

Abstract
Cobalt chloride has been used as a hypoxia mimetic because it stabilizes hypoxia inducible factor-1α (HIF1-α) and activates gene transcription through a hypoxia responsive element (HRE). However, differences between hypoxia and hypoxia mimetic cobalt chloride in gene regulation remain elusive. Expression of ferritin, the major iron storage protein, is regulated at the transcriptional and posttranscriptional levels through DNA and RNA regulatory elements. Here we demonstrate that hypoxia and cobalt chloride regulate ferritin heavy chain (ferritin H) expression by two distinct mechanisms. Both hypoxia and cobalt chloride increased HIF1-α but a putative HRE in the human ferritin H gene was not activated. Instead, cobalt chloride but not hypoxia activated ferritin H transcription through an antioxidant responsive element (ARE), to which Nrf2 was recruited. Intriguingly, cobalt chloride downregulated ferritin H protein expression while upregulated other ARE-regulated antioxidant genes in K562 cells. Further characterization demonstrated that cobalt chloride increased interaction between iron regulatory proteins (IRP1 and IRP2) and iron responsive element (IRE) in the 5′UTR of ferritin H mRNA, resulting in translational block of the accumulated ferritin H mRNA. In contrast, hypoxia had marginal effect on ferritin H transcription but increased its translation through decreased IRP1-IRE interaction. These results suggest that hypoxia and hypoxia mimetic cobalt chloride employ distinct regulatory mechanisms through the interplay between DNA and mRNA elements at the transcriptional and post-transcriptional levels.
Keywords: hypoxia, cobalt, hypoxia inducible factor (HIF), iron regulatory protein (IRP), Nrf2, ferritin
1. INTRODUCTION
All organisms from bacteria to human beings need oxygen for ATP generation and survival. Maintaining oxygen homeostasis is crucial; failure to regulate oxygen levels leading to hypoxia results in insufficient generation of ATP or paradoxical production of reactive oxygen species (ROS) (1), which damage proteins, lipids, and DNA (2,3). Therefore, cells evolved various mechanisms to maintain oxygen homeostasis, in which hypoxia-inducible factors (HIFs) serve as the master regulator of oxygen homeostasis (3). HIFs sense oxygen change and mediate developmental and physiological pathways that either deliver O2 to cells or allow cells to survive under O2 deprivation (4). The HIF family contains 3 members, HIF-1, 2, and 3; each HIF member consists of α- and β-subunits (4,5). The β-subunit is constitutively expressed but the α-subunit is regulated in an oxygen-dependent manner (4,5). In normoxia, the α-subunit expression is low through a degradation system that recognizes hydroxyl prolines located in the oxygen-dependent degradation domain (ODD). Hypoxic conditions inhibit prolyl hydroxylation that allows the α-subunit to be stabilized and associated with the β-subunit through a basic helix-loop-helix (bHLH) domain and regulate HIF-targeted genes via the hypoxia-responsive element (HRE) (4,5). Not restricted to the pathways involved in regulation of oxygen homeostasis, HIFs are multifaceted transcription factors that can mediate a diverse array of physiological pathways such as cell proliferation, differentiation, survival, angiogenesis, energy metabolism, and iron homeostasis (6).
Iron is a vital element for the maintenance of a plethora of physiological functions, serving as a cofactor of various enzymes involved in metabolisms and hemoglobin synthesis for oxygen transfer (7,8). In conjunction with tightly regulated iron transport system (9,10), ferritin plays a crucial role as a major intracellular iron reservoir in storing excess iron and releasing it when needed (11). Ferritin is a cage-like protein composed of 24 subunits of heavy (ferritin H) and light (ferritin L) chains. Ferritin H harbors ferroxidase activity to oxidize Fe2+ to Fe3+ for storage, while ferritin L supports the overall ferritin structure (11,12). Ferritin gene expression is regulated at the transcriptional and post-transcriptional levels. Iron is the major post-transcriptional regulator of ferritin; iron inhibits the interaction between iron regulatory proteins (IRPs) and the iron-responsive element (IRE) in the 5′-untranslated region (UTR) of both ferritin H and L mRNAs that facilitates recruitment of the translational machinery for increased ferritin protein synthesis. Conversely, iron chelators increase the IRP-IRE interaction and suppress ferritin translation (13,14). Therefore, high iron upregulates and low iron downregulates ferritin translation. At the transcriptional level, oxidative stressors such as arsenic (15), rotenone (16), hemin (17–19) as well as electrophiles including t-BHQ (20) and resveratrol (21) activate ferritin gene transcription. NFE2-related factor 2 (Nrf2) and JunD along with p300/CBP are recruited to an antioxidant-responsive element (ARE) located 4.5kb and 4.1kb upstream of the human and mouse ferritin H transcription start sites (22–25), respectively.
Cobalt chloride has been used as a hypoxia mimetic because it stabilizes HIF-1α through inhibition of prolyl hydroxylase and activates hypoxia-regulated genes through the HRE (26). However, similarities and differences between hypoxia and cobalt chloride in gene regulation remain elusive, particularly other genetic elements specifically regulated by cobalt chloride or hypoxia have not been fully elucidated. In this study we investigated this issue in K562 human erythroleukemic cells by elucidating coordinated regulation of the human ferritin H gene through three genetic DNA and RNA regulatory elements (the ARE and HRE in the ferritin H 5′-promoter region, and the IRE in the 5′-UTR of ferritin H mRNA). Our results demonstrated that 1) cobalt chloride, but not hypoxia, activates transcription of the human ferritin H gene through the Nrf2-ARE pathway, 2) both cobalt chloride and hypoxia increased HIF-1α but a putative HRE in the ferritin H gene was not activated, 3) cobalt chloride decreased but hypoxia increased ferritin H protein expression via their opposite effects on IRP and IRE interaction. These results suggest that cobalt chloride is a hypoxia mimetic in regard to HIF-1α activation; however, cobalt chloride and hypoxia regulate gene expression through distinct DNA and mRNA regulatory elements.
2. MATERIALS AND METHODS
2.1. Cell culture and chemical reagents
K562 human erythroleukemia cells (American Type Culture Collection) were cultured in RPMI1640 medium (Mediatech) supplemented with 25 mM Hepes, 0.3g/liter L-glutamine, and 10% FBS (Mediatech), and incubated at 37°C in a humidified 5% carbon dioxide atmosphere. For hypoxic stimulation, the culture plates were incubated in a modular incubator chamber (Billups-Rothenburg, Del Mar, CA) and pumped with a gas mixture containing 1% O2, 5% CO2, and 94% nitrogen (Airgas National Welders) for the indicated time. Cobalt chloride (CoCl2·6H2O) was purchased from Mallinckrodt Chemicals and dissolved in water.
2.2. Plasmids and Antibodies
The luciferase reporter plasmids containing ARE(+)/HRE(+), ARE(−)/HRE(+), ARE(−)/HRE(−), and TATA box of the human ferritin H enhancer/promoter regions fused to a firefly luciferase gene were described previously (22). 5′-UTR sequence containing the IRE of ferritin H mRNA was described (27), and the IRE-luciferase plasmid was constructed by cloning the IRE into ferritin H TATA box luciferase reporter. Antibodies utilized in Western blotting were purchased from the following companies: anti-HIF-1α (NB100-479), Novus Biologicals; anti-lamin B (Ab-1), Oncogene; anti-ferritin H (SC-25617), anti-NQO1 (SC-32793), anti-thioredoxin (SC-20146), anti-thioredoxin reductase 1 (SC-28321), anti-glutamate-cysteine ligase, modifier subunit (SC-22754), anti-IRP2 (SC-33682), anti-Nrf2 (SC-13032), all from Santa Cruz Biotechnology; anti-IRP1 (ab126595), Abcam; and anti-β-actin (A5441), Sigma-Aldrich.
2.3. Cell extracts and Western blotting
Whole cell lysates (WCL) were prepared by washing cells with ice-cold Phosphate Buffered Saline (PBS) and lysed with cell lysis Buffer (150mM NaCl, 10mM Na2HPO4, 1% Triton-X, 0.5% Deoxycholic Acid, 0.1% SDS, 0.2% Sodium Azide; pH 7.4). Nuclear extracts were prepared using a nuclear extraction kit (Active Motif) according to manufacturer’s instructions. Cell lysates were electrophoretically separated on sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) followed by protein transfer to polyvinylidene difluoride (PVDF) membranes (Thermo Fisher Scientific). After overnight incubation with primary antibodies, membranes were washed and incubated at room temperature with a horseradish peroxidase conjugated secondary antibody (Calbiochem) and detected by enhanced chemiluminescence (ECL) with HyGlo reagent (Denville Scientific) and visualized by exposure to x-ray film (Denville Scientific).
2.4. DNA transfection and luciferase reporter assay
K562 cells were transiently transfected with human ferritin H luciferase reporter plasmids by electroporation (Bio-Rad Gene Pulser X-Cell). After overnight incubation, cells were treated with either cobalt chloride or hypoxia and subjected to luciferase assays with GloMax 20/20 luminometer (Promega).
2.5. Small interfering RNA (siRNA) transfection
K562 cells electroporated with 100 pmol of non-targeting siRNA (siControl; D-001210-01) or siNrf2 (J-003755-11) from Thermo Fisher Scientific in 100 μl of siRNA transfection medium (sc-36868; Santa Cruz Biotechnology) were incubated at room temperature for 10 min, transferred into cell culture dishes with growth medium, and incubated at 37°C for 48 h. Cells were then treated with cobalt chloride and analyzed expression of mRNAs in RT-qPCR.
2.6. Real-time quantitative PCR (RT-qPCR)
RNA was reverse transcribed to cDNA by following manufacture’s instruction for iScript cDNA Synthesis Kit (Bio-Rad). cDNA was used for RT-qPCR with iQ SYBR Green Supermix (Bio-Rad) and primer pairs for detecting the following target genes (ferritin H: forward, 5′-ACTGATGAAGCTGCAGAACC-3′ and reverse, 5′-GTCACCCAATTCTTTGATGG-3′; transferrin receptor-1 (TfR1): forward, 5′-CGCTGGTCAGTTCGTGATTA-3′ and reverse, 5′-GCATTCCCGAAATCTGTTGT-3′; NADPH quinone oxidoreductase-1 (NQO1): forward, 5′-GAAGAGCACTGATCGTACTGGC-3′ and reverse, 5′-GGATACTGAAAGTTCGCAGGG-3′; thioredoxin: forward, 5′-GTGAAGCAGATCGAGAGCAAG-3′ and reverse, 5′-CGTGGCTGAGAAGTCAACTACTA-3′; thioredoxin reductase-1: forward, 5′-ATATGGCAAGAAGGTGATGGTCC-3′ and reverse, 5′-GGGCTTGTCCTAACAAAGCTG-3′; glutamate-cysteine ligase, modifier subunit (GCLM): forward, 5′-TGTCTTGGAATGCACTGTATCTC-3′ and reverse, 5′-CCCAGTAAGGCTGTAAATGCTC-3′; beta-2-microglobulin (B2M): forward, 5′-TGCTGTCTCCATGTTTGATGTATCT-3′ and reverse, 5′-TCTCTGCTCCCCACCTCTAAGT-3′) in the CFX96 PCR system (Bio-Rad) with the thermal cycling conditions of 95°C for 3 min followed by 40 cycles of 95°C for 10 s and 60°C for 45 s. Expression of mRNA level of each gene was normalized by β-microglobulin (ΔCt) to calculate the change of Ct in treated samples relative to untreated control or siControl (ΔΔCt) and further converted to linear values (2−ΔΔCt) for fold induction.
2.7. Chromatin Immunoprecipitation (ChIP) assay
Our ChIP assay procedure was slightly modified from the published method (28). K562 chromatin was cross-linked with 1.42% formaldehyde, quenched with 125 mM glycine, and sonicated to shear chromatin DNA as described previously (29). Cell lysates incubated with either control IgG or an Nrf2 antibody were rocked at 4°C overnight and then incubated with protein A agarose/ssDNA bead slurry (16-157; Millipore) for 45 min. After washing and decrosslinking, the genomic DNA was used for RT-qPCR with iQ SYBR Green Supermix (Bio-Rad) and primer pairs for a ferritin H ARE or non-ARE region (ferritin H ARE: forward, 5′-TCCAGGTCTTATGACTGCTC-3′ and reverse, 5′-GATGAGAGAAGAGCCAAGC-3′; ferritin H non-ARE: forward, 5′-TAGTCCCTGGCTGCTGATCT-3′ and reverse, 5′-AGTGCCTCCTCATGGAAATG-3′). The DNA in each ChIP sample was normalized by input genomic DNA (ΔCt) and defined as the change of Ct in treated samples relative to control (ΔΔCt). Exponential ΔΔCt values were converted to linear values (2−ΔΔCt) for fold induction.
2.8. Streptavidin-agarose pull-down assay
100 μg whole cell lysates were incubated with 5 μg biotinylated human ferritin H IRE RNA probe and RNaseOUT (10777-019 Invitrogen) in mid RIPA buffer (25 mM Tris-HCl (pH 7.4), 1% NP-40, 0.5% Deoxycholic acid, 15 mM NaCl) containing protease inhibitors (539131, Calbiochem) for 3 hrs at room temperature. Reactions were mixed with streptavidin-agarose beads (15942-050, Invitrogen) for additional 1 hr at room temperature. After incubation, the protein-RNA-bead complex was washed with mid RIPA buffer three times and then detected by Western blot with the anti-IRP1 or anti-IRP2 antibody. The RNA sequence of the 5′-biotinylated ferritin H IRE probe was 5′-GGU UUC CUG CUU CAA CAG UGC UUG GAC GGA AC-3′.
2.9. Statistics
One-way ANOVA with LSD post hoc test was conducted for all statistical analyses using IBM SPSS statistics ver.19 software. P < 0.05 indicates significantly different.
3. RESULTS
3.1. Cobalt chloride, but not hypoxia, activates transcription of the human ferritin H gene
Cobalt chloride can elicit cellular responses similar to hypoxia by activation of HIF-1α (30,31). According to the consensus core sequence of the hypoxia response element (HRE) characterized as A(or G)CGTG (32), a putative HRE is located in 4.3kb upstream of the human ferritin H transcription start site (GACGTGCT, Fig. 1A). To test whether both hypoxia and cobalt chloride induce ferritin H mRNA through the putative HRE, K562 cells were treated with 100 μM cobalt chloride or hypoxia for 1 to 24 hours, and ferritin H mRNA was measured by RT-qPCR. We observed that ferritin H mRNA was induced in a time-dependent manner by treatment with cobalt chloride (Fig. 1B top) but not hypoxia (Fig. 1C top). In this experiment, both hypoxia and cobalt chloride induced transferrin receptor-1 (TfR1) mRNA (Fig. 1B and 1C bottom) that is a HIF-1α-regulated iron transporter (33,34). To validate the hypoxia-mimetic effect of cobalt chloride, nuclear accumulation of HIF-1α was measured by Western blotting. Indeed, both hypoxia and cobalt chloride induced nuclear accumulation of HIF-1α (Fig. 1D and 1E), suggesting that cobalt chloride mimicked a physiological hypoxic condition in regard to the activation of HIF-1α. These results also suggest that the putative HRE in the human ferritin H gene is not functional and that the induction of ferritin H mRNA by cobalt chloride is mediated through a HIF-1α-HRE independent mechanism.
Figure 1. Cobalt chloride but not hypoxia induced human ferritin H mRNA expression.

A) A map of ARE and putative HRE enhancers in the human ferritin H gene. B, C) Total RNA was isolated from K562 cells treated with 100 μM cobalt chloride or hypoxia for 0, 1, 2, 6, 12, and 24 hrs. Ferritin H and transferrin receptor-1 (TfR1) mRNAs were measured by real-time qPCR. mRNA expression in untreated cells was set to 100%, and normalized with β2 microglobulin (B2M) gene expression. Results are means ± SE of duplicate samples from 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared to untreated sample. D, E) Nuclear protein extract was isolated from K562 cells treated with 100 μM cobalt chloride or hypoxia for 0, 0.5, 1, 2, 6, 12, and 24 hrs. The extracts were subjected to Western blotting with anti-HIF-1α antibody. Lamin B blots were used as loading controls.
We previously identified and characterized that the ferritin H gene is subject to transcriptional regulation under oxidative stress through an antioxidant response element (ARE) located far upstream from the transcription start site (Fig. 1A, (22,23)). Cobalt chloride was shown to induce generation of reactive oxygen species (1). To explore the molecular mechanism by which only cobalt chloride induces ferritin H mRNA expression, we tested whether cobalt chloride activates the ferritin H ARE. To this end, K562 cells were transiently transfected with ferritin H luciferase reporters containing the ARE and/or putative HRE (ARE(+)/HRE(+), ARE(−)/HRE(+), ARE(−)/HRE(−), and TATA box) in Fig. 2A), followed by cobalt chloride treatment and luciferase assays. As shown in Fig. 2A, cobalt chloride induced luciferase expression only driven by the ARE(+)/HRE(+) but not by others, suggesting that cobalt chloride may activate ferritin H transcription through the ARE. To directly verify the activation of the ARE by cobalt chloride, Nrf2 (NFE2-related factor 2), the major ARE transcription factor that regulates transcription of ferritin and a battery of antioxidant detoxification genes (35), was further investigated. As shown in Fig. 2B, knocking down Nrf2 in K562 cells almost completely dampened the induction of ferritin H mRNA following cobalt chloride treatment, while TfR1, a HIF-1α-regulated gene, showed marginal decrease in cobalt chloride-mediated mRNA induction. Furthermore, Nrf2 ChIP assays demonstrated that cobalt chloride induced Nrf2 binding to the ferritin H ARE but not to the non-ARE region (Fig. 2C) that preceded ferritin H mRNA upregulation (Fig. 1B). Taken together, these results indicate that cobalt chloride induces ferritin H gene transcription through the Nrf2-ARE but not HIF-1α-HRE mechanism.
Fig. 2. Cobalt chloride transcriptionally activates the human ferritin H gene through the antioxidant responsive element.

A) K562 cells were transfected with luciferase reporter plasmids containing ARE(+)/HRE(+), ARE(−)/HRE(+), ARE(−)/HRE(−), or TATA box of the human ferritin H gene, and treated with 0, 10, 100, or 250 μM cobalt chloride for 24 hrs. The Y axis represents relative luciferase units (RLU). ***p < 0.001 compared to untreated cells of ARE(+)/HRE(+). B) K562 cells were transfected with non-targeting (siControl) or Nrf2-targeting (siNrf2) siRNA for 48 hrs and then treated with 100 μM cobalt chloride for 24 hrs. Real-time qPCR was performed for ferritin H or TfR1 mRNA expression. Expression level of ferritin H or TfR1 in siControl/untreated cells was defined as 100%. C) K562 cells were treated with 100 μM cobalt chloride for 0, 3, 6, and 9 hrs, and ChIP assays were performed with control IgG or anti-Nrf2 antibody. Nrf2 binding to ferritin H ARE (top chart) or non-ARE (bottom chart) was shown compared to input (%). Mean ± SE were shown from 3 independent experiments. **p < 0.01 compared to untreated cells of anti-Nrf2.
3.2. Cobalt chloride downregulates but hypoxia upregulates ferritin H protein expression through the IRP-IRE system
Since cobalt chloride induced ferritin H transcription through the ARE (Fig. 2), we investigated whether cobalt chloride induces other antioxidant genes regulated by the Nrf2-ARE pathway. K562 cells treated with various concentrations of cobalt chloride for 12 and/or 24 hr were analyzed for protein and mRNA levels of ferritin H, thioredoxin reductase 1 (TrxR1), glutamate-cysteine ligase modifier subunit (GCLM), thioredoxin, and NAD(P)H dehydrogenase quinone 1 (NQO1). In RT-qPCR, mRNA levels of ferritin H as well as all other ARE-regulated antioxidant genes were increased after 12 hr treatment with cobalt chloride in a dose-dependent manner (Fig. 3A). However, 12 or 24 hr treatment with cobalt chloride unexpectedly suppressed only ferritin H protein expression, whereas protein expression of all other ARE-regulated antioxidant genes we tested was upregulated (Fig. 3B).
Fig. 3. Cobalt chloride specifically represses ferritin H protein expression.

A) Total RNA was isolated from K562 cells treated with 0, 20, 100, and 250 μM cobalt chloride for 12 hrs. mRNA expression of ferritin H, TrxR1, GCLM, thioredoxin and NQO1 were analyzed with real-time qPCR, and no treatment was set to 100%. Results are means ± SE of duplicate samples from 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared to untreated cells. B) Whole cell lysates were isolated from K562 cells treated with 0, 20, 100, or 250 μM cobalt chloride for 12 or 24 hrs. Protein expression was analyzed by Western blotting, and β-actin was used for loading control.
Expression of ferritin is regulated by at least two different mechanisms in response to external stimuli; transcriptional regulation by electrophiles through the Nrf2-ARE system and post-transcriptional regulation by iron or oxidative stress through interaction between IRPs and IRE located in the 5′-UTR of ferritin mRNA (18,23). As cobalt chloride activated ferritin transcription through the ARE (Fig. 2) but inhibited ferritin protein expression (Fig. 3B), we further characterized whether cobalt chloride increased the binding of IRP to the ferritin H IRE that might have caused translational repression despite the increased ferritin H mRNA. To test this possibility, K562 cells were transfected with a ferritin H luciferase reporter regulated by the 5′UTR IRE, treated with cobalt chloride or iron chelator deferoxamine (DFO) as a positive control for 24 hr, and luciferase assays were performed. Indeed, cobalt chloride repressed ferritin H IRE-driven luciferase expression in a dose dependent manner (Fig. 4A). Likewise, the iron chelator DFO repressed IRE-luciferase expression (Fig. 4A) as DFO is known to increase IRP binding to the IRE and suppress ferritin translation (14). These results suggest that cobalt chloride increases IRP binding to the ferritin H IRE thereby causes translational repression of the ferritin H gene. To verify this mechanism, IRP-IRE binding assays were performed by incubation of cell extracts from cobalt chloride-treated K562 cells with biotin-labeled ferritin H IRE RNA probe and pulled down with streptavidin-agarose, followed by Western blotting with IRP antibodies. There are two IRPs (IRP1 and IRP2) involved in IRE-mediated translational suppression. As shown in Figure 4B, both IRP1 and IRP2 were bound to ferritin H IRE prior to cobalt chloride treatment and their interactions with the IRE were significantly induced by 6 hr after the treatment, to the same extent as the iron chelator DFO treatment. Western blotting using the same extracts for the IRP-IRE binding assays showed that the expression level of IRP1 was unchanged or even slightly decreased while IRP2 was upregulated after cobalt chloride treatment (Fig. 4C). Given the fact that increased IRP1 binding to IRE is regulated by destruction of an iron-sulfur cluster in the IRP1 while IRP2 binding to IRE is regulated by the stability of the IRP2 protein (13,14), our results suggest that the increased IRP2 levels by cobalt chloride, at least in part, facilitated the IRP2 binding to the ferritin H IRE therefore suppressed ferritin H translation despite the transcriptional activation of the ferritin H gene by cobalt chloride via the ARE (Fig. 2).
Fig. 4. Cobalt chloride represses ferritin H protein expression through IRP-IRE mediated translational repression.

A) K562 cells were transfected with a luciferase reporter driven by the ferritin H IRE, followed by treatment with 0, 20, 100, or 250 μM cobalt chloride, or 100 or 250 μM of deferoxamine (DFO) for 24 hrs. The firefly luciferase activity from untreated cells was defined as 100%. Mean ± SE were shown from 3 independent experiments. *p < 0.05, ***p < 0.001 compared to untreated cells. B) K562 cells were treated with 100 μM cobalt chloride for 0, 3, or 6 hrs, or 100 μM DFO for 6 hrs. Whole cell lysates were used for pull-down assay. IRP1 or IRP2 binding to biotinylated ferritin H IRE RNA probe was detected by Western blotting using anti-IRP1 or anti-IRP2 antibody. * indicates a non-specific band. Commassie brilliant blue (CBB) staining of pulled-down proteins was used as a loading control. C) K562 whole cell lysates treated with 100 μM cobalt chloride for 0, 3, and 6 hrs, or 100 μM DFO for 6 hrs were also analyzed by Western blotting for the IRP1 and IRP2 protein expression levels. β-actin was used as loading control.
In contrast to hypoxia mimetic cobalt chloride, hypoxia failed to activate ferritin H transcription (Fig. 1) but increased ferritin H protein expression in K562 cells in a time-dependent manner (Fig. 5A), suggesting the involvement of translational regulation of ferritin H expression. To test this possibility, K562 cells were transiently transfected with a luciferase reporter containing the ferritin H 5′-IRE, followed by hypoxia for 6, 12 and 24 hr and harvested for luciferase assays. As shown in Fig. 5B, hypoxia induced luciferase expression driven by the ferritin H IRE in a time-dependent manner, to the similar extent to the treatment with ferric ammonium citrate (FAC, positive control), suggesting that hypoxia suppresses IRP-IRE interaction thereby led to translational upregulation of the ferritin H gene. Indeed, IRP binding assay showed that hypoxia significantly impaired IRP1 binding to the IRE, while IRP2 binding to the IRE sustained and rather slightly increased at 6 hrs (Fig. 5C). Similar to the effect of cobalt chloride in Fig. 4C, hypoxia did not affect the expression level of IRP1 protein but upregulated IRP2 protein expression (Fig. 5D). These results suggest that translational upregulation of ferritin H following hypoxia is mediated, at least in part, by the decrease in IRP1 binding to the IRE, being opposite to the effect of cobalt chloride on the IRP-IRE interaction (Fig. 4B).
Fig. 5. Hypoxia upregulates ferritin H protein expression through IRP-IRE mediated translational induction.

A) Whole cell lysates from K562 cells treated with hypoxia for 0, 0.5, 2, 4, 6, and 8 hrs were subjected to Western blotting using anti-ferritin H antibody. β-actin was used as loading control. B) K562 cells were transfected with luciferase reporter driven by the ferritin H IRE, treated with hypoxia or 250 μM ferric ammonium citrate (FAC) for 0, 6, 12, and 24 hrs.. The firefly luciferase activity from untreated cells was defined as 100%. Mean ± SE were shown from 3 independent experiments. **p < 0.01, ***p < 0.001 compared to untreated cells. C) K562 cells were treated with hypoxia for 0, 3, and 6 hrs or 250 μM FAC for 6 hrs.. Whole cell lysates were used for pull-down assay similar to Fig. 4B. Commassie brilliant blue (CBB) staining of pulled-down proteins was used as a loading control. D) IRP1 and IRP2 protein expression levels after hypoxia for 0, 3, and 6 hrs or 250 μM FAC for 6 hrs were measured by Western blotting.
4. DISCUSSION
Oxygen and iron are vital elements that maintain a variety of physiological functions such as oxidative phosphorylation and redox reactions in metabolism. Given the fact that cobalt chloride elicits cellular response and gene transcription similar to hypoxia (5,26,36,37), we asked in this study whether cobalt chloride mimics hypoxia in ferritin gene expression that is regulated both by DNA enhancer and mRNA regulatory elements at the transcriptional and post-transcriptional levels, respectively. Both hypoxia and cobalt chloride have been shown to regulate transcription of a common set of genes, such as erythropoietin and TfR1, through a HRE (5,33,37), to which HIF-1α stabilized by hypoxia or cobalt chloride binds and activates gene transcription (26,36). As we noted that there is a putative HRE in the human ferritin H gene, we attempted to test whether ferritin H transcription is induced by cobalt chloride and hypoxia through the HRE. Although the putative ferritin H HRE contains the ACGTG core sequence (32) and HIF-1α protein expression was increased by hypoxia as well as cobalt chloride (Fig. 1D, 1E), transcriptional activation of ferritin H through the HRE was not observed (Fig. 2A). Our ChIP assays did not show the binding of HIF-1α to the putative ferritin H HRE (unpublished observation, B.H Huang), leading us to conclude that the putative HRE in the human ferritin H gene is not functional in K562 cells. Instead, we unexpectedly found that cobalt chloride, but not hypoxia, activated ferritin H transcription through the Nrf2-ARE pathway (Fig. 2). Expression of other Nrf2-regulated genes (NQO1, thioredoxin, TrxR1, and GCLM) (38) was also induced by cobalt chloride (Fig. 3). The recent report of gene expression analyses in rat liver cell lines by microarray and mass spectrometry approaches suggested that cobalt chloride modulates expression of gene sets primarily regulated by hypoxia and oxidative stress (39). Our results are consistent with this report and provide direct evidence for upregulation of Nrf2-regulated gene expression (Fig. 3) and the recruitment of Nrf2 to the ferritin H ARE following cobalt chloride treatment (Fig. 2).
The effects of hypoxia on expression of key iron metabolism genes such as ferritin and TfR1 via the mRNA regulatory element IRE and its binding proteins IRPs (the IRP-IRE regulatory system) were previously studied (40–42). In these studies, the effects of hypoxia on binding of IRPs to IRE appear to be somewhat inconsistent or complex, or may be context-dependent including cell types and species, hypoxic oxygen percent, and duration and time points of hypoxic conditions (40–42). Subsequent studies demonstrated that the relative involvement of IRP1 and IRP2 in the IRP-IRE regulatory system was determined by tissue oxygen levels and cellular redox states (43,44), and hypoxia stabilized and activated IRP2 through destabilization of FBXL5 and SKP1-CUL1 ubiquitin ligase protein complex (45,46) while hypoxia decreased IRP1 binding to IRE (43,47). Despite the induction of ferritin H mRNA by cobalt chloride in K562 cells, we found that ferritin H protein expression was strongly suppressed (Fig. 3). The translational suppression of ferritin H by cobalt chloride can be explained by increased interaction between IRP1/2 and the ferritin H IRE (Fig. 4). A previous study on the effects of various metal ions on IRP1 and HIF-1α expression in human lung carcinoma A549 cells demonstrated that cobalt chloride as well as nickel chloride increased IRP1 binding to IRE along with HIF-1α stabilization (48). Our results are essentially consistent with these results and demonstrated the increased IRP1 and IRP2 binding to the IRE following cobalt chloride, leading to translational suppression of ferritin H in K562 cells (Fig. 4). In contrast to cobalt chloride, hypoxia increased ferritin H protein expression (Fig. 5A) without changing the ferritin H mRNA expression level in K562 cells (Fig. 1C top). Increased expression of ferritin proteins by hypoxia has also been reported in lens epithelial cells despite over 50% reduction in ferritin transcription (49). Our results showing decreased IRP1 binding to the IRE (Fig. 5C) are also supported by previous studies for ferritin protein expression by hypoxic conditions in other cell types such as human oligodendroglioma cells (50) and mouse peritoneal macrophages (51). Increased IRP2 binding to IRE (6 hrs) under decreased IRP1-IRE interaction (3 hrs) by hypoxia (Fig. 5C) also appears to be consistent with the study in HEK293 cells (42), suggesting that the effects of hypoxia observed in this study is not specific to K562 cells.
As summarized in Fig. 6, this study investigated the similarities and differences between hypoxia and hypoxia mimetic cobalt chloride, demonstrating that 1) HIF-1α was accumulated following hypoxia as well as cobalt chloride treatment but the putative HRE in the human ferritin H gene was not activated, 2) only cobalt chloride activated transcription of the human ferritin H gene through the Nrf2-ARE pathway, 3) cobalt chloride decreased but hypoxia increased ferritin H protein levels through the opposite effects on the IRP-IRE interaction for translational block. Probably a plethora of genes in cells are subject to cooperative regulation by DNA enhancers and mRNA regulatory elements including mRNA 3′UTR AU-rich elements and various microRNA target sites. Our results suggest that hypoxia and hypoxia mimetic cobalt chloride employ distinct mechanisms for expression of not only ferritin H gene but also various genes by targeting both DNA and RNA regulatory elements.
Fig. 6.
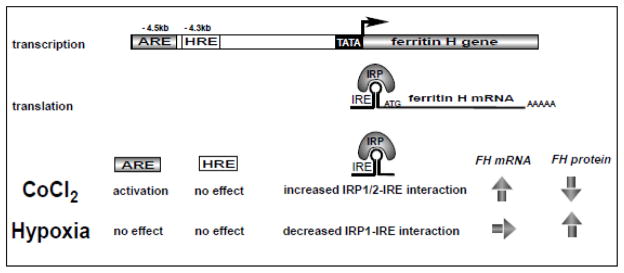
Summary of the distinct regulatory mechanism of ferritin H expression by hypoxia and hypoxia mimetic cobalt chloride.
5. CONCLUSIONS
Cobalt chloride, used as a hypoxia mimetic, and hypoxia employ distinct regulatory mechanisms for ferritin gene expression through the interplay between antioxidant response DNA enhancer and iron response mRNA elements at the transcriptional and post-transcriptional levels.
Highlights.
Cobalt chloride has been used as a hypoxia mimetic.
Hypoxia and cobalt chloride signaling mechanisms in gene regulation remain elusive.
Cobalt chloride, but not hypoxia, activates the Nrf2-ARE signaling pathway.
Cobalt chloride and hypoxia have the opposite effects on the iron-IRP-IRE system.
Hypoxia and cobalt chloride employ distinct signaling and gene regulatory mechanisms.
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) Grants RO1GM088392, and RO1GM095550 from the National Institute of General Medical Sciences (to Y.T.).
Abbreviations
- ARE
antioxidant responsive element
- ChIP
Chromatin Immunoprecipitation
- DFO
deferoxamine
- ferritin H
ferritin heavy chain
- GCLM
glutamate-cysteine ligase modifier subunit
- HIF1-α
hypoxia inducible factor-1α
- HRE
hypoxia responsive element
- IRE
iron responsive element
- IRP
iron regulatory protein
- NQO1
NAD(P)H dehydrogenase quinone 1
- Nrf2
NFE2-related factor 2
- TrxR1
thioredoxin reductase 1
- TfR1
transferrin receptor-1
- UTR
untranslated region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Experimental physiology. 2006;91:807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- 3.Galanis A, Pappa A, Giannakakis A, Lanitis E, Dangaj D, Sandaltzopoulos R. Reactive oxygen species and HIF-1 signalling in cancer. Cancer Lett. 2008;266:12–20. doi: 10.1016/j.canlet.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood. 2009;114:2015–2019. doi: 10.1182/blood-2009-05-189985. [DOI] [PubMed] [Google Scholar]
- 5.Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1) Molecular pharmacology. 2006;70:1469–1480. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- 6.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 7.Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry. 2012;51:5705–5724. doi: 10.1021/bi300752r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y, Sheftel AD, Ponka P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A. 2010;107:10775–10782. doi: 10.1073/pnas.0912925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao N, Enns CA. Iron transport machinery of human cells: players and their interactions. Current topics in membranes. 2012;69:67–93. doi: 10.1016/B978-0-12-394390-3.00003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turi JL, Yang F, Garrick MD, Piantadosi CA, Ghio AJ. The iron cycle and oxidative stress in the lung. Free Radic Biol Med. 2004;36:850–857. doi: 10.1016/j.freeradbiomed.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Theil EC. Ferritin: at the crossroads of iron and oxygen metabolism. J Nutr. 2003;133:1549S–1553S. doi: 10.1093/jn/133.5.1549S. [DOI] [PubMed] [Google Scholar]
- 12.MacKenzie EL, Iwasaki K, Tsuji Y. Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10:997–1030. doi: 10.1089/ars.2007.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 14.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 15.Huang BW, Ray PD, Iwasaki K, Tsuji Y. Transcriptional regulation of the human ferritin gene by coordinated regulation of Nrf2 and protein arginine methyltransferases PRMT1 and PRMT4. FASEB J. 2013;27:3763–3774. doi: 10.1096/fj.12-226043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacKenzie EL, Ray PD, Tsuji Y. Role and regulation of ferritin H in rotenone-mediated mitochondrial oxidative stress. Free Radic Biol Med. 2008;44:1762–1771. doi: 10.1016/j.freeradbiomed.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwasaki K, Mackenzie EL, Hailemariam K, Sakamoto K, Tsuji Y. Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and role of Ref-1 during erythroid differentiation of K562 cells. Mol Cell Biol. 2006;26:2845–2856. doi: 10.1128/MCB.26.7.2845-2856.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hintze KJ, Theil EC. DNA and mRNA elements with complementary responses to hemin, antioxidant inducers, and iron control ferritin-L expression. Proc Natl Acad Sci USA. 2005;102:15048–15052. doi: 10.1073/pnas.0505148102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyazawa M, Tsuji Y. Evidence for a novel antioxidant function and isoform-specific regulation of the human p66Shc gene. Mol Biol Cell. 2014;25:2116–2127. doi: 10.1091/mbc.E13-11-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakamoto K, Iwasaki K, Sugiyama H, Tsuji Y. Role of the tumor suppressor PTEN in antioxidant responsive element-mediated transcription and associated histone modifications. Mol Biol Cell. 2009;20:1606–1617. doi: 10.1091/mbc.E08-07-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwasaki K, Ray PD, Huang BW, Sakamoto K, Kobayashi T, Tsuji Y. Role of AMP-activated protein kinase in ferritin H gene expression by resveratrol in human T cells. Biochemistry. 2013;52:5075–5083. doi: 10.1021/bi400399f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuji Y. JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress. Oncogene. 2005;24:7567–7578. doi: 10.1038/sj.onc.1208901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, Torti FM. Coordinate transcriptional and translational regulation of ferritin inresponse to oxidative stress. Mol Cell Biol. 2000;20:5818–5827. doi: 10.1128/mcb.20.16.5818-5827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsuji Y, Moran E, Torti SV, Torti FM. Transcriptional regulation of the mouse ferritin H gene: Involvement of p300/CBP adaptor proteins in FER-1 enhancer activity. J Biol Chem. 1999;274:7501–7507. doi: 10.1074/jbc.274.11.7501. [DOI] [PubMed] [Google Scholar]
- 25.Pietsch EC, Chan JY, Torti FM, Torti SV. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J Biol Chem. 2003;278:2361–2369. doi: 10.1074/jbc.M210664200. [DOI] [PubMed] [Google Scholar]
- 26.Yuan Y, Hilliard G, Ferguson T, Millhorn DE. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J Biol Chem. 2003;278:15911–15916. doi: 10.1074/jbc.M300463200. [DOI] [PubMed] [Google Scholar]
- 27.Menotti E, Henderson BR, Kuhn LC. Translational regulation of mRNAs with distinct IRE sequences by iron regulatory proteins 1 and 2. J Biol Chem. 1998;273:1821–1824. doi: 10.1074/jbc.273.3.1821. [DOI] [PubMed] [Google Scholar]
- 28.Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- 29.Iwasaki K, Hailemariam K, Tsuji Y. PIAS3 interacts with ATF1 and regulates the human ferritin H gene through an antioxidant-responsive element. J Biol Chem. 2007;282:22335–22343. doi: 10.1074/jbc.M701477200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maxwell P, Salnikow K. HIF-1: an oxygen and metal responsive transcription factor. Cancer biology & therapy. 2004;3:29–35. doi: 10.4161/cbt.3.1.547. [DOI] [PubMed] [Google Scholar]
- 32.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Science’s STKE: signal transduction knowledge environment. 2005;2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 33.Tacchini L, Bianchi L, Bernelli-Zazzera A, Cairo G. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J Biol Chem. 1999;274:24142–24146. doi: 10.1074/jbc.274.34.24142. [DOI] [PubMed] [Google Scholar]
- 34.Lok CN, Ponka P. Identification of a hypoxia response element in the transferrin receptor gene. J Biol Chem. 1999;274:24147–24152. doi: 10.1074/jbc.274.34.24147. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi A, Ohta T, Yamamoto M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods in enzymology. 2004;378:273–286. doi: 10.1016/S0076-6879(04)78021-0. [DOI] [PubMed] [Google Scholar]
- 36.Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem. 2004;279:40337–40344. doi: 10.1074/jbc.M403057200. [DOI] [PubMed] [Google Scholar]
- 37.Goldberg MA, Gaut CC, Bunn HF. Erythropoietin mRNA levels are governed by both the rate of gene transcription and posttranscriptional events. Blood. 1991;77:271–277. [PubMed] [Google Scholar]
- 38.Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, Xue P, Pi J, Kleeberger SR, Bell DA. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40:7416–7429. doi: 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Permenter MG, Dennis WE, Sutto TE, Jackson DA, Lewis JA, Stallings JD. Exposure to cobalt causes transcriptomic and proteomic changes in two rat liver derived cell lines. PLoS One. 2013;8:e83751. doi: 10.1371/journal.pone.0083751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanson ES, Leibold EA. Regulation of iron regulatory protein 1 during hypoxia and hypoxia/reoxygenation. J Biol Chem. 1998;273:7588–7593. doi: 10.1074/jbc.273.13.7588. [DOI] [PubMed] [Google Scholar]
- 41.Toth I, Yuan L, Rogers JT, Boyce H, Bridges KR. Hypoxia alters iron-regulatory protein-1 binding capacity and modulates cellular iron homeostasis in human hepatoma and erythroleukemia cells. J Biol Chem. 1999;274:4467–4473. doi: 10.1074/jbc.274.7.4467. [DOI] [PubMed] [Google Scholar]
- 42.Schneider BD, Leibold EA. Effects of iron regulatory protein regulation on iron homeostasis during hypoxia. Blood. 2003;102:3404–3411. doi: 10.1182/blood-2003-02-0433. [DOI] [PubMed] [Google Scholar]
- 43.Meyron-Holtz EG, Ghosh MC, Rouault TA. Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science. 2004;306:2087–2090. doi: 10.1126/science.1103786. [DOI] [PubMed] [Google Scholar]
- 44.Hausmann A, Lee J, Pantopoulos K. Redox control of iron regulatory protein 2 stability. FEBS Lett. 2011;585:687–692. doi: 10.1016/j.febslet.2011.01.036. [DOI] [PubMed] [Google Scholar]
- 45.Salahudeen AA, Thompson JW, Ruiz JC, Ma HW, Kinch LN, Li Q, Grishin NV, Bruick RK. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science. 2009;326:722–726. doi: 10.1126/science.1176326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, Bhaskaran N, Persson A, Uhlen M, Sangfelt O, Spruck C, Leibold EA, Wohlschlegel JA. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science. 2009;326:718–721. doi: 10.1126/science.1176333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanson ES, Foot LM, Leibold EA. Hypoxia post-translationally activates iron-regulatory protein 2. J Biol Chem. 1999;274:5047–5052. doi: 10.1074/jbc.274.8.5047. [DOI] [PubMed] [Google Scholar]
- 48.Li Q, Chen H, Huang X, Costa M. Effects of 12 metal ions on iron regulatory protein 1 (IRP-1) and hypoxia-inducible factor-1 alpha (HIF-1alpha) and HIF-regulated genes. Toxicol Appl Pharmacol. 2006;213:245–255. doi: 10.1016/j.taap.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goralska M, Fleisher LN, McGahan MC. Hypoxia induced changes in expression of proteins involved in iron uptake and storage in cultured lens epithelial cells. Exp. Eye Res. 2014;125:135–141. doi: 10.1016/j.exer.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qi Y, Dawson G. Hypoxia specifically and reversibly induces the synthesis of ferritin in oligodendrocytes and human oligodendrogliomas. Journal of neurochemistry. 1994;63:1485–1490. doi: 10.1046/j.1471-4159.1994.63041485.x. [DOI] [PubMed] [Google Scholar]
- 51.Kuriyama-Matsumura K, Sato H, Yamaguchi M, Bannai S. Regulation of ferritin synthesis and iron regulatory protein 1 by oxygen in mouse peritoneal macrophages. Biochem Biophys Res Commun. 1998;249:241–246. doi: 10.1006/bbrc.1998.9046. [DOI] [PubMed] [Google Scholar]