

Abstract
Nicotinamide adenine dinucleotide, NAD+, is a small metabolite coenzyme that is essential for the progress of crucial cellular pathways including glycolysis, the tricarboxylic acid cycle (TCA) and mitochondrial respiration. These processes consume and produce both oxidative and reduced forms of NAD (NAD+ and NADH). NAD+ is also important for ADP(ribosyl)ation reactions mediated by the ADP-ribosyltransferase enzymes (ARTDs) or deacetylation reactions catalysed by the sirtuins (SIRTs) which use NAD+ as a substrate. In this review, we highlight the significance of NAD+ catabolism in DNA repair and cell death through its utilization by ARTDs and SIRTs. We summarize the current findings on the involvement of ARTD1 activity in DNA repair and most specifically its involvement in the trigger of cell death mediated by energy depletion. By sharing the same substrate, the activities of ARTDs and SIRTs are tightly linked and dependent on each other and are thereby involved in the same cellular processes that play an important role in cancer biology, inflammatory diseases and ischemia/reperfusion.
Keywords: NAD+, ADP(Ribosyl)ation, energy depletion, metabolic defects
1. NAD+/NADH coenzyme
1.1. NAD+ production pathways
Nicotinamide adenine dinucleotide is an essential metabolite and coenzyme present in cells in both an oxidatized form (NAD+) and reduced form (NADH). It is composed of two nucleotides, an adenine monophosphate (AMP) and a nicotinamide mononucleotide (NMN) (Figure 1A). NAD is an electron carrier and is involved in redox reactions occurring during cell metabolism. It is synthesized de novo from the amino acid tryptophan [1] but can also be produced by salvage pathways from three precursors: nicotinic acid (Na) (vitamin B3 or niacin), nicotinamide (Nam) and nicotinamide riboside (NR). Nam is also directly released from the ADP-ribose transfer reaction that consumes NAD+ (Figure 1B). This recycling pathway is mostly activated under cellular stress when the exogenous precursors are not directly available.
Figure 1.

A. Structure of NAD. Nicotinamide adenine dinucleotide (NAD+) is composed of two nucleotides, an adenine monophosphate (AMP) and a nicotinamide mononucleotide (NMN) linked together by two phosphate groups. During the ADP-ribosylation reaction the ADP-ribose unit is transferred to target proteins by ARTD enzymes and a unit of nicotinamide (Nam) is released.
B. Pathways of NAD+ synthesis. NAD+ is synthesized by a de novo pathway from the amino acid tryptophan (black arrows) and by a salvage pathway (Blue arrows).
1.2. NAD+ catabolism pathways
1.2.1. Redox roles of NAD+
NAD+ and NADH have an essential role in glycolysis, in the mitochondrial electron transport chain and in the tricarboxylic acid (TCA) cycle. These three important cellular pathways are inter-connected through a process of provider/consumer of both forms of NAD and towards the goal of ATP production (Figure 2). Therefore, cells maintain a high concentration of both NAD+ and NADH.
Figure 2.

Catabolism of NAD+/NADH. NAD+ is used by glycolysis in the cytoplasm to produce NADH which can migrate into the mitochondria to be used as a coenzyme by the respiratory chain which in turn releases NAD+. In the mitochondria, the TCA cycle is also a consumer of NAD+ and provides NADH to the respiratory chain to produce ATP and release NAD+. The NAD+ produced is used by ARTDs and SIRTs to assure their catalytic activity.
Glycolysis produces pyruvate from glucose through several different steps including the reduction of NAD+ to NADH in the cytosol. NADH can be transferred into the mitochondria through NADH shuttles [2, 3] and affects oxidative phosphorylation. The mitochondrial TCA cycle is also a consumer of NAD+ and the main provider of reduced NAD to the respiratory chain. NADH is the electron donor and its oxidation to NAD+ is mediated by complex I of the respiratory chain. The electron is then transferred through the different respiratory chain complexes until ATP synthase, or complex V, generates ATP through oxidative phosphorylation [4].
1.2.2. Non-redox roles of NAD+
Beside its role in energy production, the NAD+ coenzyme is a substrate of the ADP-ribosyltransferase enzymes (ARTD or PARP) [5] and of the sirtuins, NAD-dependent deacetylases [6].
1.2.2.1 Poly(ADP-ribosylation)
Poly(ADP-ribosyl)ation is a posttranslational modification of proteins catalysed by the poly(ADP-ribose) polymerases (PARPs), newly named ADP-ribosyltransferases diphtheria toxin-like proteins (ARTDs) [5]. The ARTD family is composed of 17 enzymes involved in multiple cellular pathways such as DNA repair response, chromatin remodelling, transcription, telomere homeostasis or cell death [7–9]. Some ARTDs are able to catalyse the poly(ADP-ribosyl)ation reaction while others are responsible for mono(ADP-ribosyl)ation [5]. In both cases, ARTDs transfer the ADR-ribose moiety of NAD+ to the carboxyl group of lysine, aspartic and glutamic acid residues of the acceptor proteins [10, 11] and release the nicotinamide unit (Figure 1B). ARTD activity is the main NAD+ catabolic process in the cell and forces the cell to continuously synthesize NAD+ from the de novo pathway or recycling pathway in the case of cellular stress, mostly during the DNA repair process. The founding member of the ARTD family, ARTD1, as well as ARTD2 and ARTD3, are involved in DNA repair and are referred to as DNA dependent-ARTDs. ARTD3 has been shown to play a major role in the repair of DNA double-strand breaks [12, 13], while ARTD1 and ARTD2 activation is triggered by DNA single-strand breaks occurring during Base Excision Repair [14].
1.2.2.2. Deacetylation
Sirtuins belong to a seven member family of NAD+ dependent deacetylases. The budding yeast gene Sir2 (Silencing information regulator 2) was the first sirtuin gene discovered [15]. The mammalian sirtuin family is composed of 7 isoforms (SIRT1-7) and each have functions with distinct subcellular localisation [16]. They have been shown to play pivotal roles in genome stability, mitochondrial and oxidative metabolism [17] and lifespan regulation. The sirtuins catalyse the removal of an acetyl group present on proteins by using NAD+ and releasing nicotinamide and acetyl-ADP-ribose units [18, 19]. As an NAD+ consumer, sirtuin activity is greatly dependent on the availability of the coenzyme and several studies have shown that the other NAD+ consuming enzymes such as ARTDs or cADP-ribose synthase might influence sirtuin activity by reducing NAD+ availability [20].
2. Role of NAD+ catabolism in DNA repair and cell death
ARTD1 (PARP1) involvement in DNA repair has been the first and the most extensively described role of the enzyme in past decades. However, this role of ARTD1 in DNA repair is primarily following moderate levels of DNA damage. Conversely, in response to excessive DNA damage, pADPr can become a death signal for the cell enduring the genotoxic insult.
2.1 Poly(ADP-ribosyl)ation in DNA repair
The involvement of ARTD1 and ARTD2 in DNA repair has been known for decades. The first studies reported that over-expression of the DNA binding domain of ARTD1 in human fibroblasts or its constitutive expression in HeLa cells allowed the discovery of a role for this enzyme in the repair of DNA damage, in particular in single-strand break and Base Excision Repair [21, 22]. In fact, over-expression of the dominant-negative mutant of ARTD1, or depletion of ARTD1 via RNA interference does not alter cell proliferation under normal conditions. However, upon alkylating agent treatment (MNNG, MMS) or gamma irradiation, triggers cell hypersensitivity, a decrease in cell survival and eventually apoptotic cell death [22–24]. Moreover, knockout of ARTD1 in mice and in fibroblasts leads to genomic instability after gamma irradiation and alkylating agent treatment, an increase in sister chromatin exchange events and cell cycle arrest in G2/M [25–28]. The mechanism of ARTD1 and ARTD2 activation by single-strand and double-strand DNA breaks is now well established. ARTD1 and ARTD2 activation, following their binding to DNA damage site, leads to chromatin relaxation by modification of histones H1 and H2B which helps in the recruitment of DNA repair proteins such as XRCC1 and DNA Polymerase β which have a strong affinity for poly(ADP-ribose) (pADPR) [29]. ARTD1 has been indicated as the main NAD+ consuming enzyme responsible for more than 99% of pADPr synthesis during genotoxic stress [30–32]. ARTD1 also undergoes auto-modification which triggers its release from the chromatin and its recycling by the degradation of pADPr by Poly(ADP-Ribose) Glycohydrolase, PARG. PARG is present in the cells in mainly 4 isoforms, coded from one gene: one in the nucleus, 2 in the cytosol and 1 in the mitochondria [29, 33, 34]. Hence, ARTD1 and PARG activities are responsible for a tight balance between pADPr production and degradation. Any dis-regulation of this balance toward an over production of pADPr is detrimental for the cell, in particular due to NAD+ over consumption. This aspect of NAD+ catabolism is further addressed in the following sections.
2.2 Poly(ADP-ribosylation) mediated cell death
Poly(ADPRibosyl)ation mediated cell death has been named Parthanatos after the fusion of PAR for Poly(ADP-Ribose) and Thanatos, the personification of death in greek mythology and has been designated as pADPr mediated cell death [35, 36]. Dawson and co-workers reported for the first time the toxicity of free pADPr [35]. They injected in vitro synthesized pADPr into living cells and observed cell death dependent on both the size and dose of the pADPr. This cell death was caspase-independent and was prevented by the pre-treatment of pADPr with PARG, supposedly by the lack of the more toxic long pADPR polymers. Two main mechanisms responsible for ARTD1 dependent cell death have been described, both requiring excessive pADPr production.
2.2.1. Programmed necrosis
The pADPr, when produced in large quantities, is able to migrate from the nucleus into the cytosol and trigger the release of the mitochondrial protein Apoptosis Inducing Factor (AIF), a flavoprotein which in turn is transferred to the nucleus where it condenses the chromatin and activates endonucleases leading to apoptosis [37, 38]. In parallel, AIF is responsible for the loss of mitochondrial membrane potential and release of cytochrome c, amplifying the apoptotic cascade [39]. Interestingly, AIF encodes a PAR Binding Motif (PBM) and its mutation diverts its role in cell death in favour of its primarily mitochondrial role of cell survival after treatment by the alkylating agent MNNG or the neurotoxin NMDA [40]. The role of PARG in this process has been reported as essential but dual and is still being evaluated. The absence of the PARG catalytic domain leads to a larval lethality in Drosophila at a normal temperature of 25°C, while a PARG knockout in mice triggers early embryonic death [41, 42]. The stem cells that originated from these mice present with a high basal level of pADPr and die by apoptosis. The absence of the nuclear isoform of PARG in mice leads to pADPr accumulation in the brain and worsens the damage created after brain ischemia [43]. Consequently, PARG over-expression protects mice from neuronal cell death [36]. Conversely, other studies reported that the absence of nuclear PARG in mice protects against renal and intestinal injury after ischemia/reperfusion, while sensitizes them after alkylating agent or ionizing radiation treatment [44–47]. Moreover, it has been shown that siRNA depletion of PARG in HeLa cells is beneficial for undamaged cells, protecting them from spontaneous DNA single-strand breaks and telomeric abnormalities while increasing radiosenstivity [48]. More recently, it has been demonstrated that PARG activities are essential for the release of AIF in MEFs exposed to H2O2 and in which ARH3, another enzyme responsible for pADPr hydrolysis, has been knocked out [49]. The authors showed that PARG activity is essential to generate free pADPr which can migrate into the cytosol and interact with AIF (Figure 3). In contrast, ARH3 would hydrolyse the pADPr to ADPr units and protect the cell against AIF dependent cell death.
Figure 3.
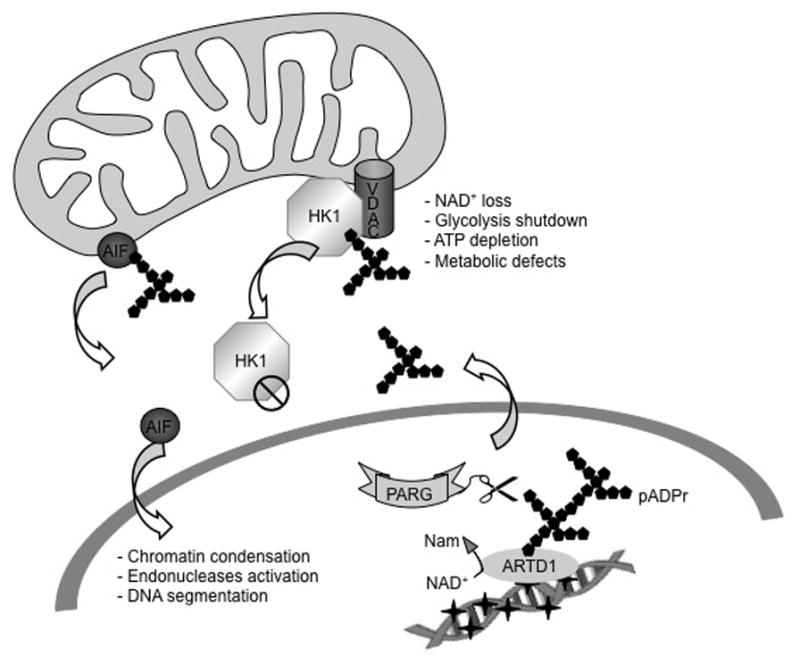
pADPr mediated cell death. Upon excessive DNA damage, the hyper-activation of ARTD1 triggers cell death via two different mechanisms. Programmed necrosis involves the release of pADPr chains by PARG into the cytosol that can then bind the mitochondrial protein AIF. AIF is released from the mitochondria and reaches the nucleus where it triggers DNA condensation, endonuclease activation and DNA segmentation. The second mechanism describes the nuclear hyper-activation of ARTD1 triggers NAD+ depletion in the mitochondria followed by loss in the cytosol and in the nucleus. Rapid ATP depletion is observed by the shutdown of the metabolic pathways relying on NAD+ such as glycolysis, the TCA cycle and mitochondrial respiration. However, a new role of ARTD1 and PARG in this process has been discovered in which the release of pADPr chains into the cytosol by PARG activity leads to the inactivation of HK1 and its release in the cytosol [56, 65].
2.2.2. ARTD suicide mechanism
The second mechanism, named “ARTD suicide” by Berger, is described as a passive form of cell death resulting from excessive energetic and metabolic depletion due to ARTD1 hyper-activation [50–52]. It has been widely observed by numerous studies that ARTD1 hyper-activation, upon massive DNA damage, is responsible for as much as 80 to 90% loss of the coenzyme NAD+ pool [53–56]. Some reports demonstrate that the loss of NAD+ precedes the induction of mitochondrial depolarization and mitochondria outer membrane permeability transition [57, 58]. Moreover, the metabolic pathways relying on NAD availability, such as glycolysis, the TCA cycle and oxidative phosphorylation, are largely impaired leading to a loss of ATP production and thereby amplifying the energetic depletion phenotype [59]. This ATP depletion occurs first in the mitochondria and is followed by a loss in both the cytosol and the nucleus [56]. The ATP depletion can also occur in an effort to re-synthesize the NAD+ pool by the ATP consuming enzymes Phophoribosyl Pyrophosphate Synthase (PRT) and nicotinamide mononucleotide adenyl transferase (NMNAT), from the nicotinamide released by the poly(ADP-ribosyl)ation reaction [52]. The role of ARTD1 activation and an impact on the cellular metabolic pathways, in the mechanism of the ARTD suicide, has become clearer. Two reports demonstrated that in astrocytes, cytosolic NAD+ depletion, resulting from ARTD1 activation, triggers a glycolytic block that can be rescued by NAD+ or TCA cycle substrates, such as α-ketoglutarate and pyruvate [53, 60] as well as by a treatment with the NAD+ biosynthesis precursor nicotinamide riboside (NR) preceding MNNG treatment [56]. Moreover, in apoptosis-deficient cells, it is suggested that only cells relying on glycolysis are sensitive to DNA damage-mediated ARTD1 hyper-activation and necrotic cell death [61].
2.2.3 A direct role of ARTD1 in glycolysis regulation after DNA damage
As mentioned above, ARTD1 or PARP1 mediated cell suicide has been attributed to a form of passive necrotic cell death resulting from bio-energetic catastrophe, NAD+ depletion, metabolic pathway shutdown and ATP loss [62]. However, recent studies have revealed a more direct role for ARTD1-activation in the process of glycolysis inhibition in response to DNA damage. A mass spectrometry analyses performed in HeLa cells [63] as well as in glioblastoma cells [56], in an attempt to identify the pADPr binding partners after alkylating agent treatment, identified the glycolytic enzyme hexokinase 1 (HK1). HK1 catalyses the first step of glycolysis in which a molecule of glucose is phosphorylated to yield glucose-6-phosphate. HK1 is one of the three major iso-enzymes of the hexokinase family. HK1 is mainly associated with the outer mitochondrial membrane through an interaction with the channel protein VDAC [64]. This interaction is known to facilitate the exchange of ATP/ADP between the mitochondria and the cytosol. Two recent studies have shown that HK1 is able to bind pADPr through an HK1-encoded pADPr Binding Domain (PBM), leading to the inhibition of its activity [56, 65]. More importantly, Fouquerel et al suggested a mechanism in which, like AIF, HK1 is released from the mitochondria upon ARTD1 hyper-activation by MNNG treatment in glioblastoma cells unable to complete BER. This release has been reported to be responsible for HK1 inactivation [66]. Thus pADPr binding to HK1 and its release into the cytosol constitutes a mechanism that would explain the glycolytic block observed after DNA damage-mediated ARTD1 hyper-activation (Figure 3). Importantly, it was demonstrated that the glycolytic block is dependent on PARG activity [56]. These reports bring a new level of complexity to pADPr-mediated cell death and describe a more active role of ARTD1, PARG and pADPr in the metabolic shutdown observed after excessive DNA damage or aborted/failed DNA repair.
2.3. Sirtuins in DNA repair and cell death
Sirtuins are a family of protein deacetylases composed of seven members, named SIRT1-SIRT7, and are expressed in various subcellular locations. SIRT1, SIRT6 and SIRT7 are present in the nucleus, SIRT2 plays its role in the cytoplasm and SIRT3, SIRT4 and SIRT5 are mitochondrial proteins [67]. As NAD+ consumers, the activities of Sirtuins and ARTDs are inter-dependent and are involved in many of the same biological processes [68]. Thus, some Sirtuin members, more particularly SIRT1 and SIRT6, have been shown to play an important role in DNA repair and cell metabolism after DNA damage.
SIRT1 is the most characterized sirtuin family member in mammals. Its involvement in DNA repair and cell death has been extensively reported. Deletion (knockout) of SIRT1 induced early postnatal lethality in mice that present with chromosomal and DNA repair defects [69]. Moreover, SIRT1 inhibition or SIRT1 knockdown by siRNA inhibits the growth of acute myeloid leukaemia cells and sensitizes them to chemotherapy [70]. This effect is explained by the observation that SIRT1 is able to deacetylate the pro-apoptotic protein protein p53 to reduce its transcriptional activity leading to a reduction of apoptosis induction following DNA damage [71, 72]. In this context, NAD+ depletion mediated by ARTD1 activation after oxidative DNA damage, leads to SIRT1 activity impairment and stimulates apoptosis [73]. Interestingly, the absence of SIRT1 enhances ARTD1 activity that is further emphasized by oxidative damage and triggers AIF release from the mitochondria [74]. SIRT1 is also directly responsible, upon H2O2 treatment, for the deacetylation of the transcription factor FOXO, leading to the activation of genes responsible for the control of cell cycle arrest, detoxification of ROS, DNA damage repair and inhibiting those involved in apoptosis [75].
SIRT6 has been recently reported to be involved in DNA repair regulation. SIRT6 is able to translocate to DNA damage sites and promote DNA repair and the loss of SIRT6 (mouse KO cells) triggers sensitivity of fibroblasts to ionizing radiation [4, 76]. More importantly, SIRT6 is directly involved in Base Excision Repair and DNA double-strand break repair by interacting with and mono-ADP-ribosylating ARTD1, thereby stimulating its activity [4]. These studies revealed the complexity of the relationship between the sirtuins and ARTD1 as they are capable of regulating each other either directly or indirectly by the consumption of their common coenzyme, the NAD+.
3. Concluding remarks
In this review, we highlight the crucial role of the coenzyme NAD+ in DNA repair, cellular metabolism and cell death through its utilization as a coenzyme for essential proteins, ARTDs and SIRTs. The balance and cooperation between ARTD1 and PARG activities as well as between ARTD1 and SIRT1 or SIRT6 are key to the control of cell fate. More importantly, the dual role of pADPr in both DNA repair and cell death demonstrates how pADPr production and degradation can promote a choice between cell survival and cell death. Following minimal DNA damage, the moderate production of pADPr will allow the recruitment of DNA damage response and DNA repair proteins to promote DNA repair and cell survival. Conversely, in the face of excessive DNA damage, the over-consumption of NAD+, due to the over-activation of ARTD1, contributes, along with the direct targeting of key proteins such as AIF or HK1 by pADPr, to the energy and metabolic defect, eventually leading to cell death. Understanding these mechanisms is of great importance in cancer therapy in which ARTD1 activity regulates DNA repair but also in the treatment of acute or chronic inflammatory diseases and brain, cardiac or kidney ischemia/reperfusion characterized by ARTD1 activation due to oxidative stress, for which the ARTD1 inhibition will lead to cell survival.
Acknowledgments
This work was supported by grants from the National Institute of Health (NIH) [CA148629] to RWS.
Abbreviations
- AMP
Adenine Monophophate
- ARTD
ADP-ribosyltransferase diphtheria toxin-like
- HK1
Hexokinase 1
- NAD
Nicotinamide Andenine Dinucleotide
- Nam
Nicotinamide
- NR
Nicotinamide Riboside
- Na
Nicotinic acid
- MNNG
N-methyl-N′-nitro-N-nitrosoguanidine
- PBM
Poly(ADP-ribose)AR Binding Motif
- pADPr
Poly(ADP-ribose)
- PARG
Poly(ADP-Ribosyl) Glycohydroalse
- SIRT
Silent Information Regulator 1
- TCA
Tricarboxylic Acid
- VDAC
Voltage Dependent Anion Chanel
Footnotes
5. Conflict of interest
RWS is a scientific consultant for Trevigen, Inc. The remaining authors state that there is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pollak N, Dolle C, Ziegler M. The power to reduce: pyridine nucleotides--small molecules with a multitude of functions. Biochem J. 2007;402:205–218. doi: 10.1042/BJ20061638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marzulli D, La Piana G, Fransvea E, Lofrumento NE. Modulation of cytochrome c-mediated extramitochondrial NADH oxidation by contact site density. Biochem Biophys Res Commun. 1999;259:325–330. doi: 10.1006/bbrc.1999.0787. [DOI] [PubMed] [Google Scholar]
- 3.Bakker BM, Overkamp KM, van Maris AJ, Kotter P, Luttik MA, van Dijken JP, Pronk JT. Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol Rev. 2001;25:15–37. doi: 10.1111/j.1574-6976.2001.tb00570.x. [DOI] [PubMed] [Google Scholar]
- 4.Rich PR. The molecular machinery of Keilin’s respiratory chain. Biochem Soc Trans. 2003;31:1095–1105. doi: 10.1042/bst0311095. [DOI] [PubMed] [Google Scholar]
- 5.Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35:208–219. doi: 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Yu J, Auwerx J. The role of sirtuins in the control of metabolic homeostasis. Ann N Y Acad Sci. 2009;1173(Suppl 1):E10–19. doi: 10.1111/j.1749-6632.2009.04952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hakme A, Wong HK, Dantzer F, Schreiber V. The expanding field of poly(ADP-ribosyl)ation reactions. ‘Protein Modifications: Beyond the Usual Suspects’ Review Series. EMBO reports. 2008;9:1094–1100. doi: 10.1038/embor.2008.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wacker DA, Frizzell KM, Zhang T, Kraus WL. Regulation of chromatin structure and chromatin-dependent transcription by poly(ADP-ribose) polymerase-1: possible targets for drug-based therapies. Subcell Biochem. 2007;41:45–69. doi: 10.1007/1-4020-5466-1_3. [DOI] [PubMed] [Google Scholar]
- 9.Kraus WL. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr Opin Cell Biol. 2008;20:294–302. doi: 10.1016/j.ceb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altmeyer M, Messner S, Hassa PO, Fey M, Hottiger MO. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009;37:3723–3738. doi: 10.1093/nar/gkp229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008;13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 12.Boehler C, Gauthier LR, Mortusewicz O, Biard DS, Saliou JM, Bresson A, Sanglier-Cianferani S, Smith S, Schreiber V, Boussin F, Dantzer F. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc Natl Acad Sci U S A. 2011;108:2783–2788. doi: 10.1073/pnas.1016574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rulten SL, Fisher AE, Robert I, Zuma MC, Rouleau M, Ju L, Poirier G, Reina-San-Martin B, Caldecott KW. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell. 2011;41:33–45. doi: 10.1016/j.molcel.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 14.Caldecott KW. Mammalian single-strand break repair: mechanisms and links with chromatin. DNA Repair (Amst) 2007;6:443–453. doi: 10.1016/j.dnarep.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Rine J, Herskowitz I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics. 1987;116:9–22. doi: 10.1093/genetics/116.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi JE, Mostoslavsky R. Sirtuins, metabolism, and DNA repair. Curr Opin Genet Dev. 2014;26C:24–32. doi: 10.1016/j.gde.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 19.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 20.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molinete M, Vermeulen W, Burkle A, Menissier-de Murcia J, Kupper JH, Hoeijmakers JH, de Murcia G. Overproduction of the poly(ADP-ribose) polymerase DNA-binding domain blocks alkylation-induced DNA repair synthesis in mammalian cells. EMBO J. 1993;12:2109–2117. doi: 10.1002/j.1460-2075.1993.tb05859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schreiber V, Hunting D, Trucco C, Gowans B, Grunwald D, De Murcia G, De Murcia JM. A dominant-negative mutant of human poly(ADP-ribose) polymerase affects cell recovery, apoptosis, and sister chromatid exchange following DNA damage. Proc Natl Acad Sci U S A. 1995;92:4753–4757. doi: 10.1073/pnas.92.11.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kupper JH, van Gool L, Burkle A. Molecular genetic systems to study the role of poly(ADP-ribosyl)ation in the cellular response to DNA damage. Biochimie. 1995;77:450–455. doi: 10.1016/0300-9084(96)88159-4. [DOI] [PubMed] [Google Scholar]
- 24.Ding R, Smulson M. Depletion of nuclear poly(ADP-ribose) polymerase by antisense RNA expression: influences on genomic stability, chromatin organization, and carcinogen cytotoxicity. Cancer Res. 1994;54:4627–4634. [PubMed] [Google Scholar]
- 25.Masutani M, Nozaki T, Nakamoto K, Nakagama H, Suzuki H, Kusuoka O, Tsutsumi M, Sugimura T. The response of Parp knockout mice against DNA damaging agents. Mutat Res. 2000;462:159–166. doi: 10.1016/s1383-5742(00)00033-8. [DOI] [PubMed] [Google Scholar]
- 26.Simbulan-Rosenthal CM, Haddad BR, Rosenthal DS, Weaver Z, Coleman A, Luo R, Young HM, Wang ZQ, Ried T, Smulson ME. Chromosomal aberrations in PARP(−/−) mice: genome stabilization in immortalized cells by reintroduction of poly(ADP-ribose) polymerase cDNA. Proc Natl Acad Sci U S A. 1999;96:13191–13196. doi: 10.1073/pnas.96.23.13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trucco C, Oliver FJ, de Murcia G, Menissier-de Murcia J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998;26:2644–2649. doi: 10.1093/nar/26.11.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang ZQ, Stingl L, Morrison C, Jantsch M, Los M, Schulze-Osthoff K, Wagner EF. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997;11:2347–2358. doi: 10.1101/gad.11.18.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 30.Vodenicharov MD, Sallmann FR, Satoh MS, Poirier GG. Base excision repair is efficient in cells lacking poly(ADP-ribose) polymerase 1. Nucleic Acids Res. 2000;28:3887–3896. doi: 10.1093/nar/28.20.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siegel C, McCullough LD. NAD+ depletion or PAR polymer formation: which plays the role of executioner in ischaemic cell death? Acta Physiol (Oxf) 2011;203:225–234. doi: 10.1111/j.1748-1716.2010.02229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK. Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp Cell Res. 2004;297:521–532. doi: 10.1016/j.yexcr.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 34.Meyer RG, Meyer-Ficca ML, Whatcott CJ, Jacobson EL, Jacobson MK. Two small enzyme isoforms mediate mammalian mitochondrial poly(ADP-ribose) glycohydrolase (PARG) activity. Exp Cell Res. 2007;313:2920–2936. doi: 10.1016/j.yexcr.2007.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci. 2009;14:1116–1128. doi: 10.2741/3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 38.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, Shimoji M, Yu SW, Dawson TM, Dawson VL. Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson’s disease. Ann N Y Acad Sci. 2003;991:132–139. doi: 10.1111/j.1749-6632.2003.tb07471.x. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos) Sci Signal. 2011;4:ra20. doi: 10.1126/scisignal.2000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanai S, Kanai M, Ohashi S, Okamoto K, Yamada M, Takahashi H, Miwa M. Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2004;101:82–86. doi: 10.1073/pnas.2237114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci U S A. 2004;101:17699–17704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cozzi A, Cipriani G, Fossati S, Faraco G, Formentini L, Min W, Cortes U, Wang ZQ, Moroni F, Chiarugi A. Poly(ADP-ribose) accumulation and enhancement of postischemic brain damage in 110-kDa poly(ADP-ribose) glycohydrolase null mice. J Cereb Blood Flow Metab. 2006;26:684–695. doi: 10.1038/sj.jcbfm.9600222. [DOI] [PubMed] [Google Scholar]
- 44.Cuzzocrea S, Wang ZQ. Role of poly(ADP-ribose) glycohydrolase (PARG) in shock, ischemia and reperfusion. Pharmacol Res. 2005;52:100–108. doi: 10.1016/j.phrs.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 45.Wang H, Wang M, Wang H, Bocker W, Iliakis G. Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. Journal of cellular physiology. 2005;202:492–502. doi: 10.1002/jcp.20141. [DOI] [PubMed] [Google Scholar]
- 46.Patel NS, Cortes U, Di Poala R, Mazzon E, Mota-Filipe H, Cuzzocrea S, Wang ZQ, Thiemermann C. Mice lacking the 110-kD isoform of poly(ADP-ribose) glycohydrolase are protected against renal ischemia/reperfusion injury. J Am Soc Nephrol. 2005;16:712–719. doi: 10.1681/ASN.2004080677. [DOI] [PubMed] [Google Scholar]
- 47.Cortez D, Glick G, Elledge SJ. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci U S A. 2004;101:10078–10083. doi: 10.1073/pnas.0403410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ame JC, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, Schreiber V. Radiation-induced mitotic catastrophe in PARG-deficient cells. J Cell Sci. 2009;122:1990–2002. doi: 10.1242/jcs.039115. [DOI] [PubMed] [Google Scholar]
- 49.Mashimo M, Kato J, Moss J. ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc Natl Acad Sci U S A. 2013;110:18964–18969. doi: 10.1073/pnas.1312783110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berger NA, Sims JL, Catino DM, Berger SJ. Poly(ADP-ribose) polymerase mediates the suicide response to massive DNA damage: studies in normal and DNA-repair defective cells. Princess Takamatsu Symp. 1983;13:219–226. [PubMed] [Google Scholar]
- 51.Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101:4–15. [PubMed] [Google Scholar]
- 52.Chiarugi A. Poly(ADP-ribose) polymerase: killer or conspirator? The ‘suicide hypothesis’ revisited. Trends in pharmacological sciences. 2002;23:122–129. doi: 10.1016/S0165-6147(00)01902-7. [DOI] [PubMed] [Google Scholar]
- 53.Ying W, Chen Y, Alano CC, Swanson RA. Tricarboxylic acid cycle substrates prevent PARP-mediated death of neurons and astrocytes. J Cereb Blood Flow Metab. 2002;22:774–779. doi: 10.1097/00004647-200207000-00002. [DOI] [PubMed] [Google Scholar]
- 54.Goellner EM, Grimme B, Brown AR, Lin YC, Wang XH, Sugrue KF, Mitchell L, Trivedi RN, Tang JB, Sobol RW. Overcoming Temozolomide Resistance in Glioblastoma via Dual Inhibition of NAD+ Biosynthesis and Base Excision Repair. Cancer Res. 2011;71:2308–2317. doi: 10.1158/0008-5472.CAN-10-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang J, Goellner EM, Wang XW, Trivedi RN, St Croix CM, Jelezcova E, Svilar D, Brown AR, Sobol RW. Bioenergetic Metabolites Regulate Base Excision Repair-Dependent Cell Death in Response to DNA Damage. Molecular Cancer Research. 2010;8:67–79. doi: 10.1158/1541-7786.MCR-09-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fouquerel E, Goellner EM, Yu Z, Gagne JP, Barbi de Moura M, Feinstein T, Wheeler D, Redpath P, Li J, Romero G, Migaud M, Van Houten B, Poirier G, Sobol RW. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell reports. 2014 doi: 10.1016/j.celrep.2014.08.036. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–18902. doi: 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- 58.Cipriani G, Rapizzi E, Vannacci A, Rizzuto R, Moroni F, Chiarugi A. Nuclear poly(ADP-ribose) polymerase-1 rapidly triggers mitochondrial dysfunction. J Biol Chem. 2005;280:17227–17234. doi: 10.1074/jbc.M414526200. [DOI] [PubMed] [Google Scholar]
- 59.Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome - a key determinant of cancer cell biology. Nat Rev Cancer. 2012 doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- 60.Ying W, Garnier P, Swanson RA. NAD+ repletion prevents PARP-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochem Biophys Res Commun. 2003;308:809–813. doi: 10.1016/s0006-291x(03)01483-9. [DOI] [PubMed] [Google Scholar]
- 61.Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes & Development. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A. 1999;96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gagne JP, Isabelle M, Lo KS, Bourassa S, Hendzel MJ, Dawson VL, Dawson TM, Poirier GG. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008;36:6959–6976. doi: 10.1093/nar/gkn771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shoshan-Barmatz V, Golan M. Mitochondrial VDAC1: function in cell life and death and a target for cancer therapy. Curr Med Chem. 2012;19:714–735. doi: 10.2174/092986712798992110. [DOI] [PubMed] [Google Scholar]
- 65.Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagne JP, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A. 2014;111:10209–10214. doi: 10.1073/pnas.1405158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saraiva LM, Seixas da Silva GS, Galina A, da-Silva WS, Klein WL, Ferreira ST, De Felice FG. Amyloid-beta triggers the release of neuronal hexokinase 1 from mitochondria. PLoS ONE. 2010;5:e15230. doi: 10.1371/journal.pone.0015230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Canto C, Sauve AA, Bai P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol Aspects Med. 2013;34:1168–1201. doi: 10.1016/j.mam.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.El Ramy R, Magroun N, Messadecq N, Gauthier LR, Boussin FD, Kolthur-Seetharam U, Schreiber V, McBurney MW, Sassone-Corsi P, Dantzer F. Functional interplay between Parp-1 and SirT1 in genome integrity and chromatin-based processes. Cell Mol Life Sci. 2009;66:3219–3234. doi: 10.1007/s00018-009-0105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sasca D, Hahnel PS, Szybinski J, Khawaja K, Kriege O, Pante SV, Bullinger L, Strand S, Strand D, Theobald M, Kindler T. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood. 2014;124:121–133. doi: 10.1182/blood-2013-11-538819. [DOI] [PubMed] [Google Scholar]
- 71.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 72.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 73.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kolthur-Seetharam U, Dantzer F, McBurney MW, de Murcia G, Sassone-Corsi P. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle. 2006;5:873–877. doi: 10.4161/cc.5.8.2690. [DOI] [PubMed] [Google Scholar]
- 75.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 76.Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD, Alt FW. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]