

Abstract
Background & Aims
New drug targets are urgently needed for the treatment of pancreatic ductal adenocarcinoma (PDA). Nearly all PDAs contain oncogenic mutations in the KRAS gene. Pharmacologic inhibition of KRAS has been unsuccessful, leading to a focus on downstream effectors that are more easily targeted with small molecule inhibitors. We investigated the contributions of phosphoinositide 3 kinase (PI3K) to KRAS-initiated tumorigenesis.
Methods
Tumorigenesis was measured in the KrasG12D/+;Ptf1aCre/+ mouse model of PDA; these mice were crossed with mice with pancreas-specific disruption of genes encoding PI3K p110α (Pik3ca), p110β (Pik3cb), or RAC1 (Rac1).Pancreatitis was induced by 5 daily intraperitoneal injections of cerulein. Pancreata and primary acinar cells were isolated; acinar cells were incubated with an inhibitor of p110α (PIK75) followed by a broad spectrum PI3K inhibitor (GDC0941). PDA cell lines (NB490 and MiaPaCa2) were incubated PIK75 followed by GDC0941. Tissues and cells were analyzed by histology, immunohistochemistry, quantitative reverse transcription PCR, and immunofluorescence analyses for factors involved in the PI3K signaling pathway. We also examined human pancreas tissue microarrays for levels of p110α and other PI3K pathway components.
Results
Pancreas-specific disruption of Pik3ca or Rac1, but not Pik3cb, prevented development of pancreatic tumors in KrasG12D/+ ;Ptf1aCre/+ mice. Loss of transformation was independent of AKT regulation. Preneoplastic ductal metaplasia developed in the mice lacking pancreatic p110α, but regressed. Levels of activated and total RAC1 were higher in pancreatic tissues from Kras G12D/+ ;Ptf1aCre/+ mice, compared with controls. Loss of p110α reduced RAC1 activity and expression in these tissues. p110α was required for the upregulation and activity of the RAC guanine exchange factor (encoded by Rasgrf1) during tumorigenesis. Levels of p110α and RAC1 were increased in human pancreatic intraepithelial neoplasias and PDAs, compared with healthy pancreata.
Conclusions
KRAS signaling, via p110α to activate RAC1, is required for transformation in Kras G12D/+;Ptf1a Cre/+ mice.
Keywords: pancreatic cancer, signal transduction, cytoskeleton, oncogene
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) is the fourth leading cause of cancer-related death worldwide, with a 6% five year survival rate1. Chemotherapy and radiation therapy have little impact on survival, leaving surgery as the most effective treatment2. Unfortunately, only 15% of patients who undergo surgery survive past five years3, 4. This bleak outcome makes understanding the molecular mechanisms of PDA formation and progression so that new “druggable” targets can be identified of paramount importance.
Nearly all PDAs harbor oncogenic mutations in the KRAS gene5, and pancreatic tumor maintenance is compromised when KRAS signaling is extinguished6, 7. While pharmacological inhibition of KRAS has thus far been unsuccessful8, oncogenic KRAS activates downstream kinase effectors such as RAF-MEK and PI3K-AKT that are more easily targeted with small molecule inhibitors. Inhibition of one downstream target, MEK, blocks formation of tumor precursors known as metaplastic ducts, thus limiting subsequent transformation9–11 and can regress mPanINs11. In this study, we focused on phosphoinositide 3-kinase (PI3K)12, 13 and its role in Kras-induced pancreatic tumorigenesis.
Class I PI3Ks are heterodimers consisting of a p110 catalytic subunit (α, β, δ or γ ) bound to a regulatory subunit. Upon activation, they phosphorylate phosphatidylinositol 4,5-bisphosphate (PIP2) to generate the lipid second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3)14. PIP3 binds to adaptor and effector proteins containing pleckstrin homology (PH) domains, driving several downstream signaling cascades. Two well-understood PI3K downstream effectors are the protein kinase AKT15 and the small G-protein RAC16. AKT and phosphoinositide-dependent kinase 1 (PDK1) are relocalized to PIP3-containing membranes where PDK1 phosphorylates and activates AKT17. PI3K primarily activates RAC by relocalization of PH-domain containing guanine nucleotide exchange factors (GEFs) to the plasma membrane16.
While it is accepted that Ras proteins activate PI3K, the precise role of PI3K signaling in Kras-induced PDA is controversial. Eser et al., demonstrate that transgenic expression of a constitutively active mutant of p110α (H1047R) is sufficient to induce pancreatic tumorigenesis18, contrary to results reported in a similar mouse model19. PDK1 is essential for Kras-induced PDA, attributed to a dependency of requisite acinar-to-ductal metaplasia (ADM) prior to tumorigenesis18. RAC1 is also required for pancreatic tumorigenesis by regulating morphological changes that accompany ADM20.
Of the catalytic isoforms, p110γ is expressed primarily by hematopoietic cells21, but is expressed in pancreatic acinar cells, where it controls cellular responses in experimental pancreatitis22. p110δ is considered hematopoietic cell specific21. p110α and p110β are ubiquitously expressed21, and have been shown to be critical for tumorigenesis in other glandular tissues, such as breast23, lung24 and prostate25. Here, we test whether p110α or p110β are involved in Kras-induced pancreatic tumorigenesis using isoform specific knockout mice. We demonstrate that the p110α catalytic subunit is required for pancreatic tumorigenesis in a PDA mouse model, whereas the p110β subunit is unnecessary. Remarkably, p110α was not required for AKT activation due to compensation by other p110 catalytic subunits. However, p110α-null pancreata were incapable of forming stable metaplastic ducts in vivo, which phenocopies Rac1-null mice. Consistent with this, we found that p110α was required for robust RAC1 activation, in part by regulating acinar cell RAC GEF activity.
MATERIALS AND METHODS
Animal Models
KrasLSL-G12D/+, Ptf1aCre/+, Trp53+/R172H, p110αFlox/Flox, p110βFlox/Flox and Rac1Flox/Flox strains have been described26–29. Experiments were conducted in accordance with the Office of Laboratory Animal Welfare and approved by the Institutional Animal Care and Use Committees of Stony Brook University and the Mayo Clinic.
Antibodies
A complete list of antibodies used is provided in the Supplemental Information.
Cerulein Treatment
250μg/kg cerulein (American Peptide Company) were administered daily, intraperitoneally (i.p.) for 5 days, with 24 hours or 7 days recovery. To assess cytoskeletal rearrangement, 30 day old mice were injected i.p. with 250μg/kg cerulein for 3 days and sacrificed 1 hour following the last injection.
Immunofluorescence Staining
Immunofluorescence was performed on frozen sections of PFA perfused pancreata as described previously30. Images were acquired on a Zeiss 510LS Meta confocal microscope (Carl Zeiss). Colocalization was determined by Zen 2012 v8.0 software (Carl Zeiss).
Histology and IHC
Pancreata were fixed overnight in 4% PFA. Tissues were processed in a Leica ASP300S Tissue Processor, paraffin-embedded and cut into 5 μm sections. IHC for phospho-Akt (T308), p110α, Vav1, Tiam1 and CK19 was performed on a Discovery Ultra XT autostainer (Ventana Medical Systems) and counterstained with hematoxylin. Dual IHC for CK19 and Amylase was performed without counterstain. IHC staining for RAC1 was performed on a DAKO Autostainer Plus (DAKO North America, Inc.) and counterstained with hematoxylin. Picrosirius red staining was performed per manufacturer’s instructions (Polysciences, Inc,). Hemotoxylin and Eosin staining was performed using Mayer’s Hemotoxylin solution (Sigma) and Eosin Y (Fisher).
Immunoblotting
For lysate preparation, the pancreas was removed from 12–14-week old mice and immediately homogenized in MLB buffer (25mM HEPES, pH 7.5, 150mM NaCl, 10mM MgCl2, 1mM EDTA, 1% NP-40, and 10% glycerol) containing protease and phosphatase inhibitors (Roche) using a Pro 250 Homogenizer (Pro Scientific). For pancreas lysate immunoblots, 50 μg of protein was separated by denaturing SDS-PAGE, transferred to Immobilon P (Millipore) and visualized by chemiluminescence. Isolated acinar cells were washed once in PBS then lysed in MLB buffer containing protease inhibitors. 75 μg of protein was separated by SDS-PAGE, transferred to Immobilon P (Millipore) and visualized by chemiluminescence. Cell lines were washed once in PBS then lysed in RIPA buffer (150mM NaCl, 10mM Tris pH 7.2, 1% sodium deoxycholate, 1% Triton X-100, and 0.1% SDS) containing protease and phosphatase inhibitors. 25 μg of protein was separated by SDS-PAGE and visualized by chemiluminescence.
Active Ras, Rac and Rac-GEF Assays
Ras-binding domain (RBD) protein for active Ras pulldowns was prepared by subcloning human cRaf (a.a. 1–149) into pGEX-5x-3 (GE Healthcare). p21-binding domain (PBD) protein for active RAC pulldowns was prepared by subcloning human PAK1 (a.a. 70–117) into the pGEX2TK vector. Rac1G15A Agarose (Cell BioLabs) was used for active RAC-GEF pulldowns. For a detailed protocol see Supplemental Information.
Primary Acinar Cell Isolation
Pancreata were harvested from 6- to 8-week old mice, washed twice in Hanks balanced salt solution (HBSS), minced and incubated with 0.2 mg/mL collagenase-P (Roche, Indianapolis, IN) at 37°C for 15 min. Tissue was wa shed 3x in HBSS containing 5% fetal bovine serum (FBS) and filtered through 500μm and 105μm polypropelene mesh (Spectrum Laboratories). Filtrate was centrifuged through 30% FBS in HBSS and resuspended in Waymouth MB 752/1 medium (Sigma-Aldrich) supplemented with 50 μg/mL gentamycin (Life Technologies), 0.4 mg/mL soybean trypsin inhibitor (Life Technologies), and 1 μg/mL dexamethasone (Sigma-Aldrich). Suspension was plated in non-adherent plates and maintained at 37°C in a 5% CO2 atmosphere. Transdifferentiation assays performed as described9.
Adenoviral Infection and Drug Treatment of Primary Acinar Cells
Isolated acinar cells were infected with either adenoviral GFP or adenoviral Cre and maintained at 37°C in a 5% CO 2 atmosphere for 3 days. Cells were washed once in PBS, lysed in MLB buffer containing protease and phosphatase inhibitors and analyzed by immunoblot.
Quantitative RT-PCR
Acinar cells were washed once in PBS, lysed in RLT plus Buffer (Qiagen) and passed 5 times through a 22 gauge needle. RNA was purified using RNeasy plus kit (Qiagen) according to the manufacturer’s instructions. All real-time PCR reactions were performed in triplicate. qRT-PCR on mouse acinar cells was performed using Fast SYBR Green master mix (Applied Biosystems) on a ViiA 7 Real-Time PCR system (Applied Biosystems) and analyzed with ViiA 7 version 1.1 software. The relative amount of each gene was normalized to HPRT1 and RPL13A for mouse acinar cells by a ΔΔCt method. See Supplemental Methods for primer sequences.
RESULTS
PI3K p110α is required for Kras-induced pancreatic tumorigenesis
The KrasLSL-G12D/+;Pft1aCre/+ mouse model of pancreas tumorigenesis (referred to hereinafter as KrasG12D), displays all stages of pancreatic intraepithelial neoplasia (PanIN) found in human patients26. To determine the role of the two ubiquitously expressed Type I PI3K isoforms, we bred p110αFlox/Flox and p110βFlox/Flox mice into the KrasG12D background, resulting in concomitant pancreatic KrasG12D expression and deletion of the desired PI3K catalytic subunit. Immunoblot analysis of pancreatic extracts showed that recombination depleted the targeted p110 protein with no effect on the other (Supplemental Fig. 1A). Mice lacking p110α (α−/−) or p110β (β−/−) in the pancreas were viable and fertile. Pancreata appeared normal, although p110α-null pancreata were consistently smaller (Supplemental Fig. 1B and C) due to smaller acinar cells (Supplemental Fig 1D). KrasG12D pancreata showed multiple areas of metaplasia and mPanIN at 8-weeks of age (Fig. 1). However, when p110α was ablated from KrasG12D mice, no lesions were found by gross or histological examination at 8-weeks (Fig. 1) or 16-weeks of age (data not shown). Fibrosis was also muted as determined by Picrosirius red stain. Six 12-month-old α−/−;KrasG12D mice also showed no tumors, suggesting that p110α ablation introduces a long-term blockade in tumor initiation (data not shown). In contrast, pancreas mass increased significantly and tumors formed efficiently in β−/−;KrasG12D mice (Fig. 1 and Supplemental Fig. 1C), indicating that the p110β isoform does not play an essential role in pancreatic tumorigenesis. The lack of tumorigenesis was not due to insufficient levels of active RAS as KrasG12D and α−/−;KrasG12D lysates contained equivalent amounts of GTP-bound RAS (Supplemental Fig. 2A). MAPK activity was also not affected, as indicated by equivalent levels of pERK1/2 in KrasG12D and α−/−;KrasG12D pancreatic lysates (Supplemental Fig. 2B). A similar tumor-blocking effect resulting from EGF receptor (EGFR) ablation is circumvented by ablating Trp539. However, p110αFlox/Flox mice crossed into the KrasLSL-G12D/+;Trp53LSL-R172H/+;Ptf1aCre/+ background28 did not develop tumors (Supplemental Fig. 3), indicating that p110α ablation creates an insurmountable blockade in tumorigenesis.
Fig. 1. Ablation of class IA PI3K p110α, but not p110β, blocks Kras-induced tumor formation.

Spontaneous mPanIN formation in 8-week old KrasG12D, α−/−;KrasG12D, and β−/−;KrasG12D shown by hematoxyin and eosin staining (H&E), dual IHC for the ductal marker CK19 (brown) and the acinar marker amylase (blue) and picrosirius red stain (Sirius red). Scale bar=100 μm.
p110α ablation and chronic inhibition upregulates AKT signaling
The oncogenic effects of PI3K are generally attributed to its regulation of AKT. However, we find a substantial elevation of phospho-AKT in both α−/− and α−/−;KrasG12D pancreatic lysates (Fig. 2A) and high levels in α−/−;KrasG12D acinar cells (Fig. 2B), compared to KrasG12D pancreata. While pAKT was elevated in α−/− mice compared to control, it was higher still when KrasG12D was expressed, demonstrating that Kras signals to AKT in the absence of p110α. To determine if elevated AKT activation was dependent on signaling from other PI3K isoforms, we isolated primary acinar cells from KrasG12D and α−/−;KrasG12D mice and treated them with GDC0941, a broad spectrum PI3K inhibitor. In cells from both genotypes, GDC0941 eliminated AKT activity (Fig. 2C). This effect was analogous to upregulation of pAKT observed when PDA cell lines from mouse (NB490) and human (MiaPaCa2) were chronically treated with the p110α selective inhibitor PIK75. With 1 day of treatment, PIK75 reduced pAKT levels (data not shown). However, when treatment was extended to 3 days, pAKT levels increased by 2–3 fold, which was reversed by treatment with GDC0941 (Fig. 2D). Immunoblotting for each p110 isoform in wild-type, KrasG12D and α−/−;KrasG12D acinar cells revealed that all p110 isoforms were expressed. p110γ was expressed under all conditions whereas p110α and p110β were substantially higher in KrasG12D cells (Fig. 2E). We conclude that elevated levels of pAKT upon p110α ablation results from compensatory activity from other PI3K isoforms. Most importantly, these data revealed that elevated AKT activity is not, by itself, sufficient to initiate pancreatic tumorigenesis.
Fig. 2. pAkt is elevated in p110α-null pancreata.

(A) Pancreas extracts from WT, α−/−, KrasG12D, and α−/−;KrasG12D were analyzed by immunoblot with the indicated antibodies. (B) IHC for phospho-AKT(T308) on sections from KrasG12D, and α−/−;KrasG12D pancreata. Scale bar=100μm for panels and 50μm for insets. Metaplasia (M) and mPanIN (P) are indicated. (C) Isolated acinar cells from the indicated genotypes were treated with DMSO or the pan PI3K inhibitor GDC0941 (500nM) for 1 hour. Total cell lysates were analyzed by immunoblot for AKT and p-AKT (T308). (D) MiaPaCa2 and NB490 cells were treated with either DMSO or the p110α selective inhibitor PIK75 (50nM) for 3 days followed by treatment with the pan-p110 inhibitor GDC0941 (500nM) for 1 hour. Cell lysates were analyzed by immunoblot for AKT and p-AKT(T308). Numbers indicate densitometric quantitation of p-AKT/AKT ratios. (E) Immunoblots for the indicated p110 isoforms in lysates from wild-type (WT), KrasG12D, and α−/−;KrasG12D acinar cells.
PI3K p110α-null acinar cells resist actin cytoskeleton rearrangement and cannot sustain metaplasia
PI3K has been proposed to support PDA progression by promoting acinar cell transdifferentiation to transformation-susceptible metaplastic ducts18. However, we found that in vitro ADM was efficient in primary cultures of acinar cells lacking p110α (Supplemental Fig. 4). Nevertheless, the complete absence of metaplasia in α−/−;KrasG12D pancreata in vivo still suggested an effect on metaplastic ducts. To explore this, we examined the effects of acute cerulein treatment, which accelerates tumorigenesis by promoting ADM31. KrasG12D and α−/−;KrasG12D mice were treated with supraphysiological doses of cerulein sufficient to induce widespread metaplasia and neoplasia in 30-day old KrasG12D mice by 7 days after treatment9. With this protocol, α−/−;KrasG12D mice showed complete protection from tumorigenesis and metaplasia (Fig. 3). To reconcile this observation with our in vitro ADM results, we reduced recovery time and examined the pancreata histologically. With only 1 day of recovery, we found isolated areas of metaplastic tissue, complete with acinar cell dropout, dilated acini and reactive stroma (Fig. 3). CK19 IHC confirmed the identity of bona fide metaplastic ducts within these regions. These data suggested that, together with our in vitro results, p110α activity is not required for metaplastic duct formation, but is instead required for their maintenance in vivo.
Fig. 3. PI3K p110α-null or Rac1-null acinar cells cannot sustain ductal metaplasia.

KrasG12D, α−/−;KrasG12D, and Rac1−/−;KrasG12D mice were treated once daily for 5 days with cerulein and allowed to recover for 1 day or 7 days. Sections were H&E stained and IHC was performed for CK19 (inset). Asterisks indicate areas depicted in insets. Scale bar=100 μm for panels,32.5 μm for insets.
Given the elevated pAKT levels in the α−/−;KrasG12D mice, we asked if compromised RAC1 function could explain the tumorigenesis blockade. We first confirmed that Rac1−/−;KrasG12D mice formed few to no tumors with age20, an essential phenocopy of α−/−;KrasG12D mice (Supplemental Fig. 5). We then tested if Rac1−/−;KrasG12D mice were resistant to cerulein-induced tumorigenesis. Just as in α−/−;KrasG12D mice, Rac1−/−;KrasG12D mice were completely protected from ADM and tumorigenesis with 7 days recovery, but contained areas of CK19-positive metaplastic tissue with 1 day recovery (Fig. 3).
Inflammation has been shown to be involved in ADM via production of specific cytokines32. To test if the inability to maintain metaplastic structures could be related to the inflammatory response, we stained pancreata from cerulein-treated mice for CD45 (Supplemental Fig. 6). All genotypes showed significant upregulation of inflammatory infiltration upon cerulein treatment. Consistent with a possible role of reduced inflammatory signaling, α−/−;KrasG12D mice had significantly fewer CD45 positive cells than KrasG12D mice. However, Rac1−/−;KrasG12D mice showed no such reduction, indicating that other cell autonomous mechanisms were likely to be responsible for this specific phenotype.
We then examined actin rearrangement during early tumorigenesis, a process dependent on RAC1 activity20. Taking advantage once again of the acute nature of cerulein-induced tumorigenesis, 30-day old KrasG12D mice were treated with cerulein for 3 days and sacrificed 1 hour after the final cerulein injection to highlight early changes within the acinar cell compartment, prior to significant ADM. Sections were co-stained with phalloidin to monitor the actin cytoskeleton, and anti-ZO-1, to highlight the acinar cell apical surface. In KrasG12D saline-injected controls, phalloidin and ZO-1 strongly co-localized. In cerulein treated KrasG12D pancreata, phalloidin staining highlighted the basolateral surface, indicating active actin cytoskeleton rearrangement (Fig. 4). In contrast, acinar cells in α−/−;KrasG12D pancreatic sections resembled saline-treated controls, maintaining phalloidin/ZO-1 colocalization at the apical surface. This was essentially identical to Rac1−/−;KrasG12D pancreata (Fig. 4), where some mobilization to the basolateral surface was apparent, possibly due to activity of other small G-proteins.
Fig. 4. Actin cytoskeleton rearrangement requires both p110α and Rac1.
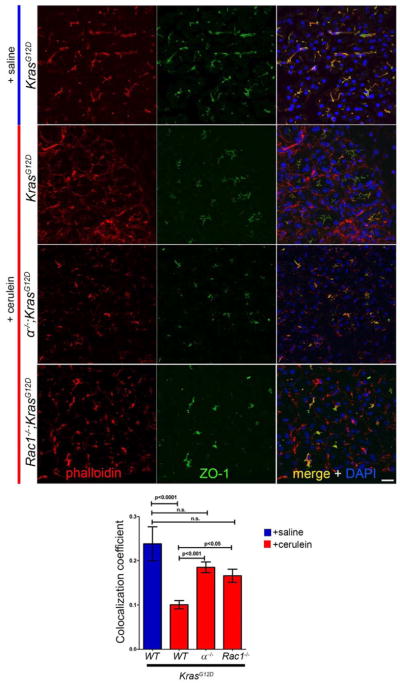
Immunofluorescence staining for ZO-1 (green) and phalloidin (red) on saline-treated KrasG12D and cerulein-treated KrasG12D, α−/−;KrasG12D, and Rac1−/−;KrasG12D mice. Mice were treated once daily for 3 days and sacrificed one hour following the last injection. Nuclei are counter-stained with DAPI. Scale bar=20μm. Graph shows quantitation of phalloidin and ZO-1 colocalization coefficients for the indicated genotypes. Values are means ±S.E.M. of 10 fields of view from 3 mice of each genotype or treatment regimen as indicated. Significance was determined by one-way ANOVA.
Kras signals through p110α to activate RAC1 early in transformation
The discovery that Rac1−/−;KrasG12D mice strongly phenocopied α−/−;KrasG12D mice led us to examine RAC1 activity in α−/−;KrasG12D pancreata. Both activated (GTP-bound) and total RAC1 levels were higher in KrasG12D pancreas lysates, as compared to wild-type control (Fig. 5A). p110α ablation significantly diminished both RAC1 activity and expression in KrasG12D pancreata (Fig. 5B). PI3K is known to affect RAC1 activity by regulating PIP3-dependent RAC1 GEF activity33, which promotes GDP-GTP exchange. To test if this was a mechanism of RAC regulation in the pancreas, we focused on two GEFs that seemed particularly relevant to our system. Vav1 is ectopically expressed in PDA cells and is necessary for cancer cell viability34. Tiam1 is a Kras-activated GEF overexpressed in breast and prostate cancers35, 36. We found elevated levels of active and total VAV1 and TIAM1 in KrasG12D pancreatic extracts, compared to control (Fig. 5C). Both active and total VAV1 and TIAM1 were significantly lower in α−/−;KrasG12D pancreata (Fig. 5C).
Fig. 5. Kras signals through p110α to activate Rac1 in the early stages of transformation.

(A) RAC1 activity was measured in total lysates from WT and KrasG12D pancreata by PAK-PBD pulldown and total RAC1 levels were measured by immunoblot. Protein loading was assessed by ponceau stain (Loading Control). Numbers indicate densitometric quantitation normalized to loading control (B) Total lysates from KrasG12D and α−/−;KrasG12D pancreata were assessed for RAC1 activity by PAK-PBD pulldown. RAC1 levels in the lysate were measured by immunoblotting for RAC1 with GAPDH as a loading control. Numbers indicate densitometric quantitation normalized to the GAPDH. (C) Total lysates from WT, α−/−, KrasG12D, and α−/−; KrasG12D pancreata were analyzed by the Rac1G15A pulldown to measure VAV1 and TIAM1 activities. Levels of VAV1 and TIAM1 in lysates were analyzed by immunoblot. HSP90 was the loading control. Numbers indicate densitometric quantitation normalized to HSP90.
To test if p110α-dependent upregulation of the Rac1 pathway is present in the earliest stages of tumorigenesis, we examined RAC1 and GEF levels in primary acinar cells isolated from six to eight-week old KrasG12D and α−/−;KrasG12D mice. Similar to results obtained from whole pancreas lysates, RAC1 activity was elevated in KrasG12D acinar cells and p110α ablation reduced this activity to control levels (Fig. 6A). Consistent with these results, VAV1 expression was higher in KrasG12D acinar cells but this increase was significantly attenuated in α−/−;KrasG12D samples (Fig. 6A). TIAM1 was not detectable in isolated acinar cells.
Fig. 6. Kras increases RAC-GEF expression in a p110α dependent manner.

(A) RAC1 activity was measured in WT, KrasG12D, and α−/−;KrasG12D acinar cell lysates by PAK-PBD pulldown compared to total VAV1, RAC1, and GAPDH levels. Numbers indicate densitometric quantitation normalized to GAPDH. (B) Acinar cells from KrasLSL-G12D/+ and p110αFlox/Flox;KrasLSL-G12D/+ mice (without Ptf1a-Cre) were isolated, cultured, and infected with either adeno-GFP or adeno-Cre for 3 days. Lysates were assayed for levels of VAV1 and ACTB by immunoblotting. Numbers indicate densitometric quantitation normalized to ACTB. (C) Quantitative RT-PCR for Vav1, Tiam1, Dock10, and Rac1 was performed mRNA isolated from WT, KrasG12D, and α−/−;KrasG12D acinar cells. Values are means ±S.E.M. of fold change compared to WT from 5 independent experiments. Significance was determined by one-way ANOVA. (D) 4-month old KrasG12D pancreata were stained for VAV1 (left panel). Scale bar=100μm. Immunofluorescence for Vav1 (green) and phalloidin (red) was performed on frozen sections from 4 month old KrasG12D mice (right panel). Nuclei were counterstained with DAPI (blue). Scale bar = 20μm. (E) Representative images from human TMA slides stained for p110α, VAV1, TIAM1 or RAC1 in normal pancreas (top panels) and PanINs (middle and bottom panels). Scale bar=100μm for panels, 50μm for insets.
To test whether VAV1 expression could be induced acutely upon Kras activation, we isolated acinar cells from KrasLSL-G12D/+ mice and infected them with adenovirus encoding either Cre recombinase (Ad-Cre) or GFP (Ad-GFP), as a control. 3 days after adenovirus infection, Cre-induced expression of oncogenic Kras resulted in upregulation of VAV1 (Fig. 6B). In contrast, in Ad-Cre infected p110αflox/flox; KrasLSL-G12D/+ acinar cells, VAV1 expression was attenuated (Fig. 6B).
Finding that oncogenic Kras expression was sufficient to upregulate VAV1 protein, we then tested if transcripts for Vav1 and other GEFs were affected. Using qRT-PCR, we found that Vav1 and Dock10 mRNA levels were significantly upregulated in KrasG12D primary acinar cells. Rac1 and Tiam1 mRNA levels were elevated, but not significantly (Fig. 6C). Vav1, Tiam1, Dock10 and Rac1 mRNA expression was not upregulated in α−/−;KrasG12D cells (Fig. 6C). Taken together, these results indicate that oncogenic Kras can increase Rac pathway transcripts in acinar cells in a PI3K p110α dependent manner, but this is unlikely to be the major mechanism of Rac regulation.
It was surprising to find VAV1 protein in KrasG12D acinar cells, since its expression is generally thought to be confined to inflammatory cells, though expression has been observed in PDA34. Vav1 IHC in KrasG12D pancreata revealed expression in stromal cells, as expected, but also at very high levels in what appeared to be tuft cells in KrasG12D metaplasia and mPanINs30. Tuft cell expression was confirmed by co-IF for Vav1 with phalloidin to highlight their characteristic actin-rich bundles (Fig 6D).
We then examined expression of p110α and components of the RAC pathway for which we could stain by IHC in a human pancreas tissue microarray containing normal, pancreatitis, PanIN and PDA tissue (Figure 6E, Table 1). p110α was detectable in 10/25 of normal pancreas samples, generally at low levels. p110α was detectable in the majority of chronic pancreatitis, PanIN and PDA samples, often at high levels (Table 1, Fig. 6E). As seen in mice, VAV1 expression was limited to tuft cells in normal, pancreatitis and PanIN lesions that reflected the frequency with which tuft cells were found in these sample types30, 37. VAV1 expression in PDA samples was uniform throughout the cancer epithelium (data not shown). TIAM1 was found at high levels in islets in all normal and diseased tissues but at modest levels in normal exocrine cells. TIAM1 was expressed in the epithelium of the majority of pancreatitis and PanIN samples and ~50% of PDAs within tumor epithelium. ADM adjacent to PDA universally expressed elevated TIAM1. Rac1 expression was easily detectable in all normal pancreas, metaplasia, PanINs, and PDAs. RAC1 levels were uniform throughout the epithelia in normal and pancreatitis samples. In contrast, the majority of PanIN lesions showed elevated Rac1 levels, as did about half of PDA samples, but in each case staining within the epithelium became less uniform, with several cells within the lesion being negative.
Table 1. Summary of TMA IHC for RAC1 Pathway components.
Human pancreas tissue microarrays were stained for p110α, VAV1, TIAM1 or RAC1. Expression was scored as undetectable (0), detectable (1) or high expression (2) in normal tissue, chronic panceatitis, PanIN1, PanIN2, PanIN3 and pancreatic ductal adenocarcinoma (PDA). (TC) indicates that expression is limited to tuft cells.
| Normal Pancreas | Pancreatitis | PanIN1 | PanIN2 | PanIN3 | PDA | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | |
| P110α | 15 | 10 | - | 13 | 14 | 12 | 2 | 1 | 8 | 3 | 5 | 9 | 1 | 1 | 6 | 5 | 14 | 11 |
| VAV1 | 23 | - | 2(TC) | 26 | 13(TC) | 3 | - | 8(TC) | 4 | - | 13(TC) | 6 | - | 2(TC) | 23 | - | 7 | |
| TIAM1 | - | 25 | - | 6 | 18 | 15 | 1 | 2 | 7 | - | 5 | 12 | 1 | - | 7 | 12 | 13 | 5 |
| RAC1 | - | 25 | - | - | 36 | 3 | - | 5 | 6 | - | 8 | 9 | - | 1 | 7 | - | 17 | 13 |
To test if p110α regulation of RAC1 by GEF regulation extended beyond early tumor progression, we investigated the dependency of RAC activity on p110α in human PDA cell lines. CFPAC-1 and Capan-2 cells have been reported to express VAV134. Acute knockdown using transient transfection of siRNAs targeting VAV1 or TIAM1 reduced RAC activity in both cell lines (Supplemental Fig. 7A&B). However, p110α siRNA knockdown diminished RAC activity only in Capan-2 (Supplemental Fig. 7A&B). To examine effects of chronic knockdown of p110α, shRNA lentiviruses targeting p110α were stably transfected into Capan-2 and CFPAC-1cells and transcripts of several GEFs reported to be associated with PI3K, including VAV1, DOCK10, PREX1 and TIAM1, were examined by qRTPCR. VAV1 was significantly lower in both cell lines with p110α knocked down (Supplemental Fig. 7C), whereas TIAM1 and other GEFs were reduced in one line or the other (Supplemental Fig. 7 C) and PREX1 even increased in CFPAC-1 knockdown cells. Together, our data indicate that p110α can continue to affect the RAC pathway in adenocarcinoma, but in a less consistent manner than it does during tumorigenesis.
DISCUSSION
Though previous attempts have proven ineffective, there is renewed interest in targeting oncogenic Kras, the primary initiator of PDA. Complementary to these efforts is the identification of Kras downstream effectors and their precise roles in PDA progression. Here we have conditionally ablated the ubiquitously-expressed PI3K p110 isoforms p110α and p110β from a KrasG12D pancreatic tumorigenesis model to study the importance of these endogenous effectors. In this system, these isoforms clearly have non-overlapping functions, as p110α ablation gave complete protection from tumorigenesis whereas p110β ablation provided none. Moreover, tumorigenesis blockade induced by p110α ablation was not overcome by introducing a mutant Trp53 allele. Trp53 ablation was capable of bypassing a similar level of protection conferred by EGFR deletion9, 38. Unlike EGFR ablation, which results in inhibition of AKT signaling38, p110α was dispensable for AKT activation by KrasG12D in vivo. However, p110α was required for Kras-induced activation of RAC1, a known regulator of pancreatic tumorigenesis.
Previous reports have implicated PI3K in PDA18, 39. PI3K p110γ is overexpressed in human PDA39. Pharmacological and siRNA down-regulation of p110γ confirms its involvement in proliferation of PDA cell lines39. Recently, it was found that a knock-in of a constitutively active p110αH1047R mutant into the ROSA26R locus induces pancreatic tumorigenesis18. This is in contrast to a knock-in of this mutant into the endogenous Pik3ca locus, which produced no pancreatic abnormalities19. While these conflicting results make it unclear whether activation of endogenous p110α is sufficient for pancreatic tumorigenesis, our data establish that it is necessary.
Saur and colleagues clearly establish the necessity of PDK1, the PI3K effector responsible for activating AKT, as genetic ablation of PDK1 blocks Kras-induced pancreatic tumorigenesis18. However, while p110α ablation completely protected mice from Kras-induced tumorigenesis, it did not block AKT activation by KrasG12D. Elevated AKT activity in p110α null pancreata resulted from compensation by other p110 isoforms, and clarifies that AKT activation is not the sole critical function of p110α in pancreatic tumorigenesis. A similar compensation could be induced with chronic inhibition of p110α in PDA cell lines, demonstrating that selective inhibition of this PI3K isoform may prove problematic.
Prior studies show that neoplastic lesions in the pancreas arise from acinar cells that have undergone ADM, a reprogramming event mediated by EGFR signaling40. Given the complete absence of metaplasia and neoplasia, we were surprised to find that ablation of p110α did not block ADM in vivo or in vitro. However, without p110α, KrasG12D was not capable of stabilizing and transforming ADM after cerulein stimulation, resulting in restoration of apparently normal pancreatic tissue given sufficient recovery time. Saur and colleagues show broad spectrum PI3K inhibitors can block ADM in vitro18. We find that use of a relatively selective p110α inhibitor PIK75 is ineffective at blocking in vitro ADM until used at a nonselective concentration (data not shown). Together, these results suggest that AKT activity is critical for in vitro ADM and the higher levels of pAKT after p110α ablation is sufficient to induce the process.
Finding that p110α was important for actin rearrangement in KrasG12D expressing acinar cells, we focused on regulation of RAC1, which is critical for this process20. Siveke and colleagues show that RAC1 expression increases in Kras-induced pancreas tumors, and ablation of Rac1 significantly reduces Kras-induced pancreatic tumorigenesis20. We found that deletion of p110α in KrasG12D mice led to decreased RAC1 expression and, more importantly, RAC1 activity, in pancreas tissue and acinar cells. It is well-established that several RAC GEFs contain PH domains and thus can be activated and organized within PIP3 containing membranes. We found that the activities of the RAC-GEFs VAV1 and TIAM1 were highly dependent on p110α (Fig. 5C) and these GEFS were also frequently expressed in precancerous lesions in humans (Fig. 6E). Taken together, our data suggest that p110α regulation of PH-domain-containing RAC GEFs may collectively contribute to tumor development.
In summary, we have shown that PI3K p110α is required for Kras-induced transformation of ADM via its regulation of RAC1 and could represent a target for PDA therapy. Indeed, a pan Class I PI3K inhibitor, GDC0941, effectively blocks tumor growth in KrasG12V;P53R172H;Ptf1a-Cre mice18. However, pan-PI3K inhibitors that target both p110α and p110β could have unacceptable cardiac toxicities41. While we see inhibition of growth with acute treatment of PDA cell lines with PIK75, within days of treatment, cells began to become resistant (data not shown) in a time frame consistent with the observed upregulation of pAKT (Fig. 2D). Similarly, we observed no changes in cell proliferation or apoptosis after PIK75 treatment of established tumors in KrasG12D mice (data not shown), also possibly due to compensation by other PI3K isoforms. We conclude that while p110α regulation of RAC1 is required for tumorigenesis, the maintenance of AKT activity is compensated for by other p110 family members, suggesting its promise as a drug target for PDA treatment likely lies in optimized combination therapies.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by NIH grants CA159222 (H.C.C.), DK62722 (R.Z.L.) and CA136754 (R.Z.L and H.C.C.), and a VA Merit Award (R.Z.L.). P30DK058404 for the Vanderbilt Digestive Diseases Research Center Tissue Morphology Subcore: GI SPORE TissueCore; NIH grants P50CA095103 and P50CA102701 for the Mayo Clinic SPORE in Pancreatic Cancer; ESC was supported by F30 CA167963.
The TROMA-III monoclonal antibody to CK19 developed by R. Kemler was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242. NB490 cells were the gift of Dr. Nabeel Bardeesy (Massachusetts General Hospital, Boston, MA).
Footnotes
Disclosures: None
Writing assistance: None
Author contributions:
Chia-Yen C. Wu: study design; acquisition of data; analysis and interpretation of data; drafting of the manuscript
Eileen S. Carpenter: study design; acquisition of data; analysis and interpretation of data; drafting of the manuscript
Kenneth K. Takeuchi: study design; acquisition of data; analysis and interpretation of data; drafting of the manuscript
Christopher J. Halbrook: acquisition of data
Louise V. Peverley: acquisition of data
Harold Bien: acquisition of data; drafting of the manuscript
Jason C. Hall: acquisition of data
Kathleen E. DelGiorno: acquisition of data
Debjani Pal: acquisition of data
Yan Song: acquisition of data
Chanjuan Shi: analysis of data
Richard Z. Lin: study concept and design; analysis and interpretation of data; drafting of the manuscript; obtained funding; administrative, study supervision
Howard C. Crawford: study concept and design; analysis and interpretation of data; drafting of the manuscript; obtained funding; administrative, study supervision
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Hackert T, Buchler MW, Werner J. Surgical options in the management of pancreatic cancer. Minerva Chir. 2009;64:465–76. [PubMed] [Google Scholar]
- 3.Ferrone CR, Brennan MF, Gonen M, et al. Pancreatic adenocarcinoma: the actual 5-year survivors. J Gastrointest Surg. 2008;12:701–6. doi: 10.1007/s11605-007-0384-8. [DOI] [PubMed] [Google Scholar]
- 4.Wagner M, Redaelli C, Lietz M, et al. Curative resection is the single most important factor determining outcome in patients with pancreatic adenocarcinoma. Br J Surg. 2004;91:586–94. doi: 10.1002/bjs.4484. [DOI] [PubMed] [Google Scholar]
- 5.Hruban RH, Iacobuzio-Donahue C, Wilentz RE, et al. Molecular pathology of pancreatic cancer. Cancer J. 2001;7:251–8. [PubMed] [Google Scholar]
- 6.Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–70. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–53. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saxena N, Lahiri SS, Hambarde S, et al. RAS: target for cancer therapy. Cancer Invest. 2008;26:948–55. doi: 10.1080/07357900802087275. [DOI] [PubMed] [Google Scholar]
- 9.Ardito CM, Gruner BM, Takeuchi KK, et al. EGF Receptor Is Required for KRAS-Induced Pancreatic Tumorigenesis. Cancer Cell. 2012;22:304–17. doi: 10.1016/j.ccr.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi G, DiRenzo D, Qu C, et al. Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene. 2013;32:1950–8. doi: 10.1038/onc.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins MA, Yan W, Sebolt-Leopold JS, et al. Mapk Signaling Is Required for Dedifferentiation of Acinar Cells and Development of Pancreatic Intraepithelial Neoplasia in Mice. Gastroenterology. 2013;146:822–834. doi: 10.1053/j.gastro.2013.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez-Viciana P, Warne PH, Dhand R, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–32. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 13.Sjolander A, Yamamoto K, Huber BE, et al. Association of p21ras with phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 1991;88:7908–7912. doi: 10.1073/pnas.88.18.7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–50. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stokoe D. The phosphoinositide 3-kinase pathway and cancer. Expert Rev Mol Med. 2005;7:1–22. doi: 10.1017/S1462399405009361. [DOI] [PubMed] [Google Scholar]
- 16.Vanhaesebroeck B, Leevers SJ, Ahmadi K, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 17.Mora A, Komander D, van Aalten DM, et al. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–70. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 18.Eser S, Reiff N, Messer M, et al. Selective Requirement of PI3K/PDK1 Signaling for Kras Oncogene-Driven Pancreatic Cell Plasticity and Cancer. Cancer Cell. 2013;23:406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 19.Collisson EA, Trejo CL, Silva JM, et al. A central role for RAF-->MEK-->ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012;2:685–93. doi: 10.1158/2159-8290.CD-11-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heid I, Lubeseder-Martellato C, Sipos B, et al. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology. 2011;141:719–30. 730 e1–7. doi: 10.1053/j.gastro.2011.04.043. [DOI] [PubMed] [Google Scholar]
- 21.Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci. 2009;34:115–27. doi: 10.1016/j.tibs.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Lupia E, Goffi A, De Giuli P, et al. Ablation of phosphoinositide 3-kinase-gamma reduces the severity of acute pancreatitis. Am J Path. 2004;165:2003–11. doi: 10.1016/s0002-9440(10)63251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Utermark T, Rao T, Cheng H, et al. The p110alpha and p110beta isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev. 2012;26:1573–86. doi: 10.1101/gad.191973.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castellano E, Sheridan C, Thin MZ, et al. Requirement for interaction of PI3-kinase p110alpha with RAS in lung tumor maintenance. Cancer Cell. 2013;24:617–30. doi: 10.1016/j.ccr.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia S, Liu Z, Zhang S, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–9. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 27.Lu Z, Jiang YP, Wang W, et al. Loss of cardiac phosphoinositide 3-kinase p110 alpha results in contractile dysfunction. Circulation. 2009;120:318–25. doi: 10.1161/CIRCULATIONAHA.109.873380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 29.Glogauer M, Marchal CC, Zhu F, et al. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170:5652–7. doi: 10.4049/jimmunol.170.11.5652. [DOI] [PubMed] [Google Scholar]
- 30.Delgiorno KE, Hall JC, Takeuchi KK, et al. Identification and manipulation of biliary metaplasia in pancreatic tumors. Gastroenterology. 2014;146:233–244. e5. doi: 10.1053/j.gastro.2013.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guerra C, Collado M, Navas C, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19:728–39. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liou GY, Doppler H, Necela B, et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J Cell Biol. 2013;202:563–77. doi: 10.1083/jcb.201301001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welch HC, Coadwell WJ, Stephens LR, et al. Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett. 2003;546:93–7. doi: 10.1016/s0014-5793(03)00454-x. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez-Zapico ME, Gonzalez-Paz NC, Weiss E, et al. Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell. 2005;7:39–49. doi: 10.1016/j.ccr.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 35.Engers R, Mueller M, Walter A, et al. Prognostic relevance of Tiam1 protein expression in prostate carcinomas. Br J Cancer. 2006;95:1081–6. doi: 10.1038/sj.bjc.6603385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adam L, Vadlamudi RK, McCrea P, et al. Tiam1 overexpression potentiates heregulin-induced lymphoid enhancer factor-1/beta -catenin nuclear signaling in breast cancer cells by modulating the intercellular stability. J Biol Chem. 2001;276:28443–50. doi: 10.1074/jbc.M009769200. [DOI] [PubMed] [Google Scholar]
- 37.Bailey JM, Alsina J, Rasheed ZA, et al. DCLK1 Marks a Morphologically Distinct Subpopulation of Cells With Stem Cell Properties in Preinvasive Pancreatic Cancer. Gastroenterology. 2014;146:245–56. doi: 10.1053/j.gastro.2013.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Navas C, Hernandez-Porras I, Schuhmacher AJ, et al. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–30. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edling CE, Selvaggi F, Buus R, et al. Key role of phosphoinositide 3-kinase class IB in pancreatic cancer. Clin Cancer Res. 2010;16:4928–37. doi: 10.1158/1078-0432.CCR-10-1210. [DOI] [PubMed] [Google Scholar]
- 40.De La OJ, Emerson LL, Goodman JL, et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;105:18907–12. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu CY, Jia Z, Wang W, et al. PI3Ks maintain the structural integrity of T-tubules in cardiac myocytes. PloS One. 2011;6:e24404. doi: 10.1371/journal.pone.0024404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.