

Abstract
The QseBC two-component system plays a pivotal role in regulating virulence and biofilm growth of the oral pathogen Aggregatibacter actinomycetemcomitans. We previously showed that QseBC autoregulates the ygiW–qseBC operon. In this study, we characterized the promoter that drives ygiW–qseBC expression. Using lacZ transcriptional fusion constructs and 5′-rapid amplification of cDNA ends, we showed that ygiW–qseBC expression is driven by a promoter that initiates transcription 53 bases upstream of ygiW and identified putative cis-acting promoter elements, whose function was confirmed using site-specific mutagenesis. Using electrophoretic mobility shift assays, two trans-acting proteins were shown to interact with the ygiW–qseBC promoter. The QseB response regulator bound to probes containing the direct repeat sequence CTTAA-N6-CTTAA, where the CTTAA repeats flank the −35 element of the promoter. The ygiW–qseBC expression could not be detected in A. actinomycetemcomitans ΔqseB or ΔqseBC strains, but was restored to WT levels in the ΔqseBC mutant when complemented by single copy chromosomal insertion of qseBC. Interestingly, qseB partially complemented the ΔqseBC strain, suggesting that QseB could be activated in the absence of QseC. QseB activation required its phosphorylation since complementation did not occur using qseBpho−, encoding a protein with the active site aspartate substituted with alanine. These results suggest that QseB is a strong positive regulator of ygiW–qseBC expression. In addition, integration host factor (IHF) bound to two sites in the promoter region and an additional site near the 5′ end of the ygiW ORF. The expression of ygiW–qseBC was increased by twofold in ΔihfA and ΔihfB strains of A. actinomycetemcomitans, suggesting that IHF is a negative regulator of the ygiW–qseBC operon.
Introduction
Periodontitis is a disease produced by a dental biofilm in association with anaerobic pathogenic bacteria. This severe infection causes a chronic inflammation of the gingival tissues, which leads to progressive loss of the alveolar bone that supports the teeth. Untreated periodontitis will eventually result in tooth loss. Aggregatibacter actinomycetemcomitans is a Gram-negative, facultative anaerobe that is associated with aggressive forms of periodontitis and other human infections including endocarditis and systemic infections (Haffajee & Socransky, 1994; Paturel et al., 2004; Wang et al., 2010; Murakami et al., 2013; Brady et al., 2014). A. actinomycetemcomitans expresses a variety of virulence factors that mediate colonization of oral tissues and biofilm formation, such as the autotransporter adhesins Aae and Api, outer-membrane protein Omp29, and the tad fimbriae (Fine et al., 2010; Yue et al., 2007; Kajiya et al., 2011; Saito et al., 2010). In addition, expression of the leukotoxin (LtxA) and cytolethal distending toxin (CdtABC) contribute to inflammation, tissue destruction and inactivation of the host immune response (Dietmann et al., 2013; Fernandes et al., 2008; Jinadasa et al., 2011). A. actinomycetemcomitans biofilm formation, iron acquisition and virulence are also regulated by a quorum-sensing system that responds to autoinducer AI-2 (Demuth et al., 2011; Fong et al., 2001, 2003). The QseBC two-component system is part of this quorum-sensing system and is regulated by AI-2 (Novak et al., 2010). In A. actinomycetemcomitans, the qseBC genes are co-expressed with ygiW, which codes for a polypeptide of unknown function in the OB-fold family of proteins and the operon is transcribed from a promoter that resides upstream of ygiW (Juárez-Rodríguez et al., 2013a).
The QseBC two-component system is present in several Gram-negative pathogens, and in Escherichia coli; it is a key component for intercellular and interkingdom communication by sensing both autoinducer-3 (AI-3) and host catecholamine hormones to regulate virulence gene expression (Clarke et al., 2006). Activation of QseC initiates the transcription of flhDC genes that encode the master regulator for the flagella and motility genes (Sperandio et al., 2002; Clarke & Sperandio, 2005), and genes encoding the FucRS two-component system for sensing of fucose in the host intestine (Pacheco et al., 2012). Furthermore, QseC can activate non-cognate response regulators KdpE and QseF that are involved in the formation of the attaching and effacing lesions by enterohaemorrhagic E. coli on epithelial cells (Hughes et al., 2009). QseBC also has an important role in the pathogenesis of uropathogenic E. coli (UPEC) (Kostakioti et al., 2009). In UPEC, QseC has been reported to function as both a kinase and phosphatase activity that modulates QseB phosphorylation. This is important since QseB is also phosphorylated by the PmrB histidine kinase (Kostakioti et al., 2009; Guckes et al., 2013). Deletion of qseC in UPEC causes an aberrant upregulation and overactivation of QseB by PmrB, since QseB cannot be dephosphorylated in the mutant. This leads to pleiotropic effects, such as the misregulation of nucleotide, amino acid and carbon metabolism, suggesting QseC controls the bacterial central metabolic circuit (Hadjifrangiskou et al., 2011).
In Salmonella, the QseC and QseE sensors are part of the AI-3/adrenaline/noradrenaline signalling system, which influences Salmonella serovar Typhimurium pathogenesis (Moreira et al., 2010) by regulating pathogenicity island 1 (SPI-1) genes responsible for the invasion of epithelial cells (Moreira & Sperandio, 2012). QseC has also been implicated in the regulation of flagellar genes in Salmonella (Bearson & Bearson 2008; Moreira et al., 2010), but this was not observed by Merighi et al. (2009). This discrepancy has been suggested to arise from the labile nature of the adrenaline/noradrenaline signals in vitro (Moreira et al., 2010; Moreira & Sperandio, 2012).
The QseBC system has also been found in other pathogens such as Edwardsiella tarda (Wang et al., 2011), Aeromonas hydrophila (Khajanchi et al., 2012) and Haemophilus influenzae (Steele et al., 2012), and it plays an important role in the pathogenic processes of these bacteria. However, the H. influenzae QseBC system is induced by ferrous iron, but differs from E. coli since it is unresponsive to adrenaline or noradrenaline (Steele et al., 2012). A. actinomycetemcomitans is a non-motile organism and lacks many of the genes that are regulated by QseBC in E. coli and Salmonella. Yet A. actinomycetemcomitans QseBC clearly plays a role in virulence, since deletion of qseC dramatically reduces biofilm growth and leads to a virulence attenuated phenotype (Novak et al., 2010; Juárez-Rodríguez et al., 2013a). This suggests that the function of QseBC in A. actinomycetemcomitans may differ from that in E. coli and other enteric organisms. The QseBC regulon in A. actinomycetemcomitans is currently unknown.
In addition, the expression of several genes encoding major virulence factors of A. actinomycetemcomitans, e.g. the leukotoxin, is also controlled by integration host factor (IHF) (Kolodrubetz et al., 2010). IHF is a DNA-binding and -bending protein that recognizes the asymmetrical consensus sequence YAANNNNTTGATW, where Y = T or C, N = any base and W = A or T (Craig & Nash, 1984; Friedman, 1988). IHF concentration increases throughout growth and becomes the second most abundant nucleoid protein during the stationary phase (Azam et al., 1999; Ishihama, 1999). Functionally, it plays many roles and is involved in the organization of the chromosomal DNA, DNA replication, DNA recombination and transcriptional regulation in Gram-negative bacteria. IHF is also implicated in the transcriptional regulation of genetic loci associated with virulence in many pathogenic bacteria (Mangan et al., 2006; Blomfield et al., 1997).
In this paper, we have characterized the promoter that drives the expression of qseBC in A. actinomycetemcomitans, and show that it is regulated by both QseB and IHF. The QseB response regulator is required for transcription of the operon and it is primarily activated by QseC sensor kinase. However, QseB may also be partially activated by other mechanism(s) in the absence of its cognate sensor kinase. QseB binds to a direct repeat sequence in the ygiW–qseBC promoter that resembles the PmrA DNA-binding site in E. coli and Salmonella. Three IHF binding sites were identified, two of them located in the ygiW–qseBC promoter region and the third motif in the ygiW ORF. In contrast to QseB, IHF binding negatively regulates the ygiW–qseBC operon.
Methods
Bacterial strains, plasmids and media.
The bacterial strains and plasmids used in this study are listed in Table S1 (available in the online Supplementary Material). Luria–Bertani (LB) broth, LB agar [LB broth plus 1.5 % (w/v) agar], TYE broth [1 % (w/v) tryptone, 0.5 % (w/v) yeast extract (DIFCO)], brain heart infusion (BHI) broth, and BHI agar (all from DIFCO) were routinely used for the propagation and plating of bacteria. A. actinomycetemcomitans (afimbriated, smooth-colony-morphotype strain 652) was grown at 37 °C under microaerophilic conditions in a candle jar. When required, the medium was supplemented with 25 µg kanamycin (Km) ml−1, 12.5 µg tetracycline (Tc) ml−1, 50 µg spectinomycin ml−1 or 100 µg ampicillin ml−1.
DNA procedures.
DNA manipulations were carried out as described elsewhere (Sambrook & Russell, 2001). Transformation of E. coli and A. actinomycetemcomitans was carried out by electroporation (Bio-Rad). Transformants containing plasmids were selected on LB agar plates supplemented with the appropriate antibiotics. Plasmid DNA was isolated using the QIAprep Spin miniprep kit (Qiagen). Restriction enzymes were used as recommended by the manufacturer (New England Biolabs). All primers used in this study (Integrated DNA Technology) are shown in Table S2 and restriction enzyme sites are underlined in the primer sequences. Primer sequences were designed based on the genome information of A. actinomycetemcomitans D11S-1 strain available from the PathoSystems Resource Integration Center (www.patricbrc.org). All constructs were verified by DNA sequencing (University of Louisville Core Sequencing Facilities).
Identification of ygiW–qseBC transcriptional start sites.
The transcriptional start sites of ygiW–qseBC were determined using a GeneRacer kit (Invitrogen). A. actinomycetemcomitans 652 was grown in BHI broth to exponential phase (OD600 0.3). This culture was treated with Qiagen RNAprotect bacterial reagent and total RNA was extracted using RNeasy Lipid Tissue Mini kit (Qiagen) as specified by the manufacturer. RNA was treated with RNase-free DNase I (New England Biolabs) to eliminate contaminating DNA. RNA was quantified by spectrophotometry using a Nanodrop ND-1000 (Thermo Fisher Scientific). A 44 base 5′ RNA adaptor oligonucleotide was ligated to the 5′ ends of the total RNA (5 µg) using T4 RNA ligase. Reverse transcription was performed with Avian Myeloblastosis Virus Reverse Transcriptase (AMV RT) and random hexamers that were included in the GeneRacer kit. PCR was performed using the GeneRacer 5′ primer and ygiW-specific primers MDJR-57R and MDJR-94R. The PCR products were subsequently cloned into a pCR4-TOPO vector and used to transform E. coli Top10 (Invitrogen). Twenty independent positive clones were sequenced and analysed for the ygiW–qseBC promoter(s).
Quantitative real-time PCR (qRT-PCR).
For qRT-PCR, total RNA was obtained as described above, and DNA was synthetized by using the High Capacity cDNA reverse transcriptase kit (Applied Biosystems by Life Technologies) according to the manufacturer’s instructions. Real-time PCR amplification of the cDNA was performed with SYBR Selected master mix (Applied Biosystems by Life Technologies), as specified by manufacturer, and primer pairs (Table S2) in a total volume of 20 µl. Reactions were performed in triplicate for each primer pair in an Applied Biosystems 7500 real-time PCR system, using the default thermal cycle conditions as follows: 1 cycle of 50 °C for 2 min for uracil-DNA glycosylase activation; then 1 cycle of 95 °C for 10 min for hot-start AmpliTaq Gold polymerase activation; followed by 40 cycles of denaturation at 95 °C for 15 s, and annealing and extension at 60 °C for 60 s; and including at the end a dissociation stage of 1 cycle of 95 °C for 15 s, 60 °C for 1 min and 95 °C for 15 s. Relative expression levels of the specific transcripts were calculated using the 16S rRNA expression level as the internal reference for normalization, and expressed as fold-differences by calculating 2−ΔΔCt (Winer et al., 1999). Differences in the expression levels of the target genes between the WT strain and deletion mutant strains were analysed by using ANOVA followed by Tukey’s post test.
Construction of ygiW–qseBC promoter/lacZ fusion plasmids.
Various fragments containing portions of or the entire 372 bp promoter region of ygiW–qseBC were amplified by PCR using A. actinomycetemcomitans genomic DNA as a template. The typical amplification profile used was 94 °C for 2 min for 1 cycle, and then 94 °C for 20 s, 60 °C for 30 s and 72 °C for 2 min for 25 cycles. For the ygiW–qseBC promoter fusion constructs (pDJR55, pDJR56, pDJR57, pDJR63 and pDJR58), portions of the 372 bp regulatory region were amplified using the primer sets MDJR-142F/MDJR-70R, MDJR-143F/MDJR-70R, MDJR-154F/MDJR-70R, MDJR-157F/MDJR-70R and MDJR-176F/MDJR-70R, respectively (see Table S2). The 338, 172, 157, 119 and 128 bp PCR products were then digested with KpnI–BamHI and cloned independently into KpnI–BamHI-digested pJT3 (Juárez-Rodríguez et al., 2013b) to create pDJR55, pDJR56, pDJR57, pDJR58 and pDJR63, respectively.
Site-directed mutagenesis.
Mutagenesis of the putative −10 element (TATAAG to GCCGCG) and −35 element (CAACAAA to GCAGAA) of ygiW–qseBC-P1 was carried out by annealing primers MDJR-158F/MDJR-159R and cloning the resulting double-stranded fragment into KpnI–BamHI-digested pJT3 to generate pDJR60. For mutation in ygiW–qseBC-P2, primer pair MDJR-274F/MDJR-70R was used to introduce mutations into the QseB binding site (CTTAATACTTTCTTAAC to CGCGATACTTTCTTAAC) by PCR. The resulting product was then digested with KpnI–BamHI and cloned into pJT3 to generate pDJR86. Mutations in the ygiW–qseBC-P2 −10 element (TAATAG to GCGCGG) were introduced using primer pair MDJR-175F/MDJR-70, and after verifying the correct base changes by DNA sequencing, the resulting amplicon was used as a template with primer pair MDJR-273F/MDJR-70. The resulting product was digested with KpnI–BamHI and cloned into pJT3 to create pDJR85. Similarly, primer pair MDJR-178/MDJR-70 was used to introduce mutations in both the −10 element and the QseB binding site of ygiW–qseBC-P2 by PCR to produce pDJR76. All mutations were confirmed by DNA sequencing.
The QuikChange site-directed mutagenesis kit (Stratagene) was used to alter the codon (GAT to GCG) encoding the conserved active site aspartate residue in QseB (D51) to an alanine residue using primers MDJR-99F/MDJR-100R. The mutations introduced into qseB were verified by sequencing in the resulting plasmid pDJR28-M1.
Single copy chromosomal complementation of A. actinomycetemcomitans ΔqseBC.
Integration of genes in single copy into the A. actinomycetemcomitans ΔqseBC genome was carried out as previously described (Torres-Escobar et al., 2014). To integrate a single copy of qseB, the qseB mutant allele (qseBpho−) encoding the QseBD51A mutant protein, qseBC or qseBpho−qseC independently into the chromosome of A. actinomycetemcomitans ΔqseBC strain by homologous recombination, the corresponding qseB or qseBC fragments together with ygiW were amplified by PCR from pDJR28 and pDJR28-M1, respectively, with the primer sets MDJR-124F/MDJR-125R (for qseBC constructs) or MDJR124F/MDJR/126R (for qseB constructs). The 3135 and 1645 bp PCR fragments were digested with NotI–PstI and cloned into NotI–PstI-digested suicide vector pJT1 to generate the plasmids pDJR45, pDJR46, pDJR47 and pDJR48. Each recombinant suicide plasmid (20 µg) was introduced individually into A. actinomycetemcomitans isogenic ΔqseBC mutant by electroporation. Ten Spr colonies were selected to verify by PCR the single-copy chromosomal insertion of qseB or qseBC, or qseBpho− or qseBpho−qseC. Verification was carried out using primer sets MDJR-63F/MDJR-61R and MDJR-54F/MDJR-77R. The Spr colonies of A. actinomycetemcomitans ΔqseBC : : qseBpho−qseC, ΔqseBC : : qseBC, ΔqseBC : : qseBpho− and ΔqseBC : : qseB that were PCR positive were selected for electroporation with the pDJR29 promoter/lacZ fusion plasmid.
Growth kinetics.
A single colony of A. actinomycetemcomitans harbouring each recombinant plasmid was independently inoculated into 10 ml BHI broth supplemented with 25 µg Km ml−1 and was grown standing for 24 h at 37 °C. The next day, the overnight culture (OD600 0.6) was diluted at a 1 : 30 ratio and used to inoculate 10 ml BHI broth with 25 µg Km ml−1 and grown standing at 37 °C. For the first 12 h of growth, an aliquot was removed each hour and culture density was determined by measuring the OD600. Additional aliquots were taken from each culture for analysis at different time points. β-Galactosidase (β-Gal) activity was also determined for each aliquot as described below.
β-Gal assays.
β-Gal activity was qualitatively assessed on LB agar plates that were supplemented with 50 µg X-Gal ml−1. Quantitative evaluation of β-Gal activity was carried out using permeabilized cells incubated with ONPG substrate (Sigma) as described by Miller (1972). Mean value (±sds) for activity units were routinely calculated from three independent assays.
Construction of qseB expression plasmid.
The structural qseB gene was amplified from A. actinomycetemcomitans genomic DNA using primer sets MDJR-80F and MDJR-81R, and the resulting 710 bp fragment was digested with NcoI–ApaI and cloned into NcoI–ApaI-digested pYA3883 (Torres-Escobar et al., 2010) expression vector to create pDJR36.
Expression and purification of QseB protein.
E. coli LMG194 harbouring pDJR36 was used for the synthesis of the hexahistidine fusion QseB protein. The expression and detection of the recombinant protein was performed essentially as described previously (Torres-Escobar et al., 2010). Purification was carried out by cobalt-based immobilized metal affinity chromatography under denaturing conditions. Briefly, cells from a 200 ml culture were harvested and frozen at −70 °C overnight. The cell pellet was thawed and suspended in lysis buffer (6M guanidine hydrochloride, 100 mM NaH2PO4, 10 mM Tris/HCl, pH 8), and lysed by homogenization. Lysed cells were centrifuged and the lysate was loaded onto HisPur cobalt resin (Thermo Scientific) for 1 h with shaking at 4 °C. The resin was washed with lysis buffer and sequentially washed with wash buffer containing 8 M urea, 100 mM NaH2PO4, 10 mM Tris/HCl pH 8, 6.3 and 5.9. Protein was then eluted with elution buffer (8M urea, 100 mM NaH2PO4, 10 mM Tris/HCl, pH 4.5). Eluted fractions containing the purified QseB were selected based on SDS-PAGE analysis. The QseB selected pools were dialysed in a Slyde-A-Lyser 3K cassette (Pierce) at 4 °C against refolding buffer containing 50 mM Tris/HCl (pH 8.5), 4 M urea, 0.5 M l-arginine, 264 mM NaCl, 11 mM KCl, 8 mM MgCl2, 0.1 % (v/v) Triton X-100, 20 % (v/v) glycerol for 24 h. Subsequently, samples were dialysed for 24 h against the buffer described above but containing 2 M urea, and then sequentially dialysed for 12 h each in buffer consisting of 50 mM Tris/HCl (pH 8.5), 100 mM NaCl, 0.1 mM KCl, 4 mM MgCl2, 2 mM Mg(CH3COO)2, 0.1 mM EDTA, 0.1 mM DTT, 30 % (v/v) glycerol, and 2, 1 or 0.5 M urea. A final dialysis was then carried out for 6 h against the buffer described above without urea. Aliquots of the QseB purified protein were stored at −70 °C. Protein concentration was determined by the Bradford assay, using BSA as a standard.
Electrophoretic mobility shift assay (EMSA).
The DNA fragments used for nonradioactive EMSA were obtained by PCR using the sets of primers described in Table S2. A biotin 3′ end labelling kit (Thermo Scientific) was used for labelling of DNA fragments according to the manufacturer's instructions. Binding reactions were performed with a total of 50 fmol each probe mixed with 4 µM or various amounts (2, 4, 6 and 8 µM) QseB protein, or 4 µM IHF protein purified previously (Torres-Escobar et al., 2014), in 20 µl binding buffer consisting of 10 mM Tris/HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 1 mM DTT, 1 µg poly (dI–dC) and 100 µg BSA ml−1. The reactions were incubated for 20 min at room temperature. Afterward, 5 µl gel loading buffer [0.25× Tris/borate-EDTA (TBE), 60 % (v/v); glycerol, 40 % (v/v); bromophenol, 0.2 % (w/v)] was added and mixtures were electrophoresed in a 6 % native polyacrylamide gel in 0.5× TBE buffer (45 mM Tris/borate, 1 mM EDTA, pH 8.0) and immunoblotted. DNA bands were detected using the LightShift chemiluminescent EMSA kit according to the manufacturer's instructions. For competitive EMSA, the specific competitor (unlabelled fragment III) was added in increasing amounts (0.5-, 5-, 10- and 20-fold molar excesses) to the EMSA reaction containing the biotin-labelled fragment III and 4 µM QseB as specified above.
Results
Identification of the ygiW–qseBC operon promoter
Our previous work showed that qseBC is co-expressed with the upstream ygiW gene in A. actinomycetemcomitans and that the promoter that drives expression of the operon resides in the 372 bp fdhE–ygiW intergenic region (Juárez-Rodríguez et al., 2013a). To localize the putative ygiW–qseBC promoter, DNA fragments encompassing portions of the intergenic region were introduced into a promoterless lacZ reporter plasmid pJT3 (see Fig. 1a) and β-Gal activity was determined. As shown in Fig. 1(c), pDJR55 and pDJR56, in which nucleotides −307 to −372 and −139 to −372 were deleted, respectively, exhibited similar β-Gal activity as pDJR29, indicating that no regulatory elements exist upstream from nucleotide −138. However, β-Gal activity was reduced by approximately 3-fold in pDJR63 when nucleotides −94 to −372 were deleted and a 78-fold reduction in activity occurred upon deletion of nucleotides −85 to −372. This suggests that regulatory elements responsible for ygiW–qseBC expression reside between −84 and −138.
Fig. 1.

Schematic diagrams of ygiW transcriptional fusion constructs showing the putative binding regions for QseB (white boxes), ygiW–qseBC-P1 promoter (grey boxes), ygiW–qseBC-P2 promoter (black boxes), QseB protein (white triangles) and transcriptional start sites (bent arrows). (a) Fragments derived from the 372 bp fdhE–ygiW intergenic region are represented by lines, and coding sequences are shown by large arrows. (b) ygiW transcriptional fusions derived from the orf–ygiW intergenic region. The putative promoter elements and QseB binding sites that were altered by site-specific mutagenesis are indicated with an X symbol. In both (a) and (b) the numbering of the nucleotides is relative to the ygiW start site and the lacZ gene is indicated by a solid black or grey arrow. (c) Graph depicting β-Gal activity in bacterial cells harbouring the transcriptional fusion constructs. β-Gal activity for each construct is expressed in Miller units and was measured in A. actinomycetemcomitans 652 strain transformed individually with each plasmid and grown in BHI medium as described in Methods. Measurements were made at the mid-exponential phase of growth, and values are means of results from three independent experiments±sds. Statistical analysis was performed using a two-tailed unpaired t-test. ns, Not significant; **, P<0.01; ****, P<0.0001.
Previously, a transcriptional start site (TSS1) was mapped to nucleotide −15 upstream of the ygiW start codon and putative −10 and −35 elements were identified at nucleotides −26 to −31 and −48 to −54, respectively (Juárez-Rodríguez et al., 2013a). For this study, this putative promoter was designated ygiW–qseBC-P1. However, the results in Fig. 1 clearly show that an additional promoter or regulatory element(s) exists between nucleotides −85 to −138, upstream from the putative −10 and −35 elements that were previously identified. To further characterize these regulatory element(s), 5′-rapid amplification of cDNA ends (5′-RACE) was repeated and a 10-fold higher number of clones was analysed. As shown in Table 1, TSS1 was again identified, but two additional putative start sites at nucleotides −21 and −53 (TSS2 and TSS3, respectively) were also identified. TSS1 and TSS2 represented 10 and 20 % of the positive clones identified by 5′-RACE, respectively. The main start site, TSS3, mapped to nucleotide −53 and represented 70 % of the positive clones identified by 5′-RACE. Associated with TSS3 are putative −10 and −35 elements located at nucleotides −59 to −64 and −83 to −88, respectively, and this putative promoter was designated ygiW–qseBC-P2. Consistent with this, a significant decrease in β-Gal activity was observed when nucleotides −85 to −93 were deleted (compare pDJR63 and pDJR58 in Fig. 1c) which disrupts the −35 element of ygiW–qseBC-P2 (nucleotides −83 to −88).
Table 1. Transcriptional start sites identified by 5′-RACE.
| ygiW transcriptional start site | Position (nucleotide)* | No. (%) of 5′-RACE clones identified |
| TSS1 | −15 (T) | 2 (10) |
| TSS2 | −21 (A) | 4 (20) |
| TSS3 | −53 (A) | 14 (70) |
The letters in brackets indicate the nucleotide determined for that 5′ end.
To further confirm that the regulatory elements in ygiW–qseBC-P2 are functional, the putative −10 element was altered by site-specific mutagenesis (TAATAG changed to GCGCGG) to generate pDJR85 (Fig. 1b). As shown in Fig. 1(c), this construct exhibited a 57-fold reduction in β-Gal activity. Finally, to determine whether ygiW–qseBC-P1 is functional, mutations were introduced into the putative −10 and −35 elements (see pDJR60 in Fig. 1b). As shown in Fig. 1(c), these mutations did not significantly reduce β-Gal activity (compare pDJR60 with pDJR58 and pDJR85), suggesting that ygiW–qseBC-P1 is non-functional. Together, these results suggest that ygiW–qseBC-P2 is the primary promoter that transcribes the ygiW–qseBC operon under the growth conditions that were used.
QseC is the primary but not the only phosphodonor for QseB
In our previous work, we showed that the QseC sensor and QseB response regulator were required for expression of the ygiW–qseBC operon, since deletion of each gene or a qseBC double deletion resulted in significant downregulation of the operon (Juárez-Rodríguez et al., 2013a). To determine whether QseC is required for activation of QseB, we first examined the effect of qseC deletion on the transcription of the ygiW–qseBC operon in A. actinomycetemcomitans by qRT-PCR using primers for the qseB gene. As shown in Fig. 2(a), transcription of the operon was not detected in the A. actinomycetemcomitans ΔqseB or ΔqseBC strains. However, transcription still occurred in the ΔqseC strain but was reduced by 58 % relative to WT. This suggests that QseB may be activated even in the absence of its cognate sensor kinase. This was further demonstrated by complementing the ΔqseBC mutant with single copy chromosomal insertions of either qseB or qseBC. As shown in Fig. 2(b), introducing qseBC in single copy into the ΔqseBC strain restored β-Gal activity to near WT levels. Complementation of the ΔqseBC mutant with qseB alone also significantly increased β-Gal activity, but the overall levels were still approximately 7.5-fold lower than the qseBC complemented strain. Together, these results indicate that QseB may be partially activated in the absence of QseC. Finally, to determine whether QseB activation in the absence of its cognate sensor requires phosphorylation of the response regulator, the ΔqseBC strain was complemented with a mutated copy of qseB (qseBpho−) encoding an inactive protein in which the functional aspartate residue was substituted with alanine (QseBD51A). As shown in Fig. 2(b), β-Gal activity produced by strains complemented with qseBpho− alone or with qseBpho−qseC was similar to the parent ΔqseBC strain. Thus, lacZ transcription in the absence of QseC requires phosphorylation of QseB. Together, these results suggest that QseC is the primary phosphodonor for QseB but that phosphorylation of QseB can also occur via another mechanism in the absence of the cognate kinase resulting in partial activation of the response regulator.
Fig. 2.

Role of QseC and QseB in the expression of the ygiW–qseBC operon. (a) Expression of the ygiW–qseBC operon in gene deletion strains of A. actinomycetemcomitans. Cultures of WT 652, ΔqseB mutant (652-JR37), ΔqseC mutant (652-JR38) and ΔqseBC mutant (652-JR39) were grown in BHI medium to mid-exponential phase, and expression of the operon was measured by qRT-PCR and normalized to the expression determined in the WT strain. The data represent the means and sds of three independent experiments. Significant differences in expression between WT and mutant strains were determined using one-way ANOVA. ***, P<0.001; ****, P<0.0001. (b) Graph depicting β-Gal activity in WT 652 strain, ΔqseBC mutant and ΔqseBC mutant strains harbouring pDJR29 (Juárez-Rodríguez et al., 2013a) and complemented with a single copy chromosomal insertion of qseBC, qseB, qseBpho−qseC or qseBpho−. β-Gal activity is expressed in Miller units and was measured at the mid-exponential phase of growth in BHI medium. Values are means of results from three independent experiments± sds. Statistical analysis was performed using a two-tailed unpaired t-test. ****, P<0.0001.
Identification of the QseB binding site in the ygiW–qseBC promoter
To determine whether QseB interacts directly with the ygiW–qseBC promoter region, a QseB-hexahistidine fusion protein was purified (Fig. 3a) and used in mobility shift reactions with a family of DNA fragments spanning the 372 bp regulatory region between ygiW–qseBC and the upstream fdhE gene (Fig. 3b). Preliminary experiments showed that the protein and probe concentrations required for optimal formation of DNA–protein complexes were 4 µM and 50 fmol, respectively (data not shown). As shown in Fig. 3(c), QseB bound only to probes I and III within the regulatory region, suggesting that the QseB binding site resides between nucleotides −13 and −156. Increasing the concentration of QseB in reactions with probe III converted all of the free probe into DNA–protein complexes (Fig. 3d), and QseB binding was inhibited by the addition of increasing amounts of unlabelled probe III DNA (Fig. 3e). QseB did not produce DNA–protein complexes with a 200 bp DNA fragment derived from the Yersinia pseudotuberculosis psaA gene, which represents a negative control (Fig. 3f).
Fig. 3.

Binding of purified QseB to the ygiW–qseBC promoter region. (a) SDS-PAGE of purified QseB protein. Lane 1, molecular-mass marker (M); lane 2, hexa-histidine QseB purified by cobalt-based immobilized metal affinity chromatography (indicated by the asterisk). (b) Schematic representation of the ygiW–qseBC promoter region showing the putative binding region for QseB. PCR fragments used for EMSA reactions are numbered from I to V and the nucleotides contained in each fragment are indicated to the right, numbered relative to the ygiW start codon. (c) PCR probes I–V were incubated in the absence (−) or presence (+) of 4 µM QseB protein. (d) Probe III was incubated in the absence (−) or in the presence of the indicated concentrations (µM) of QseB. (e) Probe III was incubated with 4 µM QseB in the absence (−) or presence of 0.5-, 5-, 10- and 20-fold molar excesses of competitive specific unlabelled fragment III (DNAc). (f) The non-specific Yp probe was incubated in the absence (−) or in the presence of the indicated concentrations (µM) of QseB. All binding reactions were performed for 30 min at room temperature and the DNA–protein complexes (indicated by an asterisk) were resolved in 6 % polyacrylamide gels. A 200 bp DNA fragment from the psa gene of Y. pseudotuberculosis (probe Yp) was used as a negative control.
To more precisely localize the QseB binding site(s) within probe III, DNA fragments spanning nucleotides −186 to+70 were tested (Fig. 4a). As shown in Fig. 4(b), only probes VI and VIII formed DNA–QseB complexes, indicating that QseB interacts with a DNA sequence between nucleotides −122 to −59 relative to the ygiW start codon. The inability of probes XII and XIII to form DNA–QseB complexes suggests that the binding site spans or resides close to nucleotides −92 and −93. Interestingly, the sequence of this region of probe VIII contains two direct repeats separated by six nucleotides (CTTAA-N6-CTTAA) at nucleotides −78 to −93 (see Fig. S1) and this sequence resembles the DNA-binding site [CTTAA(G or T)-N5-CTTAA(G or T)] for the E. coli transcriptional activator PmrA, which is related to QseB (Chen & Groisman, 2013). To confirm that this putative QseB binding site regulates ygiW–qseBC expression, pDJR57 containing nucleotides −1 to −122 (see Fig. 1b) was altered by site-directed mutagenesis from CTTAA to CGCGA to generate pDJR86. In addition, a construct in which the upstream direct repeat was deleted (pDJR58 in Fig. 1a) was tested for β-Gal activity. As shown in Table 2, mutagenesis or deletion of the upstream direct repeat resulted in a 45-fold decease in β-Gal activity, suggesting that the QseB binding site is essential for ygiW–qseBC transcription.
Fig. 4.

Localization of the QseB binding site. (a) Schematic representation of the fdhE–ygiW intergenic region showing the putative binding region for QseB. PCR fragments used for EMSA reactions are numbered from VI to XIII and the nucleotides contained by each fragment are indicated to the right, numbered relative to the ygiW start codon. (b) PCR probes were incubated in the absence (−) and presence (+) of 4 µM QseB protein for 30 min at room temperature, and the DNA–protein complexes (indicated by an asterisk) were resolved in 6 % polyacrylamide gels. A 200 bp DNA fragment from Y. pseudotuberculosis psa (probe Yp) was used as a negative control.
Table 2. Mutagenesis of the putative QseB binding site.
| Strain/plasmid | Promoter fragment | Promoter fragment |
| 652/pDJR57 | −1 to −122 | 584.6±77.9 |
| 652/pJDR86 | −1 to −122 (mutated CTTAA) | 13.0±0.7 |
| 652/pDJR58 | −1 to −84 (deleted CTTAA) | 10.5±1.1 |
Identification and function of IHF binding sites in the ygiW–qseBC operon
The results shown in Fig. 1(c) also suggest that additional regulatory elements exist upstream of the ygiW–qseBC–P2 promoter and QseB binding site (compare pDJR63 with pDJR56), between nucleotides −93 and −138. Further examination of the 372 bp intergenic region and the ygiW coding sequence identified three putative sequences that closely resemble the E. coli consensus recognition sequence of the IHF protein (YAANNNNTTGATW, where Y = T or C, N = is any base, W = A or T) (Mangan et al., 2006). The first putative IHF binding sequence resides in the ygiW ORF at nucleotides +17 to +32. The second and third sequences reside at nucleotides −31 to –49 and –96 to −111, respectively, relative to the ygiW start codon. These sequences were designated IHFs1, IHFs2 and IHFs3, respectively (see Fig. S1).
To determine whether the A. actinomycetemcomitans IHF protein interacts with these putative binding sites, A. actinomycetemcomitans IHF heterodimer was produced as previously described (Torres-Escobar et al., 2014) and used in EMSA reactions with a family of DNA fragments spanning nucleotides −372 to+70 (see Fig. 6). As shown in Fig. 5, IHF bound only to probes that contained one or more of the putative binding sites described above (i.e. probes III, VI, VIII, IX, X, XI, XII and XIII), whereas probes comprising other regions did not form DNA–IHF complexes (i.e. II and VII). IHF binding was not detected using a 200 bp DNA fragment derived from the Y. pseudotuberculosis psaA gene, which represented a negative control. To determine whether IHF regulates the expression of ygiW–qseBC, the transcriptional levels of qseB were determined in A. actinomycetemcomitans strains that lacked either the IHFα (ΔihfA) or IHFβ (ΔihfB) subunit. Total RNA was analysed by qRT-PCR from cultures of WT 652, ΔihfA mutant (652-TE78) and ΔihfB mutant (652-TE79), and as shown in Fig. 6, the transcription of ygiW–qseBC was significantly increased (P<0.05) in both the ΔihfA and ΔihfB strains compared with the WT strain. The ltxC gene was used as a positive control in these experiments, since ltxC was previously shown to be negatively regulated by IHF in A. actinomycetemcomitans (Kolodrubetz et al., 2010). As expected, ltxC expression increased in 652-TE78 and 652-TE79 (Fig. 6). Thus, IHF binds to sites within the ygiW–qseBC promoter and within the coding sequence ygiW, and results in a net reduction of ygiW–qseBC expression.
Fig. 6.
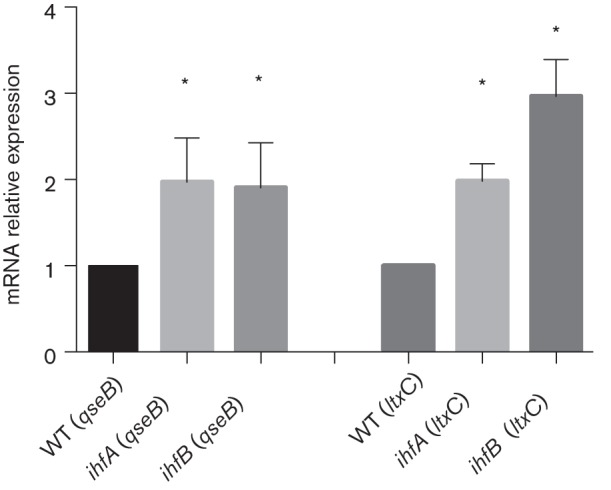
qseB and ltxC transcriptional levels in IHF mutant strains. Cultures of WT 652, ΔihfA mutant (652-TE78) and ΔihfB mutant (652-TE79) were grown in BHI medium to mid-exponential phase, and expression of qseB and ltxC (positive control) was measured by qRT-PCR and normalized to the WT strain. The data represent the means and sds of three independent experiments. Significant differences in transcription between WT and mutant strains were determined using one-way ANOVA. *, P<0.05.
Fig. 5.

Binding of purified IHF heterodimer to the ygiW–qseBC promoter. (a) Schematic representation of the fdhE–ygiW intergenic region showing the putative binding sites for IHF. PCR fragments used for EMSA reactions are numbered from II to XIII, and the nucleotides contained by each fragment are indicated to the right, and numbered relative to the ygiW start codon. (b) PCR probes were incubated in the absence (−) and presence (+) of 4 µM IHF protein for 30 min at room temperature, and the DNA–protein complexes (indicated by an asterisk) were resolved in 6 % polyacrylamide gels. A 200 bp DNA fragment from Y. pseudotuberculosis psa (probe Yp) was used as a negative control.
Discussion
In this study, we characterized the regulatory region of the ygiW–qseBC operon that encodes a two-component system that is essential for biofilm growth and virulence of A. actinomycetemcomitans. Transcription of ygiW–qseBC was directed by sequences between nucleotides −1 to −138 relative to the ygiW start codon, suggesting that the promoter resides within the intergenic region of ygiW and the small upstream ORF(D11S_1134) encoding a hypothetical protein. Two putative promoters, ygiW–qseBC-P1 and ygiW–qseBC-P2, were initially identified in this region, but our results strongly suggest that expression of the operon is primarily driven by ygiW–qseBC-P2 under the growth conditions used. This promoter comprises −10 and −35 elements as well as CTTAA direct repeat elements that flank the −35 sequence (between nucleotides −78 and −93) and function to interact with the QseB response regulator, a strong positive regulator of operon expression. Site-specific mutation of the P2 −10 sequence and the distal CTAA repeat reduced β-Gal activity almost to the level of the promoterless lacZ control construct, suggesting that ygiW–qseBC-P1 is not functional. Consistent with this, mutation of the P1 −10 and −35 elements had little effect on promoter activity. However, we cannot exclude the possibility that this promoter may be active under growth conditions different from those used in this study. In addition, deletion of nucleotides −94 to −138, upstream from the QseB binding CTAA repeats in ygiW–qseBC-P2, resulted in a threefold reduction in expression, suggesting that additional regulatory elements exist upstream from the −35 sequence and the direct repeat sequences. One possibility is that IHF interaction with its binding site that is immediately upstream from the distal CTTAA repeat (nucleotides −96 to −111) induces a DNA conformation that facilitates QseB binding and promotes ygiW–qseBC expression.
QseB belongs to the OmpR/PhoB subfamily of response regulator transcriptional factors that are distinguished by a C-terminal winged helix–turn–helix DNA binding motif (Mizuno, 1997; Martínez-Hackert & Stock, 1997). As shown in Fig. S2, the A. actinomycetemcomitans QseB is structurally highly similar to other members of the OmpR/PhoB response regulators, and shares the phosphorylation domain, dimer interface and DNA contact motifs that are present in these transcriptional regulators. The DNA sequences recognized by the OmpR/PhoB subfamily have been determined for many members and all bind to specific DNA direct repeat sequences (Chen & Groisman, 2013; Liu & De Wulf, 2004; Yamamoto & Ishihama, 2006; Zwir et al., 2012; Rhee et al., 2008; Ritzefeld et al., 2013; Tung & McMahon, 2012), consistent with our finding that CTTAA direct repeats represent the QseB binding site of A. actinomycetemcomitans. Phosphorylation of this subfamily of response regulators induces the formation of dimers, which increase the binding affinity of the C-terminal DNA-binding domain with the tandem DNA direct repeat sequences on the promoters of target genes (Toro-Roman et al., 2005; Creager-Allen et al., 2013; Barbieri et al., 2013). As expected, the primary phosphodonor for QseB is its cognate sensor, QseC, but interestingly QseB could be partially activated and induce transcription of ygiW–qseBC in the absence of QseC. The absence of QseC in E. coli results in constitutively high qseB transcription that arises from bidirectional cross-regulation between the structurally related QseBC and PmrAB two-component systems (Guckes et al., 2013). Without QseC, the non-cognate PmrB kinase phosphorylated QseB and transcription of qseBC was also activated by PmrA resulting in constitutive expression of qseB. However, the A. actinomycetemcomitans genome does not encode the PmrAB two-component system, which may explain why transcription of ygiW–qseBC is not constitutively high in the absence of QseC. Partial activation of QseB in the absence of QseC in A. actinomycetemcomitans may instead result from inefficient phosphorylation by other non-cognate kinases, or alternatively from the transfer of a phosphate from the small phosphate donor acetylphosphate (Wolfe, 2005). Consistent with latter possibility, the genes that encode enzymes required for the production of acetylphosphate are present in the A. actinomycetemcomitans genome [D11S-1227 (pta) and D11S-1228 (ackA)]. Interestingly, the PmrAB two-component system of E. coli senses ferric iron and induces the expression of qseBC in response to this signal (Chen & Groisman, 2013; Guckes et al., 2013). In contrast, in the absence of PmrAB in A. actinomycetemcomitans, the induction of ygiW–qseBC expression in response to ferrous and ferric iron is dependent on QseBC itself, and appears to be similar to ferrous iron induction of H. influenzae ygiW–fisRS expression (Steele et al., 2012). Indeed, QseC of A. actinomycetemcomitans may sense both iron and catecholamine hormones since the QseBC operon is synergistically induced when both signals are present (unpublished data).
Our data also show that IHF binds to two sites in the ygiW–qseBC-P2 promoter and a third site near the 5′ end of the ygiW ORF. Overall, IHF functions as a net negative regulator of ygiW–qseBC expression, since transcription of ygiW–qseBC increased approximately twofold in ΔihfA and ΔihfB strains of A. actinomycetemcomitans. Consistent with this, IHF binding to the IHFS1 site appears to downregulate operon expression since the β-Gal activity of the 372 bp ygiW–qseBC promoter–lacZ fusion in pDJR29 was previously shown to be higher in an A. actinomycetemcomitans strain containing an in-frame deletion in ygiW (in which IHFS1 was lost) than in the WT (Juárez-Rodríguez et al., 2013a). However, our previous work characterizing the lsr locus of A. actinomycetemcomitans showed that lsr expression is differentially regulated by IHF interaction at each of the two binding sites that are present in this promoter (Torres-Escobar et al., 2014). At present, the outcomes of IHF interaction at IHFS2 and IHFS3 are not known, although, as suggested above, it is possible that the interaction with IHFS3 may facilitate QseB binding to ygiW–qseBC-P2.
In summary, we have shown that transcription of the ygiW–qseBC operon of A. actinomycetemcomitans is primarily driven by the QseB-dependent ygiW–qseBC-P2 promoter. This promoter interacts with two trans-acting proteins; the QseB response regulator activates transcription of ygiW–qseBC by binding to CTTAA direct repeat sequences that flank the −35 element, and IHF interacts with three distinct binding sites in the promoter and functions as a net negative regulator of transcription. Finally, although QseB is mainly activated by the cognate QseC sensor, it can be partially activated by other mechanisms in the absence of QseC activity.
Acknowledgements
This research was supported by a Public Health Service grant from the National Institute for Dental and Craniofacial Research (no. RO1DE14605).
Abbreviations:
- 5′-RACE
5′-rapid amplification of cDNA ends
- β-Gal
β-galactosidase
- EMSA
electrophoretic mobility shift assay
- Km
kanamycin
- qRT-PCR
quantitative real-time PCR
- UPEC
uropathogenic Escherichia coli
Footnotes
Two supplementary tables and two supplementary figures are available with the online Supplementary Material.
References
- Azam T., Iwata A., Nishimura A., Ueda S., Ishihama A. (1999). Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J Bacteriol 181, 6361–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri C. M., Wu T., Stock A. M. (2013). Comprehensive analysis of OmpR phosphorylation, dimerization, and DNA binding supports a canonical model for activation. J Mol Biol 425, 1612–1626. 10.1016/j.jmb.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearson B. L., Bearson S. M. (2008). The role of the QseC quorum-sensing sensor kinase in colonization and norepinephrine-enhanced motility of Salmonella enterica serovar Typhimurium. Microb Pathog 44, 271–278. 10.1016/j.micpath.2007.10.001 [DOI] [PubMed] [Google Scholar]
- Blomfield I. C., Kulasekara D. H., Eisenstein B. I. (1997). Integration host factor stimulates both FimB- and FimE-mediated site-specific DNA inversion that controls phase variation of type 1 fimbriae expression in Escherichia coli. Mol Microbiol 23, 705–707. 10.1046/j.1365-2958.1997.2241615.x [DOI] [PubMed] [Google Scholar]
- Brady P., Bergin S., Cryan B., Flanagan O. (2014). Intracranial abscess secondary to dental infection. J Ir Dent Assoc 60, 32–34. [PubMed] [Google Scholar]
- Chen H. D., Groisman E. A. (2013). The biology of the PmrA/PmrB two-component system: the major regulator of lipopolysaccharide modifications. Annu Rev Microbiol 67, 83–112. 10.1146/annurev-micro-092412-155751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke M. B., Sperandio V. (2005). Transcriptional regulation of flhDC by QseBC and σ (FliA) in enterohaemorrhagic Escherichia coli. Mol Microbiol 57, 1734–1749. 10.1111/j.1365-2958.2005.04792.x [DOI] [PubMed] [Google Scholar]
- Clarke M. B., Hughes D. T., Zhu C., Boedeker E. C., Sperandio V. (2006). The QseC sensor kinase: a bacterial adrenergic receptor. Proc Natl Acad Sci U S A 103, 10420–10425. 10.1073/pnas.0604343103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig N. L., Nash H. A. (1984). E. coli integration host factor binds to specific sites in DNA. Cell 39, 707–716. 10.1016/0092-8674(84)90478-1 [DOI] [PubMed] [Google Scholar]
- Creager-Allen R. L., Silversmith R. E., Bourret R. B. (2013). A link between dimerization and autophosphorylation of the response regulator PhoB. J Biol Chem 288, 21755–21769. 10.1074/jbc.M113.471763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuth D. R., Novak E. A., Shao H. (2011). Alternative autoinducer-2 quorum sensing response circuits: impact on microbial community development. In Oral Microbial Communities: Genomic Inquiry and Interspecies Communication, pp. 263–282. Edited by Kolenbrander P. E. Washington DC: American Society for Microbiology; 10.1128/9781555817107.ch18 [DOI] [Google Scholar]
- Dietmann A., Millonig A., Combes V., Couraud P. O., Kachlany S. C., Grau G. E. (2013). Effects of Aggregatibacter actinomycetemcomitans leukotoxin on endothelial cells. Microb Pathog 61–62, 43–50. 10.1016/j.micpath.2013.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes K. P., Mayer M. P., Ando E. S., Ulbrich A. G., Amarente-Mendes J. G., Russo M. (2008). Inhibition of interferon-gamma-induced nitric oxide production in endotoxin-activated macrophages by cytolethal distending toxin. Oral Microbiol Immunol 23, 360–366. 10.1111/j.1399-302X.2008.00434.x [DOI] [PubMed] [Google Scholar]
- Fine D. H., Kaplan J. B., Furgang D., Karched M., Velliyagounder K., Yue G. (2010). Mapping the epithelial-cell-binding domain of the Aggregatibacter actinomycetemcomitans autotransporter adhesin Aae. Microbiology 156, 3412–3420. 10.1099/mic.0.037606-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong K. P., Chung W. O., Lamont R. J., Demuth D. R. (2001). Intra- and interspecies regulation of gene expression by Actinobacillus actinomycetemcomitans LuxS. Infect Immun 69, 7625–7634. 10.1128/IAI.69.12.7625-7634.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong K. P., Gao L., Demuth D. R. (2003). luxS and arcB control aerobic growth of Actinobacillus actinomycetemcomitans under iron limitation. Infect Immun 71, 298–308. 10.1128/IAI.71.1.298-308.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman D. I. (1988). Integration host factor: a protein for all reasons. Cell 55, 545–554. 10.1016/0092-8674(88)90213-9 [DOI] [PubMed] [Google Scholar]
- Guckes K. R., Kostakioti M., Breland E. J., Gu A. P., Shaffer C. L., Martinez C. R., III, Hultgren S. J., Hadjifrangiskou M. (2013). Strong cross-system interactions drive the activation of the QseB response regulator in the absence of its cognate sensor. Proc Natl Acad Sci U S A 110, 16592–16597. 10.1073/pnas.1315320110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjifrangiskou M., Kostakioti M., Chen S. L., Henderson J. P., Greene S. E., Hultgren S. J. (2011). A central metabolic circuit controlled by QseC in pathogenic Escherichia coli. Mol Microbiol 80, 1516–1529. 10.1111/j.1365-2958.2011.07660.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffajee A. D., Socransky S. S. (1994). Microbial etiological agents of destructive periodontal diseases. Periodontol 2000 5, 78–111. 10.1111/j.1600-0757.1994.tb00020.x [DOI] [PubMed] [Google Scholar]
- Hughes D. T., Clarke M. B., Yamamoto K., Rasko D. A., Sperandio V. (2009). The QseC adrenergic signaling cascade in enterohemorrhagic E. coli (EHEC). PLoS Pathog 5, e1000553. 10.1371/journal.ppat.1000553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihama A. (1999). Modulation of the nucleoid, the transcription apparatus, and the translation machinery in bacteria for stationary phase survival. Genes Cells 4, 135–143. 10.1046/j.1365-2443.1999.00247.x [DOI] [PubMed] [Google Scholar]
- Jinadasa R. N., Bloom S. E., Weiss R. S. G., Duhamel G. E. (2011). Cytolethal distending toxin: a conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 157, 1851–1875. 10.1099/mic.0.049536-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juárez-Rodríguez M. D., Torres-Escobar A., Demuth D. R. (2013a). ygiW and qseBC are co-expressed in Aggregatibacter actinomycetemcomitans and regulate biofilm growth. Microbiology 159, 989–1001. 10.1099/mic.0.066183-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juárez-Rodríguez M. D., Torres-Escobar A., Demuth D. R. (2013b). Construction of new cloning, lacZ reporter and scarless-markerless suicide vectors for genetic studies in Aggregatibacter actinomycetemcomitans. Plasmid 69, 211–222. 10.1016/j.plasmid.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiya M., Komatsuzawa H., Papantonakis A., Seki M., Makihira S., Ouhara K., Kusumoto Y., Murakami S., Taubman M. A., Kawai T. (2011). Aggregatibacter actinomycetemcomitans Omp29 is associated with bacterial entry to gingival epithelial cells by F-actin rearrangement. PLoS ONE 6, e18287. 10.1371/journal.pone.0018287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajanchi B. K., Kozlova E. V., Sha J., Popov V. L., Chopra A. K. (2012). The two-component QseBC signalling system regulates in vitro and in vivo virulence of Aeromonas hydrophila. Microbiology 158, 259–271. 10.1099/mic.0.051805-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodrubetz D., Phillips L., Burgum A. (2010). Repression of aerobic leukotoxin transcription by integration host factor in Aggregatibacter actinomycetemcomitans. Res Microbiol 161, 541–548. 10.1016/j.resmic.2010.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostakioti M., Hadjifrangiskou M., Pinkner J. S., Hultgren S. J. (2009). QseC-mediated dephosphorylation of QseB is required for expression of genes associated with virulence in uropathogenic Escherichia coli. Mol Microbiol 73, 1020–1031. 10.1111/j.1365-2958.2009.06826.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., De Wulf P. (2004). Probing the ArcA-P modulon of Escherichia coli by whole genome transcriptional analysis and sequence recognition profiling. J Biol Chem 279, 12588–12597. 10.1074/jbc.M313454200 [DOI] [PubMed] [Google Scholar]
- Mangan M. W., Lucchini S., Danino V., Cróinín T. O., Hinton J. C., Dorman C. J. (2006). The integration host factor (IHF) integrates stationary-phase and virulence gene expression in Salmonella enterica serovar Typhimurium. Mol Microbiol 59, 1831–1847. 10.1111/j.1365-2958.2006.05062.x [DOI] [PubMed] [Google Scholar]
- Martínez-Hackert E., Stock A. M. (1997). Structural relationships in the OmpR family of winged-helix transcription factors. J Mol Biol 269, 301–312. 10.1006/jmbi.1997.1065 [DOI] [PubMed] [Google Scholar]
- Merighi M., Septer A. N., Carroll-Portillo A., Bhatiya A., Porwollik S., McClelland M., Gunn J. S. (2009). Genome-wide analysis of the PreA/PreB (QseB/QseC) regulon of Salmonella enterica serovar Typhimurium. BMC Microbiol 9, 42. 10.1186/1471-2180-9-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. H. (1972). Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Mizuno T. (1997). Compilation of all genes encoding two-component phosphotransfer signal transducers in the genome of Escherichia coli. DNA Res 4, 161–168. 10.1093/dnares/4.2.161 [DOI] [PubMed] [Google Scholar]
- Moreira C. G., Sperandio V. (2012). Interplay between the QseC and QseE bacterial adrenergic sensor kinases in Salmonella enterica serovar Typhimurium pathogenesis. Infect Immun 80, 4344–4353. 10.1128/IAI.00803-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira C. G., Weinshenker D., Sperandio V. (2010). QseC mediates Salmonella enterica serovar Typhimurium virulence in vitro and in vivo. Infect Immun 78, 914–926. 10.1128/IAI.01038-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M., Suzuki J., Yamazaki S., Ikezoe M., Matsushima R., Ashigaki N., Aoyama N., Kobayashi N., Wakayama K. & other authors (2013). High incidence of Aggregatibacter actinomycetemcomitans infection in patients with cerebral infarction and diabetic renal failure: a cross-sectional study. BMC Infect Dis 13, 557–567. 10.1186/1471-2334-13-557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak E. A., Shao H., Daep C. A., Demuth D. R. (2010). Autoinducer-2 and QseC control biofilm formation and in vivo virulence of Aggregatibacter actinomycetemcomitans. Infect Immun 78, 2919–2926. 10.1128/IAI.01376-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco A. R., Curtis M. M., Ritchie J. M., Munera D., Waldor M. K., Moreira C. G., Sperandio V. (2012). Fucose sensing regulates bacterial intestinal colonization. Nature 492, 113–117. 10.1038/nature11623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paturel L., Casalta J. P., Habib G., Nezri M., Raoult D. (2004). Actinobacillus actinomycetemcomitans endocarditis. Clin Microbiol Infect 10, 98–118. 10.1111/j.1469-0691.2004.00794.x [DOI] [PubMed] [Google Scholar]
- Rhee J. E., Sheng W., Morgan L. K., Nolet R., Liao X., Kenney L. J. (2008). Amino acids important for DNA recognition by the response regulator OmpR. J Biol Chem 283, 8664–8677. 10.1074/jbc.M705550200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzefeld M., Walhorn V., Kleineberg C., Bieker A., Kock K., Herrmann C., Anselmetti D., Sewald N. (2013). Cooperative binding of PhoBDBD to its cognate DNA sequence–a combined application of single-molecule and ensemble methods. Biochemistry 52, 8177–8186. 10.1021/bi400718r [DOI] [PubMed] [Google Scholar]
- Saito T., Ishihara K. M., Ryu M., Okuda K., Sakurai K. (2010). Fimbriae-associated genes are biofilm-forming factors in Aggregatibacter actinomycetemcomitans strains. Bull Tokyo Dent Coll 51, 145–150. 10.2209/tdcpublication.51.145 [DOI] [PubMed] [Google Scholar]
- Sambrook J., Russell D. W. (2001). Molecular Cloning: a Laboratory Manual, 3rd edn Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Sperandio V., Torres A. G., Kaper J. B. (2002). Quorum sensing Escherichia coli regulators B and C (QseBC): a novel two-component regulatory system involved in the regulation of flagella and motility by quorum sensing in E. coli. Mol Microbiol 43, 809–821. 10.1046/j.1365-2958.2002.02803.x [DOI] [PubMed] [Google Scholar]
- Steele K. H., O’Connor L. H., Burpo N., Kohler K., Johnston J. W. (2012). Characterization of a ferrous iron-responsive two-component system in nontypeable Haemophilus influenzae. J Bacteriol 194, 6162–6173. 10.1128/JB.01465-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro-Roman A., Mack T. R., Stock A. M. (2005). Structural analysis and solution studies of the activated regulatory domain of the response regulator ArcA: a symmetric dimer mediated by the α4-β5-α5 face. J Mol Biol 349, 11–26. 10.1016/j.jmb.2005.03.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Escobar A., Juárez-Rodríguez M. D., Curtiss R., III (2010). Biogenesis of Yersinia pestis PsaA in recombinant attenuated Salmonella Typhimurium vaccine (RASV) strain. FEMS Microbiol Lett 302, 106–113. 10.1111/j.1574-6968.2009.01827.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Escobar A., Juárez-Rodríguez M. D., Demuth D. R. (2014). Differential transcriptional regulation of Aggregatibacter actinomycetemcomitans lsrACDBFG and lsrRK operons by integration host factor protein. J Bacteriol 196, 1597–1607. 10.1128/JB.00006-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung C. S., McMahon B. H. (2012). A structural model of the E. coli PhoB dimer in the transcription initiation complex. BMC Struct Biol 12, 3. 10.1186/1472-6807-12-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. Y., Wang H. C., Li J. M., Wang J. Y., Yang K. C., Ho Y. K., Lin P. Y., Lee L. N., Yu C. J. & other authors (2010). Invasive infections of Aggregatibacter (Actinobacillus) actinomycetemcomitans. J Microbiol Immunol Infect 43, 491–497. 10.1016/S1684-1182(10)60076-X [DOI] [PubMed] [Google Scholar]
- Wang X., Wang Q., Yang M., Xiao J., Liu Q., Wu H., Zhang Y. (2011). QseBC controls flagellar motility, fimbrial hemagglutination and intracellular virulence in fish pathogen Edwardsiella tarda. Fish Shellfish Immunol 30, 944–953. 10.1016/j.fsi.2011.01.019 [DOI] [PubMed] [Google Scholar]
- Winer J., Jung C. K. S., Shackel I., Williams P. M. (1999). Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem 270, 41–49. 10.1006/abio.1999.4085 [DOI] [PubMed] [Google Scholar]
- Wolfe A. J. (2005). The acetate switch. Microbiol Mol Biol Rev 69, 12–50. 10.1128/MMBR.69.1.12-50.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K., Ishihama A. (2006). Characterization of copper-inducible promoters regulated by CpxA/CpxR in Escherichia coli. Biosci Biotechnol Biochem 70, 1688–1695. 10.1271/bbb.60024 [DOI] [PubMed] [Google Scholar]
- Yue G., Kaplan J. B., Furgang D., Mansfield K. G., Fine D. H. (2007). A second Aggregatibacter actinomycetemcomitans autotransporter adhesin exhibits specificity for buccal epithelial cells in humans and Old World primates. Infect Immun 75, 4440–4448. 10.1128/IAI.02020-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwir I., Latifi T., Perez J. Ch., Huang H., Groisman E. A. (2012). The promoter architectural landscape of the Salmonella PhoP regulon. Mol Microbiol 84, 463–485. 10.1111/j.1365-2958.2012.08036.x [DOI] [PMC free article] [PubMed] [Google Scholar]