

Abstract
Protein arginine methyltransferase 5 (PRMT5) symmetrically methylates arginine residues of histones and non-histone protein substrates and regulates a variety of cellular processes through epigenetic control of target gene expression or post-translational modification of signaling molecules. Recent evidence suggests that PRMT5 may function as an oncogene and its overexpression contributes to the development and progression of several human cancers. However, the mechanism underlying the regulation of PRMT5 expression in cancer cells remains largely unknown. In the present study, we have mapped the proximal promoter of PRMT5 to the −240 bp region and identified nuclear transcription factor Y (NF-Y) as a critical transcription factor that binds to the two inverted CCAAT boxes and regulates PRMT5 expression in multiple cancer cell lines. Further, we present evidence that loss of PRMT5 is responsible for cell growth inhibition induced by knockdown of NF-YA, a subunit of NF-Y that forms a heterotrimeric complex with NF-YB and NF-YC for function. Significantly, we have found that activation of protein kinase C (PKC) by phorbol 12-myristate 13-acetate (PMA) in LNCaP prostate cancer cells down-regulates the expression of NF-YA and PRMT5 at the transcription level in a c-Fos-dependent manner. Given that down-regulation of several PKC isozymes is implicated in the development and progression of several human cancers, our findings suggest that the PKC-c-Fos-NF-Y signaling pathway may be responsible for PRMT5 overexpression in a subset of human cancer patients.
Keywords: NF-Y, PRMT5, PKC, prostate cancer, lung cancer, c-Fos
1. Introduction
Protein arginine methyltransferase 5 (PRMT5), a type II methyltransferase that symmetrically methylates arginine residues of histones and non-histone protein substrates [1, 2], regulates a variety of cellular processes by epigenetic regulation of target gene expression and by post-translational modification of critical signaling molecules [1]. Recently, several studies have shown that PRMT5 is overexpressed in human cancers such as lung cancer [3, 4], ovarian cancer [5], colorectal cancer [6], breast cancer [7], melanoma [8], leukemia and lymphoma [9, 10], and glioblastoma [11]. The overexpression of PRMT5 correlates with disease progression and poor prognosis. Importantly, these studies also present evidence that silencing PRMT5 expression in these cancer cells inhibits cell proliferation and/or induces apoptosis, suggesting that PRMT5 overexpression in cancer cells plays an important role in the development and progression of human cancers. However, how PRMT5 expression is transcriptionally regulated in cancer cells has not yet been investigated.
Nuclear transcription factor Y (NF-Y) is an important transcription factor that is highly conserved across the species [12–14]. NF-Y is composed of three subunits, NF-YA, NF-YB and NF-YC, and functions as a heterotrimeric complex to bind the CCAAT box in promoter regions to regulate gene transcription. CCAAT boxes are usually positioned in either orientation between −60 and −100, and are present in almost 30% of human promoters, particularly those that drive expression of oncogenes in human cancers [15–17]. In addition, NF-Y binding sites overlap with binding sites of several other transcription factors, such as SP1, E2F1, GATA, and c-Fos, to cooperatively regulate cell growth [12, 15, 18]. The NF-Y transcriptional activity can be modulated by increasing DNA binding to the CCAAT boxes [19, 20] or by increasing expression of the NF-YA subunit [12, 21–23]. However, whether the cancer signaling regulates NF-YA expression remains unknown.
Protein kinase C (PKC) is a family of serine/threonine protein kinases that regulates a wide range of cellular processes [24]. PKC isozymes can be classified into three groups including calcium-dependent “classical” cPKCs (α, βI, βII and γ), calcium-independent “novel” nPKCs (δ, ε, η and θ), and calcium-independent “atypical” aPKCs (ζ and ι/λ). Classical and novel PKC isozymes, but not atypical PKC isozymes, can be activated by diacylglycerol (DAG) and phorbol 12-myristate 13-acetate (PMA). Although it is generally thought that most PKC isozymes are overexpressed in human cancers and promote cellular transformation, proliferation, and migration, the opposite effects have also been reported [24]. This is exemplified by the use of prostate cancer cells as a model system to study distinct roles of PKC isozymes in apoptosis in prostate cancer cells [25], in which treatment of LNCaP, but not DU 145 and PC-3 cells, with PMA induces apoptosis [26]. Consistent with their differential roles in cell-based studies, the expression level of several PKC isozymes in some human cancers inversely correlates with the aggressiveness of the disease [27, 28]. However, the mechanism by which down-regulation of PKC isozymes regulates cancer cell growth remains unknown.
Activator protein 1 (AP-1) is a family of dimeric transcription factors which includes c-Jun and c-Fos [29]. AP-1 was discovered as a complex of c-Fos/c-Jun that can be induced by serum and PMA [30–32]. Although activation or overexpression of AP-1 proteins is implicated in the development and progression of many human cancers, distinct roles of AP-1 proteins have also been observed [29, 33, 34]. For example, reduced expression of c-Fos and c-Jun has been observed in a subset of human prostate cancer patients [35–38], though the clinical significance of reduced AP-1 protein expression remains unclear. Recently, we have demonstrated that c-Jun acts as a transcriptional repressor of the androgen receptor (AR) signaling, and that silencing c-Jun promotes the growth of both androgen-dependent LNCaP cells and castration-resistant C4-2 cells [39], providing evidence that down-regulation of c-Jun expression in a subset of human prostate cancer patients may promote disease progression by enhancing the AR signaling. In the present study, we demonstrate that NF-Y is a major transcription factor to drive PRMT5 transcription in several cancer cell lines, and knockdown of NF-YA leads to down-regulation of PRMT5 expression and suppression of cell growth. Further, we show that PMA treatment in LNCaP cells down-regulates the expression of NF-YA and PRMT5 in a PKC− and c-Fos-dependent manner.
2. Materials and methods
2.1. Cell culture and treatment
The prostate cancer cell lines LNCaP and PC-3 cells were cultured as described previously [40, 41]. Lung cancer cell line A549 was kindly provided by Wanqing Liu, and cells were cultured in F-K12 medium containing 10% fetal bovine serum and 1% penicillin/streptomycin. PMA was purchased from Sigma (P 1585), and bisindolylmaleimide I (GF109203X, GFX), a pan-PKC inhibitor, was purchased from Tocris Bioscience (a gift of the Val Watts lab). For PMA treatment, cells were seeded into 6 cm dishes for 24 hours (approximately 80~90% confluence), and then treated with different doses of PMA for the indicated times in the presence or absence of GFX.
2.2. Plasmid construction
Two distinct types of the PRMT5 promoters (−3461/+75 bp and −3474/+75 bp) were amplified from LNCaP cell genomic DNA by PCR with Phusion High-Fidelity DNA Polymerase (NEB) using primers 5′-CGGGGTACCCTGGGCACAACTAGGGCAGAGAAC-3′ and 5′-GAAGATCTTCCACGCCGGGATTCCTTGATAC-3′. The PCR products were then cloned into pGL4.10 [luc2]-Basic Vector (Promega). To construct a series of luciferase reporter genes (A1: −1723/+75, A2: −1156/+75, A3: −459/+75, A4: −323/+75, A5: −240/+75, B1: −1736/+75, B2: −1169/+75, B3: −472/+75, B4: −323/+75, B5: −240/+75, B6: −68/+75, B7: +8/+75), the same methods were used for PCR amplification by using two types of PRMT5 promoters as templates. For mutagenesis, nucleotide substitutions in putative binding motifs were introduced by ligation PCR [42]. The expression plasmids pFLAG-c-Fos and pFLAG-c-Jun were previously constructed [39, 43, 44]. The cDNA encoding PRMT5 was amplified by PCR using primers 5′-CTGAATTCGGATGGCGGCGATGGCGGT-3′ and 5′-GCCTCGAGAGAGGCCAATGGTATATGAGCG-3′ and cloned into pCMV-Myc vector (Clontech). All plasmid constructs were verified with DNA sequencing.
2.3. Luciferase reporter gene assay
Prostate cancer cells were plated in 12-well plates at a density of 2×105/well, and A549 cells were plated at a density of 1×105/well. After 24 hours, 1 μg of a short-hairpin RNA (shRNA) plasmid targeting NF-YA was transiently co-transfected with 0.5 μg of a PRMT5 reporter gene, along with 0.1 μg of pRL-TK (Promega) by FuGENE HD or FuGENE 6 (Promega). Forty-eight hours after transfection, Firefly and Renilla luciferase activities were determined by a TopCount NXT microplate luminescence counter (Packard) using Dual-luciferase Reporter Assay Kit (Promega) according to the manufacture’s instruction with minor modifications as described previously [43, 44].
2.4. Immunoblotting
Preparation of total cell lysate (TCL) and immunoblotting were performed as described before [41]. Densitometric quantification was performed with Image J software (NIH, Rockville, MD, USA). The antibodies used for immunoblotting analysis were: anti-β-actin (A1978, Sigma), anti-NF-YA (H-209, sc-10779, Santa Cruz) [45], anti-c-Jun (H-79, sc-1694, Santa Cruz), anti-c-Fos (H125, sc-9202, Santa Cruz), anti-PRMT5 (07-405, Millipore), anti-FLAG M2 (F3165, Sigma), anti-Myc (631206, Clontech), and anti-cyclin A2 (CCNA2, BF683, cell signaling). Secondary HRP-conjugated antibodies were purchased from GE Healthcare UK Ltd (Buckinghamshire, UK).
2.5. RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from cells by using TRIzol reagent (Invitrogen) according to the manufacturer’s instruction and verified for integrity by agarose gel electrophoresis. One μg of RNA was used for reverse-transcription using random primers (100 ng) and MMLV reverse transcriptase (Promega). The mRNA level of PRMT5, NF-YA, NF-YB, NF-YC and GAPDH was quantified using qRT-PCR with gene specific primers. PRMT5 forward, 5′-CAGAGAAGGAGTTCTGCTCCTAC-3′ and PRMT5 reverse, 5′-ATGGCCTGCTGGTACTGAGAGT-3′; NF-YA forward, 5′-CTGTGACACTACCAGTGGCAG-3′ and NF-YA reverse, 5′-TGCCTCCTCTTAAGAATACGG-3′; NF-YB forward, 5′-GCAAGTGAAAGGTGCCATCAAGAG-3′ and NF-YB reverse, 5′-CTGCTCCACCAATTCCCTTTTCTC-3′; NF-YC forward, 5′-GAACTGAAACCTCCAAAGCGTC-3′ and NF-YC reverse, 5′-TGTGCGATGATGATCTGCCCAG-3′. GAPDH forward, 5′-CTGACTTCAACAGCGACACC-3′ and GAPDH reverse, 5′-CCCTGTTGCTGTAGCCAAAT-3′. qRT-PCR was performed with SYBR@GREEN PCR Master Mix (Roche) by using a ViiA7 Real-Time PCR system (Applied Biosystems) for 40 cycles. The relative expression of each individual gene was normalized to GAPDH and was calculated using the comparative 2−ΔΔCT method [46].
2.6. Chromatin immunoprecipitation (ChIP)
Cells cultured in 10 cm dishes were cross-linked with 1% formaldehyde for 10 minutes and then stopped by adding 125 mM Glycine. Chromatin from 2-dish cells was sheared by a Branson Digital Sonifier 250 to an average size of approximately 0.5 kb in 1 ml immunoprecipitation (IP) buffer (50 mM Tris Cl, pH 7.4, 0.5% NP-40, 1% Triton X-100, 150 mM NaCl, 5 mM EDTA, and 0.5 mM DTT). The sheared chromatin (DNA-protein complexes) was incubated with anti-NF-YA (G-2, sc-17753X, Santa Cruz) [47], or the control IgG (sc-2025, Santa Cruz) at 4°C for overnight and the DNA-protein complexes were recovered by protein G-agarose beads (Santa Cruz, sc-2002). The immunoprecipitated DNA was isolated by 10% Chelex-100 using the fast ChIP method [48], and then subjected to qRT-PCR. The relative fold enrichment was calculated by normalizing to IgG control. A non-target region in the PRMT5 distal promoter and a region containing a validated NF-Y binding site in the CCNA2 promoter were amplified from the same IP sample, and used as negative control and positive control, respectively. The primers used for ChIP are listed as follow: the region containing two NF-Y binding sites in the PRMT5 proximal promoter (5′-CACTGTTTCTCTCCGTGATGGTAC-3′ and 5′-GCGTCTGCCACAGCTCCCGAAC-3′); and a non-target region in the PRMT5 distal promoter (5′-CTGGGCACAACTAGGGCAGAGAAC-3′ and 5′-TTAGTAGAGACGGGGTTTCAC-3′); the region containing one validated NF-Y binding site in the CCNA2 promoter (5′-GCCCCTGCTCAGTTTCCTTTG-3′ and 5′-CGGCGGCTGTTCTTGCAGTTCA-3′)
2.7. Lentivirus production and establishment of stable cell lines
For the construction of shRNA expressing plasmids, the pLKO-Tet-On inducible lentiviral RNAi system was used [49]. Several targeting sequences were selected from the RNAi Consortium (Sigma) as follow: NF-YA (shYA#1), 5′-CCATCGTCTATCAACCAGTTA-3′ (TRCN0000014930); NF-YA (shYA#2), 5′-CCATCATGCAAGTACCTGTTT-3′ (TRCN0000014932); and c-Fos, 5′-GCGGAGACAGACCAACTAGAA-3′ (TRCN0000273941). Scrambled control (SC), 5′-AACAAGATGAAGAGCACCAA-3′, was used as a negative control for all knockdown experiments. Annealed oligonucleotides were cloned into pLKO-Tet-On. To generate viral particles, HEK 293T cells were cultured in a 10-cm dish without antibiotics for 24 hours, and then co-transfected with 2 μg of pLKO.1-Tet-On shRNA vector, 1.5 μg of pHR’-CMV-ΔR8.2Δvpr packaging plasmid, and 0.5 μg of pHR’-CMV-VSVG envelope plasmid using FuGENE HD reagent. The supernatant containing viruses was harvested 3 days post-transfection, and then filtered through a 0.45 μm filter to remove cell debris. Prostate cancer cells and lung cancer cells were then infected by applying 6 ml viral supernatant in 10 ml complete medium. Polybrene was added to a final concentration of 8 μg/ml to facilitate the infection. Cells were selected with 2 μg/ml of puromycin (for PC-3, 3.5 μg/ml) for 3 days for stable integration of the shRNA plasmids, and surviving cells were maintained in the presence of 1 μg/ml of puromycin. To knock down NF-YA or c-Fos, cells were induced with 1 μg/ml of doxycycline (Dox) for at least 3 days.
2.8. Cell growth analysis and Trypan blue exclusion assay
LNCaP and PC-3, or A549 stable cell lines were seeded in 6-well plates in triplicate at a density of 1×105 cells/well or 2×104 cells/well, respectively. Cells were then induced with or without Dox (1 μg/ml) for various times, and medium and Dox were changed every 3 days during culture. The number of viable and dead cells from each well was determined by Trypan blue staining. To determine the effect of NF-YA knockdown on cell proliferation, the indicated stable cell lines were seeded and grew on coverslips in 6-well plates at a cell density of 1×105 cells/well or 2×104 cells/well, followed by treatment with or without Dox (1 μg/ml) for 84 hours. Bromodeoxyuridine (BrdU, Calbiochem Cat#QIA58) was then added to each well for incubation of another 8 hours and cells were processed as described previously [39]. For quantification of BrdU-incorporated cells, at least 1000 cells from 10 fields were counted for each cell line under a Nikon TE2000-U inverted fluorescence microscope. Fluorescent images were taken at 200× magnification and the percentage of BrdU positive cells was shown.
2.9. Statistical analyses
Statistical analyses were performed with the GraphPad Prism 6 Software (Graphpad Software, San Diego, CA, USA). Briefly, Student’s t test was used to compare means of two different groups, while one-way Analysis of Variance (ANOVA) was used for multiple group comparison, followed by Tukey’s post-hoc test or Dunnett’s test. Two-way ANOVA was used to compare the means of two independent variables, followed by Tukey’s post-hoc test. All data were expressed as mean ± SEM, and p values less than 0.05 between groups were considered statistically significant. To analyze the correlation between the expression of PRMT5 and NF-YA in prostate cancer, we searched the Oncomine database (www.oncomine.org) and included each study that has more than 60 samples. A total of six independent studies met this criterion, and the results from these studies were pooled for correlation analysis. For each pair, the statistic Q was calculated to test the homogeneity of effect sizes across studies [50]. It turns out that, for each pair, the effect sizes across studies are not homogeneous (all with p value<0.0001). Therefore, we employed a random-effects model for the meta-analysis of each pair [51].
3. Results
3.1. Identification of the proximal promoter of PRMT5
To investigate how PRMT5 expression is transcriptionally regulated, we cloned a 3.5-kb PRMT5 promoter from LNCaP cells and found that there were two distinct types of promoters that harbor 6 single nucleotide polymorphisms (SNPs) and one 13 bp insertion/deletion polymorphism (indel) within 1.8 kb (Fig. 1A). To know whether these SNPs may impact the promoter activity, we used the 1.8 kb of the promoter to construct a series of truncated luciferase reporter genes (Fig. 1A). Transfection of these reporter genes into LNCaP cells resulted in at least a 7-fold increase in the promoter activity when compared with the vector control, with the B3 showing the highest activity (Fig. 1B). Similar results were obtained in PC-3 cells (Fig. 1C). However, mutations of all SNPs did not show any significant impact on the reporter gene activity (data not shown). Taken together, these results suggest that these SNPs have negligible effect on the 1.8 kb promoter activity.
Fig. 1.
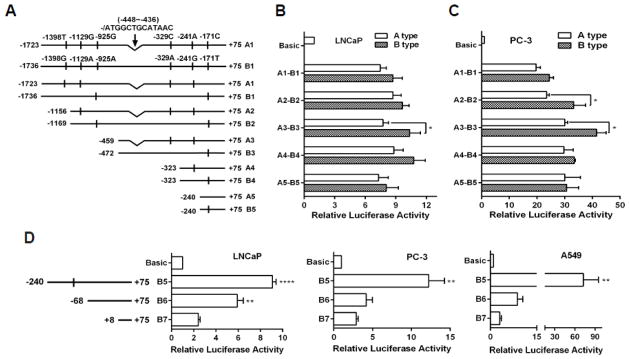
Identification of the proximal promoter of PRMT5. (A) Two types of PRMT5 promoters cloned from LNCaP genomic DNA with indicated SNPs and an indel, as well as a series of 5′-truncated promoters were used to construct luciferase reporter genes. (B and C) The indicated reporter genes in A were co-transfected with pRL-TK into LNCaP and PC-3 cells for 24 hours for measurement of the luciferase activities. Results were obtained from at least three independent experiments in triplicate, and were normalized to the vector control (Basic). (*, p<0.05; Student’s t test). (D) Luciferase activities of 5′-truncated reporter genes (B6 and B7) in LNCaP, PC-3 and A549 cells. Results from 4–6 independent experiments are presented as mean ± SEM. Statistical significance (**, p<0.01 and ****, p<0.0001) was determined when compared with B7 by one-way ANOVA followed by Dunnett’s test.
To identify a proximal promoter region, we constructed two other reporter genes (B6: −68/+75; B7: +8/+75) (Fig. 1D) and found that further deletions (B6 and B7) dramatically decreased the reporter gene activity in LNCaP cells (Fig. 1D), indicating that the region −240 to +75 is critical for the PRMT5 promoter activity. Similar results were observed in PC-3 cells (Fig. 1D). Since PRMT5 expression is also required for the growth of lung cancer cells (A549) [3], we transfected these reporter genes into A549 cells and observed that the reporter gene activity of B5 in A549 was 2-fold higher than that in LNCaP and PC-3 cells, though a comparable reporter gene activity of B6 and B7 was observed in all three cell lines (Fig. 1D). These results demonstrate that the proximal −240 region is important for PRMT5 transcription in a cell context-dependent manner.
3.2. The two inverted CCAAT boxes are critical for the proximal promoter activity of PRMT5
We next used AliBaba2.1 and TFSEARCH online software to search for putative cis-regulatory elements and identified one consensus GATA binding site for GATA binding, one GC box for SP1 binding, and three identical inverted CCAAT boxes for NF-Y binding in the proximal promoter region (Fig. 2A). In order to determine whether these putative binding sites contribute to the proximal promoter activity, we mutated these consensus motifs by site-directed mutagenesis (Fig. 2B), and examined their activities by using the luciferase reporter gene assays. In LNCaP cells, mutation of Y1 or Y2 (from CCAAT to CAGAA) [52], decreased the reporter gene activity by 33% and 21%, respectively (Fig. 2C). Significantly, mutations of both NF-Y binding sites resulted in 70% reduction in the reporter gene activity. Contrary to the two CCAAT box binding sites, single mutation introduced into the SP1 (GGGCGG to GGAAAG) or GATA (GATA to GCAA) binding site, which was demonstrated previously to abolish their binding [53, 54], increased the promoter activity by 36% or 27%, respectively (Fig. 2C). However, mutation of both SP1 and GATA binding sites did not show any further increase in the promoter activity. Similar effect of mutations in NF-Y sites was observed in PC-3 (Fig. 2D) and A549 cells (Fig. 2E), though single mutation of the first NF-Y site (Y1) had a more profound effect compared with the second NF-Y site (Y2). These results suggest that the two NF-Y binding sites may positively regulate PRMT5 transcription in all three cell lines whereas the SP1 and GATA binding sites may negatively regulate PRMT5 transcription in LNCaP cells but not in PC-3 and A549 cells. To know how these binding sites cooperatively contribute to the PRMT5 promoter activity, we mutated these binding sites in combination (Fig. 2B), and observed an overall inhibitory effect on the luciferase reporter gene activity, which is similar to the effect of mutations in the first two NF-Y binding sites (mY1,2). Note that a third NF-Y binding site (Y3) is located at +42, however, mutation of Y3 did not decrease the reporter gene activity in all three cell lines. Instead, a slight increase was observed (Fig. 2F–H). When all three NF-Y binding sites were mutated, a comparable suppression of the reporter gene activity to that with Y1/Y2 mutated was observed in all three cell lines (Fig. 2F–H). Taken together, these results suggest that the first two putative NF-Y binding sites are the major cis-regulatory elements to drive PRMT5 transcription.
Fig. 2.

The two CCAAT boxes are critical for the proximal promoter activity of PRMT5. (A) Sequences of the proximal promoter region from −240 to +75 with predicted cis-regulatory elements. The transcription start site was indicated by arrow. Y1, Y2, or Y3 indicates the first, second or third NF-Y binding site. (B) Illustration of a series of B5-based luciferase reporter gene constructs. Triangle indicates the corresponding cis-regulatory element was mutated. (C–E) CCAAT boxes are critical for luciferase activity driven by the PRMT5 promoter. The luciferase activity of the indicated reporter gene constructs in B was determined in the indicated cancer cell lines. (F–H) The third NF-Y binding site has little effect on the PRMT5 promoter activity. The indicated luciferase reporter gene was co-transfected with pRL-TK into LNCaP (F), PC-3 (G) and A549 (H) cells for 24 hours, and the relative luciferase activity was determined. Results in C-H were from at least three independent experiments, and were normalized to the vector control and are presented as mean ± SEM. Statistical significance (*, p<0.05, **, p<0.01, ***, p<0.001 and ****, p<0.0001) was determined when compared with WT (wild-type) by one-way ANOVA followed by Dunnett’s test.
3.3. NF-Y regulates PRMT5 expression in LNCaP cells via binding to the two CCAAT boxes
Unlike NF-YB and NF-YC, whose expression is relatively stable, NF-YA is the limited subunit for specific binding to CCAAT boxes in cells [12, 21–23]. To confirm the role of NF-Y in PRMT5 transcription at the endogenous level, we established two stable cell lines that inducibly express shRNAs targeting two different sequences in the coding region of NF-YA to evaluate the effect of NF-YA knockdown on PRMT5 expression. As shown in Fig. 3A, the two shRNAs knocked down the expression of NF-YA-S, the shorter isoform of NF-YA that is predominantly expressed in LNCaP cells, by more than 65%. The reduction of PRMT5 expression at protein level was similar to that of NF-YA. We confirmed that the expression of a well-known NF-Y target gene CCNA2 was also reduced, demonstrating the specificity of the two NF-YA shRNAs. Since the shYA#1 showed higher knockdown efficiency in LNCaP, it was chosen for the following experiments. We found that knockdown of NF-YA decreased the PRMT5 mRNA level (Fig. 3B), suggesting that the reduction of PRMT5 by NF-YA knockdown likely occurs at the transcriptional level. Transient knockdown of NF-YA significantly inhibited the WT reporter gene activity, but had no effect on the mutant reporter gene activity (Fig. 3C), suggesting that the two CCAAT boxes in the proximal promoter region likely mediates the effect of NF-Y on PRMT5 transcription. We next performed ChIP assays and confirmed that NF-YA bound to the region containing the two CCAAT boxes (P2 in Fig. 3D), but not the distal promoter region that does not contain CCAAT box (P1 in Fig. 3D). As a positive control, NF-Y also bound to the proximal promoter of CCNA2 [55]. These results demonstrate that NF-Y indeed binds to the two CCAAT boxes in the proximal promoter of PRMT5 and regulates PRMT5 transcription in LNCaP cells. To know whether NF-Y may regulate PRMT5 expression in human prostate cancer tissues, we searched Oncomine database and found that there was a strong positive correlation between the transcript level of NF-YA and PRMT5 (Fig. 3E), as evidenced by a meta-analysis from six independent studies. This result further supports our finding that NF-Y regulates PRMT5 expression in prostate cancer cells.
Fig. 3.

NF-Y is essential for PRMT5 expression in LNCaP cells. (A) NF-YA knockdown inhibits PRMT5 expression. Doxycycline (Dox) was added at 1 μg/ml for 96 hours to induce NF-YA knockdown in ShYA#1 and shYA#2 stable cell lines, and total cell lysate was used for immunoblotting analysis of PRMT5, shorter isoform of NF-YA (NF-YA-S), CCNA2 and β-actin. Shown are representative blots from three independent experiments, and the numbers indicate relative fold changes analyzed by Image J. (B) Knockdown of NF-YA inhibits PRMT5 mRNA expression. shRNA expression was induced by Dox for 72 hours, and qRT-PCR was performed to determine the mRNA level of PRMT5. Results are mean ± SEM from 4 independent experiments, and student’s t test was used for statistical analysis (**, p<0.01). (C) Knockdown of NF-YA decreases the PRMT5 proximal promoter activity. One μg of plasmids encoding SC or shYA1# (with Dox induction) was co-transfected with 0.5 μg of the B5 reporter gene plasmid (WT) or the mutant reporter gene (mY1,2,3), along with 100 ng of pRL-TK into LNCaP for 48 hours, and Dual-Luciferase Reporter Assays were performed and analyzed. Luciferase activities are presented as percentage from at least three independent experiments. **, p<0.01. (D) NF-YA binds to the two inverted CCAAT boxes. Shown (top) is a schematic of the two regions (P1 and P2) in the PRMT5 promoter for ChIP analysis. Results (bottom) are mean ± SEM from four independent experiments (**, p<0.01). The binding of NF-YA to the CCNA2 promoter was used as a positive control. (E) The transcript level of NF-YA positively correlates with the transcript level of PRMT5 in prostate cancer. Data shown are a meta-analysis from six independent studies deposited in Oncomine database.
3.4. NF-Y regulation of PRMT5 expression is required for prostate cancer cell growth
Given that NF-Y is critical for PRMT5 expression in several cancer cell lines, we next sought to determine the importance of NF-Y regulation of PRMT5 expression in cell growth. Using the two shRNA constructs, we were able to establish a stable cell line in A549 to knockdown NF-YA by 50%, accompanied by a 39% reduction in PRMT5 expression (Supplementary Fig. S1A). However, the two shRNAs did not exhibit acceptable knockdown efficiency in PC-3 (Supplementary Fig. S1B). We then examined the effect of NF-YA knockdown on cell growth and cell death in LNCaP and A549. Knockdown of NF-YA inhibited cell growth in LNCaP and A549 cells (Fig. 4, A and B). The inhibition of cell growth in both LNCaP and A549 by NF-YA knockdown was attributable to the inhibition of cell proliferation (Fig. 4, C and D; Supplementary Fig. S1 C and D) and the induction of cell death (Fig. 4, E and F), in agreement with previous findings that NF-Y plays a role in regulating cell proliferation and cell death [12]. Because NF-Y may influence growth of these cancer cells by controlling expression of many other genes [12, 15, 17], we next performed a PRMT5 rescue experiment to determine to what extent PRMT5 down-regulation is responsible for cell growth inhibition induced by NF-YA knockdown. As shown in Fig. 4, G and H, transient expression of PRMT5 partially rescued cell growth inhibition only in LNCaP cells, but not in A549 cells. Taken together, these results suggest that the regulation of cell growth by NF-Y may be partially mediated through up-regulation of PRMT5 expression in a cell context-dependent manner.
Fig. 4.

NF-Y regulation of PRMT5 expression is required for prostate cancer cell growth. (A and B) Knockdown of NF-YA inhibits cell growth in LNCaP (A) and A549 (B). Stable cell lines SC and shYA#1 were induced with 1 μg/ml of doxycycline (Dox+) to express shRNAs or without treatment (Dox-) for the indicated times, and cell numbers were counted using haemocytometer. Results from 4 independent experiments in duplicate are presented as mean ± SEM. Statistical significance (*, p<0.05; ***, p<0.001; ****, p<0.0001) was determined by two-way ANOVA followed by Tukey’s test. (C and D) Knockdown of NF-YA decreases BrdU-incorporated positive cells in LNCaP and A549 cells. SC and shYA#1 stable cell lines were induced with 1 μg/ml of Dox (Dox+) or without treatment (Dox-) for 84 hours, followed by BrdU treatment for another 8 hours. Number of BrdU-positive cells was determined using Image J software (total cell number > 1000, n=10). Results obtained from 4 independent experiments in duplicate are presented as mean ± SEM. Statistical significance (**, p<0.01; ***, p<0.001) when compared with SC was determined by two-way ANOVA followed by Dunnett’s test. (E and F) Effect of NF-YA knockdown on cell death. Stable and inducible cell lines targeting NF-YA (shYA#1) or the SC control were cultured in 6 cm dishes, and induced with 1 μg/ml of Dox (Dox+) or without treatment (Dox−) for 72 hours. Cells were trypsinized and counted to determine the percentage of dead cells by Trypan blue exclusion method. (G and H) Overexpression of PRMT5 rescues cell growth inhibition induced by NF-YA knockdown in LNCaP cells, but not in A549. LNCaP and A549 stable cell lines expressing shYA#1 were induced with 1 μg/ml of Dox (Dox+) or without induction (Dox−) for 48 hours, followed by transient transfection with pCMV-Myc (Myc-vector) or pCMV-Myc-PRMT5 (Myc-PRMT5) and incubation for another 48 hours. Top, results are presented as mean ± SEM from three independent experiments. Statistical significance was determined by two-way ANOVA followed by Tukey’s test. *, p<0.05; n.s., no significance. Bottom, the expression level of PRMT5 and NF-YA was determined by immunoblotting analysis. Shown are representative blots from three independent experiments. Note that the expression of both NF-YA longer isoform (NF-YA-L) and shorter isoform (NF-YA-S) was detectable in A549 cells whereas the expression of NF-YA-S was detectable in LNCaP cells only.
3.5. The PKC signaling negatively regulates PRMT5 expression in LNCaP cells
We next searched for possible cell signaling that may regulate PRMT5 expression in LNCaP cells by treating cells with various protein kinase inhibitors or agents that activate cell signaling pathways, and observed that treatment of cells with PMA resulted in a dramatic decrease of PRMT5 expression in a dose- and time-dependent manner (Fig. 5, A and B). Interestingly, NF-YA expression was similarly inhibited (Fig. 5, A and B). Significantly, the mRNA level of PRMT5 (Fig. 5C) and NF-YA, but not NF-YB and NF-YC (Fig. 5D), was inhibited by PMA treatment as well. Because PMA-induced PKC activation contributes to cell growth inhibition and apoptosis in LNCaP cells [26], we examined whether inhibition of PKC can restore the expression of NF-YA and PRMT5 in LNCaP cells, and found that treatment of cells with a pan-PKC inhibitor GFX completely restored the expression of NF-YA and PRMT5 at mRNA and protein level (Fig. 5C–E). The observed increase in NF-YB mRNA in cells treated with PMA plus GFX was likely due to the effect of GFX alone, because GFX treatment only increased NF-YB expression at the mRNA level but had no effect on the expression of PRMT5, NF-YA, and NF-YC (Supplementary Fig. S2). Consistent with a role for NF-Y in regulating PRMT5 transcription via the NF-Y binding sites in the proximal promoter region, PMA treatment resulted in almost 75% reduction of the NF-YA binding to the proximal promoter region of PRMT5 (Fig. 5F). In agreement with previous findings that PMA inhibits cell growth and induces apoptosis only in LNCaP, but not in DU 145 and PC-3 cells [25, 26], PMA treatment did not cause any significant change in NF-YA and PRMT5 expression in PC-3 cells (Fig. 5G). Additionally, PMA did not have any effect on NF-YA and PRMT5 expression in A549 cells (Fig. 5H). Thus, PMA treatment appears to have a specific effect on the expression of NF-YA and PRMT5 in LNCaP cells.
Fig. 5.

PKC negatively regulates PRMT5 expression in LNCaP. (A and B) The PKC activator PMA inhibits NF-YA and PRMT5 expression in a dose- and time-dependent manner. LNCaP cells were treated with PMA at the indicated doses (A) for 24 hours or treated with 100 nM of PMA for the indicated time points (B), and total cell lysate was used for immunoblotting analysis of PRMT5 and NF-YA expression. (C and D) A pan-PKC inhibitor inhibits PMA-induced down-regulation of PRMT5 and NF-YA at the mRNA level. LNCaP cells were treated with 100 nM of PMA in the presence or absence of a pan-PKC inhibitor GFX (200 nM) for 24 hours, and relative mRNA level of PRMT5 (C) or NF-YA, NF-YB and NF-YC (D) was determined by qRT-PCR. Results from three independent experiments are presented as mean ± SEM in C and D, and statistical significance (*, p<0.05, ***, p<0.001) was determined by one-way ANOVA followed by Tukey’s test. (E) PKC inhibition restores NF-YA and PRMT5 expression at the protein level in cells treated with PMA. LNCaP cells were treated with 100 nM of PMA in the presence or absence of GFX (200 nM) for 24 hours, then NF-YA and PRMT5 expression was analyzed by immunoblotting. Representative blots from three independent experiments are shown. (F) PMA treatment decreases NF-YA binding to the PRMT5 promoter. ChIP analysis was conducted using anti-NF-YA antibody to determine the binding of NF-YA to the two CCAAT boxes in the proximal promoter region of PRMT5. ***, p<0.001 (Student’s t test). (G and H) PMA does not significantly affect the expression of NF-YA and PRMT5 in PC-3 and A549. PC-3 and A549 cells were treated with PMA at the indicated concentration for 24 hours, and total cell lysate was used for immunoblotting detection of NF-YA and PRMT5 expression. PMA−, DMSO treatment (Fig. 5C–F).
3.6. c-Fos mediates the PKC signaling to regulate PRMT5 transcription via down-regulation of NF-YA expression
As AP-1 proteins c-Fos and c-Jun are downstream transcription factors of PKC that can be induced by PMA [30–32], we confirmed that PMA treatment indeed induced expression of c-Fos and c-Jun in LNCaP cells (Fig. 6A). However, overexpression of c-Fos, but not c-Jun, inhibited the PRMT5 reporter gene activity (Fig. 6B). Consistent with its effect on the PRMT5 reporter gene, overexpressed c-Fos, but not c-Jun, decreased PRMT5 mRNA (Fig. 6C) and protein expression (Fig. 6D). We found that NF-YA expression at both mRNA and protein levels was also inhibited by c-Fos (Fig. 6, C and D). These results suggest that c-Fos may mediate the PKC signaling to down-regulate the expression of NF-YA and PRMT5. To test this, we generated a shRNA construct targeting c-Fos and observed that knockdown of c-Fos increased the PRMT5 reporter gene activity by 54% (Fig. 6E). Further, we used the shRNA construct to establish an inducible stable cell line to knock down c-Fos, and observed that PMA-induced NF-YA and PRMT5 down-regulation was partially restored when c-Fos was knocked down (Fig. 6, F and G). Since the ENCODE ChIP-seq data from the UCSC database (http://genome.ucsc.edu/ENCODE/) show that c-Fos also binds to the proximal promoter region in HeLa-S3 and K562 cells, we were interesting to know whether c-Fos has any direct impact on the PRMT5 promoter activity in LNCaP cells. To this end, we examined the effect of c-Fos overexpression or knockdown on the WT and the mutant PRMT5 reporter gene activity. As shown in Fig. 6H and I, we found that overexpression of c-Fos decreased WT PRMT5 reporter gene activity by 62.3%, but had no effect on the mutant reporter gene activity in which all three NF-Y binding sites were mutated (mY1,2,3). In contrast, transient knockdown of c-Fos remarkably increased WT PRMT5 reporter gene activity, but had no effect on the mutant reporter gene activity. These results provide evidence that c-Fos indeed mediates, at least partially, the PKC signaling to negatively regulate PRMT5 transcription via down-regulation of NF-YA in LNCaP cells.
Fig. 6.

c-Fos mediates the PKC signaling to down-regulate PRMT5 expression via NF-YA. (A) PMA increases c-Jun and c-Fos expression in LNCaP. LNCaP cells were treated with 100 nM of PMA in the presence or absence of GFX (200 nM) for 24 hours, and the expression of c-Fos and c-Jun was determined by immunoblotting. (B) Overexpression of c-Fos, but not c-Jun, inhibits the PRMT5 promoter activity. One μg of pCMV-FLAG (Vector), pFLAG-c-Fos (c-Fos) or pFLAG-c-Jun (c-Jun) was co-transfected with 0.5 μg of the wild-type (B5) reporter gene, along with 0.1 μg of pRL-TK into LNCaP cells. The luciferase activity was determined 24 hours after the transfection. Results from six independent experiments in triplicate are presented as mean ± SEM, and statistical significance (***, p<0.001, ****, p<0.0001) was determined using one-way ANOVA followed by Dunnett’s test. (C and D) Overexpression of c-Fos, but not c-Jun, inhibits NF-YA and PRMT5 expression. LNCaP cells were transfected with 3 μg of the indicated plasmids as described in B. The mRNA and protein expression of NF-YA and PRMT5 was determined by qRT-PCR (C) and immunobloting (D), respectively. Results from at least three independent experiments are presented as mean ± SEM. Statistical significance (**, p<0.01) was determined by using one-way ANOVA followed by Dunnett’s test. (E) Knockdown of c-Fos increases the PRMT5 promoter activity. The SC or c-Fos shRNA (shFos) was co-transfected with 0.5 μg of the wild-type (B5) reporter gene, along with 0.1 μg of pRL-TK into LNCaP cells. The luciferase activity was determined 48 hours after the transfection. **, p<0.01 versus SC (Student’s t test). (F and G) Knockdown of c-Fos partially rescues NF-YA and PRMT5 expression. Stable cell line that can inducibly express a c-Fos shRNA was induced with 1 μg/ml of doxycycline (Dox+) or without treatment (Dox−) for 48 hours. Cells then were treated with 100 nM of PMA (PMA+) or DMSO (PMA−) for another 24 hours, followed by determination of the mRNA expression (F) and protein expression (G) of NF-YA and PRMT5. Statistical significance (*, p<0.05, **, p<0.01) was determined by two-way ANOVA followed by Tukey’s test. The numbers in G indicate the relative expression level of each protein analyzed by Image J software. (H and I) c-Fos decreases PRMT5 promoter activity mainly through CCAAT boxes. The indicated plasmids were transfected into LNCaP cells, and the luciferase assays were performed following the same procedure as described in B and E, respectively. Results from three independent experiments in triplicate are presented as mean ± SEM, and statistical significance (**, p<0.01, ****, p<0.0001).
4. Discussion
It has been reported that PRMT5 may function as an oncogene to promote cancer cell growth [1–3, 5–7, 9, 10]. Although NF-Y directly regulates transcription of many target genes to control cell cycle progression, cell proliferation and cell survival [12, 13, 15, 17], our finding that NF-Y transcriptionally activates PRMT5 expression suggests that NF-Y may also regulate cancer cell growth by controlling the expression level of PRMT5, an emerging epigenetic enzyme that functions as an oncogene in human cancers [1]. For example, E2F1 is a member of the E2F family transcription factor required for transactivation of target genes involved in cell cycle progression in cancer cells [56]. Because the transcriptional activity of E2F1 is under the control of the tumor suppressor Rb, loss of Rb leads to constitutive activation of E2F1 and cancer development [57]. Interestingly, PRMT5 can epigenetically silence transcription of Rb [9]. Thus, activation or overexpression of NF-Y may lead to PRMT5 overexpression, by which Rb is silenced and E2F1 is activated, providing another pathway to promote cell cycle progression in cancer cells that harbor the wild-type Rb gene [9]. As NF-Y also regulates the transcription of the same target genes such as E2F1 [58], future studies of how NF-Y coordinates the regulation of PRMT5 expression and other target genes will likely provide novel insights into the oncogenic role of both NF-Y and PRMT5 in cancer cells.
Recent evidence indicates that PRMT5 is overexpressed in multiple human cancers [3–11], though it is unknown how PRMT5 expression is regulated by cancer signaling. In leukemia and lymphoma cells, down-regulation of several miRNAs contributes to PRMT5 overexpression [9, 10]. We have provided several lines of evidence that NF-Y regulates PRMT5 transcription via the binding to the two CCAAT boxes in the proximal promoter region of PRMT5. First, mutagenesis analyses showed that mutation of the two CCAAT boxes in the proximal promoter region resulted in 70% reduction in the luciferase reporter gene activity in three different cancer cell lines (Fig. 2C–E). Second, endogenous NF-YA also specifically bound to the proximal promoter region containing the two CCAAT boxes in LNCaP cells (Fig. 3D). Third, knockdown of NF-YA not only inhibited the PRMT5 promoter-driven luciferase report gene activity but also decreased the expression of PRMT5 at both mRNA and protein levels (Fig. 3A–C). We also show that the PKC/c-Fos signaling negatively regulates PRMT5 expression via down-regulation of NF-YA transcription in LNCaP prostate cancer cells (Fig. 5 and Fig. 7A). Although the mechanism by which c-Fos represses NF-YA transcription remains to be investigated, it is interesting to note that our preliminary analysis of the NF-Y promoter identified 3 consensus AP-1 binding sites within the 6 kb promoter region. It is therefore possible that c-Fos may directly repress NF-YA transcription by binding to these consensus AP-1 binding sites. Alternatively, c-Fos may indirectly repress NF-YA transcription through a secondary effect (e.g., up-regulation of a transcriptional repressor of NF-YA). Nevertheless, our findings suggest that cell signaling may up-regulate PRMT5 expression by down-regulation of PKC or by direct up-regulation of NF-YA to promote cancer cell growth (Fig. 7B). This is further supported by the fact that several isozymes of PKC are down-regulated in human cancers [59]. Indeed, a preliminary analysis of the Oncomine database shows that the transcript level of several PKC isozymes inversely correlates with the transcript level of PRMT5 in prostate cancer and lung cancer (Supplementary Fig. S3). It will be interesting to see whether down-regulation of these PKC isozymes correlates with PRMT5 overexpression at the protein level in human cancer tissues.
Fig. 7.

Model for the regulation of PRMT5 expression by the PKC-c-Fos-NF-Y signaling in human cancer. (A) The PKC signaling negatively regulates PRMT5 expression in a c-Fos- and NF-Y-dependent manner in LNCaP cells. In response to PMA treatment, activation of PKC leads to the induction of c-Fos, which in turn suppresses NF-YA transcription and results in down-regulation of PRMT5. As a result, cell growth is inhibited. (B) Proposed mechanisms underlying up-regulation of PRMT5 expression in cancer cells. Two possible mechanisms may underlie PRMT5 overexpression in human cancers. One is the inactivation or down-regulation of PKC by cell signaling, and the other is direct activation or up-regulation of NF-YA by cell signaling that remains to be identified (X). Dashed lines indicate unknown factors that remain to be identified. Thick solid arrows illustrate the up-regulation or down-regulation of the indicated protein.
The cell growth-promoting role of PRMT5 is mediated by controlling the expression of target genes or by post-translational modification of signaling molecules that are involved in cell cycle progression, apoptosis and DNA repair [1]. Although knockdown of PRMT5 in LNCaP cells inhibits cell proliferation [60], the downstream signaling mediating this effect remains unknown. A previous study suggests that PRMT5 may be required for the transcriptional activity of AR in a luciferase reporter gene assay [61]. Given that PMA-induced down-regulation of PRMT5 is mainly observed in AR positive LNCaP cells, but not in AR negative DU 145 and PC-3 cells, it is plausible to hypothesize that down-regulation of PRMT5 by PMA in LNCaP cells may contribute to the suppression of LNCaP cell growth and induction of apoptosis by attenuating the AR activity [61]. As a recent report shows that PMA treatment in LNCaP cells can down-regulate AR expression [62], it would be interesting to examine whether PRMT5 has any effect on AR expression. Alternatively, PMA-induced PRMT5 down-regulation may contribute to PMA-induced apoptosis by enhancing the activity of p38δ, a major serine/threonine protein kinase mediating PMA-induced apoptosis in LNCaP cells [26]. Support for this notion comes from a recent observation that PRMT5 forms a complex with p38δ and suppresses PKCδ- and p38δ-dependent signaling in keratinocytes [63]. Future studies to distinguish these possibilities will provide a novel insight into the regulatory role of PRMT5 in prostate cancer cells.
In summary, we have identified NF-Y as the major transcriptional activator of PRMT5 in multiple cancer cell lines, and demonstrated that the PKC/c-Fos signaling negatively regulates PRMT5 expression in LNCaP prostate cancer cells through down-regulation of NF-YA transcription. Because down-regulation of several PKC isozymes correlates with human cancer development and progression [59], further analysis of the interplay between PRMT5 and the PKC/c-Fos signaling in human cancer will provide novel insights into the oncogenic role of RPMT5 in human cancers.
Supplementary Material
Highlights.
The two inverted CCAAT boxes are critical for the PRMT5 promoter activity.
NF-Y binds to the two inverted CCAAT boxes and activates PRMT5 transcription.
PRMT5 mediates the effect of NF-Y to regulate cell grown in LNCaP cells.
The PKC/c-Fos signaling negatively regulates PRMT5 expression via NF-Y.
Acknowledgments
This study was partially supported by grants from U.S. Army Medical Research Acquisition Activity, Prostate Cancer Research Program (PC11190 and PC120512), and Purdue University Center for Cancer Research Small Grants. DNA sequencing was conducted in the Purdue University Center for Cancer Research Genomic Core Facility supported by NCI CCSG CA23168 to Purdue University Center for Cancer Research. We thank members of the Hu lab for helpful suggestions and discussions. Huan-Tian Zhang was supported by a scholarship from China Scholarship Council and a Graduate Fellowship from Jinan University and Institute of Orthopedic Diseases of Jinan University for his study in the Hu lab at Purdue University.
The Abbreviations used are
- PRMT5
protein arginine methyltransferase 5
- NF-Y
nuclear transcription factor Y
- PKC
protein kinase C
- AP-1
activator protein-1
- PMA
phorbol 12-myristate 13-acetate
- GFX
bisindolylmaleimide I
- TCL
total cell lysate
- CCNA2
cyclin A2
- Dox
doxycycline
- SC
scrambled control
- shRNA
short hairpin RNA
- BrdU
Bromodeoxyuridine
- indel
insertion-deletion
- SNPs
single nucleotides polymorphisms
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Karkhanis V, Hu YJ, Baiocchi RA, Imbalzano AN, Sif S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem Sci. 2011;36:633–641. doi: 10.1016/j.tibs.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, Pestka S. Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther. 2007;113:50–87. doi: 10.1016/j.pharmthera.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Gu Z, Gao S, Zhang F, Wang Z, Ma W, Davis RE, Wang Z. Protein arginine methyltransferase 5 is essential for growth of lung cancer cells. Biochem J. 2012;446:235–241. doi: 10.1042/BJ20120768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei TY, Juan CC, Hisa JY, Su LJ, Lee YC, Chou HY, Chen JM, Wu YC, Chiu SC, Hsu CP, Liu KL, Yu CT. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci. 2012;103:1640–1650. doi: 10.1111/j.1349-7006.2012.02367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bao X, Zhao S, Liu T, Liu Y, Yang X. Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J Histochem Cytochem. 2013;61:206–217. doi: 10.1369/0022155413475452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho EC, Zheng S, Munro S, Liu G, Carr SM, Moehlenbrink J, Lu YC, Stimson L, Khan O, Konietzny R, McGouran J, Coutts AS, Kessler B, Kerr DJ, Thangue NB. Arginine methylation controls growth regulation by E2F-1. EMBO J. 2012;31:1785–1797. doi: 10.1038/emboj.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powers MA, Fay MM, Factor RE, Welm AL, Ullman KS. Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Res. 2011;71:5579–5587. doi: 10.1158/0008-5472.CAN-11-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicholas C, Yang J, Peters SB, Bill MA, Baiocchi RA, Yan F, Sif S, Tae S, Gaudio E, Wu X, Grever MR, Young GS, Lesinski GB. PRMT5 is upregulated in malignant and metastatic melanoma and regulates expression of MITF and p27(Kip1.) PLoS One. 2013;8:e74710. doi: 10.1371/journal.pone.0074710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L, Pal S, Sif S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol Cell Biol. 2008;28:6262–6277. doi: 10.1128/MCB.00923-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pal S, Baiocchi RA, Byrd JC, Grever MR, Jacob ST, Sif S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007;26:3558–3569. doi: 10.1038/sj.emboj.7601794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan F, Alinari L, Lustberg ME, Katherine Martin L, Cordero-Nieves HM, Banasavadi-Siddegowda Y, Virk S, Barnholtz-Sloan J, Bell EH, Wojton J, Jacob NK, Chakravarti A, Nowicki MO, Wu X, Lapalombella R, Datta J, Yu B, Gordon K, Haseley A, Patton JT, Smith PL, Ryu J, Zhang X, Mo X, Marcucci G, Nuovo G, Kwon CH, Byrd JC, Chiocca EA, Li C, Sif S, Jacob S, Lawler S, Kaur B, Baiocchi RA. Genetic Validation of the Protein Arginine Methyltransferase PRMT5 as a Candidate Therapeutic Target in Glioblastoma. Cancer Res. 2014;74:1752–1765. doi: 10.1158/0008-5472.CAN-13-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dolfini D, Gatta R, Mantovani R. NF-Y and the transcriptional activation of CCAAT promoters. Crit Rev Biochem Mol Biol. 2012;47:29–49. doi: 10.3109/10409238.2011.628970. [DOI] [PubMed] [Google Scholar]
- 13.Mantovani R. The molecular biology of the CCAAT-binding factor NF-Y. Gene. 1999;239:15–27. doi: 10.1016/s0378-1119(99)00368-6. [DOI] [PubMed] [Google Scholar]
- 14.Li XY, Mantovani R, Hooft van Huijsduijnen R, Andre I, Benoist C, Mathis D. Evolutionary variation of the CCAAT-binding transcription factor NF-Y. Nucleic Acids Res. 1992;20:1087–1091. doi: 10.1093/nar/20.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolfini D, Zambelli F, Pavesi G, Mantovani R. A perspective of promoter architecture from the CCAAT box. Cell Cycle. 2009;8:4127–4137. doi: 10.4161/cc.8.24.10240. [DOI] [PubMed] [Google Scholar]
- 16.Mantovani R. A survey of 178 NF-Y binding CCAAT boxes. Nucleic Acids Res. 1998;26:1135–1143. doi: 10.1093/nar/26.5.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolfini D, Mantovani R. Targeting the Y/CCAAT box in cancer: YB-1 (YBX1) or NF-Y? Cell Death Differ. 2013;20:676–685. doi: 10.1038/cdd.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fleming JD, Pavesi G, Benatti P, Imbriano C, Mantovani R, Struhl K. NF-Y coassociates with FOS at promoters, enhancers, repetitive elements, and inactive chromatin regions, and is stereo-positioned with growth-controlling transcription factors. Genome Res. 2013;23:1195–1209. doi: 10.1101/gr.148080.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SR, Park JH, Park EK, Chung CH, Kang SS, Bang OS. Akt-induced promotion of cell-cycle progression at G2/M phase involves upregulation of NF-Y binding activity in PC12 cells. J Cell Physiol. 2005;205:270–277. doi: 10.1002/jcp.20395. [DOI] [PubMed] [Google Scholar]
- 20.Yokota S, Okabayashi T, Rehli M, Fujii N, Amano K. Helicobacter pylori lipopolysaccharides upregulate toll-like receptor 4 expression and proliferation of gastric epithelial cells via the MEK1/2-ERK1/2 mitogen-activated protein kinase pathway. Infect Immun. 2010;78:468–476. doi: 10.1128/IAI.00903-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manni I, Caretti G, Artuso S, Gurtner A, Emiliozzi V, Sacchi A, Mantovani R, Piaggio G. Posttranslational regulation of NF-YA modulates NF-Y transcriptional activity. Mol Biol Cell. 2008;19:5203–5213. doi: 10.1091/mbc.E08-03-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farina A, Manni I, Fontemaggi G, Tiainen M, Cenciarelli C, Bellorini M, Mantovani R, Sacchi A, Piaggio G. Down-regulation of cyclin B1 gene transcription in terminally differentiated skeletal muscle cells is associated with loss of functional CCAAT-binding NF-Y complex. Oncogene. 1999;18:2818–2827. doi: 10.1038/sj.onc.1202472. [DOI] [PubMed] [Google Scholar]
- 23.Chang ZF, Liu CJ. Human thymidine kinase CCAAT-binding protein is NF-Y, whose A subunit expression is serum-dependent in human IMR-90 diploid fibroblasts. J Biol Chem. 1994;269:17893–17898. [PubMed] [Google Scholar]
- 24.Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nature reviews Drug Discov. 2012;11:937–957. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao L, Caino MC, von Burstin VA, Oliva JL, Kazanietz MG. Phorbol ester-induced apoptosis and senescence in cancer cell models. Methods Enzymol. 2008;446:123–139. doi: 10.1016/S0076-6879(08)01607-8. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez-Guerrico AM, Meshki J, Xiao L, Benavides F, Conti CJ, Kazanietz MG. Molecular mechanisms of protein kinase C-induced apoptosis in prostate cancer cells. J Biochem Mol Biol. 2005;38:639–645. doi: 10.5483/bmbrep.2005.38.6.639. [DOI] [PubMed] [Google Scholar]
- 27.Fukase N, Kawamoto T, Kishimoto K, Hara H, Okada Y, Onishi Y, Toda M, Kurosaka M, Akisue T. Protein kinase Cdelta in tumorigenesis of human malignant fibrous histiocytoma. Oncol Rep. 2011;26:1221–1226. doi: 10.3892/or.2011.1415. [DOI] [PubMed] [Google Scholar]
- 28.Gwak J, Jung SJ, Kang DI, Kim EY, Kim DE, Chung YH, Shin JG, Oh S. Stimulation of protein kinase C-alpha suppresses colon cancer cell proliferation by down-regulation of beta-catenin. J Cell Mol Med. 2009;13:2171–2180. doi: 10.1111/j.1582-4934.2008.00683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 30.Angel P, Imagawa M, Chiu R, Stein B, Imbra RJ, Rahmsdorf HJ, Jonat C, Herrlich P, Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- 31.Lee W, Mitchell P, Tjian R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell. 1987;49:741–752. doi: 10.1016/0092-8674(87)90612-x. [DOI] [PubMed] [Google Scholar]
- 32.Lamph WW, Wamsley P, Sassone-Corsi P, Verma IM. Induction of proto-oncogene JUN/AP-1 by serum and TPA. Nature. 1988;334:629–631. doi: 10.1038/334629a0. [DOI] [PubMed] [Google Scholar]
- 33.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 34.van Dam H, Castellazzi M. Distinct roles of Jun : Fos and Jun : ATF dimers in oncogenesis. Oncogene. 2001;20:2453–2464. doi: 10.1038/sj.onc.1204239. [DOI] [PubMed] [Google Scholar]
- 35.Ouyang X, Jessen WJ, Al-Ahmadie H, Serio AM, Lin Y, Shih WJ, Reuter VE, Scardino PT, Shen MM, Aronow BJ, Vickers AJ, Gerald WL, Abate-Shen C. Activator protein-1 transcription factors are associated with progression and recurrence of prostate cancer. Cancer Res. 2008;68:2132–2144. doi: 10.1158/0008-5472.CAN-07-6055. [DOI] [PubMed] [Google Scholar]
- 36.Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, Michalopoulos G, Becich M, Monzon FA. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edwards J, Krishna NS, Mukherjee R, Bartlett JM. The role of c-Jun and c-Fos expression in androgen-independent prostate cancer. J Pathol. 2004;204:153–158. doi: 10.1002/path.1605. [DOI] [PubMed] [Google Scholar]
- 38.Tamura K, Furihata M, Tsunoda T, Ashida S, Takata R, Obara W, Yoshioka H, Daigo Y, Nasu Y, Kumon H, Konaka H, Namiki M, Tozawa K, Kohri K, Tanji N, Yokoyama M, Shimazui T, Akaza H, Mizutani Y, Miki T, Fujioka T, Shuin T, Nakamura Y, Nakagawa H. Molecular features of hormone-refractory prostate cancer cells by genome-wide gene expression profiles. Cancer Res. 2007;67:5117–5125. doi: 10.1158/0008-5472.CAN-06-4040. [DOI] [PubMed] [Google Scholar]
- 39.Hsu CC, Hu CD. Transcriptional activity of c-Jun is critical for the suppression of AR function. Mol Cell Endocrinol. 2013;372:12–22. doi: 10.1016/j.mce.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng X, Elzey BD, Poulson JM, Morrison WB, Ko SC, Hahn NM, Ratliff TL, Hu CD. Ionizing radiation induces neuroendocrine differentiation of prostate cancer cells in vitro, in vivo and in prostate cancer patients. Am J Cancer Res. 2011;1:834–844. [PMC free article] [PubMed] [Google Scholar]
- 41.Deng X, Liu H, Huang J, Cheng L, Keller ET, Parsons SJ, Hu CD. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer Res. 2008;68:9663–9670. doi: 10.1158/0008-5472.CAN-08-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ali SA, Steinkasserer A. PCR-ligation-PCR mutagenesis: a protocol for creating gene fusions and mutations. BioTechniques. 1995;18:746–750. [PubMed] [Google Scholar]
- 43.Liu H, Deng X, Shyu YJ, Li JJ, Taparowsky EJ, Hu CD. Mutual Regulation of c-Jun and ATF2 by Transcriptional Activation and Subcellular Localization. EMBO J. 2006;25:1058–1069. doi: 10.1038/sj.emboj.7601020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsu CC, Hu CD. Critical role of N-terminal end-localized nuclear export signal in regulation of activating transcription factor 2 (ATF2) subcellular localization and transcriptional activity. J Biol Chem. 2012;287:8621–8632. doi: 10.1074/jbc.M111.294272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benatti P, Dolfini D, Vigano A, Ravo M, Weisz A, Imbriano C. Specific inhibition of NF-Y subunits triggers different cell proliferation defects. Nucleic Acids Res. 2011;39:5356–5368. doi: 10.1093/nar/gkr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 47.Kelly TK, Miranda TB, Liang G, Berman BP, Lin JC, Tanay A, Jones PA. H2A.Z maintenance during mitosis reveals nucleosome shifting on mitotically silenced genes. Mol Cell. 2010;39:901–911. doi: 10.1016/j.molcel.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nelson JD, Denisenko O, Sova P, Bomsztyk K. Fast chromatin immunoprecipitation assay. Nucleic Acids Res. 2006;34:e2. doi: 10.1093/nar/gnj004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao YM, Lengauer C, Sellers WR, Benson JD. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle. 2009;8:498–504. doi: 10.4161/cc.8.3.7701. [DOI] [PubMed] [Google Scholar]
- 50.Hedges LV, Olkin I. Statistical methods for meta-analysis. Academic Press; Orlando, FL: 1985. [Google Scholar]
- 51.Hedges LV, Vevea JL. Fixed- and random-effects models in meta-analysis. Psychological Methods. 1998;3:485–504. [Google Scholar]
- 52.Bi W, Wu L, Coustry F, de Crombrugghe B, Maity SN. DNA binding specificity of the CCAAT-binding factor CBF/NF-Y. J Biol Chem. 1997;272:26562–26572. doi: 10.1074/jbc.272.42.26562. [DOI] [PubMed] [Google Scholar]
- 53.Gidoni D, Kadonaga JT, Barrera-Saldana H, Takahashi K, Chambon P, Tjian R. Bidirectional SV40 transcription mediated by tandem Sp1 binding interactions. Science. 1985;230:511–517. doi: 10.1126/science.2996137. [DOI] [PubMed] [Google Scholar]
- 54.Merika M, Orkin SH. DNA-binding specificity of GATA family transcription factors. Mol Cell Biol. 1993;13:3999–4010. doi: 10.1128/mcb.13.7.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garipov A, Li H, Bitler BG, Thapa RJ, Balachandran S, Zhang R. NF-YA underlies EZH2 upregulation and is essential for proliferation of human epithelial ovarian cancer cells. Mol Cancer Res. 2013;11:360–369. doi: 10.1158/1541-7786.MCR-12-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer. 2009;9:738–748. doi: 10.1038/nrc2718. [DOI] [PubMed] [Google Scholar]
- 57.Engelmann D, Putzer BM. The dark side of E2F1: in transit beyond apoptosis. Cancer Res. 2012;72:571–575. doi: 10.1158/0008-5472.CAN-11-2575. [DOI] [PubMed] [Google Scholar]
- 58.Kabe Y, Yamada J, Uga H, Yamaguchi Y, Wada T, Handa H. NF-Y is essential for the recruitment of RNA polymerase II and inducible transcription of several CCAAT box-containing genes. Mol Cell Biol. 2005;25:512–522. doi: 10.1128/MCB.25.1.512-522.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ali AS, Ali S, El-Rayes BF, Philip PA, Sarkar FH. Exploitation of protein kinase C: a useful target for cancer therapy. Cancer Treat Rev. 2009;35:1–8. doi: 10.1016/j.ctrv.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 60.Gu Z, Li Y, Lee P, Liu T, Wan C, Wang Z. Protein arginine methyltransferase 5 functions in opposite ways in the cytoplasm and nucleus of prostate cancer cells. PLoS One. 2012;7:e44033. doi: 10.1371/journal.pone.0044033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hosohata K, Li P, Hosohata Y, Qin J, Roeder RG, Wang Z. Purification and identification of a novel complex which is involved in androgen receptor-dependent transcription. Mol Cell Biol. 2003;23:7019–7029. doi: 10.1128/MCB.23.19.7019-7029.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Itsumi M, Shiota M, Yokomizo A, Takeuchi A, Kashiwagi E, Dejima T, Inokuchi J, Tatsugami K, Uchiumi T, Naito S. PMA induces androgen receptor downregulation and cellular apoptosis in prostate cancer cells. J Mol Endocrinol. 2014;53:31–41. doi: 10.1530/JME-13-0303. [DOI] [PubMed] [Google Scholar]
- 63.Kanade SR, Eckert RL. Protein arginine methyltransferase 5 (PRMT5) signaling suppresses protein kinase Cdelta- and p38delta-dependent signaling and keratinocyte differentiation. J Biol Chem. 2012;287:7313–7323. doi: 10.1074/jbc.M111.331660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.