

Abstract
NAD+ metabolism is an essential regulator of cellular redox reactions, energy pathways, and a substrate provider for NAD+ consuming enzymes. We recently demonstrated that enhancement of NAD+/NADH levels in breast cancer cells with impaired mitochondrial NADH dehydrogenase activity, through augmentation of complex I or by supplementing tumor cell nutrients with NAD+ precursors, inhibits tumorigenicity and metastasis. To more fully understand how aberrantly low NAD+ levels promote tumor cell dissemination, we here asked whether inhibition of NAD+ salvage pathway activity by reduction in nicotinamide phosphoribosyltransferase (NAMPT) expression can impact metastasis and tumor cell adhesive functions. We show that knockdown of NAMPT, the enzyme catalyzing the rate-limiting step of the NAD+ salvage pathway, enhances metastatic aggressiveness in human breast cancer cells and involves modulation of integrin expression and function. Reduction in NAMPT expression is associated with upregulation of select adhesion receptors, particularly αvβ3 and β1 integrins, and results in increased breast cancer cell attachment to extracellular matrix proteins, a key function in tumor cell dissemination. Interestingly, NAMPT downregulation prompts expression of integrin αvβ3 in a high affinity conformation, known to promote tumor cell adhesive interactions during hematogenous metastasis. NAMPT has been selected as a therapeutic target for cancer therapy based on the essential functions of this enzyme in NAD+ metabolism, cellular redox, DNA repair and energy pathways. Notably, our results indicate that incomplete inhibition of NAMPT, which impedes NAD+ metabolism but does not kill a tumor cell can alter its phenotype to be more aggressive and metastatic. This phenomenon could promote cancer recurrence, even if NAMPT inhibition initially reduces tumor growth.
Keywords: NAMPT, NAD+ salvage pathway, integrins, αvβ3, breast cancer, metastasis
1. Introduction
Nicotinamide adenine dinucleotide (NAD+) metabolism is an essential regulator of cellular redox reactions, energy pathways, and a substrate provider for NAD+ consuming enzymes 1–3. These include ADP-ribose transferases, poly(ADP-ribose) polymerases (PARPs), and NADases such as CD38 and sirtuins. While NAD+ consuming enzymes affect NAD+ availability and typically degrade it to nicotinamide, redox reactions involve reduction of NAD+ to NADH, which can be converted back to NAD+ by NADH dehydrogenase activity of mitochondrial complex I1–9.
NAD+ can be synthesized de novo from tryptophan, nicotinamide, nicotinic acid or nicotinamide riboside, or be derived via the salvage pathway. The initial and rate-limiting step within the NAD+ salvage pathway is mediated by nicotinamide phosphoribosyltransferase (NAMPT), the enzyme that catalyzes conversion of nicotinamide to nicotinamide mononucleotide (NMN+) using phosphoribosylpyrophosphate as a co-substrate. NMN+ is then converted to NAD+ by nicotinamide nucleotide adenylyltransferases (NMNAT)1,2,10–12.
Cancer cells, especially highly proliferative cells in fast growing tumors such as triple negative breast cancers, generally accumulate high levels of DNA damage and genomic instability13–16. These cells can have increased NAD+ degrading PARP activity for DNA damage repair, and thus a high need for NAD+ to maintain cell viability. Therefore, fast growing cells often have low NAD+ levels which sensitize them to further NAD+ reduction1,17,18. Thus, it has been suggested that high NAMPT expression should enhance tumor cell survival by elevating NAD+ levels19–21, while chemical inhibition of NAMPT to reduce cellular NAD+ levels should inhibit tumor cell viability, especially when used in combination with PARP inhibitors. This strategy has been proposed as a therapeutic approach against breast cancer11,20,22,23.
In contrast to the concept that inhibition of NAMPT activity might eliminate tumor cells, we showed earlier that impaired NAD+ metabolism activity and resulting decrease in NAD+/NADH redox levels in human breast cancer cells can actually significantly stimulate their metastatic properties7. Mechanisms underlying the enhanced metastatic aggressiveness were found associated with aberrant mitochondrial complex I and poor NADH dehydrogenase activity. Metastatic aggressiveness could be suppressed by enhancing complex I function through expression of yeast NADH dehydrogenase Ndi1 in the tumor cells. Augmentation of complex I activity enhanced the cellular NAD+/NADH balance and supported autophagy, while suppressing the mTORC1 pathway7. To more fully understand how reduced NAD+ levels promote tumor cell dissemination, we here asked whether inhibition of NAD+ salvage pathway activity by reduction in NAMPT expression can impact tumor cell adhesive properties. Aberrant cell adhesion supports the metastatic process by mediating tumor cell interaction with vascular cells or the lymphatic system, as well as with tissues and matrices in target organs of metastasis24,25. As major adhesive, migratory and invasive tumor cell functions are mediated by adhesion receptors, we here asked whether interference with NAMPT affects adhesion receptor expression and function.
2. Materials and methods
2.1 Cell culture
MDA-MB-231 human breast cancer cells and their variants were stably transduced with Firefly luciferase (F-luc) using lentiviral expression vector pTacoma (CMV promoter) (BE Torbett, TSRI) to analyze metastasis by non-invasive bioluminescence imaging (55). Cells were grown in EMEM supplemented with nonessential amino acids, vitamins, 2 mM L-glutamine, 1 mM pyruvate, and 10% FBS.
2.2 NAMPT expression and knockdown
Lentiviral vector containing small hairpin RNA (shRNA) against NAMPT (shNAMPT) (TRCN0000116180) or non-mammalian targeting control shRNA (SHC0002) (shCT) were from Sigma-Aldrich, MO. Knockdown efficiency was quantified by real time PCR using FastStart Universal SYBR Green Master (Rox) (Roche) and the following primers: human NAMPT-F (GCCAGCAGGGAATTTTGTTA), human NAMPT-R (TGATGTGCTGCTTCCAGTTC), human GAPDH-F (GGGAAGGTGAAGGTCGGAGT), and human GAPDH-R (TCCACTTTACCAGAGTTAAAAGCAG). The same primers were used to analyze NAMPT gene expression in lung metastases developing in SCID mice after i.v. injecting shNAMPT versus shRNA control cells. Data were recorded and analyzed using an ABI-PRISM 7700 Sequence Detection System (Applied Biosystems) and Sequence Detector Software (SDS v2.0). Reduction of NAMPT protein expression in shNAMPT knockdown cells was confirmed by Western blot using cells lysed in Laemmli’s buffer and analyzing 20 μg of total protein, probed with anti-NAMPT (ab45890, abcam, MA) or anti β-tubulin (Sigma-Aldrich, MO), and followed with secondary antibodies conjugated to IRDye 800 (NAMPT) or IRDye 680 (β-tubulin) using an Odyssey infrared imaging system (LI-COR Biosciences). Data were analyzed and quantified using Odyssey infrared imaging system application software v3.0.
Moreover, NAMPT Knockdown was confirmed by western blot. Cells were lysed with Laemmli’s buffer and 20uG of total protein was loaded in a SDS-page gel. Western blots were incubated with antibodies against: NAMPT (ab45890, abcam, MA) and β-tubulin (Sigma-Aldrich, MO). Antibody binding was detected following incubation with secondary antibodies conjugated to IRDye 680 (β-tubulin) 800 (NAMPT), using an Odyssey infrared imaging system (LI-COR Biosciences). Data were analyzed and quantified using Odyssey infrared imaging system application software v3.0.
2.3 NAD+/NADH measurement
NAD+ and NADH were analyzed independently in extracts of whole cells (1×106) prepared as previously described7. Concentrations were determined using a fluorescence NAD+/NADH detection kit according to the manufacturer’s protocol (Cell Technology, Inc).
2.4 Metastatic activity
For experimental metastasis, female 6–8 week-old C.B-17/SCID mice were injected intravenously (i.v.) with F-luc–tagged cancer cells: 2.5×105 MDA-MB-231 shCT or MDA-MB-231 shNAMPT cells. Mice were imaged weekly (IVIS 200, Xenogen) 10 min after i.p. injection of D-luciferin (100 mg/kg). Bioluminescence was quantified as photons/second/cm2 in defined regions of interest using Living Image software. Animal work complied with National Institutes of Health and institutional guidelines (TSRI is AAALAC accredited).
2.5 Cell-adhesion assay
Cell-adhesion assays were performed in 96- well plates coated with 50 μl of one of the following matix proteins: human vitronectin (10 μg/ml), fibrinogen (20 μg/ml), fibronectin (10 μg/ml) or collagen I (10 μg/ml), in PBS overnight at 4 °C. The plates were washed twice with TBS and blocked with 2% BSA in TBS overnight at 4 °C. Cells were harvested with versene and washed with adhesion buffer (serum free EMEM medium containing 0.5% BSA) and then pre-incubated with or without 10 μg/ml function blocking monoclonal anti-integrin antibodies: β3 (M21.3), αv (HB8448), αvβ5 (P1F6), β1 (P5D2) and α5 (P1D6)26–29 for 15 min at RT. Cells were then seeded at 4×104/well in the presence of the antibodies. Adhesion time was 35 min at 37°C. Non-adhered cells were removed by adding floatation medium (0.9% NaCl, 80% Percoll) as described previously30. Attached cells were fixed with 3.3% glutaraldehyde and 20% Percoll. The attached cells were stained with crystal violet, lysed with 0.5% Triton X-100 in water, and quantified by measuring optical absorbance at 585 nm.
2.6 Integrin expression
Surface integrin expression was analyzed by flow cytometry as follows: cells were harvested with versene, washed with PBS, and resuspended in ice cold binding buffer (1% BSA in TBS pH 7.4). 50 μl containing 105 cells were then incubated with 30 μg/ml of anti-integrin antibodies: αvβ3 (VNR1),αvβ5(P1F6), α5 (P1D6) and β1 (P5D2)26–29, in binding buffer for 45 min on ice. Cells were washed with cold PBS and incubated with 50 μl of 1:100 APC-conjugated goat anti- mouse polyclonal antibody in binding buffer for 45 min on ice. After washing, cells were analyzed by flow cytometry using a FACS-Calibur 2 in FL-4 (Becton-Dickinson).
2.7 Integrin affinity based on binding of ligand mimetic antibody
The affinity state of integrin αvβ3 was assessed based on binding of soluble ligand mimetic RGD-containing human antibody ScFv126 which was here re-engineered into an intact IgG (EW84-IgG). Tumor cells were harvested with versene, washed first in ice cold cation free TBS and then in TBS with 2mM Ca2+, before incubation with EW84-IgG15 at μg/ml in TBS/Ca2+ on ice for 45 min. After washing in TBS/Ca2+, cells were incubated with AP-conjugated anti-human Fc in TBS/Ca2+ for 45 min on ice, then washed as above and read in FL-4 on a FACS-Calibur 2. β3 integrin expression in these experiments was verified with non-cation dependent and non-ligand mimetic mAb AV-10 detected with goat-anti mouse APC conjugate31.
3. Results
3.1 NAMPT expression affects metastatic activity of human breast cancer cells
To further understand how changes in cellular NAD+ levels and NAD+/NADH ratios affect the ability of breast cancer cells to metastasize, we knocked-down expression of NAMPT, the rate-limiting enzyme in NAD+ synthesis. NAMPT converts NAD+ precursor nicotinamide to nicotinamide mononucleotide (NMN+) which is then coupled with the AMP moiety of ATP to form NAD+ 1,8,10. Using an shRNA approach, NAMPT gene and protein expression was significantly reduced in MDA-MB-231 human breast cancer cells (Fig. 1A, B). This resulted in reduced NAD+ and slightly reduced NADH concentrations in these cells, and significantly lowered cellular NAD+/NADH ratio (Fig. 1C). Upon injection of the tumor cells into the venous circulation of SCID mice, NAMPT knock-down cells showed significantly enhanced metastatic activity compared to control shRNA transduced cells (Fig. 1D,E). Reduced levels of NAMPT expression in the highly metastatic knockdown cells were confirmed to be stable even in the metastatic niche (Fig. 1F). NAMPT Knockdown was confirmed to be stable even in the metastatic niche. Thus, reduction in NAD+ synthesis caused enhanced aggressiveness in the already highly metastatic MDA-MB-231 breast cancer cells.
Figure 1. Down regulation of NAMPT expression reduces NAD+/NADH ratios and enhances metastatic activity in human breast cancer cells.

(A) NAMPT knockdown (shNAMPT) reduced NAMPT expression by 97 % in MDA-MB-231 cells, compared to controls transduced with scrambled shRNA (shCT). NAMPT mRNA levels were analyzed by real time PCR and are expressed relative to GAPDH (***P<0.001) (n = 3). (B) NAMPT knockdown (shNAMPT) reduced NAMPT protein expression in MDA-MB-231 cells, shown by Western blot analysis. Quantification of NAMPT protein was related to β-tubulin expression. (C) Interference with NAD+ synthesis and salvage pathways reduced absolute amounts of NAD+ and NADH, and cellular NAD+/NADH ratios. NAD+ and NADH were analyzed independently in whole cell extracts of 1×106 cells. Metabolite concentrations were determined using a NAD+/NADH fluorescence detection kit (Cell Technology, Inc). Groups were compared by unpaired two-tailed Student’s t-test (*P<0.05). Data are expressed as mean ± SEM. (D) NAMPT knockdown increased lung colonization activity in MDA-MB-231 cells. Metastatic growth was measured by repeated non-invasive bioluminescence imaging (x-axes: time in weeks). Groups were compared by unpaired two-tailed Student’s t-test (*P<0.05). (*P<0.05, n = 6/group). (E) Representative bioluminescence images of mice (n=5), 6 weeks after i.v. injection of 2.5×105 MDA-MB-231 control or shNAMPT expressing cells. (F) Down regulation of NAMPT expression in NAMPT knockdown cells (shNAMPT) was maintained in lung metastases. Lung lesions developing in SCID mice after injection of shNAMPT cells versus control cells (shCT) as shown in (E) were analyzed for NAMPT gene expression as detailed in (A).
3.2 NAMPT expression modulates breast cancer cell adhesive properties
Important cellular functions that help mediate tumor cell dissemination during metastasis are adhesive properties that support cell interaction with matrix proteins during cell adhesion, migration, and invasion. To investigate whether breast cancer cell binding to the extracellular matrix is altered when NAD+ metabolism is changed, we analyzed MDA-MB-231 cell attachment to major matrix proteins, comparing NAMPT knock-down cells versus controls. We found that cell adhesion to vitronectin, fibrinogen, and type I collagen were clearly enhanced when NAMPT expression was reduced, while attachment to fibronectin was not significantly altered (Fig. 2A). Cell attachment to these extracellular matrix proteins is primarily mediated by adhesion receptors of the integrin family25. We therefore analyzed if down-regulation of NAD+ metabolism through NAMPT knock-down affected expression of integrins known to support cell interaction with the tested matrix proteins. Two major changes were observed. NAMPT knock-down was associated with expression of integrin αvβ3 which is largely missing MDA-MB-231 parental or knock-down control cells, and an upregulation of β1 integrins (Fig. 2B, C). Attachment of MDA-MB-231 parental cells to vitronectin is known to be mediated primarily by integrin αvβ532. Expression of this integrin was not changed in response to NAMPT knock-down. The upregulation of αvβ3 expression upon NAMPT knock-down is therefore likely to contribute to the enhanced vitronectin adhesion of these cells (Fig. 2A). Attachment of MDA-MB-231 parental cells to fibronectin is mediated predominantly by integrin α5β132. We found no change in α5 integrin subunit expression in NAMPT knock-down cells, and this correlated with their unchanged adhesion to fibronectin (Fig. 2A,B). In contrast, attachment to collagen I was upregulated in NAMPT knock-down cells, consistent with increased expression of β1 integrins as seen by flow cytometry (Fig. 2B). The predominant collagen receptor of MDA-MB-231 cells is integrin α2β133, and this receptor may be affected by NAMPT knock-down induced alteration in NAD+ metabolism.
Figure 2. NAMPT knockdown increases breast cancer cell adhesion to select substrates and upregulates β3 and β1 integrin expression.

(A) NAMPT knockdown (shNAMPT) enhanced MDA-MB-231 cell adhesion to vitronectin (VN), fibrinogen (Fgn), and collagen type I (Col I) compared to controls transduced with scrambled shRNA (shCT). (***P<0.001) (n = 3) (B) NAMPT knockdown (shNAMPT) enhanced expression of integrin αvβ3 and β1 integrins in MDA-MB-231 cells, measured by flow cytometry based on fluorescence intensity (FI) using mAbs directed against the αvβ3 complex, αvβ5 complex, or the α5 and β1 integrin subunits (*P<0.05) (n = 3). (C) Representative histogram of cell surface expression of integrin αvβ3 in NAMPT knockdown (shNAMPT) MDA-MB-231 cells or their controls transduced with scrambled shRNA (shCT).
Together, our results indicate that interference with NAD+ metabolism by reduction of NAMPT expression in breast cancer cells is associated with upregulation of specific integrins, and results in alterations in cell adhesion to major extracellular matrix proteins.
3.3 NAMPT expression specifically modifies β3 and β1 integrin adhesion receptors in breast cancer cells
Inhibition of NAD+ generation through NAMPT mediated NAD+ synthesis from NAD+ precursor nicotinamide most prominently impacts expression of αvβ3 and β1 integrins in the examined breast cancer cells. To analyze the contribution of upregulated αvβ3 and β1 integrin expression to tumor cell adhesion, we used function blocking anti-integrin antibodies. NAMPT knock-down cells were compared to shRNA control treated cells for adhesion to key extracellular matrix proteins in the presence or absence of antibodies directed to β3, αvβ5, αv, α5 or β1, alone or in combination (Fig. 3).
Figure 3. NAMPT knockdown increases αvβ3 and β1 integrin function in mediating breast cancer cell adhesion.

Adhesion of MDA-MB-231 NAMPT knockdown cells (shNAMPT) or controls transduced with scrambled shRNA (shCT) to vitronectin (A), fibrinogen (B), fibronectin (C) and collagen type I (D) in the absence (no Ab) or presence of function blocking antibodies directed to the αvβ3 complex, the αvβ5 complex, or to integrin subunits αv, α5, or β1, alone or in combination. (*P<0.05, ***P<0.001) (n = 3) (E) Overview of integrin contributions toAMD-MB-231 cell adhesion to the indicated substrates in response to NAMPT knockdown in the tumor cells. Values reflecting cell adhesion of control compared to NAMPT knockdown cells are listed in the top row of the table (shCT/shNAMPT) (***p< 0.001) (n=3). Yes = this integrin was found to contribute to adhesion of both shNAMPT and shCT MDA-MB-231 cells as a neutralizing antibody against this integrin significantly inhibited cell attachment (>50%); Partial = adhesion to this integrin was partially inhibited, No = no significant inhibition. - = no measured; (n=3).
Increased MDA-MB-231 cell adhesion to vitronectin upon NAMPT knock-down was confirmed, and a clear reduction of this enhanced adhesion was seen in the presence of blocking anti-αvβ5 and anti-αv antibodies (Fig. 3A, E). These results identify αvβ5 as the predominant receptor for vitronectin in control as well as in NAMPT knock-down cells where αvβ3 is upregulated in the entire cell population (Fig. 2C). Failure of the anti-β3 antibody to markedly reduce the enhanced vitronectin adhesion of NAMPT knock-down cells, and the strong inhibition by anti-αvβ5 and anti-αv could indicate a cooperative effect of αvβ3 and αvβ5 in these cells, which strengthens vitronectin adhesion that cannot be abolished by the anti-β3 antibody alone. Furthermore, NAD+ metabolism may modulate αvβ5 activity rather than expression. β1 integrin targeting had no effect, confirming that no β1 integrin such as α8β1, a known vitronectin receptor34, is involved (Fig. 3A, E).
To directly address a functional role of αvβ3 in the adhesion of NAMPT knock-down cells, fibrinogen was included as a substrate. Parental MDA-MB-231 cells almost completely lack αvβ3 and cannot attach to fibrinogen, as this protein is not recognized by any other adhesion receptor expressed by these cells. The functional contribution of integrin αvβ3, upregulated in NAMPT knock-down cells, was evident in the fibrinogen adhesion analysis. The role of this receptor in supporting fibrinogen attachment was confirmed, as β3- and αv-blocking antibodies completely inhibited adhesion to this substrate, while anti-αvβ5 had no effect (Fig. 3B,E).
Supporting the results on fibronectin adhesion in Fig. 2, use of function blocking anti-integrin antibodies demonstrated that α5β1 is the predominant adhesion receptor on MDA-MB-231 cells for this substrate, regardless of the NAMPT knock-down effect on NAD+ metabolism (Fig. 3C,E). Blocking β1 integrin function reduced adhesion to fibronectin to the same level in control and NAMPT knock-down cells. This finding, and the strong inhibition of adhesion in both cell types by combining antibodies against αv and β1, could indicate that MDA-MB-231 cells express and utilize integrin αvβ1 for adhesion to fibronectin. This receptor has not been previously reported as mediating fibronectin adhesion in these breast cancer cells. There are no function blocking antibodies to αvβ1, and it is therefore difficult to definitively demonstrate its role here. It is possible that αvβ1 is slightly upregulated in NAMPT knock-down cell and contributes to the observed slight enhancement of fibronectin adhesion in these cells.
Tumor cell attachment to collagen I was found enhanced when NAMPT expression was reduced (Fig. 2A). Treatment with neutralizing anti-β1 antibody blocked collagen I adhesion nearly completely in control and NAMPT knock-down cells (Fig. 3D,E). This indicates that the observed upregulation of β1 integrin expression upon NAMPT knock-down (Fig. 2B) is responsible for the increased ability of the cells to interact with collagen I, a major substrate found in tissues and the vessel wall.
3.4 NAMPT expression influences the β3 integrin affinity state in breast cancer cells
It is well known that the affinity state of integrins expressed at the cell surface influences ligand recognition and the ability of the receptors to bind soluble ligand35. Classical examples are platelet and leukocyte integrins that can be activated and require activation to support platelet cohesion during thrombus formation36, and to mediate leukocyte arrest within the vasculature at sites of inflammation37. We showed earlier that integrin αvβ3 can also exist in distinct states of activation in tumor cells, and that expression of the high affinity receptor in breast cancer cells supports their metastatic activity38,39. To analyze the affinity state of integrin αvβ3, which we found upregulated in MDA-MB-231 breast cancer cells upon NAMPT knock-down (Fig. 2B), we studied the ability of the cells to bind a ligand mimetic antibody that we developed earlier26. This antibody mimics ligands of αvβ3 that express the RGD integrin-binding motif by displaying an RGD sequence within CDR3 of the heavy chain26. Even though multiple integrins are known to recognize the RGD motif in multiple natural ligands, our ligand mimetic antibody is specific for integrin αvβ3 only26. Originally isolated from a phage displayed human cancer patient scFv antibody library, we now re-engineered the scFv into an intact IgG and analyzed soluble IgG binding to the NAMPT knock-down tumor cells as a measure of integrin αvβ3 affinity in these cells. We found that NAMPT knock-down cells, but not their control cells, bound the ligand mimetic IgG in a cation dependent manner (Fig. 4A). In the presence of Ca2+, antibody binding was observed (Fig. 4A) proportionate to binding of non-cation and non-activation dependent anti-β3 mAb AV10 (Fig. 4B). This finding - first - confirms upregulated expression of αvβ3 integrin upon NAMPT knock-down in the tumor cells, and - second - indicates that most of the expressed αvβ3 is present on the tumor cells in a high affinity state. This notion is supported by our result that binding of the ligand mimetic antibody seen with Ca2+ did not increase in the presence of Mn2+, a cation known to induce a high affinity state in most integrins (not shown)26.
Figure 4. NAMPT knockdown leads to expression of integrin αvβ3 in its high affinity conformation on human breast cancer cells.
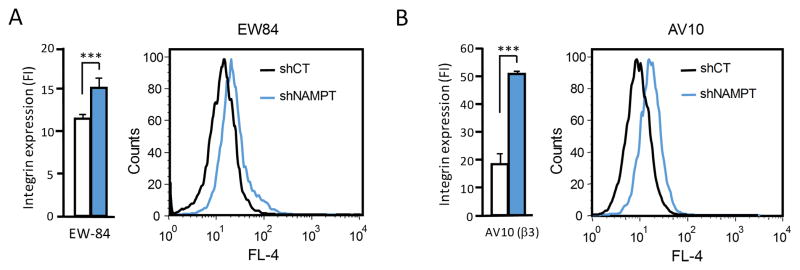
(A) Measurement of high affinity αvβ3 expression: binding average (left panel) and representative histogram (right panel) of ligand mimetic αvβ3-specific human monoclonal antibody (IgG EW84) to MDA-MB-231 NAMPT knockdown cells (shNAMPT) or controls transduced with scrambled shRNA (shCT) in the presence of calcium, measured by flow cytometry. (B) Measurement of overall β3 integrin expression: binding average (left panel) and representative histogram (right panel) of integrin αvβ3 reported on MDA-MB-231 NAMPT knockdown cells (shNAMPT) or controls transduced with scrambled shRNA (shCT) by staining with non-activation dependent anti-β3 mAb AV10, measured by flow cytometry.
Together, our results indicate that inhibition of NAD+ synthesis from NAD+ precursor nicotinamide by interference with NAMPT expression affects the expression of certain integrins on tumor cells and thereby influences their adhesive and ligand binding properties. Most notably, integrin αvβ3 is upregulated in association with NAMPT knock-down from a nearly undetectable level in the parental and control tumor cells. Importantly, not only the expression of the integrin is induced but also the presentation of the receptor at the cell surface in a conformation that is compatible with a high affinity state, as evidenced by the ability of the receptor to bind ligand from solution. This result is supported by our finding that the induced expression of αvβ3 in NAMPT knock-down cells supports adhesion of the tumor cells to immobilized fibrinogen, a protein that requires αvβ3 activation for support of cell adhesion31.
4. Discussion
This study demonstrates that NAMPT, the enzyme catalyzing the initial and rate-limiting step in the NAD+ salvage pathway, can regulate metastatic activity in tumor cells, and that this can involve modulation of integrin expression and function which alter tumor cell adhesive functions associated with tumor cell dissemination.
4.1 Involvement of NAD+ metabolism in regulating metastatic activity
We showed earlier that impaired NAD+ metabolism activity and resulting decrease in NAD+/NADH redox levels in human breast cancer cells can significantly stimulate their metastatic properties7. Mechanisms underlying enhanced metastatic aggressiveness were found associated with aberrant mitochondrial complex I and poor NADH dehydrogenase activity, resulting in altered autophagy functions and enhanced mTORC1 activity7. Furthermore, we showed that therapeutic enhancement of the NAD+/NADH ratio through treatment with NAD+ precursors can inhibit breast cancer metastasis and interfere with oncogene driven breast cancer progression in the MMTV-PyMT mouse model7. Detailed mechanisms through which enhancement of NAD+ metabolism may impact tumor progression can be complex. NAD+ and its reduced form NADH are cofactors of more than 200 metabolic reactions in mammalian cells, including regulation of NAD+ degrading enzymes, such as PARPs, Sirtuins, and CD38. These enzymes and NAD+/NADH levels contribute to tumor suppressor functions of p53, BRCA1, and BRCA2, and can affect chromatin structure and genomic stability2,5,9,40–43,44,45,46,47. Our current results indicate a role of NAD+ metabolism in regulating tumor cell adhesive functions that may have direct impact on metastatic behavior.
4.2 Regulation of NAD+ metabolism through NAMPT in cancer cells
Within the NAD+ salvage pathway, catalytic activity of NAMPT is significantly lower than NMN adenylyl tranferase (NMNAT) activity which converts NMN+ to NAD+, and therefore NAMPT limits the rate of this pathway. In human cancer, intracellular as well as extracellular NAMPT (iNAMPT and eNAMPT) can be upregulated 12,23,48,49 and associate with a more aggressive phenotype and poor prognosis50. While iNAMPT is the primary regulator of NAD+ salvage from NAD+ precursor NAM, eNAMPT is secreted by specific cell types including adipocytes, macrophages and hepatocytes, and apparently has very low catalytic activity in NAD+ synthesis12,49. However eNAMPT may regulate intracellular pathways and cell survival by affecting cellular glucose metabolism through insulin-mimetic activity. Rapid tumor cell growth further demands increased functionality of NAD+ degrading enzymes that support high transcriptional activity. Compensatory upregulation of NAMPT expression in these cells could therefore support sustained growth and cell viability. Thus, NAMPT has been suggested as a target for anti-cancer therapy1,10–12,20,23. Surprisingly, our results demonstrate that inhibition of NAMPT expression and function, at levels that do not kill tumor cells can lead to a significantly more aggressive phenotype 7. Integrin alterations, such as upregulation of αvβ3 and β1 integrin expression and modulation of functional activity as demonstrated in this study, likely contribute to the enhanced metastatic activity which we found associated with NAMPT downregulation in breast cancer cells.
4.3 NAD+ metabolism induced modulation of cell adhesion, integrin expression and activity
Among the cell adhesion receptors found upregulated in response to NAMPT knock-down in this study, is integrin αvβ3 which is not significantly expressed by the parental tumor cell population. This integrin is known to be associated with a highly aggressive phenotype in breast cancer38 where it critically supports tumor cell interaction with a number of Arg-Gly-Asp (RGD) containing extracellular matrix proteins in tissues, the vessel wall, and matrices of the lymphatic system. Furthermore, our results show that inhibition of NAD+ synthesis through NAMPT knock-down is accompanied by increased tumor cell adhesion to collagen I, another prominent component of the vascular basement membrane and connective tissue. Enhanced collagen adhesion is primarily mediated by β1 integrins which we also found upregulated in response to NAMPT knock-down, but in part by de novo expressed αvβ3. While αvβ3 does not support cell interaction with native collagen, RGD sequences within this matrix protein are exposed when it is degraded, and αvβ3 can now recognize collagen fragments51. Reported association of uPAR with αvβ3 in cell membrane lipid rafts can foster activation of metalloproteinases that efficiently degrade collagen52,53 and may thereby contribute to αvβ3 mediated collagen adhesion.
It is still an open question why and through which mechanisms only select integrins are modified in the breast cancer cell model we studied, while other integrins are apparently not affected. In a glioma model, it has been reported that genetic and pharmacologic inhibition of NAMPT and resulting fluctuations in intracellular NAD+/NADH levels affect cell motility and invasion54. Another important consideration is that nicotinamide riboside kinases (Nrk) within the NAD+ pathway may impact cell adhesion. In particular, Nrk2 which catalyzes phosphorylation of nicotinamide riboside (NR) and nicotinic acid riboside (NaR) to form NMN and nicotinic acid mononucleotide (NaMN), can regulate deposition of the extracellular matrix protein laminin and cell adhesion to this substrate55. This process apparently involves the ability of Nrk2 to directly interact with the β1 integrin subunit.
4.4 Impact of integrin affinity state on tumor cell adhesive properties and metastatic activity
We discovered earlier that tumor cells can carry integrin adhesion receptors in distinct states of affinity, and that constitutive expression of the high affinity conformer of certain integrins strongly promotes an invasive, metastatic phenotype24. Our previous studies on mechanisms mediating blood-borne tumor cell interaction with vascular cells during metastasis specifically revealed high affinity tumor cell integrin αvβ3 as a critical receptor for disseminating cells56. We found that high affinity αvβ3 supports tumor cell interaction with platelets, transendothelial migration, and invasion of matrices found in target tissues57. Importantly, targeting of the high affinity conformer of αvβ3 with ligand-mimetic antibodies that we derived from the cancer patient immune repertoire, prevented hematogenous metastasis26 and also interfered with advanced metastatic lesions58.
In the present study, interference with tumor cell NAD+ metabolism by knock-down of NAMPT expression was found associated with upregulation of integrin αvβ3 in MDA-MB-231 breast cancer cells and with expression of this receptor in its high affinity form. This result is remarkable in two major ways. First, MDA-MB-231 cells largely lack αvβ3 indicating that alterations in NAMPT levels can induce an adhesion receptor that is not otherwise expressed. Second, we determined the affinity state of NAMPT knock-down associated tumor cell αvβ3 based on binding of a soluble ligand-mimetic antibody which we isolated and identified as a metastasis blocking reagent59. Binding of this αvβ3-specific soluble ligand to NAMPT knock-down cells indicates that the cells can likely interact with soluble ligands, such as plasma proteins, that circulating tumor cells encounter and use for retention within the target organ vasculature and extravasation from the blood stream. Our results show that alteration in NAD+ levels, through genetic modification of NAMPT expression, modify integrin expression and function through conformational changes in the receptor at the tumor cell surface. This finding conceptually agrees with a previous study showing that mono-ADP-ribosylation of an integrin subunit affected structure and function of the adhesion receptor60. This principle might also apply to integrin αvβ5, as our current study shows that NAMPT knockdown increased αvβ5 dependent cell adhesion to vitronectin without modifying αvβ5 surface expression.
4.5 Implications for clinical cancer progression and therapy
NAMPT inhibitors such as FK866/APO866 and CHS828/GMX1777 are being evaluated for anti-cancer effects in the clinic61, based on the rationale that NAMPT supports cell viability and growth by promoting DNA repair and pro-survival pathways such as ERK1/2 and p38 activity, expression of matrix degrading metalloproteinases such as MMP-2/9, and angiogenesis19–21,62–64. While some of these processes involve NAMPT mediated regulation of the NAD+ salvage pathway, others apparently depend on the less well understood cytokine activity of the enzyme65. Notably, NAMPT was suggested to play a role in development of chronic inflammation which is known to promote tumor progression and chemoresistance62,66,67. However, only few studies correlated NAMPT expression in tumors with patient outcome48.
Clinically used NAMPT inhibitor FK866 is cytotoxic for a number of different tumor cells and noncompetitively inhibits NAMPT activity. The drug has been used in phase I and II clinical trials. CHS828, another NAMPT inhibitor originally identified as an antihypertensive agent, inhibits NAD+ synthesis, can kill cancer cells, and has also been tested in phase I and II trials11. Despite the promise of in vitro and mouse model studies, no objective tumor remission could be documented in the clinic, even though transient reductions in VEGF levels associated with angiogenesis were observed with NAMPT inhibitors given as single agents11. Next generation NAMPT antagonists are being developed, mainly to improve selectivity and pharmacologic characteristics such as plasma half-life11. Targeting of NAMPT, a rate-limiting enzyme of the vital NAD+ salvage pathway required for cellular energy metabolism, appears logical when the goal is to kill a cell. However, there are two major implications that need to be considered and more fully investigated to unambiguously identify NAMPT as a target for effective cancer therapy. First, the specificity of NAMPT inhibitors for cancer cells has not been sufficiently addressed. Second, as documented in our present study and previous report7, incomplete inhibition of NAMPT which does not kill a tumor cell can alter its phenotype and make it more aggressive and metastatic. This phenomenon could associate with enhanced cancer recurrence, even if NAMPT inhibitor treatment reduces the size of an existing tumor or metastatic lesion.
Highlights.
Inhibition of NAD+ salvage pathway activity by reduction in nicotinamide phosphoribosyltransferase (NAMPT) expression enhances metastatic aggressiveness in human breast cancer cells, and this involves modulation of integrin adhesion receptor expression and function.
Reduced NAMPT is associated with upregulation of β1 integrins and αvβ3 in tumor cells, and with receptor expression in a high affinity form that supports enhanced adhesion to extracellular matrix proteins.
The results indicate that incomplete inhibition of NAMPT in tumor cells that fails to kill all cells, may render remaining cells more aggressive and thereby promote cancer recurrence.
Acknowledgments
This study was supported by NIH grants R01CA170737, and R01CA170140 (to BFH), UL1RR025774 (to Eric J Topol, Pilot award to BFH), CDMRP DoD grant BC123479 (to BFH), and California Breast Cancer Research Program grants 17NB-0058, 16IB-0052, 12NB-0176, and 18NB-0022 (to BFH), as well as gifts from the Plotkin-Weiss Foundation and Las Patronas to the Felding lab. AFS received a postdoctoral fellowship from the Susan G. Komen Foundation.
Abbreviations
- AMP
Adenosine monophosphate
- ATP
Adenosine triphosphate
- BRCA1
Breast cancer 1
- BRCA2
Breast cancer 2
- eNAMPT
Extracellular nicotinamide phosphoribosyltransferase
- ERK
Extracellular signal-regulated kinases
- iNAMPT
Intracellular nicotinamide phosphoribosyltransferase
- MMTV-PyMT
Mouse mammary tumor virus - polyoma middle T antigen
- mTORC1
Mammalian target of rapamycin complex 1
- MMP
Matrix metalloproteinases
- NAD+
Nicotinamide adenine dinucleotide
- NADH
Reduced nicotinamide adenine dinucleotide
- NaMN
Nicotinic acid mononucleotide
- NAMPT
Nicotinamide phosphoribosyltransferase
- NaR
Nicotinic acid riboside
- Ndi1
NADH-ubiquinone reductase (H(+)-translocating
- NMN+
Nicotinamide mononucleotide
- NMNAT
Nicotinamide nucleotide adenylyltransferases
- NR
Nicotinamide riboside
- NRK
Nicotinamide riboside kinases
- PARPs
Poly (adenosine diphosphate-ribose) polymerases
- SCID
Severe Combined Immunodeficiency
- shRNA
Small hairpin RNA
- uPAR
urokinase Plasminogen activator receptor
- VEGF
Vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Antonio F. Santidrian, Email: afsant@scripps.edu.
Sarah E. LeBoeuf, Email: sleboeuf@scripps.edu.
Erik D. Wold, Email: wolde@scripps.edu.
Melissa Ritland, Email: melissa@scripps.edu.
Jane S. Forsyth, Email: jsfrio@yahoo.com.
References
- 1.Chiarugi A, Dölle C, Felici R, Ziegler M. The NAD metabolome--a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–52. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- 2.Xu P, Sauve Aa. Vitamin B3, the nicotinamide adenine dinucleotides and aging. Mech Ageing Dev. 2010;131:287–98. doi: 10.1016/j.mad.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Evans C, et al. NAD+ metabolite levels as a function of vitamins and calorie restriction: evidence for different mechanisms of longevity. BMC Chem Biol. 2010;10:2. doi: 10.1186/1472-6769-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115–30. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- 5.Imai S, Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab. 2013;15 (Suppl 3):26–33. doi: 10.1111/dom.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirkland JB. Niacin status, NAD distribution and ADP-ribose metabolism. Curr Pharm Des. 2009;15:3–11. doi: 10.2174/138161209787185823. [DOI] [PubMed] [Google Scholar]
- 7.Santidrian AF, et al. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013;3 doi: 10.1172/JCI64264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denu JM. Vitamins and aging: pathways to NAD+ synthesis. Cell. 2007;129:453–4. doi: 10.1016/j.cell.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 9.Feldman JL, Dittenhafer-Reed KE, Denu JM. Sirtuin catalysis and regulation. J Biol Chem. 2012;287:42419–27. doi: 10.1074/jbc.R112.378877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallí M, Van Gool F, Rongvaux A, Andris F, Leo O. The nicotinamide phosphoribosyltransferase: a molecular link between metabolism, inflammation, and cancer. Cancer Res. 2010;70:8–11. doi: 10.1158/0008-5472.CAN-09-2465. [DOI] [PubMed] [Google Scholar]
- 11.Galli U, et al. Medicinal chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. J Med Chem. 2013;56:6279–96. doi: 10.1021/jm4001049. [DOI] [PubMed] [Google Scholar]
- 12.Garten A, Petzold S, Körner A, Imai SI, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab. 2009;20:130–8. doi: 10.1016/j.tem.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergamaschi A, et al. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. 2006;45:1033–40. doi: 10.1002/gcc.20366. [DOI] [PubMed] [Google Scholar]
- 14.Sato T, Akiyama F, Sakamoto G, Kasumi F, Nakamura Y. Accumulation of genetic alterations and progression of primary breast cancer. Cancer Res. 1991;51:5794–9. [PubMed] [Google Scholar]
- 15.Fang M, et al. Genomic differences between estrogen receptor (ER)-positive and ER-negative human breast carcinoma identified by single nucleotide polymorphism array comparative genome hybridization analysis. Cancer. 2011;117:2024–34. doi: 10.1002/cncr.25770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loo LWM, et al. Array comparative genomic hybridization analysis of genomic alterations in breast cancer subtypes. Cancer Res. 2004;64:8541–9. doi: 10.1158/0008-5472.CAN-04-1992. [DOI] [PubMed] [Google Scholar]
- 17.MORTON RK. Enzymic synthesis of coenzyme I in relation to chemical control of cell growth. Nature. 1958;181:540–2. doi: 10.1038/181540a0. [DOI] [PubMed] [Google Scholar]
- 18.Branster MV, Morton R. Comparative rates of synthesis in diphosphopyridine nucleotide by normal and tumour tissue from mouse mammary gland: studies with isolated nuclei. Biochem J. 1956;63:640. doi: 10.1042/bj0630640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bajrami I, et al. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol Med. 2012;4:1087–96. doi: 10.1002/emmm.201201250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu CP, Hariharan N, Alcendor RR, Oka S, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through autophagy in cardiomyocytes. Autophagy. 2009;5:1229–1231. doi: 10.4161/auto.5.8.10275. [DOI] [PubMed] [Google Scholar]
- 22.Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–42. [PubMed] [Google Scholar]
- 23.Wang B, et al. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene. 2011;30:907–21. doi: 10.1038/onc.2010.468. [DOI] [PubMed] [Google Scholar]
- 24.Felding-Habermann B. Integrin adhesion receptors in tumor metastasis. Clin Exp Metastasis. 2003;20:203–13. doi: 10.1023/a:1022983000355. [DOI] [PubMed] [Google Scholar]
- 25.Hynes RO. The emergence of integrins: a personal and historical perspective. Matrix Biol. 2004;23:333–40. doi: 10.1016/j.matbio.2004.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Felding-Habermann B, et al. Combinatorial antibody libraries from cancer patients yield ligand-mimetic Arg-Gly-Asp-containing immunoglobulins that inhibit breast cancer metastasis. Proc Natl Acad Sci U S A. 2004;101:17210–5. doi: 10.1073/pnas.0407869101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houghton AN, Eisinger M, Albino AP, Cairncross JG, Old LJ. Surface antigens of melanocytes and melanomas. Markers of melanocyte differentiation and melanoma subsets. J Exp Med. 1982;156:1755–66. doi: 10.1084/jem.156.6.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowditch RD, et al. Identification of a novel integrin binding site in fibronectin. Differential utilization by beta 3 integrins. J Biol Chem. 1994;269:10856–63. [PubMed] [Google Scholar]
- 29.Wayner EA, Orlando RA, Cheresh DA. Integrins alpha v beta 3 and alpha v beta 5 contribute to cell attachment to vitronectin but differentially distribute on the cell surface. J Cell Biol. 1991;113:919–29. doi: 10.1083/jcb.113.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodwin AE, Pauli BU. A new adhesion assay using buoyancy to remove non-adherent cells. J Immunol Methods. 1995;187:213–9. doi: 10.1016/0022-1759(95)00187-6. [DOI] [PubMed] [Google Scholar]
- 31.Pilch J, Habermann R, Felding-Habermann B. Unique ability of integrin alpha(v)beta 3 to support tumor cell arrest under dynamic flow conditions. J Biol Chem. 2002;277:21930–8. doi: 10.1074/jbc.M201630200. [DOI] [PubMed] [Google Scholar]
- 32.Wong NC, et al. Alphav integrins mediate adhesion and migration of breast carcinoma cell lines. Clin Exp Metastasis. 1998;16:50–61. doi: 10.1023/a:1006512018609. [DOI] [PubMed] [Google Scholar]
- 33.Lundström A, Holmbom J, Lindqvist C, Nordström T. The role of alpha2 beta1 and alpha3 beta1 integrin receptors in the initial anchoring of MDA-MB-231 human breast cancer cells to cortical bone matrix. Biochem Biophys Res Commun. 1998;250:735–40. doi: 10.1006/bbrc.1998.9389. [DOI] [PubMed] [Google Scholar]
- 34.Denda S, Müller U, Crossin KL, Erickson HP, Reichardt LF. Utilization of a soluble integrin-alkaline phosphatase chimera to characterize integrin alpha 8 beta 1 receptor interactions with tenascin: murine alpha 8 beta 1 binds to the RGD site in tenascin-C fragments, but not to native tenascin-C. Biochemistry. 1998;37:5464–74. doi: 10.1021/bi9727489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ginsberg MH, Partridge A, Shattil SJ. Integrin regulation. Curr Opin Cell Biol. 2005;17:509–16. doi: 10.1016/j.ceb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 36.Liddington RC, Ginsberg MH. Integrin activation takes shape. J Cell Biol. 2002;158:833–9. doi: 10.1083/jcb.200206011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johansson MW, Mosher DF. Integrin activation States and eosinophil recruitment in asthma. Front Pharmacol. 2013;4:33. doi: 10.3389/fphar.2013.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Felding-Habermann B, et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci U S A. 2001;98:1853–8. doi: 10.1073/pnas.98.4.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferguson MA, Williams AF. Cell-surface anchoring of proteins via glycosyl-phosphatidylinositol structures. Annu Rev Biochem. 1988;57:285–320. doi: 10.1146/annurev.bi.57.070188.001441. [DOI] [PubMed] [Google Scholar]
- 40.Koch-Nolte F, Fischer S, Haag F, Ziegler M. Compartmentation of NAD+-dependent signalling. FEBS Lett. 2011;585:1651–6. doi: 10.1016/j.febslet.2011.03.045. [DOI] [PubMed] [Google Scholar]
- 41.Valenzuela MT, et al. PARP-1 modifies the effectiveness of p53-mediated DNA damage response. Oncogene. 2002;21:1108–16. doi: 10.1038/sj.onc.1205169. [DOI] [PubMed] [Google Scholar]
- 42.Vaziri H, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–59. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 43.Sauve Aa, Youn DY. Sirtuins: NAD(+)-dependent deacetylase mechanism and regulation. Curr Opin Chem Biol. 2012;16:535–43. doi: 10.1016/j.cbpa.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 44.McLure KG, Takagi M, Kastan MB. NAD+ modulates p53 DNA binding specificity and function. Mol Cell Biol. 2004;24:9958–67. doi: 10.1128/MCB.24.22.9958-9967.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng Y, et al. Redox-dependent Brca1 transcriptional regulation by an NADH-sensor CtBP1. Oncogene. 2010;29:6603–8. doi: 10.1038/onc.2010.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di LJ, Fernandez AG, De Siervi A, Longo DL, Gardner K. Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat Struct Mol Biol. 2010;17:1406–13. doi: 10.1038/nsmb.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tripathi MK, et al. Regulation of BRCA2 gene expression by the SLUG repressor protein in human breast cells. J Biol Chem. 2005;280:17163–71. doi: 10.1074/jbc.M501375200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee YC, et al. High visfatin expression in breast cancer tissue is associated with poor survival. Cancer Epidemiol Biomarkers Prev. 2011;20:1892–901. doi: 10.1158/1055-9965.EPI-11-0399. [DOI] [PubMed] [Google Scholar]
- 49.Shackelford RE, Mayhall K, Maxwell NM, Kandil E, Coppola D. Nicotinamide Phosphoribosyltransferase in Malignancy: A Review. Genes Cancer. 2013;4:447–456. doi: 10.1177/1947601913507576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Folgueira MAAK, et al. Gene expression profile associated with response to doxorubicin-based therapy in breast cancer. Clin Cancer Res. 2005;11:7434–43. doi: 10.1158/1078-0432.CCR-04-0548. [DOI] [PubMed] [Google Scholar]
- 51.Von Wnuck Lipinski K, et al. Integrin-mediated transcriptional activation of inhibitor of apoptosis proteins protects smooth muscle cells against apoptosis induced by degraded collagen. Circ Res. 2006;98:1490–7. doi: 10.1161/01.RES.0000229267.77982.0d. [DOI] [PubMed] [Google Scholar]
- 52.Wu Y, et al. Kininostatin associates with membrane rafts and inhibits alpha(v)beta3 integrin activation in human umbilical vein endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:1968–75. doi: 10.1161/ATVBAHA.107.148759. [DOI] [PubMed] [Google Scholar]
- 53.Margheri F, et al. The receptor for urokinase-plasminogen activator (uPAR) controls plasticity of cancer cell movement in mesenchymal and amoeboid migration style. Oncotarget. 2014;5:1538–53. doi: 10.18632/oncotarget.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Horssen R, et al. Intracellular NAD(H) levels control motility and invasion of glioma cells. Cell Mol Life Sci. 2013;70:2175–90. doi: 10.1007/s00018-012-1249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- 56.Lorger M, Krueger JS, O’Neal M, Staflin K, Felding-Habermann B. Activation of tumor cell integrin alphavbeta3 controls angiogenesis and metastatic growth in the brain. Proc Natl Acad Sci U S A. 2009;106:10666–71. doi: 10.1073/pnas.0903035106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11:123–34. doi: 10.1038/nrc3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Staflin K, et al. Targeting activated integrin alphavbeta3 with patient-derived antibodies impacts late-stage multiorgan metastasis. Clin Exp Metastasis. 2010;27:217–31. doi: 10.1007/s10585-010-9320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Felding-Habermann B, et al. Combinatorial antibody libraries from cancer patients yield ligand-mimetic Arg-Gly-Asp-containing immunoglobulins that inhibit breast cancer metastasis. Proc Natl Acad Sci U S A. 2004;101:17210–5. doi: 10.1073/pnas.0407869101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao Z, Gruszczynska-Biegala J, Zolkiewska A. ADP-ribosylation of integrin alpha7 modulates the binding of integrin alpha7beta1 to laminin. Biochem J. 2005;385:309–17. doi: 10.1042/BJ20040590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bi T, Che X. Nampt/PBEF/visfatin and cancer. Cancer Biol Ther. 2010;10:119–25. doi: 10.4161/cbt.10.2.12581. [DOI] [PubMed] [Google Scholar]
- 62.Moschen aR, et al. Visfatin, an Adipocytokine with Proinflammatory and Immunomodulating Properties. J Immunol. 2007;178:1748–1758. doi: 10.4049/jimmunol.178.3.1748. [DOI] [PubMed] [Google Scholar]
- 63.Cea M, et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood. 2012;120:3519–29. doi: 10.1182/blood-2012-03-416776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adya R, Tan BK, Punn A, Chen J, Randeva HS. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc Res. 2008;78:356–65. doi: 10.1093/cvr/cvm111. [DOI] [PubMed] [Google Scholar]
- 65.Li Y, et al. Extracellular Nampt promotes macrophage survival via a nonenzymatic interleukin-6/STAT3 signaling mechanism. J Biol Chem. 2008;283:34833–43. doi: 10.1074/jbc.M805866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen R, Alvero AB, Silasi DA, Mor G. Inflammation, cancer and chemoresistance: taking advantage of the toll-like receptor signaling pathway. Am J Reprod Immunol. 2007;57:93–107. doi: 10.1111/j.1600-0897.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- 67.Jacques C, et al. Proinflammatory actions of visfatin/nicotinamide phosphoribosyltransferase (Nampt) involve regulation of insulin signaling pathway and Nampt enzymatic activity. J Biol Chem. 2012;287:15100–8. doi: 10.1074/jbc.M112.350215. [DOI] [PMC free article] [PubMed] [Google Scholar]