

Abstract
Botulinum neurotoxins (BoNTs) are among the most poisonous biological substances known. They assemble with non-toxic non-hemagglutinin (NTNHA) protein to form the minimally functional progenitor toxin complexes (M-PTC), which protects BoNT in the gastrointestinal tract and release it upon entry into the circulation. Here we provide molecular insight into the assembly between BoNT/A and NTNHA-A using small-angle X-ray scattering. We found that the free form BoNT/A maintains a pH-independent conformation with limited domain flexibility. Intriguingly, the free form NTNHA-A adopts pH-dependent conformational changes due to a torsional motion of its C-terminal domain. Once forming a complex at acidic pH, they each adopt a stable conformation that is similar to that observed in the crystal structure of the M-PTC. Our results suggest that assembly of the M-PTC depends on the environmental pH, and that the complex form of BoNT/A is induced by interacting with NTNHA-A at acidic pH.
Keywords: botulinum neurotoxin, bacterial toxin, neurotoxin-associated proteins, non-toxic non-hemagglutinin, progenitor toxin complex, protein complex, X-ray scattering
Introduction
Botulinum neurotoxins (BoNTs) are produced by the anaerobic bacterium Clostridium botulinum, and are the cause of the often fatal disease botulism. Eight BoNT serotypes have been identified that are termed BoNT/A–H [1, 2], whereas human botulism is usually caused by BoNT/A, B, or E, rarely type F [3, 4]. Food-borne botulism is the major naturally occurring botulism, which happens when BoNTs are ingested together with contaminated food [5]. One of the remarkable features of BoNTs is that they are capable of surviving transit through the gastrointestinal (GI) tract before being absorbed into the general circulation. The high stability of BoNTs in extremely acidic and protease-rich GI environment is attributed to the formation of progenitor toxin complexes (PTCs), in which BoNTs are protected by the non-toxic neurotoxin-associated proteins (NAPs) [6-8]. Naturally, BoNT/A, B, C, D and G are produced in bacteria together with four NAPs, which include non-toxic non-hemagglutinin (NTNHA) and three hemagglutinins (HAs: HA70, HA33 and HA17). Three major types of PTCs with different molecular sizes have been identified, including the M-PTC (12S), L-PTC (16S), and LL-PTC (19S). The M-PTC is composed of BoNT and NTNHA. The L-PTC is a bimodular complex composed of the M-PTC and the HA complex, which is assembled by HA70, HA17, and HA33 and facilitates toxin absorption in the small intestine [9-14]. The structure of the LL-PTC is still largely unknown [15]. Animal toxicity studies suggest that the oral toxicity of BoNTs is highly related to size of the PTCs, decreasing in the order of L-PTC, M-PTC, and free BoNT [6, 16].
BoNT is synthesized as a single polypeptide chain, which is post-translationally nicked into two segments, light chain (LC) and heavy chain (HC), which are covalently linked by a disulfide bond. LC is a zinc-containing protease; the N-terminal domain of HC (HN) is involved in membrane translocation of the toxin and the C-terminal domain of HC (HC) is responsible for host cell surface binding (Fig. 1B) [3, 17]. Interestingly, the overall structure of NTNHA is highly similar to that of BoNT despite ∼20% amino acid sequence identity. It is composed of three domains nLC, nHN, and nHC that are homologous to LC, HN, and HC of BoNT, respectively (Fig. 1) [8]. BoNT/A adopts a large conformational change upon NTNHA-A binding. The HC domain rotates about 140° around a peptide linker connecting HN and HC while the LC–HN moiety maintains a rigid conformation similar to that of the free form of BoNT/A (Fig. 1B and 1D) [8, 18]. In contrast, the crystal structure of NTNHA-A in complex with BoNT/A and the free form of NTNHA-D display a similar conformation [8, 19].
Fig. 1. Structures of the M-PTC, BoNT/Ai, and NTNHA-A.
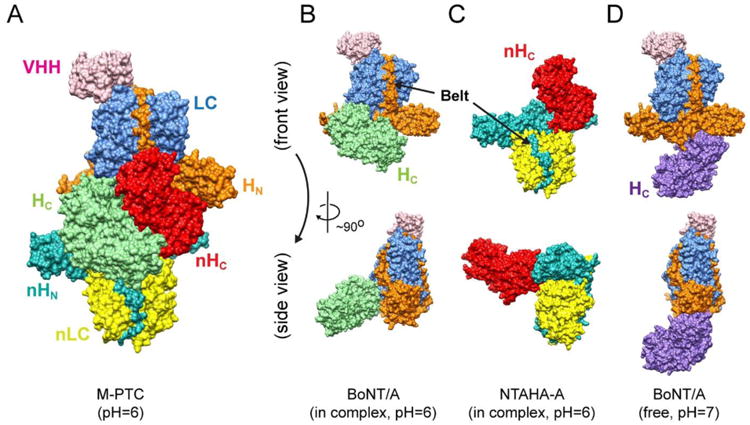
(A) Surface representation of the M-PTC with each domain labeled in a distinct color (PDB ID: 3V0A) [8]. The same color code and view angles are used in all the following figures. (B–C) The structures of BoNT/Ai and NTNHA-A in the context of the M-PTC. (D) BoNT/A in the free form (PDB ID: 3BTA) [18]. Note that the HC domain is labeled in light green or purple in the complex form or the free form, respectively.
Recent studies suggested that BoNT/A and NTNHA-A assemble into the M-PTC only at acidic pH, which protects BoNT/A against the low pH environment and digestive proteases in the GI tract. BoNT/A is released upon entry into the circulation, triggered by environmental pH change [8]. However, despite high-resolution static pictures of BoNT and NTNHA, their dynamic solution conformations and mechanism underlying the pH-dependent assembly and disassembly of the M-PTC are largely unknown. Here we performed comprehensive small angle X-ray scattering (SAXS) studies to examine the solution structures of BoNT/A and NTNHA-A as well as the dynamic assembly of the M-PTC. Our data suggested that the free form BoNT/A maintains a pH-independent conformation in solution, whereas NTNHA-A adopts distinct conformations at pH 6.0 and 8.0. The M-PTC is assembled through induced fit between BoNT/A and NTNHA-A at acidic pH, and its solution structure is indistinguishable from its crystal structure.
Results
BoNT/A adopts an indistinguishable conformation at pH 8.0 and 6.0
The enzymatically inactive recombinant BoNT/A1 (termed BoNT/Ai), which carries three point mutations in LC (E224Q/R363A/Y366F), was used throughout our studies [8]. Highly homogeneous recombinant BoNT/Ai with a heavy-chain-only antibody (VHH-F12) bound in LC was prepared as previously described [8]. VHH is used as a BoNT/Ai-specific structural marker throughout this study, because BoNT and NTNHA have similar structures and adopt a pseudo 2-fold symmetry in the M-PTC. It has been shown that the three point mutations in LC that inactivate BoNT/A and the bound VHH do not affect BoNT/Ai structure or its interaction with NTNHA-A. To investigate the solution structure of the free BoNT/Ai, we performed SAXS studies at pH 8.0 and 6.0, representing two conditions that have opposite effects on regulating the assembly of the M-PTC [8]. BoNT/Ai at concentrations ranging from 0.51 to 6.10 mg/ml was used for experiments at pH 8.0, and they were 0.58 to 6.90 mg/ml at pH 6.0. At both conditions, no protein aggregation was observed except for interparticle interactions in several high concentration samples. Since interparticle interactions were not observed at protein concentrations less than 1.0 mg/ml, those concentration curves were used for the extrapolation to zero concentration and a composite SAXS profile was carefully generated by scaling and merging with a curve at the highest concentration (Figs. 2A and S1).
Fig. 2. SAXS studies of BoNT/Ai.
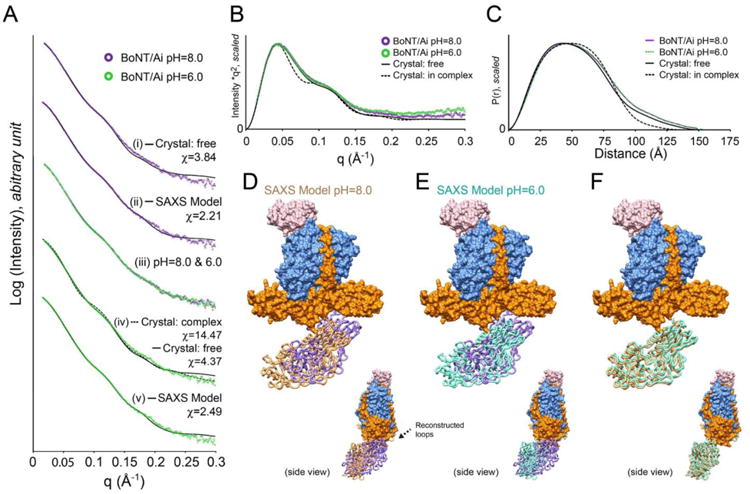
(A) Scattering profiles of BoNT/Ai at pH 8.0 (purple) and 6.0 (green). (i) Curve fitting of data at pH 8.0 with the crystal structure of free form BoNT/A. (ii) Curve fitting of data at pH 8.0 with the SAXS model determined by rigid body refinement. (iii) Superimposition of SAXS profiles at pH 8.0 and 6.0. Constant subtractions used in the rigid body refinements were taken into account. (iv) Curve fitting of data at pH 6.0 with the crystal structures of BoNT/A in the free (solid line) and the complex (dash line) forms. (v) Curve fitting of data at pH 6.0 with the SAXS model determined by rigid body refinement. All curve fittings were performed using the program CORAL [20]. (B) Kratky plots of the experimental profiles and theoretical curve of BoNT/Ai. (C) Pair distance distributions P(r) of BoNT/Ai at pH 8.0 and 6.0. Theoretical curves are shown as well. (D) The SAXS model of BoNT/Ai at pH 8.0 determined by rigid body refinement. The HC domain was refined as a rigid body (shown as brown tubes). Crystal structure of the free form BoNT/A is superimposed onto the LC–HN domain, with the HC domain labeled in purple tubes. The position of reconstructed loop is indicated by an arrow. (E) The SAXS model of BoNT/Ai at pH 6.0 with the HC domain labeled in cyan. (F) Comparison of the SAXS models between pH 8.0 and 6.0.
To analyze the solution structures of BoNT/Ai, two different types of structural “flexibility” are taken into consideration in advance of rigid body refinement modeling. One is related to the intrinsically disordered structures; the electron densities in such regions are poorly defined in the crystal structures (e.g. flexible termini or disordered loops). The other is domain flexibility in multi-domain proteins where each domain is well folded but inter-domain conformation is flexible, which is usually induced by flexible inter-domain linkers. Kratky plot can be used to judge the degree of both types of flexibility. The Kratky plots of scattering curves at both pH 8.0 and 6.0 were examined. They converge well to the baseline, suggesting that BoNT/Ai does not have significant disordered local structures. Furthermore, both Kratky plots are very similar to the theoretical curve independently calculated based on the crystal structure of BoNT/A in the free form (PDB ID: 3BTA) but not the complex form (PDB ID: 3V0A) (Fig. 2B). This suggests that the elongated shape of Kratky plot does not indicate domain flexibility but reflects its multi-domain organization.
At pH 8.0, BoNT/Ai displays a distance distribution function P(r) (real space Rg = 44.7 Å and Dmax = 150.0 Å), which is slightly different from the theoretical curve generated from the crystal structure of the free form (Rg = 43.4 Å and Dmax = 156.2 Å) that was crystallized at pH 7.0 [18]. The fit of the theoretical curve to the experimental profile yields a χ value of 3.84 (Fig. 2A (i)). Together with a slightly larger tail of P(r) function compared to the crystal structure, the data suggest that the solution structure of BoNT/Ai at pH 8.0 is similar to the crystal structure of the free form, albeit adopting a relatively more extended conformation (Fig. 2C). The estimated molecular weight suggests a monomeric state in solution (Table S1), which is consistent with the previous analytic ultracentrifugation (AUC) analysis [8].
Previous crystallographic studies suggest that the LC and HN domains of BoNT/A are tightly linked by the belt region and display an overall rigid conformation, and that VHH-F12 tightly binds to LC [8]. Therefore, we treated the VHH–LC–HN moiety and the HC domain as two independent rigid bodies and performed the rigid body refinement using the program CORAL with a scattering profile up to q = 0.3 Å-1 [20]. The structure of the free form BoNT/A was used as an initial refinement model due to their structural similarity to the solution conformation as suggested by curve-fitting analysis described above. In addition, eighteen residues between LC and HN (Arg432–Ala449) that were disordered in the crystal structure were assigned as a lacking fragment and reconstructed as a chain of dummy residues. Five residues between HN and HC (Lys871–Asn875) were re-assigned as a flexible linker to be reconstructed. Ten independent refinements were performed and an average position of the HC domain was then determined (Fig. 2 and S2). Those reconstructed fragments were further refined with fixing the resulting HC position. The rigid body refinement significantly improved the agreement between the SAXS model and the experimental profile (χ = 2.21) (Fig. 2A (ii)). Compared to the crystal structure of the free form, the HC domain of BoNT/Ai demonstrates a more extended conformation relative to the LC–HN moiety with a mobility of 16.6 Å (between the domain centers) (Fig. 2D and Table S2).
The SAXS profile of BoNT/Ai at pH 6.0 is almost identical to that at pH 8.0 (Fig. 2A (iii)), although a slightly higher background is observed in Kratky plot (q > 0.15 Å-1 on Fig. 2B). Specifically, the real space Rg (∼44.9 Å-1), Dmax (∼153.0 Å), and the P(r) functions are highly similar for data collected at pH 6.0 and pH 8.0 (Fig. 2C). The SAXS experimental profile at pH 6.0 does not agree well with the crystal structure of the free form (χ = 4.37); it is even worse when compared with the complex form (χ = 14.47) (Fig. 2A (iv)). The difference is further emphasized by different shapes in Kratky plot and P(r) function (Figs. 2B–C). The rigid body refinement was performed in the same manner as that at pH 8.0. Consistent with the model at pH 8.0, the SAXS model of BoNT/Ai at pH 6.0 also shows a slightly extended conformation with a mobility of HC at 15.0 Å (Fig. 2E). The final refined HC conformation turned out to be nearly identical to that observed at pH 8.0. The two independently refined SAXS models at pH 8.0 and 6.0 suggest that the conformation of the HC domain is slightly shifted but without any large torsional motion compared to the crystal structure of the free BoNT/A. The SAXS models of BoNT/Ai also explain the more elongated experimental P(r) functions than the theoretical curve derived from the crystal structure of the free form (Fig. 2C).
To rule out experimental and analytical systematic error (e.g. signal to noise ratio, over-refinement, etc.), several BoNT/Ai models with different conformations of the HC domain were constructed and tested as initial models for rigid body refinement with different resolution ranges. For example, one of the HC conformation tested was similar to that observed in the complex form. We found that the resulting HC positions were converged to a position that was nearly identical to the SAXS models as shown in Figures 2 and S2. No significant difference was observed for HC positions from ten independent refinements at pH 8.0 and pH=6.0 (Fig. S2). The subtle difference between the models at pH 8.0 and pH 6.0 (Fig. 2F) could be explained by slightly different signal-to-noise ratios of scattering intensity due to random noise, error of sample concentration, marginally different background level caused by buffer subtraction, etc.
Taken together, we conclude that BoNT/Ai maintains an indistinguishable conformation at pH 8.0 and 6.0, which is similar to the crystal structure of the free form but with a more extended HC domain. Therefore, the conformation of BoNT/Ai observed in the M-PTC is not directly induced by environmental pH change.
NTNHA-A has different conformations at pH 6.0 and 8.0
The solution structure of NTNHA-A was also investigated at pH 6.0 and 8.0. SAXS experiments were performed with protein concentrations ranging from 0.6 to 10.0 mg/ml. Intriguingly, NTNHA-A showed different behaviors at different pHs: while concentration-dependent interparticle interactions were only observed at protein concentrations higher than 1.3 mg/ml at pH 6.0 (Fig. S1), radiation-induced aggregations were observed at lower concentrations at pH 8.0. The composite SAXS profiles were carefully generated in the same manner as that of BoNT/Ai. The tails of Kratky plots are well converged to a baseline at higher q range with comparable levels of a theoretical curve that was independently calculated from the crystal structure (Fig. 3B). These data indicate that NTNHA-A in solution is well folded without significant disordered regions or domain flexibility.
Fig. 3. SAXS studies of NTNHA-A.
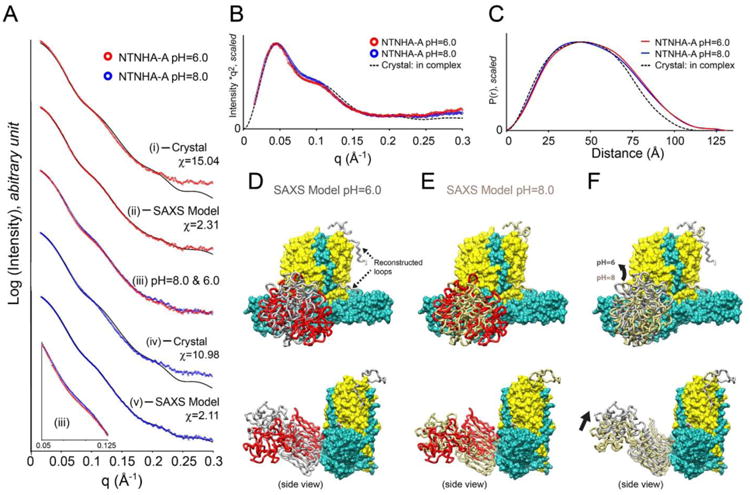
(A) Scattering profiles of NTNHA-A at pH 6.0 (red) and 8.0 (blue). (i) Curve fitting with the crystal structure in the M-PTC (PDB ID: 3V0A, chain B). (ii) Curve fitting with the SAXS model determined by rigid body refinement. (iii) Superimposition of SAXS profiles between pH 8.0 and 6.0. Constant subtractions used at the rigid body refinements were taken into account. A close up view at q = 0.05–0.125 Å-1 is shown in the inset. (iv) Curve fitting with the crystal structure (PDB ID: 3V0A, chain B). (v) Curve fitting with the SAXS model determined by rigid body refinement. (B) Kratky plots of NTNHA-A at pH 8.0 and 6.0. Theoretical curve of the crystal structure is also shown. (C) Pair distance distributions P(r) of NTNHA-A at pH 8.0 and 6.0. Theoretical curve is shown as well. Peak positions are significantly different. (D) The SAXS model of NTNHA-A at pH 6.0 determined by rigid body refinement (shown as gray tubes). The nHC domain was refined as rigid body while two disordered loops in the crystal structure were reconstructed. The crystal structure is superimposed onto the nLC–nHN domain and the nHC domain is shown in red tube. The positions of reconstructed loops are indicated by arrows. (E) The SAXS model of NTNHA-A at pH 8.0 (gold tube). (F) Comparison of SAXS models between pH 6.0 and 8.0.
The SAXS profile of NTNHA-A at pH 6.0 yields real space Rg of 40.3 Å-1 and Dmax of 131.0 Å, which are slightly different from the theoretical values (Rg = 38.4 Å-1 and Dmax = 125.4Å) derived from the crystal structure. The subtle difference is reasonable considering that there are 44 disordered residues not observed in the crystal structure (Fig. 3C). The estimated molecular weight suggests a monomeric state for NTNHA-A in solution that is consistent with the previous AUC analysis [8]. However, the SAXS profile of NTNHA-A at pH 6.0 does not agree with the crystal structure of its complex form (χ = 15.04), suggesting a large conformational change (Fig. 3A (i)).
We next performed rigid body refinements on NTNHA-A using its crystal structure as the model. Similar to the strategy described above for analyzing BoNT/Ai, the nLC-nHN and nHC domains were treated as rigid bodies (Fig. 1C). Five residues (Glu832–Try836) between nHN and nHC domains were assigned as an inter-domain linker, and two disordered regions (Gly114– Ala148, also termed the nLoop, and Asn439–Asn447) not observed in the crystal structure were assigned as lacking fragments to be reconstructed.
The rigid body refinement and reconstruction of the two disordered loops remarkably improved the fit between NTNHA-A's SAXS model and the experimental profile at pH 6.0 (χ = 2.31) (Fig. A (ii)). Compared to the crystal structure, SAXS model adopts a different conformation due to a hinge motion of the nHC domain. The C-terminus of nHC domain is slightly splayed out from the position of the complex form (Fig. 3D). In comparison to the HC domain of BoNT/Ai, the nHC domain demonstrates less mobility (4.17 Å) but an obvious torsional motion. This motion appears to slightly open the interface between the nHC and nHN domains, which presumably exposes the interface to bind and accommodate the HC domain of BoNT/Ai.
The SAXS studies of NTNHA-A at pH 8.0 demonstrates a scattering profile different to that of pH 6.0 at the middle q region, q = ∼0.08 Å-1 (Fig. 3A (iii)), which is emphasized by Kratky plot (Fig. 3B), resulting in a P(r) function with a distinct peak shift (Fig. 3C). Its real space Rg (39.9 Å-1) and Dmax (127.5 Å) are closer to those of the crystal structures. However, the data derived from the crystal structure of NTNHA-A does not fit well the experimental profile (χ = 10.98) (Fig. 3A (iv)).
The rigid body refinement significantly improved the agreement between the SAXS data and the experimental profile at pH 8.0 (χ = 2.11). Intriguingly, the nHC domain was also splayed out but in a conformation different from that at pH 6.0 (Fig. 3E). The mobility of the center of the nHC domain (3.74 Å) is very similar to that at pH 6.0. We notice that the conformation of the reconstructed nLoop is structurally different between the SAXS models of pH 6.0 and pH 8.0 (Fig. 3F). In order to verify whether the results are potentially biased by experimental or systematic errors, different initial models that have different nHC conformations were tested in rigid body refinement. For example, one of the tested nHC conformation mimics that of the HC domain in free form BoNT/A. No distinctive nHC conformation was observed compared to the SAXS models shown in Fig. 3.
A discrepancy between data collected at pH 6.0 and 8.0 was observed at the mid-angle range of scattering profiles (Figs. 3A and 3B), which could be caused by conformational change of the nHC domain or the reconstructed loops. In general, the middle angle scattering region is associated with domain orientation, whereas the low angle scattering provides information on size and oligomeric state [21]. In fact, repetitions of the rigid body refinement at pH= 6.0 and 8.0 clearly showed different distributions of nHC position (Fig. S2). The theoretical scattering profiles derived from the crystal models with or without the reconstructed loops obtained from the SAXS models are indistinguishable at the middle angle part (Fig. S3). It thus suggests that the differences observed at the middle angle between pH 6.0 and pH 8.0 (Fig. 3A (iii)) is caused by conformational change of the nHC domain rather than the addition of the reconstructed loops. To further verify this, we simulated the sensitivity of the scattering profile on detecting the positioning of the nHC domain (Fig. S4). We found that even a slight torsional motion of the nHC domain is sufficient to alter the scattering profile at middle q angles. Therefore, we conclude that NTNHA-A adopts different conformations at pH 6.0 and 8.0 with different nHC orientation (Fig. 3F).
Solution structure of the M-PTC at pH 6.0
The SAXS studies of the M-PTC were performed at pH 6.0, because it is formed only at the acidic environment [8]. The preliminary equilibrium SAXS experiments of the M-PTC showed excessive concentration dependence. Therefore, we performed the FPLC-SAXS experiments that used an online size-exclusion chromatography to obtain more monodispersed sample. M-PTC was eluted as a single peak, around which the scattering profiles were investigated for interparticle interactions (Fig. 4A). Reciprocal and real space Rg indicated that concentration-dependent interparticle interactions still existed in the data set, but it could be alleviated by averaging profiles in three different regions (Fig. S5). Kratky plot showed no significant flexibility and was consistent with the theoretical curve calculated from the crystal structure (Fig. 4B). Ab-initio shape reconstruction up to q = 0.3 Å-1 was performed for the M-PTC in solution using the program GASBOR [22]. The models were reproducible with an average normalized spatial discrepancy (NSD) of 1.583 and variation of 0.047. The most populated volume computed from 15 models is shown in Figure 4C. The crystal structure of the M-PTC fits well to the envelope, clearly indicating a similar structure of the M-PTC in crystal or in solution.
Fig. 4. FPLC-SAXS studies of the M-PTC.
(A) Gel-filtration profile and Rg values in the online FPLC-SAXS experiment. The profile was made by plotting X-ray intensity of each image. The real and reciprocal space Rgs were estimated by the program AUTOGNOM [36]. (B) SAXS profile of the M-PTC (yellow) and curve fitting with the crystal structure. Kratky plot is shown in inset. (C) An ab-initio model derived from the SAXS profile. The most populated volume in solution, which was filtered using the program DAMFILT in DAMAVER suite [38] is displayed in gray.
Discussion
Since BoNT/Ai and NTNHA-A have a non-globular multi-domain configuration, it is technically challenging to judge their structural flexibility (disordered structures and/or domain flexibility) without atomic coordinates due to the difficulty to apply Porod-Debey plot [23]. We have successfully ruled out such flexibility of BoNT/Ai and NTNHA-A by comparing the experimental Kratky plots and the ones calculated from the known crystal structures.
Furthermore, the missing fragments in the crystal structures were reconstructed during SAXS-based rigid body refinement, taking advantage of the program CORAL. The good agreement between the SAXS model and the experimental profile also ruled out the multimodality of the domain conformation. In particular, the novel pH-dependent conformational changes of NTNHA-A at pH 8.0 and 6.0 were precisely characterized based on the following achievement and verifications. (1) High flux synchrotron radiation combined with high signal-to-noise ratio detector made it possible to measure low concentration samples, allowing to extrapolate zero-concentration scattering profile. (2) Since the overall shape of the nHC domain is elongated and asymmetric, the movement of the nHC domain in the non-globular multi-domain conformation of NTNHA-A readily alters scattering profile at middle q due to dramatic rearrangement of interatomic distance distribution (P(r) function) of the whole protein (Fig. S4). (3) The flexible nLoop located at the tip of the nLC domain mainly compensates size information at low q (Guinier region) but hardly disturbs scattering profile at middle q range associated with nHC orientation (Figs. S3–S4).
Our data on the solution structure of free BoNT/Ai show that the HC domain is coordinated rigidly with limited flexibility relative to the LC–HN moiety, regardless the environmental pH. Compared to the crystal structure of the free form, the SAXS model of free BoNT/Ai displays a more extended conformation caused by a small reorientation of the HC domain. This subtle difference is likely caused by the effect of crystal packing. We suggest that the SAXS model of BoNT/Ai represents its dominant conformation in solution, which can be subsequently induced into various conformations depending on its functional needs. For example, the large reorientation of the HC domain of BoNT/Ai observed in the M-PTC is likely induced by interactions with NTNHA-A to achieve the optimal protection in the GI tract. We and other groups have also found that reorientation of the HC domain through the HN–HC linker is crucial to coordinate receptor binding and LC translocation of BoNT when attacking motoneurons [8, 24, 25].
Despite adopting similar structures in the M-PTC, BoNT/Ai and NTNHA-A in the free form behaves differently in solution. The free NTNHA-A displays a pH-dependent conformational change, which is largely due to spatial re-orientation of nHC (Fig. 5). We used the structure of the complex form NTNHA-A to generate a theoretical scattering curve and the structures of its three individual domains were employed as initial models for rigid body refinement of the SAXS models. Interestingly, the resulting solution conformations of NTNHA-A at pH 6.0 and pH 8.0 both differ from its crystal structures. These data suggest that NTNHA-A is capable of sensing the environmental pH change and its conformation in the M-PTC is a result of mutual induced fit with BoNT/Ai (Fig. 5).
Fig. 5. A schematic representation of the assembly of the M-PTC.
BoNT/A and NTNHA-A are predominantly monomeric and each adopts a relatively rigid conformation at pH 8.0, which is unable to form a complex. Decreasing the environmental pH to 6.0 does not significantly change the conformation of BoNT/A. However, it induces a conformational change of the nHC domain of NTNHA-A, which is then slightly splayed out to initiate interactions with BoNT/A. At the meantime, the acidic pH protonates pH-sensing residues on BoNT/A and NTNHA-A, which strengthen the inter-molecule interactions. Subsequently, a mutual induced fit triggers a large-reorientation of the HC domain of BoNT/A while the nHC domain of NTNHA-A undergoes a further conformational change to tightly lock with BoNT/A.
It is noted that our solution models of NTNHA-A disagree with a previously reported SAXS structure of NTNHA-D that displays a different hinge motion of the nHC domain [19]. Our data of NTNHA-A showed that concentration-dependent interparticle interactions occurs at pH 6.0 (Fig. S1), which were successfully minimized by examining samples at multiple concentrations as described above. In contrast, if only data collected at protein concentrations at 1.3 mg/ml or higher had been used for ab-initio modeling of NTNHA-A at pH 6.0, we would have observed an artificial conformation of NTNHA-A (data not shown) that was similar to the reported SAXS structure of NTNHA-D, which was derived from a single protein concentration at 1 mg/ml [19]. NTNHA-A did not show interparticle interactions at pH 8.0. We speculate that the pH-dependent electrostatic change of surface property of NTNHA-A presumably accounts for not only its concentration-dependent interparticle interactions but also its domain re-orientation (Fig. 5).
In summary, our SAXS studies reveal a dynamic pH-dependent assembly of the M-PTC (Fig. 5). In comparison to the relatively rigid conformation of BoNT/A, NTNHA-A is able to sense the change of environmental pH. Under acidic condition, NTNHA-A adopts a specific conformation that initiates its interaction with BoNT/A. At the same time, the pH-sensing residues in BoNT/A and NTNHA-A are protonated to allow favorable local electrostatic interactions between them [8]. The inter-locked conformation of the M-PTC is subsequently formed through a mutual induced fit between these two proteins. These studies have provided new insight into the mechanism that regulates the assembly of the M-PTC, which may ultimately lead to new strategies to counteract this toxin self-protecting process.
Material and Methods
Protein preparation
The recombinant BoNT/Ai, VHH-F12, and NTNHA-A were prepared as described previously [8]. Protein samples were then exchanged into a buffer containing 50 mM Mes, pH 6.0, and 250 mM NaCl (Buffer A) or 50 mM Tris, pH 8.0, and 250 mM NaCl (Buffer B) using centrifugal filters (Amicon Ultracel, EMD Millipore) for the subsequent experiments. BoNT/Ai with bound VHH and NTNHA-A were concentrated to ∼10.0 mg/ml and 7.0 mg/ml, respectively, before being diluted to various experimental conditions (Table S1).
SAXS data collection and initial data treatment
All SAXS experiments were performed using Bio-SAXS beam line BL4-2 at Stanford Synchrotron Radiation Lightsource (SSRL) [26]. Data were collected on a Rayonix MX225-HE CCD detector (Rayonix, Evanston, IL) with a 1.7 m sample-to-detector distance and a beam energy of 11 keV (wavelength, λ = 1.127 Å) was used. The momentum transfer (scattering vector) q was defined as q = 4πsin (θ)/λ, where 2θ was scattering angle. The q scale was calibrated by silver behenate powder diffraction [27].
A series of protein samples (concentration series) and its equivalent buffer were placed on a 96-well plate of the automatic sample-loading robot, which was operated by the data acquisition program Blu-ICE [28, 29]. A 1.5 mm quartz capillary cell (Hampton Research, Aliso Viejo, CA) was maintained at 20 °C that kept sample aliquot in the x-ray beam. The sample aliquot in the capillary oscillated during exposures to reduce radiation damage. The 15 scattering images with 1 second exposure were obtained from a 30 μl sample or buffer aliquots.
The data processing program SasTool (http://ssrl.slac.stanford.edu/∼saxs/analysis/sastool.htm) was used for scaling, azimuthal integration, and averaging of individual scattering images after inspection for any variations that were potentially caused by radiation damage or sample heterogeneity during oscillation. The averaged buffer curve was then subtracted from the averaged protein curves. The subtracted curves were used for further analysis. The program PRIMUS was employed to investigate concentration-dependent interparticle interactions and to merge curves in cases when those interactions were observed [30]. Pairwise distribution function P(r) were calculated up to q = 0.3 Å-1 using the program GNOM [31]. All theoretical Rg, Dmax and P(r) were also calculated using GNOM. Molecular weight was estimated by two ways: one was based on water scattering [32] and the other was to use online SAXS MoW applet (http://www.if.sc.usp.br/∼saxs/) [33]. All experimental and structural parameters are summarized in the supplemental table (Table S1).
Rigid body refinement
SAXS-based rigid body modeling of BoNT/Ai and NTNHA-A was performed using the program CORAL [20]. For BoNT/Ai, the crystal structure of the free form was employed as an initial model whereas VHH–LC–HN and HC were treated as individual rigid bodies, as described in the result section. A disordered region in the crystal structure (Arg432–Ala449) was assigned as lack fragments to be reconstructed. The 5 residues between HN and HC domains (Lys871–Asn875) were assigned as a flexible linker. The crystal structure of NTNHA-A in the complex form was employed as an initial model when nLC–nHN and nHC domains were treated as individual rigid bodies. Two disordered regions (Gly114–Ala148 and Asn439–Asn447) that were not observed in the crystal were assigned as lack fragments. Furthermore, the nLoop was assigned as a nicked loop between Lys133 and Lys134 as reported previously [8]. Similarly, the 5 residues between nHN and nHC domains (Glu832–Try836) were assigned as a flexible linker. SAXS data up to q = 0.3 Å-1 was used for all refinements. Ten independent refinements were performed and the average HC or nHC position was obtained using the program AVEPDB [34]. The final lack fragments and a flexible linker were further refined with fixing the averaged Hc or nHC position. The program CORAL was also used for the crystal structures in order to obtain χ value in the same way. Results are summarized in Tables S1 and S2.
Online FPLC-SAXS
Experimental setup for the online FPLC-SAXS was similar to that described previously [35]. Briefly, the beam line setup was identical to what described above and a 120 μl of the M-PTC at 10.9 mg/ml was applied to a Superdex-200 PC 3.2/30 size-exclusion column (GE Healthcare, WI, USA) with a flow rate of 0.08 ml/min. The first 100 images were scaled and averaged to create a buffer-scattering profile, which was then subtracted from each of the subsequent images to produce the final scattering curve for each exposure. A series of real space and reciprocal space Rg values were systematically estimated using the program AUTOGNOM [36]. The composite profile was generated by scaling and merging 3 averaged profiles (images No.132– 141, 167–178, and 180–190, excluding images having outlier Rg values) at q = 0.0609–0.0720 Å-1, which were then used for curve fitting with data derived from the crystal structure using the program FoXS [37].
Ab-initio modeling
Ab-initio modeling was performed using the composite profile generated from the FPLC-SAXS data. Fifteen molecular shapes were calculated using the program GASBOR with no symmetry constrains [22]. They were averaged and then filtered using programs in the DAMAVER suite [38]. Chiral model of the filtered model was also generated using the program MOLEMAN [34] and the crystal structure was docked into those models using “molmap” and ‘map-fit” functions in the UCSF Chimera [39]. The chirality was carefully examined and the final position was slightly aligned and determined based on visual inspection.
Supplementary Material
Highlights.
Assembly of BoNT/A and NTNHA-A is a pH-dependent process.
The free form BoNT/A maintains a pH-independent conformation in solution.
Environmental pH induces a torsional motion of the C-terminal domain of NTNHA-A.
The conformation of BoNT/A in the complex is induced by NTNHA-A at acidic pH.
Acknowledgments
This work was supported in part by National Institute of Allergy and Infectious Diseases (NIAID) grants (R01AI091823 to R.J.) and by the Swiss Federal Office for Civil Protection (BABS #353003325 to A.R.). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209), and National Institute of General Medical Sciences (NIGMS). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
Abbreviations footnote
- BoNT
botulinum neurotoxin
- HA
hemagglutinin
- NAP
neurotoxin-associated protein
- NTNHA
non-toxic non-hemagglutinin
- PTC
progenitor toxin complex
- SAXS
small-angle X-ray scattering
- FPLC
fast protein liquid chromatography
- NSD
normalized spatial discrepancy
- SSRL
Stanford Synchrotron Radiation Lightsource
Footnotes
All graphical representations in the present paper were produced using UCSF Chimera.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hill KK, Smith TJ. Genetic diversity within Clostridium botulinum serotypes, botulinum neurotoxin gene clusters and toxin subtypes. Curr Top Microbiol Immunol. 2013;364:1–20. doi: 10.1007/978-3-642-33570-9_1. [DOI] [PubMed] [Google Scholar]
- 2.Dover N, Barash JR, Hill KK, Xie G, Arnon SS. Molecular characterization of a novel botulinum neurotoxin type H gene. The Journal of infectious diseases. 2014;209:192–202. doi: 10.1093/infdis/jit450. [DOI] [PubMed] [Google Scholar]
- 3.Montecucco C, Schiavo G. Structure and function of tetanus and botulinum neurotoxins. Q Rev Biophys. 1995;28:423–72. doi: 10.1017/s0033583500003292. [DOI] [PubMed] [Google Scholar]
- 4.Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, et al. Botulinum toxin as a biological weapon: medical and public health management. JAMA. 2001;285:1059–70. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- 5.Sugiyama H. Clostridium botulinum neurotoxin. Microbiol Rev. 1980;44:419–48. doi: 10.1128/mr.44.3.419-448.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohishi I, Sugii S, Sakaguchi G. Oral toxicities of Clostridium botulinum toxins in response to molecular size. Infect Immun. 1977;16:107–9. doi: 10.1128/iai.16.1.107-109.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu S, Jin R. Assembly and function of the botulinum neurotoxin progenitor complex. Curr Top Microbiol Immunol. 2013;364:21–44. doi: 10.1007/978-3-642-33570-9_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu S, Rumpel S, Zhou J, Strotmeier J, Bigalke H, Perry K, et al. Botulinum neurotoxin is shielded by NTNHA in an interlocked complex. Science. 2012;335:977–81. doi: 10.1126/science.1214270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee K, Gu S, Jin L, Le TT, Cheng LW, Strotmeier J, et al. Structure of a bimodular botulinum neurotoxin complex provides insights into its oral toxicity. PLoS Pathog. 2013;9:e1003690. doi: 10.1371/journal.ppat.1003690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee K, Lam KH, Kruel AM, Perry K, Rummel A, Jin R. High-resolution crystal structure of HA33 of botulinum neurotoxin type B progenitor toxin complex. Biochem Biophys Res Commun. 2014;446:568–73. doi: 10.1016/j.bbrc.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benefield DA, Dessain SK, Shine N, Ohi MD, Lacy DB. Molecular assembly of botulinum neurotoxin progenitor complexes. Proc Natl Acad Sci U S A. 2013;110:5630–5. doi: 10.1073/pnas.1222139110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amatsu S, Sugawara Y, Matsumura T, Kitadokoro K, Fujinaga Y. Crystal structure of Clostridium botulinum whole hemagglutinin reveals a huge triskelion-shaped molecular complex. The Journal of biological chemistry. 2013;288:35617–25. doi: 10.1074/jbc.M113.521179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao G, Lee K, Gu S, Lam KH, Jin R. Botulinum neurotoxin A complex recognizes host carbohydrates through its hemagglutinin component. Toxins. 2014;6:624–35. doi: 10.3390/toxins6020624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee K, Zhong X, Gu S, Kruel AM, Dorner MB, Perry K, et al. Molecular basis for disruption of E-cadherin adhesion by botulinum neurotoxin A complex. Science. 2014;344:1405–10. doi: 10.1126/science.1253823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugii S, Sakaguchi G. Molecular construction of Clostridium botulinum type A toxins. Infect Immun. 1975;12:1262–70. doi: 10.1128/iai.12.6.1262-1270.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng LW, Onisko B, Johnson EA, Reader JR, Griffey SM, Larson AE, et al. Effects of purification on the bioavailability of botulinum neurotoxin type A. Toxicology. 2008;249:123–9. doi: 10.1016/j.tox.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 17.Montal M. Botulinum neurotoxin: a marvel of protein design. Annu Rev Biochem. 2010;79:591–617. doi: 10.1146/annurev.biochem.051908.125345. [DOI] [PubMed] [Google Scholar]
- 18.Lacy DB, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat Struct Biol. 1998;5:898–902. doi: 10.1038/2338. [DOI] [PubMed] [Google Scholar]
- 19.Sagane Y, Miyashita SI, Miyata K, Matsumoto T, Inui K, Hayashi S, et al. Small-angle X-ray scattering reveals structural dynamics of the botulinum neurotoxin associating protein, nontoxic nonhemagglutinin. Biochemical and Biophysical Research Communications. 2012;425:256–60. doi: 10.1016/j.bbrc.2012.07.077. [DOI] [PubMed] [Google Scholar]
- 20.Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophysical Journal. 2005;89:1237–50. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly Reviews of Biophysics. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 22.Svergun DI, Petoukhov MV, Koch MHJ. Determination of domain structure of proteins from X-ray solution scattering. Biophysical Journal. 2001;80:2946–53. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rambo RP, Tainer JA. Characterizing Flexible and Intrinsically Unstructured Biological Macromolecules by SAS Using the Porod-Debye Law. Biopolymers. 2011;95:559–71. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumaran D, Eswaramoorthy S, Furey W, Navaza J, Sax M, Swaminathan S. Domain organization in Clostridium botulinum neurotoxin type E is unique: its implication in faster translocation. J Mol Biol. 2009;386:233–45. doi: 10.1016/j.jmb.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 25.Wang JF, Meng JH, Lawrence GW, Zurawski TH, Sasse A, Bodeker MO, et al. Novel chimeras of botulinum neurotoxins A and E unveil contributions from the binding, translocation, and protease domains to their functional characteristics. Journal of Biological Chemistry. 2008;283:16993–7002. doi: 10.1074/jbc.M710442200. [DOI] [PubMed] [Google Scholar]
- 26.Smolsky IL, Liu P, Niebuhr M, Ito K, Weiss TM, Tsuruta H. Biological small-angle x-ray scattering facility at the Stanford synchrotron radiation laboratory. Journal of Applied Crystallography. 2007;40:s453–s8. [Google Scholar]
- 27.Huang TC, Toraya H, Blanton TN, Wu Y. X-ray powder diffraction analysis of silver behenate, a possible low-angle diffraction standard. Journal of Applied Crystallography. 1993;26:180–4. [Google Scholar]
- 28.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, et al. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. Journal of Synchrotron Radiation. 2002;9:401–6. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 29.Martel A, Liu P, Weiss TM, Niebuhr M, Tsuruta H. An integrated high-throughput data acquisition system for biological solution X-ray scattering studies. Journal of Synchrotron Radiation. 2012;19:431–4. doi: 10.1107/S0909049512008072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography. 2003;36:1277–82. [Google Scholar]
- 31.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. Journal of Applied Crystallography. 1992;25:495–503. [Google Scholar]
- 32.Orthaber D, Bergmann A, Glatter O. SAXS experiments on absolute scale with Kratky systems using water as a secondary standard. Journal of Applied Crystallography. 2000;33:218–25. [Google Scholar]
- 33.Fischer H, de Oliveira Neto M, Napolitano HB, Polikarpov I, Craievich AF. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. Journal of Applied Crystallography. 2010;43:101–9. [Google Scholar]
- 34.Kleywegt GJ, Zou JY, Kjeldgaard M, Jones TA, Around O. In: International Tables for Crystallography, Volume F Crystallography of Biological Macromolecules. Rossmann MG, Arnold E, editors. Dordrecht, The Netherlands: Kluwer Academic; 2001. pp. 353–356.pp. 366–367. [Google Scholar]
- 35.West AL, Evans SE, Gonzalez JM, Carter LG, Tsuruta H, Pozharski E, et al. Ni(II) coordination to mixed sites modulates DNA binding of HpNikR via a long- range effect. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5633–8. doi: 10.1073/pnas.1120283109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1 - towards automated and web-supported small-angle scattering data analysis. Journal of Applied Crystallography. 2007;40:s223–s8. [Google Scholar]
- 37.Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Research. 2010;38:W540–W4. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2003;36:860–4. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF chimera -A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.