–83. doi: 10.1097/WAD.0b013e31824a7df4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, Collins TL, Standaert DG, Dunah AW. Biochemical fractionation of brain tissue for studies of receptor distribution and trafficking. Curr Protoc Neurosci Chapter. 2008;1 doi: 10.1002/0471142301.ns0116s42. Unit 1.16. [DOI] [PubMed] [Google Scholar]
- Hofmann SG, Pollack MH, Otto MW. Augmentation treatment of psychotherapy for anxiety disorders with D-cycloserine. CNS Drug Rev. 2006;12:208–217. doi: 10.1111/j.1527-3458.2006.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Jack CR., Jr Anatomic correlates of stereotypies in frontotemporal lobar degeneration. Neurobiol Aging. 2008;29:1859–1863. doi: 10.1016/j.neurobiolaging.2007.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphzan H, Buffington SA, Ramaraj AB, Lingrel JB, Rasband MN, Santini E, Klann E. Genetic reduction of the α1 subunit of Na/K-ATPase corrects multiple hippocampal phenotypes in Angelman syndrome. Cell Rep. 2013;4:405–412. doi: 10.1016/j.celrep.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JN, Plenz D. Dendritic calcium encodes striatal neuron output during up-states. J Neurosci. 2002;22:1499–1512. doi: 10.1523/JNEUROSCI.22-05-01499.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Sidhu M, Gaus SE, Huang EJ, Hof PR, Miller BL, DeArmond SJ, Seeley WW. Selective frontoinsular von Economo neuron and fork cell loss in early behavioral variant frontotemporal dementia. Cereb Cortex. 2012;22:251–259. doi: 10.1093/cercor/bhr004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Whitcomb DJ, Jo J, Regan P, Piers T, Heo S, Brown C, Hashikawa T, Murayama M, Seok H, Sotiropoulos I, Kim E, Collingridge GL, Takashima A, Cho K. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos Trans R Soc Lond B. 2014;369:20130144. doi: 10.1098/rstb.2013.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan P, Korvatska E, Poorkaj P, Evstafjeva Z, Robinson L, Greenup L, Leverenz J, Schellenberg GD, D'Souza I. Tau isoform regulation is region- and cell-specific in mouse brain. J Comp Neurol. 2008;511:788–803. doi: 10.1002/cne.21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez MF, Perryman KM. Neuropsychiatric features of frontotemporal dementia: evaluation of consensus criteria and review. J Neuropsychiatry Clin Neurosci. 2002;14:424–429. doi: 10.1176/appi.neuropsych.14.4.424. [DOI] [PubMed] [Google Scholar]
- Mendez MF, Shapira JS. The spectrum of recurrent thoughts and behaviors in frontotemporal dementia. CNS Spectr. 2008;13:202–208. doi: 10.1017/s1092852900028443. [DOI] [PubMed] [Google Scholar]
- Mendez MF, Perryman KM, Miller BL, Swartz JR, Cummings JL. Compulsive behaviors as presenting symptoms of frontotemporal dementia. J Geriatr Psychiatry Neurol. 1997;10:154–157. doi: 10.1177/089198879701000405. [DOI] [PubMed] [Google Scholar]
- Mondragón-Rodríguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, Boehm J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-d-aspartate receptor-dependent tau phosphorylation. J Biol Chem. 2012;287:32040–32053. doi: 10.1074/jbc.M112.401240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/WNL.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Otto MW, Tolin DF, Simon NM, Pearlson GD, Basden S, Meunier SA, Hofmann SG, Eisenmenger K, Krystal JH, Pollack MH. Efficacy of D-cycloserine for enhancing response to cognitive-behavior therapy for panic disorder. Biol Psychiatry. 2010;67:365–370. doi: 10.1016/j.biopsych.2009.07.036. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L, Roberson ED. Quantifying biomarkers of cognitive dysfunction and neuronal network hyperexcitability in mouse models of Alzheimer's disease: depletion of calcium-dependent proteins and inhibitory hippocampal remodeling. Methods Mol Biol. 2011;670:245–262. doi: 10.1007/978-1-60761-744-0_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- Posey DJ, Kem DL, Swiezy NB, Sweeten TL, Wiegand RE, McDougle CJ. A pilot study of D-cycloserine in subjects with autistic disorder. Am J Psychiatry. 2004;161:2115–2117. doi: 10.1176/appi.ajp.161.11.2115. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Rabinovici GD, Seeley WW, Kim EJ, Gorno-Tempini ML, Rascovsky K, Pagliaro TA, Allison SC, Halabi C, Kramer JH, Johnson JK, Weiner MW, Forman MS, Trojanowski JQ, Dearmond SJ, Miller BL, Rosen HJ. Distinct MRI atrophy patterns in autopsy-proven Alzheimer's disease and frontotemporal lobar degeneration. Am J Alzheimers Dis Other Demen. 2007;22:474–488. doi: 10.1177/1533317507308779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gorno-Tempini ML, Rosen H, Prioleau-Latham CE, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED. Mouse models of frontotemporal dementia. Ann Neurol. 2012;72:837–849. doi: 10.1002/ana.23722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santa Ana EJ, Rounsaville BJ, Frankforter TL, Nich C, Babuscio T, Poling J, Gonsai K, Hill KP, Carroll KM. D-Cycloserine attenuates reactivity to smoking cues in nicotine dependent smokers: a pilot investigation. Drug Alcohol Depend. 2009;104:220–227. doi: 10.1016/j.drugalcdep.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW. Anterior insula degeneration in frontotemporal dementia. Brain Struct Funct. 2010;214:465–475. doi: 10.1007/s00429-010-0263-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62:42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson GB, Bolanowski MA, Baganoff MP, Deppeler CL, Lanthorn TH. D-cycloserine acts as a partial agonist at the glycine modulatory site of the NMDA receptor expressed in Xenopus oocytes. Brain Res. 1990;510:158–160. doi: 10.1016/0006-8993(90)90745-W. [DOI] [PubMed] [Google Scholar]
- Wilhelm S, Buhlmann U, Tolin DF, Meunier SA, Pearlson GD, Reese HE, Cannistraro P, Jenike MA, Rauch SL. Augmentation of behavior therapy with D-cycloserine for obsessive-compulsive disorder. Am J Psychiatry. 2008;165:335–341. doi: 10.1176/appi.ajp.2007.07050776. quiz 409. [DOI] [PubMed] [Google Scholar]
- Wolf JA, Moyer JT, Lazarewicz MT, Contreras D, Benoit-Marand M, O'Donnell P, Finkel LH. NMDA/AMPA ratio impacts state transitions and entrainment to oscillations in a computational model of the nucleus accumbens medium spiny projection neuron. J Neurosci. 2005;25:9080–9095. doi: 10.1523/JNEUROSCI.2220-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. Aβ oligomers cause localized Ca2+ elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Warren RA. Postnatal development of excitatory postsynaptic currents in nucleus accumbens medium spiny neurons. Neuroscience. 2008;154:1440–1449. doi: 10.1016/j.neuroscience.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Zhou J, Greicius MD, Gennatas ED, Growdon ME, Jang JY, Rabinovici GD, Kramer JH, Weiner M, Miller BL, Seeley WW. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain. 2010;133:1352–1367. doi: 10.1093/brain/awq075. [DOI] [PMC free article] [PubMed] [Google Scholar]