

Abstract
Background
Urokinase [uPA] modulates cellular and extracellular matrix responses within the microenvironment of the vessel wall and has been shown to activate the epidermal growth factor receptor [EGFR]. This study examines the role of the protease domain of uPA during EGFR activation in human vascular smooth muscle cells [VSMC].
Methods
Human coronary VSMC were cultured in vitro. Assays of cell proliferation and EGFR phosphorylation were examined in response to the C-terminal fragment of uPA [CTF] in the presence and absence of the plasmin, metalloprotease [MMP] and A Disintegrin and Metalloproteinase [ADAM] inhibitors, heparin-bound epidermal growth factor [HB-EGF], and EGFR inhibitors, and small interfering RNA [siRNA] to EGFR and ADAMs.
Results
CTF produced a dose-dependent increase in DNA synthesis and cell proliferation in human VSMC, which was blocked in a dose-dependent manner by both plasmin inhibitors and the EGFR inhibitor, AG1478. CTF induced time-dependent EGFR phosphorylation, which was blocked by inhibitors of plasmin and MMP activity. The presence of uPAR was not required. Inhibition of ADAM-10 and -12, and of HB-EGF blocked EGFR activation in response to CTF. CTF-mediated activation of EGFR was mediated through Gβγ, src and NAD[P]H oxidase.
Conclusions
In human coronary VSMC, uPA induces uPAR-independent, domain-dependent smooth muscle cell proliferation through transactivation of EGFR by a plasmin-mediated, ADAM-induced, HB-EGF-dependent process, which is mediated by the intracellular pathways involving Gαi, Gβγ, src and NAD[P]H oxidase.
Keywords: uPA, protease domain, epidermal growth factor receptor, proliferation, cell signaling, human coronary smooth muscle cell
INTRODUCTION
Vessel remodeling occurs in response to flow and injury through an integrative program of cell proliferation, migration and extracellular matrix modulation (1). The proliferation and migration of vascular smooth muscle cells [VSMC] involves the complex regulation of proteases, integrins and extracellular molecules. Urokinase plasminogen activator [uPA] is a serine protease that is the primary plasminogen activator involved in tissue remodeling processes. Elevated uPA and decreased plasminogen activator inhibitor-1 [PAI-1] levels are predictors for angiographic coronary restenosis (2). After injury to the rat carotid artery, there is a time-dependent enhanced expression of plasminogen activators and increased plasminogen activator activity on smooth muscle cells within the vessel wall compared to the uninjured state (3). Overexpression of uPA or a deficiency of PAI-1 in transgenic mice has been shown to promote neointimal formation in response to transmural electrical injury. A lack of uPA or plasminogen leads to a reduction in neointimal hyperplasia (4, 5). uPA binds to the cell membrane through a GPI-tailed receptor, uPAR.
uPA has been shown to induce domain-dependent cell proliferation and migration, with the former due to its carboxyterminal, protease-containing domain [CTF] and the latter due to its aminoterminal, growth factor-binding domain (6). The biological data from the transgenic murine models suggests that uPA has a significant role in stimulating proliferation, and that it is plasmin dependent (7, 8). Using mutants of single chain uPA [sc-uPA], others have shown that cell proliferation is dependent on the mutant sc-uPA molecules containing CTF (9–12). CTF contains a protease domain and leads to plasmin generation, while plasmin inhibition reduces DNA synthesis. Plasmin will induce VSMC proliferation and inhibition of plasmin’s protease activity inhibits VSMC proliferation (6). Plasmin can activate metalloproteinases [MMP], which, in turn, may lead to epidermal growth factor receptor [EGFR] activation (5, 13, 14). We have recently demonstrated that uPA induces smooth muscle cell proliferation through ERK1/2 and p38MAPK-mediated pathways and is dependent on EGFR transactivation (13). CTF, through its protease domain or through generation of plasmin, may also activate G protein-coupled protease activated receptors on the cell membrane (15, 16).
Receptor transactivation is the pathway whereby activation of a given G protein-coupled receptor [GPCR] activates a heterologous receptor-linked tyrosine kinase [RTK] (17). At present, the triple membrane passing signaling [TMPS] mechanism of GPCR-induced EGFR activation is a widely accepted model of RTK transactivation. In this model, there is a sequence of three transmembrane signaling events: GPCR activation, followed by MMP activation and subsequent activation of EGFR by the tethered ligand heparin binding-EGF [HB-EGF] or other latent ligands of EGFR. Activation of the MMP step has been shown to involve one or more of the following signaling molecules: src, calcium, Pyk2 and protein kinase C [PKC]. This study tests the hypothesis that the protease domain of uPA induces cell proliferation through the triple membrane passing signaling mechanism in human VSMC, leading to transactivation of EGFR, and that this is independent of uPAR.
METHODS
Experimental Design
Human coronary arterial VSMCs were cultured in vitro [passage 3–6; Clontech, Mountain View, CA]. Assays of cellular DNA synthesis and proliferation, EGFR phosphorylation, and MAPK activation were examined in response to CTF in the presence and absence of a general protease inhibitor [1, 10-phenanthrolene], plasmin inhibitors [ε-aminocaproic acid and aprotinin], a matrix metalloproteinase [MMP] inhibitor [GM6001], ADAM inhibitors [TAPI-0 and TAPI-1], HB-EGF inhibitor [CRM197], HB-EGF antibodies and an EGFR inhibitor [AG1478]. Additional experiments were performed with small interfering RNA [siRNA] to EGFR. We further examined intracellular pathways leading to EGFR phosphorylation with specific inhibitors: pertussis toxin [PTx] and GP-2A for Gαi and Gαq, respectively; beta-adrenergic receptor kinase 1 (carboxy terminus) [βARKCT] for Gβγ; PP2 for src; H7 and GF109203X for PKC; BAPTA-AM, a chelator of intracellular calcium; N-acetylcysteine [NAC] as a free radical scavenger; and diphenyleneiodium [DPI] and apocynin as NAD[P]H inhibitors [Table 1].
Table 1.
| Action | Inhibitor |
|---|---|
| Plasmin Activation inhibitors | ε-aminocaproic acid and aprotinin, |
| Matrix Metalloproteinase [MMP] inhibitor | GM6001 |
| ADAM inhibitors | TAPI-0 |
| ADAM inhibitors | TAPI-1 |
| HB-EGF inhibitor | CRM197 |
| HB-EGF inhibitor | HB-EGF antibodies |
| EGFR inhibitor | AG1478 |
| Gαi inhibitor | pertussis toxin [PTx] |
| Gαq inhibitor | GP-2A |
| Gβγ inhibitor | βARKCT |
| src inhibitor | PP2; |
| src inhibitor control | PP3 |
| PKC inhibitor | H7 |
| PKC inhibitor | GF109203X for PKC; |
| Calcium singalling inhibitor | BAPTA-AM |
| Free radical scavenger | N-acetylcysteine [NAC] |
| NAD[P]H inhibitor | diphenyleneiodium [DPI] |
| NAD[P]H inhibitor | apocynin |
Materials
CTF was purchased from American Diagnostica, Inc. [Greenwich, CT]. EGF was purchased from Sigma Chemical Co. [St. Louis, MO]. AG1478 and CRM197 were purchased from Calbiochem [La Jolla, CA]. GM6001 was purchased from Chemicon International, Inc. [Temecula, CA]. CRM197, TAPI-0 and TAPI-1 were purchased from Biomol [Hamburg, Germany]. The anti–HB-EGF antibody was purchased from R&D Systems, Inc. [Minneapolis, MN]. The anti-EGFR antibody [151-IgG] developed by Dr. Ann Hubbard was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa [Department of Biological Sciences, Iowa City, IA]. Pertussis toxin, GP2A, AG1478, PP3, PP2, H7, GF109203X, BAPTA-AM, NAC, apocynin and DPI were purchased from Calbiochem [La Jolla, CA]. Peroxidase-conjugated anti-rabbit IgG antibody [raised in goat] was purchased from Jackson ImmunoResearch Laboratories, Inc. [West Grove, PA]. Peroxidase-conjugated anti-mouse IgG antibody [raised in goat] was purchased from Biorad Laboratories [Hercules, CA]. Phospho- EGFR [Y1068], Total EGFR antibodies were obtained from Cell Signaling Technology, Inc. [Beverley, MA]. Dulbecco’s modified Eagle minimal essential medium [DMEM] and Dulbecco’s phosphate-buffered saline were purchased from Mediatech [Herndon, VA].
[3H] Thymidine Incorporation
VSMC were allowed to reach ~70% confluence as judged by microscopic examination. The cells were then placed in DMEM for 2 days to induce a quiescent state in the cells. The experiments were initiated by adding [3H]-thymidine [1 μCi/mL; ICN, Irvine, CA] with and without pharmacological inhibitors, and stimulating the cells with CTF [10 nM]. Incorporation of [3H]-thymidine into acid-precipitable material was measured after a 24- hour incubation period [air plus 5% CO2 at 37°C]. Stimulation was expressed as fold increa se over starved cells [DMEM only], and the effects of inhibitors were expressed as percentage of the stimulated control.
Cell Proliferation
Culture wells [24-well cluster dishes, Falcon] were inoculated with 3 mL growth medium containing 5×104 VSMC and allowed to attach overnight. Cell proliferation in response to CTF [10 nM] with and without pharmacological inhibitors was determined. Cell counts were determined on days 1, 3, 5, and 7. Cells from each well were removed by trypsinization and the resultant suspension counted using a hemocytometer [Hausser Scientific, Horsham, PA]. Proliferation was expressed as fold increase over unstimulated cells [DMEM only].
Western Blotting
VSMC were allowed to grow to 80% confluence and starved for 48 hours. Cells were then stimulated with CTF or EGF alone and in the presence of pharmacological and peptide inhibitors; cells were harvested at time points from 0 to 30 minutes. Western blotting was performed as previously described (18).
siRNA Transfection
Pre-designed HPLC-purified siRNA for gene knockdown for EGFR or control siRNA was purchased from Santa Cruz Biotechnology [Santa Cruz, CA]. siRNA sequences for ADAMs were as follows: ADAM9, AAUCACUGUGGAGACAUUUGCdTdT and AAACUUCC AGUGUGUAGAUGCdTdT; ADAM10, AAUGAAGAGGGACACUUCCCUdTdT and AAGUUGCCUCCUCCUAAACCAdTdT; and ADAM12 AACCUCGCUGCAAAGAAUGUGdTdT and AAGACCUUGATACGACUGCUGdTdT. VSMC of 50% confluence in 60-mm plates were starved overnight in 4 mL Opti-MEM reduced serum medium [Gibco]. siRNA was transfected using Lipofectamine 2000 from Invitrogen, Inc. [Carlsbad, CA] following the product protocol. Briefly, 22 μL of Lipofectamine 2000 was first incubated in a total volume of 250 μL of Opti-MEM for 5 minutes at room temperature. This was then added to 250 μL of Opti-MEM containing 440 pmoles of siRNA. The solution was mixed gently and incubated for 20 minutes at room temperature, after which it was added to the starved plates. The medium can was changed after 4–6 hours of incubation. The cells were used between 24–72 hours after transfection for EGFR assays. Scrambled siRNA served as a control. Using the methodologies described, we conducted concentration-dependent experiments with siRNA against EGFR and demonstrated a concentration-dependent decrease in protein expression that was specific for the protein targeted without altering the expression of the other proteins.
Adenoviral Infection
Adenoviral vectors were constructed by Welgen, Inc. [Worcester, MA] using purified plasmid encoding βARKCT, obtained from the Guthrie cDNA Resource Center [Missouri University of Science and Technology, Rolla, MO]. VSMC were plated at 70% confluence in 100-mm dishes and allowed to grow overnight. Recombinant adenovirus was then added at the appropriate concentrations [βARKCT; 50 MOI] in a reduced volume of media [1.5 – 2 mL]. After 48-hour incubation, the media was changed and cells grown for an additional 24 hours. The cells were then used for EGFR assays. Empty vector served as control.
Data and Statistical Analysis
All data are presented as the mean ± standard error of the mean [s.e.m.] and statistical differences between groups were tested with a Kruskal-Wallis nonparametric test with post hoc Dunn’s multiple comparison correction, where appropriate. A p-value less than 0.05 was regarded as significant. Non-significant p-values were expressed as p=ns.
RESULTS
Cell proliferation
CTF produces an increase in DNA synthesis and cell proliferation of the VSMC. Responses to CTF were blocked by the EGFR kinase inhibitor, AG1478, in a dose-dependent manner, suggesting that EGFR is involved in these responses [Figure 1A and 1B]. DNA synthesis in response to EGF and HB-EGF were also inhibited by the presence of AG1478 [Figure 1A].
Figure 1.


[A] [3H]-thymidine incorporation of cultured human coronary vascular smooth muscle cells in response to CTF [10 nM], EGF [10 μg/mL] and HB-EGF [10 μg/mL] in the presence and absence of the EGFR inhibitor [AG1478]. Values are the mean±s.e.m. fold increase over control for five experiments [* p<0.05, **p<0.01].
[B] Cell proliferation of cultured human coronary vascular smooth muscle cells over a seven-day period in response to CTF [10 nM] in the presence and absence of the EGFR inhibitor [AG1478]. Values are the mean±s.e.m. fold increase over control for five experiments [**p<0.01 vs DMEM control].
EGFR activation
Given the findings that CTF induced DNA synthesis and cell proliferation in an EGFR-dependent manner, we examined the activation of EGFR by CTF. CTF induced time-dependent EGFR phosphorylation [Figure 2A], which was inhibited by AG1478 in a concentration-dependent manner [Figure 2B]. Activation of EGFR by EGF and HB-EGF was also inhibited by AG1478 [Figure 2C and 2D]. Incubation with siRNA to EGFR also inhibited the response to CTF, EGF and HB-EGF in the cells [Figure 2E and 2F]. Activation of EGFR was confirmed by immunoprecipitation of EGFR and immunoblotting for phosphotyrosine. Removal of uPAR by phosphatidylinositol-specific phospholipase C did not alter the proliferative response [Figure 2G].
Figure 2. CTF and EGFR activation.



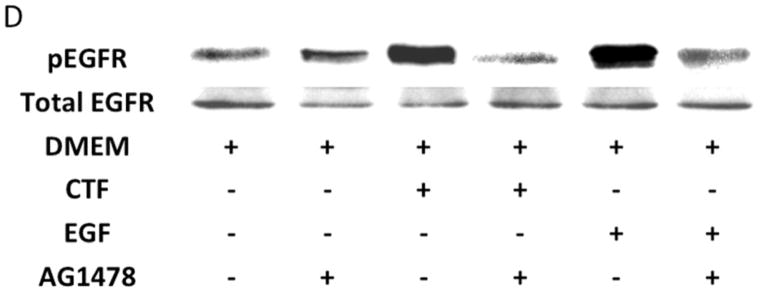



[A] Time-dependent EGFR phosphorylation in response to CTF [10 nM] in cultured human coronary vascular smooth muscle cells. Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments. Representative western blot is shown.
[B] Inhibition of EGFR phosphorylation by AG1478 in response to CTF [10 nM] occurs in a concentration-dependent manner. Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments [**p<0.01].
[C] Inhibition of EGFR phosphorylation in response to CTF [10 nM], EGF [10 μg/mL] and HB-EGF [10 μg/mL] in the presence and absence of the EGFR inhibitor [AG1478]. Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments [*p<0.05; **p<0.01]. Representative western blots are shown in [D].
[E] EGFR phosphorylation in response to CTF [10 nM], EGF [10 μg/mL] and HB-EGF [10 μg/mL] in the presence and absence of the siRNA to EGFR. Scr siRNA, scrambled siRNA control. Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments [*p<0.05; **p<0.01]. Representative western blots for CTF and EGF are shown in [F].
[G] EGFR phosphorylation in response to CTF [10 nM], EGF [10 μg/mL] and HB-EGF [10 μg/mL] in the presence and absence of the uPAR on the cell surface. Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments.
Extracellular Pathway leading to EGFR activation
Protease activity is involved in EGFR transactivation. We examined EGFR phosphorylation in the presence of a general protease inhibitor and inhibitors of plasmin and metalloproteinases. EGFR phosphorylation by CTF was blocked by the general protease inhibitor, 1–10 phenanthrolene, and by inhibitors of plasmin activation, EACA and aprotinin [Figure 3A and 3B]. The metalloproteinase inhibitor, GM6001, also blocked EGFR activation by CTF [Figure 3A and 3B]. These inhibitors have no effect on EGF-mediated EGFR phosphorylation [Figure 3C]. The non specific ADAM inhibitors TAPI-0 and TAPI-1 also reduced EGFR activation in response to CTF but did not interrupt EGF activation of EGFR [Figure 3B, 3C and 3D] (19). As CTF-stimulated phosphorylation of EGFR is inhibited by MMP and ADAM inhibitors, this suggests that EGFR activation by CTF requires MMP cleavage of a tethered ligand such as HB-EGF. Pre-incubation with CRM197, an inhibitor of HB-EGF, was able to inhibit EGFR phosphorylation in response to CTF [Figure 3E and 3F]. We confirmed this finding through the use of HB-EGF-blocking antibodies [Figure 3E and 3F]. We have already demonstrated that EGF is capable of inducing EGFR phosphorylation in the presence of each of these inhibitors (19). Incubation with antibodies against the EGFR receptor prevented both CTF and EGF-mediated EGFR phosphorylation [Figure 3E and 3F]. The protease inhibitors did not affect the activation of EGFR by HB-EGF but those inhibitors that target HB-EGF did inhibit the responses appropriately [Figure 3]. These findings suggest that EGFR phosphorylation is dependent on the protease activity and subsequent MMP-mediated shedding of HB-EGF. A likely target of these intermediate kinases is the ADAM [A Disintegrin And Metalloproteinase Domain] family of MMPs, which are transmembrane molecules. VSMC express ADAM-9, ADAM-10 and ADAM-12 (20). We examined their contribution to EGFR activation by the use of siRNA to ADAM-9, ADAM-10 and ADAM-12. siRNA reduced ADAM-9, -10, -12 proteins levels by >50% (20). Incubation with siRNA against ADAM -10 and ADAM-12 significantly reduced EGFR activation in response to CTF. Scrambled siRNA and siRNA against ADAM-9 had no effect [Figure 3H]. There was no effect on EGF and HB-EGF activation of EGFR [Figure 3H].
Figure 3. CTF and extracellular pathway.








[A] EGFR phosphorylation in response to CTF [10 nM], EGF [10 μg/mL] and HB-EGF [10 μg/mL] in the presence and absence of the protease inhibitor 1,10-phenanthrolene, the plasmin inhibitors [aprotinin and ε-aminocaproic acid, (EACA)] and the MMP inhibitor [GM6001]. Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments [**p<0.01]. Representative western blots are shown in [B] and [C].
[D] EGFR phosphorylation in response to CTF [10 nM], EGF [10 μg/mL] and HB-EGF [10 μg/mL] in the presence and absence of the ADAM inhibitors TAPI-0 and TAPI-1. Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments [*p<0.05; **p<0.01].
[E] EGFR phosphorylation in response to CTF [10 nM], EGF [10 μg/mL] and HB-EGF [10 μg/mL] in the presence and absence of the HB-EGF inhibitor CRM197, HB-EGF blocking antibody and EGFR blocking antibody. Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments [**p<0.01]. Representative western blots are shown in [F] and [G].
[H] Incubation with siRNA to ADAM-10 and -12 significantly reduced EGFR activation in response to CTF. Responses to EGF and HB-EGF were unaffected. Scrambled siRNA and siRNA against ADAM-9 had no effect. We had previously shown that siRNA to ADAM-9, -10 and -12 resulted in a ~50% decrease in their respective A Disintegrin And Metalloproteinase Domains (ADAM) protein expression compared to DMEM control or scrambled siRNA. (20). Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments [*p<0.05; **p<0.01].
Intracellular Pathway leading to EGFR activation
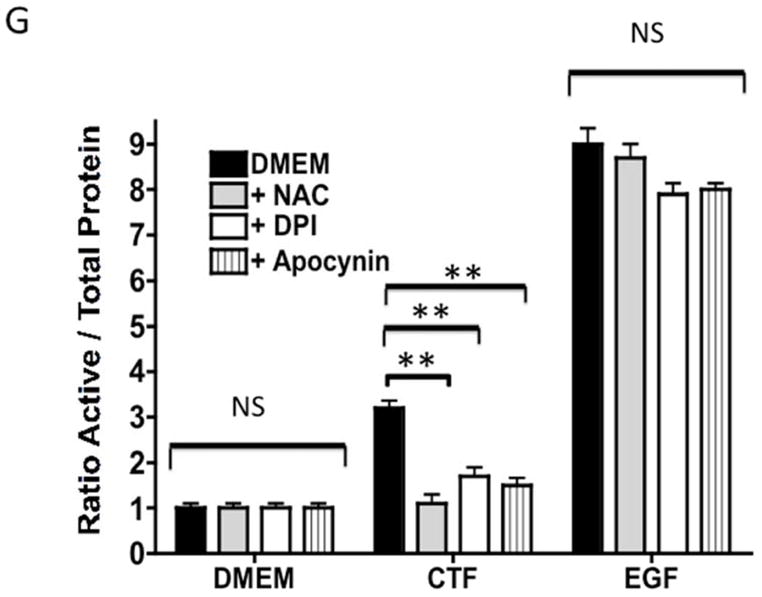
An alternative method for activation of surface MMPs and subsequent release of a tethered ligand may be through G protein-coupled receptor activation. These receptors are coupled to Gαi/ Gαq and Gβγ proteins. Pre-incubation with the Gαi inhibitor, pertussis toxin, resulted in inhibition of EGFR phosphorylation in response to CTF, while the Gαq inhibitor, GP-2A, had no effect [Figure 4A and 4B]. The activation of EGFR by EGF was unaffected by these inhibitors [Figure 4A and 4C]. Pre-incubation with βARKCT, a peptide that functions as a Gβγ protein inhibitor, resulted in a decreased EGFR phosphorylation in response to CTF [Figure 4A and 4B]. Many GPCRs utilize intracellular calcium, src, PKC and oxygen free radical generation to induce HB-EGF release and EGFR phosphorylation. Inhibition of intracellular calcium or src but not PKC blocked EGFR activation in response to CTF [Figure 4D and 4E]. The activation of EGFR by EGF was unaffected by these inhibitors [Figure 4D and 4F]. Inhibition of oxygen free radical production through either the general inhibitor NAC and the more specific inhibitors DPI and apocynin also decreased EGFR activation in response to CTF but not in response to EGF [Figure 4G and 4H]. The proposed pathway defined by these experiments is shown in Figure 5.
Figure 4. CTF and intracellular pathway.







[A] EGFR phosphorylation in response to CTF [10 nM] and EGF [10μg/dL] in the presence and absence of the pertussis toxin [PTx] or Gαq inhibitor [GP-2A]. Preincubation with bARKCT resulted in a decreased EGFR phosphorylation in response to CTF. EV, empty vector. Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments [**p<0.01]. Representative western blots are shown [B] and [C].
[D] Inhibition of intracellular calcium [BAPTA-AM], src [PP2] and PKC [H7 and GFX] blocked EGFR activation in response to CTF [10 nM] and EGF [10 μg/dL]. Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments **p<0.01]. Representative western blots are shown [E] and [F].
[G] Inhibition of oxygen free radical production through either the general inhibitor NAC and the more specific inhibitors DPI and apocynin also decreased EGFR activation in response to CTF [10 nM] and EGF [10 μg/dL]. Values are the mean±s.e.m. ratio of active /total EGFR protein for five experiments [**p<0.01]. Representative western blots are shown in [H].
Figure 5. Representative pathway for CTF transmembrane signaling.
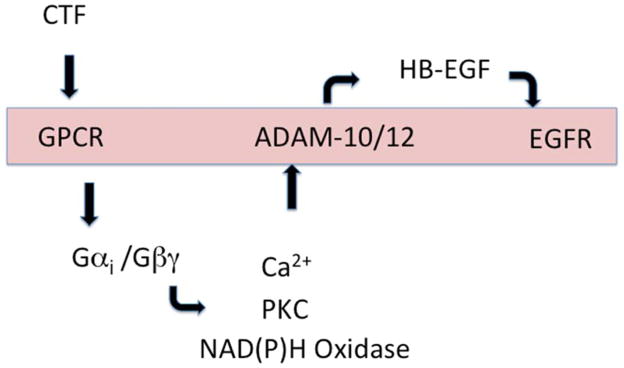
CTF interacts with a G protein-coupled receptor which activates activates both Gαi and Gβγ. In turn, a series of intracellular molecules are involved which lead to activation of A Disintegrin And Metalloproteinase Domains (ADAM) proteases (ADAM-10 and ADAM-12). Activation of ADAM-associated MMP activity leads to release of the tethered ligand, HB-EGF, which in turn activates the EGFR. Activation of EGFR is necessary for CTF-mediated proliferation.
DISCUSSION
We have shown that uPA induces smooth muscle cell proliferation through both ERK1/2 and p38MAPK-mediated pathways (6) and have demonstrated that there is a distinct difference between uPA and plasmin with regard to the signaling pathways utilized to induce cell proliferation (13). Plasmin appears only to use ERK1/2, while uPA uses both ERK1/2 and p38MAPK (13, 19, 21). In this report, we use the protease domain of uPA, CTF, to examine the problem more specifically and demonstrate for the first time that CTF-dependent smooth muscle cell proliferation occurs through transactivation of EGFR by a plasmin-mediated, ADAM-induced, HB-EGF-dependent process, which required activation of the intracellular pathways Gαi/Gβγ, src and NAD[P]H oxidase. Rather than signaling through uPAR, the CTF requires a G protein-coupled receptor to activate this cascade; this may occur through its protease domain directly or through generation of plasmin. Our results suggest these actions may activate G protein-coupled protease activated receptors on the cell membrane (15, 16) that signal through ARK1, since the ARK1 inhibitor βARKCT prevents CTF-stimulated phosphorylation of EGFR. Transmembrane molecules such as ADAM are attractive candidate molecules for the transmembrane portion of the signaling pathway. ADAMs are approximately 70 to 90 kDa [mature proteins; the unprocessed precursors are about 20 kDa larger due to their prodomain]. They feature a common modular ectodomain structure, encompassing a variable stalk region; a cysteine-rich domain; a disintegrin domain binding to integrin-class cell adhesion molecules; and a Zn-binding metalloprotease domain. ADAM-9, ADAM-10, ADAM-12 and ADAM-17 have been reported to be involved in the ectodomain shedding of proHB-EGF (22, 23). We have shown that VSMC express ADAM-9, ADAM-10 and ADAM-12. Furthermore, we have recently identified that during migration the predominant pathways that lead to HB-EGF release are ADAM-9 and ADAM-10 dependent in these particular VSMC (20). This study suggests that in proliferation ADAM-10 and ADAM-12 are involved.
The ErbB-1/EGFR consists of a glycosylated ligand-binding domain, a single transmembrane domain and a cytoplasmic tyrosine kinase domain (24, 25) and is expressed in the injured vessel wall (26–28). HB-EGF is a tethered ligand for EGFR whose cleavage is MMP dependent and can be inhibited with heparin. Our data using CRM197 and antibodies against HB-EGF and EGFR support the fact that an extracellular pathway using HB-EGF is required for CTF-mediated EGFR activation. Activation of HB-EGF requires that it be cleaved from the membrane and also requires cell surface heparan sulfate proteoglycans to act as a co-receptors; HB-EGF binding to EGFR can be antagonized in a dose-dependent manner by heparin (29). We also demonstrated that this pathway was protease dependent and in particular dependent on metalloproteinase activity. We further extended these findings to demonstrate that ADAMs were involved. This sequence of events utilizes classical extracellular pathways of receptor transactivation whereby activation of a given receptor activates a heterologous receptor (17). In this model, there is a sequence of three transmembrane signaling events: G protein-coupled receptor activation, followed by ADAM-dependent MMP activation, and then cleavage of and subsequent binding of HB-EGF to the EGFR receptor. ADAM-10, ADAM-12 and ADAM-17 [TNF-α converting enzyme] have been shown to cleave HB-EGF (30). Activation of the ADAM pathway has been shown to involve one or more of the following signaling molecules: src, intracellular calcium, and/or protein kinase C (17). An alternative mechanism may be inactivation of protein tyrosine phosphatases due to the generation of oxygen free radicals by the NAD[P]H oxidase complex.
As with all cell signaling experiments, one must realize that the in vitro analysis does not completely represent the in vivo circumstance due to the fact that the in vivo VSMC is encased in a changing extracellular matrix and is under constant fluctuating hemodynamic and inflammatory forces. When we examine the pathways leading EGFR activation, one must accept that there are redundant pathways (extracellular and intracellular) that may mediate the effects of SMC proliferation and that these could be stimulated by other growth factors/upstream pathways.
Conclusion
In human coronary VSMC, urokinase induces uPAR-independent, domain-dependent smooth muscle cell proliferation through transactivation of EGFR by a plasmin-mediated, ADAM-induced HB-EGF-dependent process, which is mediated by the intracellular pathways involving Gαi, Gβγ, src and NAD[P]H oxidase.
Acknowledgments
The authors thank Daynene Vykoukal, Ph.D. for critical reading of the manuscript.
Footnotes
Conflict of Interest: Enrico A. Duru: None
Yuyang Fu: None
Mark G. Davies: None
Presented in part at 6th Academic Surgical Congress, Huntington Beach CA, Feb 2011.
CONTRIBUTIONS:
Enrico A. Duru: concept and design, performance of the work, analysis and/or interpretation of data; critical writing or revising the intellectual content; and final approval of the version to be published
Yuyang Fu: concept and design, performance of the work, analysis and/or interpretation of data; critical writing or revising the intellectual content; and final approval of the version to be published
Mark G. Davies: concept and design, analysis and/or interpretation of data; critical writing or revising the intellectual content; and final approval of the version to be published
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davies MG, Hagen P-O. Pathobiology of Intimal Hyperplasia. Br J Surg. 1994;81:1254–69. doi: 10.1002/bjs.1800810904. [DOI] [PubMed] [Google Scholar]
- 2.Strauss BH, Lau HK, Bowman KA, Sparkes J, Chisholm RJ, Garvey MB, et al. Plasma urokinase antigen and PAI-1 antigen levels predict angiographic coronary restenosis. Circulation. 1999;100(15):1616–22. doi: 10.1161/01.cir.100.15.1616. [DOI] [PubMed] [Google Scholar]
- 3.Clowes AW, Reidy MA, Clowes MM, Belin D. Smooth muscle cells express urokinase during mitogenesis and tissue-type plasminogen activator during migration in injured rat carotid. Circ Res. 1990;67:61–7. doi: 10.1161/01.res.67.1.61. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P, Moons L, Herbert J-M, Crawley J, Lupu F, Lijnen R, et al. Urokinase but not tissue plasminogen activator mediates arterial neointima formation in mice. Circ Res. 1997;81:829–39. doi: 10.1161/01.res.81.5.829. [DOI] [PubMed] [Google Scholar]
- 5.Lijnen HR, VanHoef B, Lupu F, Moon L, Carmeliet P, Collen D. Function of the plasminogen/plasmin and MMP systems after vascular injury in mice with targeted inactivation of fibrinolytic genes. Arterioscler Thromb Vasc Biol. 1998;18(7):1035–45. doi: 10.1161/01.atv.18.7.1035. [DOI] [PubMed] [Google Scholar]
- 6.Tanski WJ, Fegley AJ, Roztocil E, Davies MG. Domain dependent actions of urokinase on smooth muscle cell responses. J Vasc Surg. 2004;39:214–22. doi: 10.1016/s0741-5214(03)01031-0. [DOI] [PubMed] [Google Scholar]
- 7.Menshikov M, Plekhanova O, Cai H, Chalupsky K, Parfyonova Y, Bashtrikov P, et al. Urokinase plasminogen activator stimulates vascular smooth muscle cell proliferation via redox-dependent pathways. Arterioscler Thromb Vasc Biol. 2006;26(4):801–7. doi: 10.1161/01.ATV.0000207277.27432.15. [DOI] [PubMed] [Google Scholar]
- 8.Plekhanova OS, Parfenova EV, Bibilashvily RS, Stepanova VV, Erne P, Bobik A, et al. Urokinase plasminogen activator enhances neointima growth and reduces lumen size in carotid arteries. J Hypertens. 2000;18(8):10065–9. doi: 10.1097/00004872-200018080-00011. [DOI] [PubMed] [Google Scholar]
- 9.Stepanova V, Bobik A, Bibilashvity R, Belogurov A, Rybalkin I, Domogatsky S, et al. Urokinase plasminogen activator induces smooth muscle cell migration: key role of growth factor-like domain. FEBS Lett. 1997;414:471–4. doi: 10.1016/s0014-5793(97)00993-9. [DOI] [PubMed] [Google Scholar]
- 10.Stepanova V, Mukhina S, Kohler E, Resink TJ, Erne P, Tkachuk VA. Urokinase plasminogen activator induces human smooth muscle cell migration and proliferation via distinct receptor dependent and proteolysis-dependent mechanisms. Mol Cell Biochem. 1999;195:199–206. doi: 10.1023/a:1006936623106. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen DH, Hussaini IM, Gonias SL. Binding of uPA to its receptor in MCF-7 cells activates ERK1 and 2 which is required for increased motility. J Biol Chem. 1998;273:8502–607. doi: 10.1074/jbc.273.14.8502. [DOI] [PubMed] [Google Scholar]
- 12.Schafer K, Konstantinides S, Riedel C, Thinnes T, Muller K, Dellas C, et al. Different mechanisms of increased luminal stenosis after arterial injury in mice deficient for urokinase or tissue-type plasminogen activator. Circulation. 2002;106:1847. doi: 10.1161/01.cir.0000031162.80988.2b. [DOI] [PubMed] [Google Scholar]
- 13.Nicholl SM, Roztocil E, Davies MG. Urokinase (uPA) induced smooth muscle cell responses require distinct signaling pathways: a role for the epidermal growth factor receptor. J Vasc Surg. 2005;41:672–81. doi: 10.1016/j.jvs.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 14.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–8. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 15.Mao Y, Jin J, Daniel JL, Kunapuli SP. Regulation of plasmin-induced protease-activated receptor 4 activation in platelets. Platelets. 2009;20(3):191–8. doi: 10.1080/09537100902803635. [DOI] [PubMed] [Google Scholar]
- 16.Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE. Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry. 1999;39(14):4572–85. doi: 10.1021/bi9824792. [DOI] [PubMed] [Google Scholar]
- 17.Wetzker R, Bohmer FD. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol. 2003;4(8):651–7. doi: 10.1038/nrm1173. [DOI] [PubMed] [Google Scholar]
- 18.Tanski WJ, Nicholl SM, Kim D, Fegley AJ, Roztocil E, Davies MG. Sphingosine-1-Phosphate-induced smooth muscle cell migration involves the Mammalian Target of Rapamycin. J Vasc Surg. 2005;39:91–8. doi: 10.1016/j.jvs.2004.08.058. [DOI] [PubMed] [Google Scholar]
- 19.Nicholl SM, Roztocil E, Galaria II, Davies MG. Plasmin induced smooth muscle cell proliferation requires epidermal growth factor activation. Surgery. 2005;138(2):180–6. doi: 10.1016/j.surg.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 20.Bakken AM, Protack CD, Roztocil E, Nicholl SM, Davies MG. Cell migration in response to the Amintoterminal Fragment of Urokinase requires epidermal growth factor receptor activation through an ADAM-mediated mechanism. J Vasc Surg. 2009;49(5):1296–303. doi: 10.1016/j.jvs.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicholl SM, Roztocil E, Galaria II, Davies MG. Plasmin induces smooth muscle cell proliferation through G[alpha]i and G[alpha]q mediated pathways. J Surg Res. 2005;127:39–45. doi: 10.1016/j.jss.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Nanba D, Higashiyama S. Dual intracellular signaling by proteolytic cleavage of membrane-anchored heparin-binding EGF-like growth factor. Cytokine Growth Factor Rev. 2004;15:13–9. doi: 10.1016/j.cytogfr.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, et al. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat Med. 2002;8:35–40. doi: 10.1038/nm0102-35. [DOI] [PubMed] [Google Scholar]
- 24.Barnes CJ, Kumar R. Biology of the epidermal growth factor receptor family. Cancer Treat Res. 2004;119:1–13. doi: 10.1007/1-4020-7847-1_1. [DOI] [PubMed] [Google Scholar]
- 25.Carpenter G. Nuclear localization and possible functions of receptor tyrosine kinases. Curr Opin Cell Biol. 2003;15(2):143–8. doi: 10.1016/s0955-0674(03)00015-2. [DOI] [PubMed] [Google Scholar]
- 26.Pastore CJ, Isner JM, Bacha PA, Kearney M, Pickering JG. Epidermal growth factor receptor-targeted cytotoxin inhibits neointimal hyperplasia in vivo: results of local versus systemic administration. Circ Res. 1995;77:519–29. doi: 10.1161/01.res.77.3.519. [DOI] [PubMed] [Google Scholar]
- 27.Trieu VN, Narla RK, Myers DE, Uckun FM. EGF-genistein inhibits neointimal hyperplasia after vascular injury in an experimental restenosis model. J Cardiovasc Pharmacol. 2000;35:595–605. doi: 10.1097/00005344-200004000-00013. [DOI] [PubMed] [Google Scholar]
- 28.Chan AK, Kalmes A, Hawkins S, Daum G, Clowes AW. Blockade of the epidermal growth factor receptor decreases intimal hyperplasia in balloon-injured rat carotid artery. J Vasc Surg. 2003;37(3):644–9. doi: 10.1067/mva.2003.92. [DOI] [PubMed] [Google Scholar]
- 29.Bresner GE, Whelton D, Crissman-Combs MA, Steffen CL, Kim GY, Brigstock DR. Interaction of heparin-binding EGF-like growth factor (HB-EGF) with the epidermal growth factor receptor: modulation by heparin, heparinase or synthetic heparin-binding HB-EGF fragments. Growth Factors. 1992;7:289–96. doi: 10.3109/08977199209046411. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki M, Raab G, Moses MA, Fernandez CA, Klagsbrun M. Matrix metalloproteinase-3 releases active heparin-binding EGF-like growth factor by cleavage at a specific juxtamembrane site. J Biol Chem. 1997;(272):31730–7. doi: 10.1074/jbc.272.50.31730. [DOI] [PubMed] [Google Scholar]
- 31.Plekhanova OS, Men’shikov MY, Bashtrykov PP, Berk BC, Tkachuk VA, Parfenova EV. Urokinase induces ROS production in vascular smooth muscle cells. Bull Exp Biol Med. 2006;142(3):304–7. doi: 10.1007/s10517-006-0352-4. [DOI] [PubMed] [Google Scholar]