

Summary
The myxovirus resistance (Mx) proteins are interferon-induced dynamin GTPases that can inhibit a variety of viruses. Recently, MxB, but not MxA, was shown to restrict HIV-1 by an unknown mechanism that likely occurs in close proximity to the host cell nucleus and involves the viral capsid. Here, we present the crystal structure of MxB and reveal determinants involved in HIV-1 restriction. MxB adopts an extended anti-parallel dimer and dimerization, but not higher-ordered oligomerization, is critical for restriction. Although MxB is structurally similar to MxA, the orientation of individual domains differs between MxA and MxB and their antiviral functions rely on separate determinants, indicating distinct mechanisms for virus inhibition. Additionally, MxB directly binds the HIV-1 capsid and this interaction depends on dimerization and the N-terminus of MxB as well as the assembled capsid lattice. These insights establish a framework for understanding the mechanism by which MxB restricts HIV-1.
Introduction
Myxovirus resistance protein 2 (MxB) is an interferon-induced inhibitor of HIV-1 infection (Goujon et al., 2013; Kane et al., 2013; Liu et al., 2013). MxB was traditionally thought to function in cell-cycle progression and regulation of nuclear import (King et al., 2004; Melen et al., 1996). This newly discovered antiviral function occurs downstream of reverse transcription, decreasing the amount of integrated viral DNA (Liu et al., 2013) and 2-long terminal repeat (2-LTR) circular DNA (Goujon et al., 2013; Kane et al., 2013) that marks translocation of the cytoplasmic reverse transcription complex into the nucleus. These results suggest that MxB inhibits HIV-1 nuclear import, or destabilizes nuclear viral DNA (Goujon et al., 2013; Kane et al., 2013). MxB is highly homologous in sequence (63% identity) to MxA, whose antiviral activities are well established (Aebi et al., 1989; Hefti et al., 1999). MxA restricts both DNA and RNA viruses, including influenza A virus (Haller and Kochs, 2011). It has been shown that MxA interferes with translocation of viral components between the cytoplasm and the nucleus, potentially via binding to and causing mislocalization of viral nucleocapsid protein (Kochs and Haller, 1999a, b; Kochs et al., 2002b; Reichelt et al., 2004).
Both MxB and MxA are guanosine triphosphatases (GTPases) that belong to the dynamin superfamily. Extensive structural, biochemical, and cellular studies have revealed the function of each MxA domain. The amino-terminal GTPase domain binds and hydrolyzes GTP, while a bundle signaling element (BSE) domain connects and transmits signals between the GTPase and the stalk domains (Gao et al., 2011). The stalk domain is critical for oligomerization (Gao et al., 2011; Gao et al., 2010; Haller et al., 2010; Kochs et al., 2002a). GTPase activity and oligomerization are critical for viral inhibition by MxA (Daumke et al., 2010; Di Paolo et al., 1999; Melen et al., 1992; Pavlovic et al., 1993; Schwemmle et al., 1995).
Despite the similarity in sequence and architecture, MxB and MxA work against different viruses and appear to have different mechanisms of action. MxB restricts HIV-1, which is not among the diverse range of viruses inhibited by MxA. MxB (715 amino acids) harbors a 43 residue N-terminal extension that contains a nuclear localization signal (NLS), which is critical for HIV-1 restriction (Kane et al., 2013; Melen et al., 1996). A shorter MxB isoform that initiates from Met26 lacks the NLS and therefore would not restrict HIV-1 (Melen et al., 1996). The MxB N-terminal region also contains anti-HIV-1 specificity determinants distinct from the NLS (Busnadiego et al., 2014; Goujon et al., 2014)(Matreyek et al., unpublished data). Besides the N-terminal differences, MxB mutants that are unable to bind or hydrolyze GTP retain the ability to restrict HIV-1 (Goujon et al., 2013; Kane et al., 2013), which is contrary to the GTPase-dependent restriction activity of MxA. Furthermore, instead of targeting the nucleocapsid protein like MxA, the antiviral activity of MxB involves the HIV-1 capsid protein (CA), as CA mutations can counteract restriction by MxB (Busnadiego et al., 2014; Goujon et al., 2014; Kane et al., 2013; Liu et al., 2013). Though HIV-1 CA interacts with many cellular factors including CypA, TRIM5α, CPSF6, and NUP153 (Ambrose and Aiken, 2014; Matreyek and Engelman, 2013), it remains to be determined if there is a direct interaction between MxB and CA, or if other cellular factors mediate the CA-dependent activity of MxB. In addition, it is unknown whether MxB functions by forming MxA-like higher order oligomers (Melen and Julkunen, 1997).
To provide insight into its mechanism of HIV-1 restriction, we determined the crystal structure of MxB. The structure shows that MxB has a similar architecture to MxA but with different domain orientations. We further performed detailed mutagenesis studies that inform about the regions of MxB that are critical for HIV-1 restriction. Our results reveal key differences between the antiviral activities of MxA and MxB, demonstrating that these closely related proteins have distinct mechanisms of action. Importantly, our study establishes that MxB binds directly to HIV-1 capsid assemblies and indicates that direct engagement of the capsid lattice by the antiparallel MxB dimer is critical to antiviral function.
Results
Crystal structure of MxB
To investigate the structural basis for HIV-1 restriction, we crystallized an N-terminal truncation of MxB. To improve the solution behavior of MxB, we deleted the first 83 amino acids, which are predicted to be unstructured, and introduced mutations into loop 2 of the stalk domain (YRGK487-490AAAA), as similar changes improved the solution behavior of MxA (Gao et al., 2011; Gao et al., 2010). This construct, MxB84YRGK, allowed for the purification of monodispersed, dimeric protein that crystallized and diffracted X-rays to 3.2 Å resolution. We solved the structure by molecular replacement using MxA as a search model and refined the structure to Rwork/Rfree of 25.6/29.6% with one MxB dimer in the asymmetric unit (Figure 1A). The detailed statistics are shown in Table 1. Two MxB protomers form an extended anti-parallel dimer (Figure 1A). The GTPase and stalk domains are located at either end of the MxB protomer, bridged by the BSE domain that is composed of three helices originating from distinct regions of the primary amino acid sequence (Figure 1B). Residues 84-92, 145-149, 231-237, 580-621, and 712-715 are disordered in the structure. The structures of the corresponding domains in the two protomers are nearly identical, with root-mean-square deviations (RMSD) ranging between 0.25 and 0.38 Å (Figure 1C). The overall RMSD of the two MxB protomers is 1.7 Å, which is substantially larger than that of the individual domains, indicating flexibility at the linker regions connecting the domains.
Figure 1. Structure of MxB84YRGK and its antiviral activity.

(A) Structure of the MxB dimer shown in two orientations, with protomers 1 and 2 colored in purple and yellow, respectively. (B) Schematic (left) and structure (right) of an MxB protomer with residues of domain boundaries denoted and colored. The arrows in the schematic denote the first and last visible residues in the structure. (C) Superposition of protomer 1 and protomer 2 in two views. (D) Cells expressing HA-tagged WT or MxBYRGK were analyzed for MxB expression. Total cellular proteins were extracted, resolved by SDS-PAGE, and visualized by western blotting with anti-HA antibody. WT MxB expression was set to 1. Results are the mean of 3 independent experiments, with error bars denoting standard error. (E) Immunofluorescent microscopy of untransduced or MxB-expressing cells. Blue, nuclear DNA. (F) Susceptibility of WT or MxBYRGK-expressing versus control (non-transduced) HOS cells to HIV-1 (dark gray), EIAV (light gray), or FIV (striped gray) infection. Error bars denote 95% confidence intervals derived from 7 independent experiments.
Table 1.
Data Collection and Refinement Statistics
| Data Collection | |
| Wavelength (Å) | 0.9792 |
| Space Group | P21 |
| Cell Dimensions | |
| a,b,c (Å) | 53.18, 80.78, 183.67 |
| α, β, γ (°) | 90.00, 95.73, 90.00 |
| Molecules/asymmetric unit | 2 |
| Resolution (Å) | 43.9-3.2 (3.26-3.20)* |
| Rmerge | 0.087 (>1.0) |
| I/σI | 16.1 (1.1) |
| Completeness (%) | 94.7 (89.9) |
| Redundancy | 5.4 (5.1) |
| Unique reflections | 24,248 |
| Refinement | |
| Number of nonhydrogen atoms | 9082 |
| Rwork/Rfree (%) | 25.6/29.6 |
| Average B factor | 148 |
| Root mean-squared deviation (rmsd) | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 1.0 |
| Ramachandran analysis | |
| Preferred regions (%) | 94.34 |
| Allowed regions (%) | 4.67 |
| Outliers (%) | 0.99 |
Values in parenthesis are for highest-resolution shell.
To characterize the physiological relevance of MxB84YRGK, we assessed the antiviral activities of wild-type (WT) and mutant MxB proteins. MxB was stably expressed with a C-terminal hemagglutinin (HA) tag to facilitate its immunodetection in human osteosarcoma (HOS) cells, which were used previously to assess the mechanism of HIV-1 restriction (Kane et al., 2013). Independent of stimulation by interferon α, HOS cells did not detectably express endogenous MxB protein (Matreyek et al., unpublished observations). Immunofluorescent staining revealed that WT MxB formed cytoplasmic puncta and localized to the nuclear rim (Figure 1E), as described previously (Kane et al., 2013; King et al., 2004; Melen et al., 1996). Although the MxB84YRGK construct is presumably inactive due to loss of the N-terminal NLS (Kane et al., 2013), we independently tested whether the YRGK487-490AAAA mutation affected the antiviral activity of full-length MxB. The mutant protein (MxBYRGK) was expressed to about 80% of the level of WT MxB (Figure 1D) and, in contrast to the punctate staining observed with the WT protein, MxBYRGK exhibited more diffuse staining within the cytoplasm (Figure 1E). WT MxB restricted HIV-1 infection about 10-fold without noticeably affecting the infectivity of equine infectious anemia virus (EIAV) or feline immunodeficiency virus (FIV) (Figure 1F). MxBYRGK also significantly inhibited HIV-1 infection (~6-fold). The mutant protein restricted EIAV infection by about 3-fold but remained inert against FIV. The gain-of-function against EIAV could be due to the altered pattern of MxBYRGK subcellular localization (Figure 1E). WT MxB moreover was reported in an independent study to restrict EIAV at a level similar to that observed here for MxBYRGK (Kane et al., 2013). In any case, the solubility-enhancing YRGK487-490AAAA mutations used for crystallographic studies did not significantly alter the ability for MxB to restrict HIV-1.
BSE hinge communication is not required for HIV restriction
Although the overall arrangement of individual protein domains is similar between the MxA and MxB structures, large differences in domain orientation are observed between the proteins (Figure 2A). The individual domain structures of the two proteins are quite similar, with corresponding RMSDs in the range of 0.8-1.1 Å, while the overall RMSD between MxA and MxB protomers is greater than 6.4 Å. The difference in domain orientations, pivoted around two hinge regions connecting the domains, is primarily responsible for the overall deviation between MxA and MxB (Figure 2A).
Figure 2. MxB hinge mutations do not abolish antiviral activity.

(A) Superposition of MxA monomer (PDBID: 3SZR; red) and MxB protomer 1 (purple), based on either GTPase domain (left) or stalk domain (right). (B) Zoomed in view of BSE domain with hinges and key residues highlighted. (C-E) Cells expressing WT, E681A, or R689A mutant MxB were analyzed for (C) total expression, (D) subcellular localization, and (E) antiviral activity as described in Figure 1. Relative expression values are an average of at least 3 independent blotting experiments, with error bars denoting standard error. Infection values are the mean of 5 independent experiments, with error bars denoting 95% confidence intervals.
We next tested the potential involvement of hinge communication in MxB function. Residues at the ends of the BSE are thought to act as hinges that transfer signals between the GTPase and stalk domains in dynamin superfamily proteins (Gao et al., 2011; Prakash et al., 2000). We compared the structures and sequences of MxA and MxB and identified key residues in each of the two MxB hinges (Figure 2B). Hinge 1 has two loops that connect the BSE to the stalk domain (residues 406-416 and 679-684). The highly conserved residue R689 on helix 3 of the BSE contacts G408 and D410 on BSE loop 1. R689 also interacts with the side chain of E681 on BSE loop 2. Hinge 2 pivots around P387, which causes a kink in the alpha helix connecting the GTPase domain and the BSE. MxB hinge mutants E681A and R689A were expressed in HOS cells at levels similar to the WT protein (Figures 2C and 2D) and significantly inhibited HIV-1 infection by ~10-fold and ~5.5-fold, respectively (Figure 2E). The corresponding MxA mutations, E632A and R640A, reduced MxA oligomerization, GTPase activity, and antiviral activity (Gao et al., 2011). These observations indicate that anti-HIV-1 activity is not dependent on the transfer of information from the GTPase domain to the stalk domain, which is consistent with data showing that MxB antiviral function is independent of GTPase activity (Goujon et al., 2013; Kane et al., 2013).
Dimerization, but not higher-order oligomerization, of MxB is required for antiviral activity
MxB forms an anti-parallel dimer with the dimer interface lying at the center of the two protomers. It is composed of residues on stalk helices 3 and 4 (Figure 3A). To be consistent with the MxA/dynamin convention, we refer to the dimer interface as interface 2 (Figure 3A). The buried surface area at this interface is 1,074 Å2, with symmetric hydrophobic contacts between M567, L570, M574, and V578 of each protomer with M567, L570, F647, and Y651 of the other protomer. The dimer is further stabilized by hydrogen bonds between Q571 of each protomer and Q644 of the other protomer (Figure 3B). We tested the importance of interface 2 in MxB dimerization and found that an M574D mutation destabilized the dimerization interface. MxB84YRGK eluted from a size exclusion column at a volume that corresponds to the molecular weight of an extended dimer based on molecular weight standards. The M574D mutation decreased dimerization, as the majority of MxB84YRGK/M574D eluted at a volume corresponding to the molecular weight of monomeric MxB (Figure 3C). This is consistent with the observation that the corresponding residue in MxA is critical for MxA dimerization (Gao et al., 2010).
Figure 3. The MxB dimer is required for antiviral activity.

(A) Dimer of MxB (top) with zoomed in view of dimer interface in surface representation (bottom). (B) Zoomed view of dimer interface with key residues shown as sticks. (C) Size exclusion chromatography analysis of MxB84YRGK (green), which elutes at a volume corresponding to a dimer, and MxB84YRGK/M574D (cyan), a majority of which elutes at a volume corresponding to a monomer. Inset, SDS-PAGE of peak fractions corresponding to monomeric (lane 2 of each set) and dimeric (lane 1) MxB. (D) Antiviral activities of WT and interface 2 mutants of MxB. Activity assays are as described in Figure 1F. Results are an average of at least 4 independent experiments, with error bars denoting 95% confidence intervals. (E) Total mutant MxB expression levels relative to WT MxB (set to 1). Results are an average of 3 independent experiments, with error bars denoting standard error. (F) WT and mutant MxB localization as determined by confocal microscopy following antibody staining.
We tested the requirement of MxB dimerization for its antiviral function. Interface 2 mutants M574D, Y651D, and M567D/L570D were ineffective against HIV-1, each inhibiting infection 1.6-fold or less, which did not differ significantly from EIAV or FIV (Figure 3D). The dimerization state of MxB appears to affect its expression pattern, as the interface 2 mutants were expressed at ~30-50% of the level of WT MxB (Figure 3E). The mutant proteins also exhibited drastically altered localization compared to WT MxB, as they stained throughout the cell, including prominent staining within the intranuclear space (Figure 3F). In a separate study we determined that the MxB GTPase mutant T151A, which was expressed at ~38% of WT MxB, displayed full restriction activity (Matreyek et al., unpublished observations). Our findings accordingly indicate that dimerization plays an important role in MxB subcellular localization and is required for viral restriction.
MxB dimers can form higher-order assemblies by interdigitating through the stalk and BSE regions as observed in MxA. This mode of higher-order oligomerization is formed through MxB crystal packing interactions (Figure 4A). Through this interaction, MxB dimers may assemble to form filaments. The MxB dimer-dimer interface revealed in the crystal structure has a buried surface area of >2500 Å2. Following the convention for MxA, we refer to the interface region at the beginning of stalk helices as interface 1. Interaction at this interface is critical for MxA function (Gao et al., 2010). Interface 1 involves the stalks from one protomer of each interacting MxB dimer (designated protomer 1 and protomer 1’, the prime symbol denotes an adjacent dimer) (Figure 4A). The interactions at this interface include the symmetric hydrophobic contacts of I423, M666, and L669 in both stalks (Figure 4A). This is further stabilized by a hydrogen bond between E424 of protomer 1 and K663 of protomer 1’.
Figure 4. Higher-order oligomerization of MxB is not required for antiviral activity.

(A) Two adjacent MxB dimers representing formation of higher-order oligomers in two views (left) and a zoomed in view of higher-order interface 1 (right). Protomer 1 and 2 of one dimer are colored as in Figure 3 while protomer 1’ and 2’ are colored in dark grey and light grey, respectively. Important interface residues are shown in sticks. (B) CryoEM images of full-length MBP-MxB WT (6 μM) and the interface mutant MBP-MxBYRGK/IKM (3 μM). Short filaments (marked by arrows) and circular structures (marked by triangles) are seen for WT MBP-MxB. (C) Size exclusion chromatography analysis of MBP-MxB1-715 (green), which elutes close to the void volume, and MBP-MxB1-715 IK/DD (red) and MBP-MxB1-715 IKM/DDD (blue), which elute at volumes corresponding to dimers. Inset, SDS-PAGE of peak fractions. (D) Antiviral activities of WT and MxB interface 1 mutants. Results are an average of at least 4 independent experiments, with error bars denoting 95% confidence intervals. (E) Total mutant MxB expression levels relative to WT MxB (set to 1). Results are an average of at least 3 independent experiments, with error bars denoting standard error. (F) WT and mutant MxB localization as determined by confocal microscopy.
To gain insight into higher-ordered oligmerization of MxB, we analyzed how mutations in these interfaces alter MxB's oligomerization state by cryo-electron microscopy (cryoEM). An assortment of structural assemblies was observed by cryoEM for the purified full-length WT MxB tagged with maltose-binding protein (MBP-MxB). Short filaments and circular structures were seen clustering together into aggregate assemblies (Figure 4B, left panel). Introduction of I423D/K663D/M666D mutations into the full-length MBP-MxBYRGK construct (MBP-MxBYRGK/IKM) improved the solution behavior and rendered the protein less prone to aggregation. Discrete particles of oligomers ~30-40 nm across were the majority among the structural assemblies observed for this interface mutant (Figure 4B, right panel). This observation is consistent with our results by size exclusion chromatography, where MBP-MxBYRGK/IKM, MBP-MxBIKM, and MBP-MxBIK primarily eluted as soluble dimers (Figure 4C and Figure 5E), while MBP-MxB migrated as a higher-order assembly or aggregate (Figure 4C).
Figure 5. Interactions with capsid assemblies by WT and mutant MxB proteins.

(A) Cell extracts containing HA-tagged MxB were tested for binding to cross-linked CA tubular assemblies. Pelleted proteins resolved by SDS-PAGE were visualized by western blotting (MxB) or stained with Coomassie Blue (CA). Results are an average of 5 independent experiments with standard errors plotted. Lane indicators above the representative western blot: i, 20% of input cell lysate in absence of CA; -, pellets from binding reactions in the absence of CA; +, reactions in the presence of CA. Lane 1 of lower panel, 20% of input CA. (B) Scatterplot of WT and mutant MxB-HA binding (x-axis) versus normalized level of HIV-1 infectivity (y-axis). Points denote the geometric mean of each data set. The comparison exhibited a negative correlation with a significant Spearman rank correlation (P = 0.028). (C) Binding of purified MxB (with or without CypA or CPSF6313-327), maltose binding protein (MBP) (negative control), or a region of TRIMCyp (CC-Cyp) (positive control) to cross-linked CA assemblies. Total (T), soluble (S), and pellet (P) fractions resolved by SDS-PAGE were visualized with Coomassie staining and quantified with ImageJ. Three CA variants were analyzed for binding, A14C/E45C (CA), A14C/E45C/G89V (G89V) and A14C/E45C/N74D (N74D). Quantification of the binding, with standard errors, from 3 independent experiments is plotted below the gels. P values from two sided, unequal variance t-tests are shown for the pelleting comparison for each MxB construct with or without CA. (D) Visualization of the interaction of MxB constructs with HIV-1 CA tubes. CryoEM images of reaction mixtures containing cross-linked CA assemblies (10 μM) and MBP-MxB1-715YRGK/IKM (5 μM) or MBP-MxB84-715YRGK/IKM (5 μM) shows additional protein density decorating the tubes (middle and right panels) that is not observed in the control tubes without MxB (left). Substantially more decoration of the CA tubes occurs with the full-length MxB construct (middle) than with the N-terminal truncation construct (right). (E) Size exclusion chromatograms of purified MBP-MxBYRGK/IKM (blue), cross-linked CA hexamers (red), and their mixture (green). CA hexamers and MBP-MxBYRGK/IKM do not bind, as the elution profile of the mixture is the exact superposition of the individual ones. The SDS-PAGE analysis of the peaks (labeled 1, 2 and 3) is shown.
We tested the requirement of higher-order oligomerization for MxB antiviral function. Interface 1 mutants I423D, K663D, M666D, and the I423D/K663D double mutant potently inhibited HIV-1 infection (greater than 11-fold; Figure 4D). WT MxB and interface 1 mutants were expressed at similar levels as assessed by western blotting (Figure 4E) and displayed similar subcellular localization (Figure 4F). These results indicate that MxB oligomerization through interface 1 is not critical for HIV-1 restriction.
MxB directly interacts with HIV-1 capsid assemblies
We initially probed MxB binding to HIV-1 capsid using HOS cell lysates and recombinant A14C/E45C CA, which forms stable, cross-linked tubular assemblies (Pornillos et al., 2009). The CA assemblies are sufficiently large to pellet in an Eppendorf centrifuge (Matreyek et al., 2013) and capsid-binding partners can be detected in a co-pelleting assay (Henning et al., 2014; Stremlau et al., 2006). MxB binding was quantified as the percent of input protein that co-pelleted in the presence of CA corrected for the level of protein that non-specifically pelleted in control reactions that lacked CA. WT MxB-HA efficiently co-pelleted with CA assemblies (Figure 5A; ~45% of input protein recovered versus ~8% non-specific pelleting). MxB interface 1 mutants K663D, I423D/K663D, and M666D also interacted efficiently with CA, yielding 27%, 40%, and 26% binding specificity, respectively (Figure 5A). Hinge 1 mutant E681A displayed reduced binding specificity compared to WT MxB (about 9.8% after background correction). Interface 2 mutants M574D and Y651D were more defective, yielding binding specificity values of about 2% and <0.1%, respectively. To assess the relevance of MxB-CA binding to restriction of HIV-1 infection, the two parameters were correlated by scatter plot analysis. The resulting negative correlation yielded a Spearman rank correlation coefficient P value of 0.028 (Figure 5B), indicating that the interaction between MxB and CA plays a role in restriction of HIV-1 infection.
To evaluate if MxB interacts directly with capsid, binding assays were performed using recombinant MxB protein purified from E. coli. The crystallization construct (MxB84YRGK) and full-length mutant MBP-MxBYRGK/IKM proteins were used in these experiments to ameliorate the aggregation tendency of the wild type protein. As before, binding specificity was assessed by correcting for the level of pelleted MxB in control reactions that lacked CA. Approximately 25% of MBP-MxBYRGK/IKM co-sedimented with CA after background (pelleted MxB without CA) correction (Figure 5C). Although the comparison of this result to the binding of WT MxB-HA in cell extracts should be approached with a note of caution, the results nevertheless indicate that cellular cofactors are not required for the binding between MxB and HIV-1 CA. In contrast, only ~9% of MxB84YRGK co-sedimented with CA (Figure 5C). Although this level of residual binding was significantly greater than the background control, significantly less MxB84YRGK as compared to full-length MBP-MxBYRGK/IKM co-pelleted with CA (P = 0.002). These data provide evidence of a direct interaction between MxB and HIV-1 capsid and indicate that the first 84 residues of MxB contribute to the interaction. This is consistent with the previously published results showing the MxB N-terminus is critical for HIV-1 restriction (Busnadiego et al., 2014; Goujon et al., 2014)(Matreyek et al., unpublished data). At this point we can only infer that the dimer is the basic CA binding unit of MxB; multiple attempts to purify recombinant dimerization mutants such as M574D failed to yield soluble protein for binding studies.
As certain CA mutations abolish MxB restriction of HIV-1, we investigated whether some of these (G89V and N74D) alter MxB's ability to bind HIV-1 CA assemblies in vitro. The G89V mutation abolishes CypA binding (Schaller et al., 2011) and eliminates MxB restriction of HIV-1 (Kane et al., 2013). The N74D mutation abolishes CA binding to the capsid cofactor CPSF6 (Lee et al., 2010; Price et al., 2012) and reduces but does not eliminate MxB's ability to restrict HIV-1 (Kane et al., 2013). Interestingly, neither of the two mutations reduced MxB binding to the CA assemblies in our assay (Figure 5C). These results show that these functionally important CA residues are not required for MxB binding.
To further probe the involvement of these CA residues in MxB binding we tested whether the presence of CypA or a capsid-binding peptide of CPSF6 (CPSF6313-327) altered the ability of MBP-MxBYRGK/IKM to bind CA. Our results show MBP-MxBYRGK/IKM binds to CA with similar affinity in the presence or absence of these two CA-binding partners (Figure 5C). Because we could not saturate the tubes with CypA and were uncertain if the CPSF6313-327 peptide occupies all available binding sites, it is not clear whether the binding events are mutually exclusive. However, as we did not see an increase in pelleted MBP-MxBYRGK/IKM in the presence of CypA or CPSF6313-327, these factors do not appear to enhance MxB binding.
The interaction between MBP-MxBYRGK/IKM or MBP-MxB84-715 YRGK/IKM and CA assemblies was additionally analyzed by cryoEM (Figure 5D). Inspection of the cryoEM images of the reaction mixtures shows distinct protein densities decorating the CA tubes that are only seen in the presence of the MxB constructs (Figure 5D). As expected, the decoration of CA tubes by MBP-MxBYRGK/IKM was much more pronounced than for MBP-MxB84-715 YRGK/IKM, confirming both direct protein binding and the importance of the MxB N-terminus in this interaction.
To probe the oligomeric state of CA required for MxB binding, we analyzed the ability of MxB to bind soluble CA hexamers. We incubated purified MBP-MxBYRGK/IKM with cross-linked CA hexamers and examined their interaction using size exclusion chromatography. No interaction was detected between the two as MBP-MxBYRGK/IKM and CA hexamers eluted from the size exclusion column as two distinct peaks at positions corresponding to those of the individual components (Figure 5E). This result, together with those from the co-pelleting and cryoEM assays, suggests that MxB does not have appreciable affinity for single CA hexamers but binds to capsid assemblies, implying that MxB may function as a capsid pattern sensor that only recognizes the assembled CA lattice.
Discussion
MxB is a recently identified HIV-1 restriction factor whose mode of inhibition is incompletely understood. The research presented herein establishes a structural and biochemical framework for understanding the mechanism by which MxB restricts HIV-1. Our results reveal characteristics of MxB that are required for its antiviral activity, uncover separate determinants for the functions of MxB and the homologous MxA, and, importantly, demonstrate a direct interaction between MxB and HIV-1 CA that requires higher-order capsid assembly. These results provide important insight into how MxB may bind viral capsid and interfere with capsid uncoating-related processes to inhibit HIV-1.
The structure and sequence of MxB and MxA are similar, but the two proteins have different antiviral characteristics. These proteins are dimers in solution and our data show that, like MxA, the dimerization of MxB is required for viral restriction. Contrary to MxA's mode of restriction that requires further multimerization, higher-order oligomerization of MxB is not critical for HIV-1 restriction. Mutation of residues at the higher-order interface 1 did not alter MxB activity (Figure 4D), while altering the corresponding residues on MxA abolished its restriction ability (Gao et al., 2010). This difference may be related to the presence of a NLS in MxB. The NLS-mediated localization of MxB to the nuclear envelope may increase its local concentration at the nuclear pore, which may mitigate the need for higher-order oligomers to exert its antiviral effects. In addition, we show that conformational coupling between the GTPase and the stalk domain, which is important for regulation of GTPase activity and antiviral function of MxA, is not necessary in MxB (Figure 2E).
Our identification of MxB as a direct HIV-1 capsid-binding protein substantially advances our understanding of the mechanism of HIV-1 restriction by MxB. Our data demonstrate that interaction between MxB and CA is dependent on the first 83 residues of MxB, and does not require other host factors (Figure 5). It has been well established that the N-terminal residues of MxB are critical for its anti-HIV-1 activity (Busnadiego et al., 2014; Goujon et al., 2014). This is partially attributed to the required nuclear localization of MxB (Kane et al., 2013)(Matreyek et al., unpublished data). Our results now show the N-terminus of MxB is also critical for its interaction with HIV-1 CA. This is consistent with the recent report that the N-terminal domain of MxB (91 amino acids), which includes residues downstream from the NLS, confers HIV-1 restriction to MxA (Goujon et al., 2014). These results suggest that the direct engagement between MxB and capsid is an important step in the restriction of HIV-1 by MxB.
During the review of this work, Fricke et al. reported that ectopically expressed MxB binds higher-order capsid assemblies in vitro and that MxB decreases the extent of HIV-1 capsid uncoating during infection (Fricke et al., 2014). These results agree with our finding that a direct interaction occurs between MxB and capsid. In contrast, Fricke et al. concluded that multimerization of MxB is important for the interaction with the HIV-1 core. Our crystal structure shows that the reported truncation mutations (Δ572-715 and Δ623-715) removed the majority of the stalk domain that is central to MxB dimer formation, and the reported point mutation (L661K) seems likely to disrupt MxB folding because it is located within the hydrophobic core of the stalk. The reported multimerization-disrupting mutations therefore likely disrupted MxB dimerization, which is consistent with the importance of dimerization revealed in our study.
Our structural and binding studies provide important insight into a HIV-1 capsid recognition. We observe binding of MxB to capsid assemblies, but not individual CA hexamers (Figure 5). This suggests that MxB, like TRIM5 restriction factors, is a capsid pattern sensor that recognizes higher-order CA assemblies (Pertel et al., 2011). This is further supported by data showing that CA residues 207, 208, and 210, which are near the trimeric interface of CA hexamers, are critical for MxB restriction (Busnadiego et al., 2014). Furthermore, we show that the N-terminus of MxB is important for the MxB-capsid interaction. These results, together with our crystal structure, allow us to create a model of an MxB dimer binding to the CA lattice (Figure 6). The stalk domains provide proper spacing over the CA lattice, as the distance is ~180 Å between the N-termini of the dimer, which matches the distance between the CA hexamer interfaces (Figure 6A). Each N-terminus is positioned near the interface between either trimers (Figure 6B) or dimers (Figure 6C) of CA hexamers, in a location amenable to binding. The predicted MxB-binding sites overlap with the binding site for the small molecule CPIPB inhibitor (referred to as compound 1 in Lemke et al., 2013), which is consistent with the report that this compound could competitively inhibit the binding of ectopically-expressed MxB to CA-nucleocapsid tubular structures in vitro (Fricke et al., 2014). The hinge regions and the potentially flexible N-terminus of MxB provide plasticity to adapt to the varying curvature of the CA core. Although the majority of binding is likely provided by the MxB N-terminus, its GTPase domain and hinge 1 region may also contact the capsid. This is consistent with our data that the N-terminal deletion construct MxB84YRGK retained some binding to capsid (Figure 5C and 5D) and that hinge 1 mutations reduced the capsid interaction (Figure 5A and 5B). While this model does not provide detailed information about the binding interfaces, it establishes a framework for understanding and guiding future studies on the interaction between MxB and HIV-1 capsid.
Figure 6. Conceptual binding model of the MxB dimer to HIV capsid.
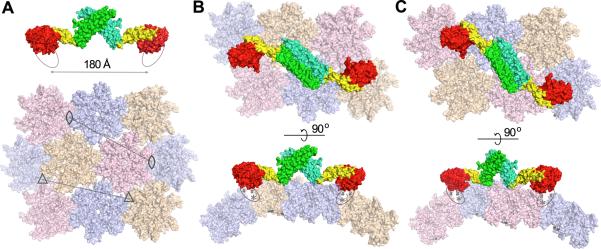
(A) The dimension of the MxB dimer matches the spacing (marked by the lines) between either trimers (3-fold axes marked by triangles) or dimers (2-fold axes marked by eye-shaped symbols) of CA hexamers. The MxB dimer (top) is colored with both the GTPase and the BSE domains in red and yellow, respectively, and with the stalk domain of each protomer in green or cyan. The N-terminus of MxB is indicated by an oval. The capsid model (bottom) was created by docking the crystal structure of HIV-1 CA hexamer (PDBID: 3H4E) to the EM map of HIV-CA helical tube (EMDB accession code: EMD-5136). (B) Two orthogonal views of a possible binding mode of the MxB dimer to the capsid at the interfaces of a trimer of CA hexamers. The GTPase domains of MxB are oriented such that the N-termini (ovals) of MxB can extend and interact with residues known to be important (207/210, asterisks) at the hexamer interfaces and the sites (marked by the # signs) where binding of the CPIPB inhibitor led to competitive inhibition of MxB binding to capsid. The flexibility of the MxB N-terminus and the hinge regions may allow MxB to adjust to the changing curvature of the HIV-1 capsid. The pink colored hexamers are removed in the side view for clarity. (B) Potential MxB binding to the interfaces of a dimer of CA hexamers. The tan colored hexamers are removed in the side view for clarity.
MxB likely functions as a critical player in a restriction pathway involving other host factors that interact with HIV-1 capsid. The capsid-interaction events of MxB and some of these factors, such as CypA and CPSF6, appear to be independent (Figure 5C). Our data show that MxB binds to CA mutants that abolish MxB's antiviral activity, demonstrating the mutated CA residues are not critical for binding MxB (Figure 5C). These CA mutations are important for binding of other cellular factors including CypA, CPSF6, NUP153 and NUP358 (Lee et al., 2010; Matreyek et al., 2013; Schaller et al., 2011). Furthermore, the presence of CypA or CPSF6313-327 does not enhance MxB binding to CA assemblies (Figure 5C). These results suggest that direct interactions between MxB and these CA-binding proteins are not required but instead MxB restriction may occur at an independent step in a pathway that is dependent on proper interaction between other cellular factors and CA. Alternatively, MxB may alter the affinity of other host factors for the capsid or the nature of their binding such that they become inhibitory. Taken together, MxB may be able to directly modulate or affect other cellular factors to control the stability of the HIV-1 capsid. This, coupled with the localization of MxB to the nuclear rim, suggests that MxB may function to interfere with the proper uncoating and translocation of the HIV-1 capsid core near the cell nucleus.
Experimental Procedures
Cloning and expression
MxBFL was cloned into pCDF Duet (Novagen) with an N-terminal MBP tag. MxB84-715 was cloned into pETDUET-1 (Novagen) with an N-terminal 6xHis tag. Proteins were overexpressed in E. coli BL21(DE3) cells at 18° C for 18 h by induction with 0.5 mM IPTG. MxB with a C-terminal HA was cloned into the retroviral transfer vector pLPCX (Clontech). Mutations were made by QuickChange site-directed mutagenesis (Stratagene) on MxB vector templates.
Protein purification
Bacterial cells were harvested by centrifugation at 5000 rpm. Cells were resuspended in lysis buffer (50 mM Tris pH 8.0, 500 mM NaCl, 20 mM imidazole, 0.1 mM TCEP) and lysed using a microfluidizer. Cell debris was clarified by centrifugation at 15,000 rpm for 45 min for 6xHis tagged construct and at 40,000 rpm for 35 min for MBP tagged constructs. MBP-MxB1-715 constructs were purified by MBP affinity and size exclusion chromatography. 6xHis MxB84-715 were purified by nickel affinity, anion exchange and size exclusion chromatography and were analyzed at each step by SDS-PAGE gel electrophoresis.
Crystallization and data collection
Crystallization of the protein was performed by the microbatch under-oil method (Chayen et al., 1990). 1 μl of the protein solution (2 mg ml−1 in 50 mM Tris, pH 8, 150 mM NaCl, 0.1 mM TCEP), 2 mM DTT and the precipitant solution (0.1 M MES, pH 6.5, 5% PEG4000, 10% 2-Propanol, 0.05 M MgCl) were mixed. Crystals formed at RT were cryoprotected using the precipitant solution with 30% glycerol before freezing in liquid nitrogen. Diffraction data were collected at the NE-CAT beamline 24ID-E at the Advanced Photon Source and the beamline X25 at the National Synchrotron Light Source. The data collection statistics are in Table 1.
Structure determination and refinement
The structure was solved using the GTPase domain and the stalk domain of MxA (Gao et al., 2011; Gao et al., 2010) as search models for molecular replacement using the CCP4 program Phaser (Collaborative Computational Project, 1994; McCoy et al., 2007; Vagin and Teplyakov, 2000). Iterative rounds of model building in COOT (Emsley and Cowtan, 2004) and refinement with REFMAC5 (Murshudov et al., 1997) and PHENIX (Adams et al., 2010) were carried out. Data sharpening was performed (Liu and Xiong, 2014) to facilitate model building. The final model has an Rwork/Rfree of 25.6%/29.6%. The refinement statistics are summarized in Table 1.
Cells and infectivity assays
HOS cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 0.1 mg/ml streptomycin. HOS cells stably transduced with LPCX transfer vectors were selected and maintained with 2 μg/ml puromycin.
Cells seeded onto 48-well plates were infected with various reporter viruses as described (Matreyek et al., 2013). Percentages of GFP-positive cells were determined 48 h post-infection using a FACSCanto flow cytometer equipped with FACSDIVA software. Virus inoculates were adjusted to yield ~40% GFP-positive cells in control samples that contained empty LPCX.
Immunofluorescence confocal microscopy
Cells cultured on Nunc Lab-Tek II chamber slides (Thermo Scientific) were fixed with 4% paraformaldehyde for 10 min, washed with phosphate buffered saline (PBS), and permeabilized with ice-cold MeOH for 10 min. The permeabilized cells were blocked with PBS containing 10% FBS for 30 min, and stained with 1:300 dilution of anti-HA antibody 16b12 (Covance). After a 30 min wash with PBS, the cells were incubated for 1 h with a 1:1,000 dilution of an Alexa Fluor 555 conjugated goat anti-mouse IgG antibody (Invitrogen), as well as Hoescht 33342 (Invitrogen) diluted to a concentration of 1 μg/ml. After an additional 30 min wash with PBS, the samples were covered with mounting medium (150 mM NaCl, 25 mM Tris pH 8.0, 0.5% N-propyl gallate, and 90% glycerol). The processed samples were analyzed on a Nikon Eclipse spinning disk confocal microscope at the Dana-Farber Cancer Institute Confocal and Light Microscopy core.
Western blotting
Cells pelleted at 300 × g were resuspended in PBS supplemented to contain 0.2% NP-40 and 10 U/ml Turbo DNAse in 1X Turbo DNAse buffer (Ambion). After 30 min on ice, mixtures were adjusted to contain 62.5 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 5% beta-mercaptoethanol, 100 mM DTT, and 0.001% bromophenol blue. Samples heated at 100 °C were separated on Tris-glycine polyacrylamide gels, transferred to polyvinylidene fluoride membrane, and MxB-HA was detected with a 1:2000 dilution of HRP-conjugated 3F10 antibody (Roche). Beta-actin was detected with a 1:10,000 dilution of HRP-conjugated antibody clone AC-15 (Sigma). The amount of MxB-HA or beta-actin signal in each sample was quantitated relative to the level of each signal in a matched WT MxB-expressing sample, which was set to 1, using Image Lab 4.1 (Bio-Rad). The MxB expression ratio was calculated by dividing the MxB-HA signal with that of beta-actin.
CA binding assay with cell lysate
Recombinant HIV-1 CA A14C/E45C was expressed in E. coli and purified as described previously (Pornillos et al., 2009). Cross-linked CA assemblies were prepared by direct dilution into assembly buffer (50 mM Tris pH 8.0, 1 M NaCl, 0.2 mM β-mercaptoethanol), incubation at 37°C for 1 h, and dilution to the final concentration of 4 mg/ml. HOS cells expressing WT or mutant MxB-HA were lysed in buffer A [50 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP40, 1x Complete protease inhibitor (Roche)], and lysates were clarified by centrifugation at 21,000 × g at 4 °C. Approximately 20 μg of lysate was mixed with assembled CA, yielding final concentrations of 64 μM CA and 5 mg/ml total cell protein, followed by incubation at RT for 30 min. Binding reactions were centrifuged through a 35% sucrose cushion in PBS at 21,000 × g at 4 °C for 30 min. The supernatant was removed, and the pellet was resuspended in buffer lacking reducing agent. MxB-HA was detected by western blotting whereas CA was detected by staining with Coomassie blue.
CA binding assays with purified proteins
Cross-linked CA (A14C/E45C) tubes were dialyzed overnight at 4°C into assembly buffer (1 M NaCl, 50 mM Tris pH 8.0), followed by dialysis into binding buffer (150 mM NaCl, 50 mM Tris pH 8.0) (Pornillos et al., 2009). MxB and control proteins were spun at 20,000 × g for 30 min at 4 °C. MxB (10 μM) and control proteins in 10.5 μL were added to 10.5 μL CA tubes and incubated at RT for 1 h. Subsequently, 7 μL aliquots were withdrawn. The remaining was pelleted at 20,000 × g for 30 min at 4 °C. Total, supernatant, and pellet samples were analyzed by SDS-PAGE. Co-pelleting experiments with CypA and CPSF6313-327 were performed as described with 30 μM CypA and 200 μM CPSF6313-327.
The interaction between MxB and cross-linked CA (A14C/E45C) hexamers was examined by size exclusion chromatography. CA hexamers were assembled as described previously (Pornillos et al., 2009). Samples (200 μL) of MBP-MxBYRGK/IKM (11.25 μM), CA hexamers (55 μM), and their mixtures were run on a Superdex 200 10/300 GL. Peak fractions were analyzed by SDS-PAGE. Gels were quantified with ImageJ.
CryoEM
Sample (3 μL) of purified MxB variants or the mixture from the binding assays was applied onto a glow-discharged Quantifoil (R2/2) grid. Excess fluid on the grid was blotted with filter paper and then the grid was rapidly frozen in liquid ethane using a homemade manual freezing device. Frozen grids were transferred into a Gatan cryo-holder and examined with a FEI Tecnai 200 kV Field Emission Gun transmission electron microscope equipped with a Gatan 4K × 4K charge-coupled device camera. Low dose (~20e−/Å2) images were recorded at nominal magnifications of either 29000× or 50000× at underfocus values of 2-4 μm.
Highlights.
- MxB forms an antiparallel dimer that is important for HIV-1 restriction.
- MxB interacts directly with HIV-1 capsid assemblies.
- MxB dimerization is important for restriction and its interaction with HIV-1 capsid.
- Higher-order MxB oligomerization is not essential for restriction or capsid binding.
Acknowledgements
We thank Q. Zhao for technical assistance and discussions. We also thank the staff at the Advanced Photon Source beamline 24-ID and the National Synchrotron Light Source beamlines X25. This work was funded in part by NIH grants AI052014 (to A.E.) and GM082251 (to A.E., C.A., and P.Z.), and a Collaboration Development Pilot Program award from the Pittsburgh Center for HIV Protein Interactions (to Y.X.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors Contributions
J.L.F., H.C.N., and K.A.M. contributed equally to this work. J.L.F. and H.C.N. performed protein expression and purification, crystallization, and data collection. J.L.F. and B.J.S. performed in vitro co-pelleting assays. H.C.N. performed structure determination and analysis. K.A.M. performed immunofluorescence confocal microscopy. K.A.M and T.G.D. performed cell-based assays and cell lysate co-pelleting assays. F.J.D.A performed cryo-EM studies and binding assays. C.R.A. provided capsid proteins. J.LF, H.C.N., K.A.M, C.A., P.Z., A.E., and Y.X. designed the experiments. J.L.F., H.C.N., K.A.M., A.E, and Y.X. wrote the manuscript with input from all authors.
Accession Numbers
The Protein Data Bank accession code for the MxB structure and diffraction data is 4WHJ.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebi M, Fah J, Hurt N, Samuel CE, Thomis D, Bazzigher L, Pavlovic J, Haller O, Staeheli P. cDNA structures and regulation of two interferon-induced human Mx proteins. Molecular and cellular biology. 1989;9:5062–5072. doi: 10.1128/mcb.9.11.5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose Z, Aiken C. HIV-1 uncoating: connection to nuclear entry and regulation by host proteins. Virology. 2014;454-455:371–379. doi: 10.1016/j.virol.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busnadiego I, Kane M, Rihn SJ, Preugschas HF, Hughes J, Blanco-Melo D, Strouvelle VP, Zang TM, Willett BJ, Boutell C, et al. Host and viral determinants of Mx2 antiretroviral activity. Journal of virology. 2014;88:7738–7752. doi: 10.1128/JVI.00214-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chayen NE, Stewart PDS, Maeder DL, Blow DM. An Automated-System for Microbatch Protein Crystallization and Screening. J Appl Crystallogr. 1990;23:297–302. [Google Scholar]
- Collaborative Computational Project, N The CCP4 suite: programs for protein crystallography. Acta crystallographica Section D, Biological crystallography. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Daumke O, Gao S, von der Malsburg A, Haller O, Kochs G. Structure of the MxA stalk elucidates the assembly of ring-like units of an antiviral module. Small GTPases. 2010;1:62–64. doi: 10.4161/sgtp.1.1.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo C, Hefti HP, Meli M, Landis H, Pavlovic J. Intramolecular backfolding of the carboxyl-terminal end of MxA protein is a prerequisite for its oligomerization. The Journal of biological chemistry. 1999;274:32071–32078. doi: 10.1074/jbc.274.45.32071. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fricke T, White TE, Schulte B, de Souza Aranha Vieira DA, Dharan A, Campbell EM, Brandariz-Nunez A, Diaz-Griffero F. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology. 2014;11:68. doi: 10.1186/s12977-014-0068-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, von der Malsburg A, Dick A, Faelber K, Schroder GF, Haller O, Kochs G, Daumke O. Structure of myxovirus resistance protein a reveals intra- and intermolecular domain interactions required for the antiviral function. Immunity. 2011;35:514–525. doi: 10.1016/j.immuni.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Gao S, von der Malsburg A, Paeschke S, Behlke J, Haller O, Kochs G, Daumke O. Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature. 2010;465:502–506. doi: 10.1038/nature08972. [DOI] [PubMed] [Google Scholar]
- Goujon C, Moncorge O, Bauby H, Doyle T, Barclay WS, Malim MH. Transfer of the Amino-Terminal Nuclear Envelope Targeting Domain of Human MX2 Converts MX1 into an HIV-1 Resistance Factor. Journal of virology. 2014;88:9017–9026. doi: 10.1128/JVI.01269-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon C, Moncorge O, Bauby H, Doyle T, Ward CC, Schaller T, Hue S, Barclay WS, Schulz R, Malim MH. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature. 2013;502:559–562. doi: 10.1038/nature12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller O, Gao S, von der Malsburg A, Daumke O, Kochs G. Dynamin-like MxA GTPase: structural insights into oligomerization and implications for antiviral activity. The Journal of biological chemistry. 2010;285:28419–28424. doi: 10.1074/jbc.R110.145839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller O, Kochs G. Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2011;31:79–87. doi: 10.1089/jir.2010.0076. [DOI] [PubMed] [Google Scholar]
- Hefti HP, Frese M, Landis H, Di Paolo C, Aguzzi A, Haller O, Pavlovic J. Human MxA protein protects mice lacking a functional alpha/beta interferon system against La crosse virus and other lethal viral infections. Journal of virology. 1999;73:6984–6991. doi: 10.1128/jvi.73.8.6984-6991.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning MS, Dubose BN, Burse MJ, Aiken C, Yamashita M. In vivo functions of CPSF6 for HIV-1 as revealed by HIV-1 capsid evolution in HLA-B27-positive subjects. PLoS pathogens. 2014;10:e1003868. doi: 10.1371/journal.ppat.1003868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature. 2013;502:563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, Raposo G, Lemmon MA. Inhibition of nuclear import and cell-cycle progression by mutated forms of the dynamin-like GTPase MxB. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8957–8962. doi: 10.1073/pnas.0403167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochs G, Haener M, Aebi U, Haller O. Self-assembly of human MxA GTPase into highly ordered dynamin-like oligomers. The Journal of biological chemistry. 2002a;277:14172–14176. doi: 10.1074/jbc.M200244200. [DOI] [PubMed] [Google Scholar]
- Kochs G, Haller O. GTP-bound human MxA protein interacts with the nucleocapsids of Thogoto virus (Orthomyxoviridae). The Journal of biological chemistry. 1999a;274:4370–4376. doi: 10.1074/jbc.274.7.4370. [DOI] [PubMed] [Google Scholar]
- Kochs G, Haller O. Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proceedings of the National Academy of Sciences of the United States of America. 1999b;96:2082–2086. doi: 10.1073/pnas.96.5.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochs G, Janzen C, Hohenberg H, Haller O. Antivirally active MxA protein sequesters La Crosse virus nucleocapsid protein into perinuclear complexes. Proceedings of the National Academy of Sciences of the United States of America. 2002b;99:3153–3158. doi: 10.1073/pnas.052430399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, et al. Flexible use of nuclear import pathways by HIV-1. Cell host & microbe. 2010;7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke CT, Titolo S, Goudreau N, Faucher AM, Mason SW, Bonneau P. A novel inhibitor-binding site on the HIV-1 capsid N-terminal domain leads to improved crystallization via compound-mediated dimerization. Acta crystallographica Section D, Biological crystallography. 2013;69:1115–1123. doi: 10.1107/S0907444913006409. [DOI] [PubMed] [Google Scholar]
- Liu C, Xiong Y. Electron density sharpening as a general technique in crystallographic studies. J Mol Biol. 2014;426:980–993. doi: 10.1016/j.jmb.2013.11.014. [DOI] [PubMed] [Google Scholar]
- Liu Z, Pan Q, Ding S, Qian J, Xu F, Zhou J, Cen S, Guo F, Liang C. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell host & microbe. 2013;14:398–410. doi: 10.1016/j.chom.2013.08.015. [DOI] [PubMed] [Google Scholar]
- Matreyek KA, Engelman A. Viral and cellular requirements for the nuclear entry of retroviral preintegration nucleoprotein complexes. Viruses. 2013;5:2483–2511. doi: 10.3390/v5102483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek KA, Yucel SS, Li X, Engelman A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS pathogens. 2013;9:e1003693. doi: 10.1371/journal.ppat.1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melen K, Julkunen I. Nuclear cotransport mechanism of cytoplasmic human MxB protein. The Journal of biological chemistry. 1997;272:32353–32359. doi: 10.1074/jbc.272.51.32353. [DOI] [PubMed] [Google Scholar]
- Melen K, Keskinen P, Ronni T, Sareneva T, Lounatmaa K, Julkunen I. Human MxB protein, an interferon-alpha-inducible GTPase, contains a nuclear targeting signal and is localized in the heterochromatin region beneath the nuclear envelope. The Journal of biological chemistry. 1996;271:23478–23486. doi: 10.1074/jbc.271.38.23478. [DOI] [PubMed] [Google Scholar]
- Melen K, Ronni T, Broni B, Krug RM, von Bonsdorff CH, Julkunen I. Interferon-induced Mx proteins form oligomers and contain a putative leucine zipper. The Journal of biological chemistry. 1992;267:25898–25907. [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta crystallographica Section D, Biological crystallography. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Pavlovic J, Schroder A, Blank A, Pitossi F, Staeheli P. Mx proteins: GTPases involved in the interferon-induced antiviral state. Ciba Foundation symposium. 1993;176:233–243. doi: 10.1002/9780470514450.ch15. discussion 243-237. [DOI] [PubMed] [Google Scholar]
- Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 2011;472:361–365. doi: 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. X-ray structures of the hexameric building block of the HIV capsid. Cell. 2009;137:1282–1292. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash B, Praefcke GJ, Renault L, Wittinghofer A, Herrmann C. Structure of human guanylate-binding protein 1 representing a unique class of GTP-binding proteins. Nature. 2000;403:567–571. doi: 10.1038/35000617. [DOI] [PubMed] [Google Scholar]
- Price AJ, Fletcher AJ, Schaller T, Elliott T, Lee K, KewalRamani VN, Chin JW, Towers GJ, James LC. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS pathogens. 2012;8:e1002896. doi: 10.1371/journal.ppat.1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt M, Stertz S, Krijnse-Locker J, Haller O, Kochs G. Missorting of LaCrosse virus nucleocapsid protein by the interferon-induced MxA GTPase involves smooth ER membranes. Traffic. 2004;5:772–784. doi: 10.1111/j.1600-0854.2004.00219.x. [DOI] [PubMed] [Google Scholar]
- Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hue S, Fletcher AJ, Lee K, KewalRamani VN, et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS pathogens. 2011;7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwemmle M, Richter MF, Herrmann C, Nassar N, Staeheli P. Unexpected structural requirements for GTPase activity of the interferon-induced MxA protein. The Journal of biological chemistry. 1995;270:13518–13523. [PubMed] [Google Scholar]
- Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5514–5519. doi: 10.1073/pnas.0509996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta crystallographica Section D, Biological crystallography. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]