

Abstract
Berardinelli-Seip congenital lipodystrophy (BSCL) is a very rare autosomal recessive disorder characterized by various dermatological and systemic manifestations such as lipoatrophy, hypertriglyceridemia, hepatomegaly, acanthosis nigricans, and acromegaloid features. BSCL type 2 is more common and severe, with onset in the neonatal period or in early infancy. The locus for BSCL2 has been identified on chromosome 11q13. Early recognition and differentiation from other congenital generalized lipodystrophies help in the initiation of appropriate preventive and therapeutic measures such as lifestyle modification and pharmacotherapy that helps postpone the onset of metabolic syndrome. We report BSCL type 2 in two siblings with several cutaneous manifestations like acanthosis nigricans, hypertrichosis, prominent subcutaneous veins, and increased lanugo hair.
Keywords: Acanthosis nigricans, acromegaloid features, Berardinelli-Seip congenital lipodystrophy, BSCL2 gene, hypertrichosis, recurrent pyoderma
INTRODUCTION
Berardinelli-Seip congenital lipodystrophy (BSCL) also described as generalized lipodystrophy, congenital lipodystrophy, Seip-Lawrence syndrome, and lipoatrophic diabetes is a rare autosomal recessive disorder, reported in approximately 250 patients of various ethnic origins.[1] The estimated worldwide prevalence is one in 10 million population.[2]
BSCL families are classified into BSCL1, BSCL2, and BSCLX.[3] This rare entity is characterized by defect in the 1-acylglycerol-3-phosphate-O-acyltransferase-2 (AGPAT2) gene on chromosome 9q34 in type 1 variant and in BSCL2 gene on chromosome 11q13 in the type 2 variant.
In all BSCL subjects, there is a near-total absence of adipose tissue resulting in aberrant storage of dietary and endogenous fat in metabolically important tissues such as muscle and liver. This leads to severe insulin resistance and ultimately diabetes mellitus. Other metabolic consequences include hypertriglyceridemia leading to hepatic steatosis and ultimately cirrhosis and finally to death due to hepatic failure. The characteristic clinical feature is the near-total absence of adipose tissue and a consequent generalized muscular appearance that is recognized easily at birth. The condition is also associated with various dermatological and systemic manifestations. The metabolic abnormalities may prove fatal and so necessitate optimal therapeutic and preventive interventions.[4]
CASE REPORT
A 6-year-old girl, product of a nonconsan guineous marriage presented to us with loss of fat all over the body, protuberant abdomen since early months, poor weight gain despite having voracious appetite, hyperpigmentation of the neck and the body flexures, hypertrichosis, recurrent pyodermas, and upper respiratory tract infections. Family history revealed similar features in her 3-year-old sister with loss of fat all over the body, protuberant abdomen since early months, poor weight gain, hyperpig mentation of the neck and the body flexures, and hypertrichosis [Figure 1].
Figure 1.
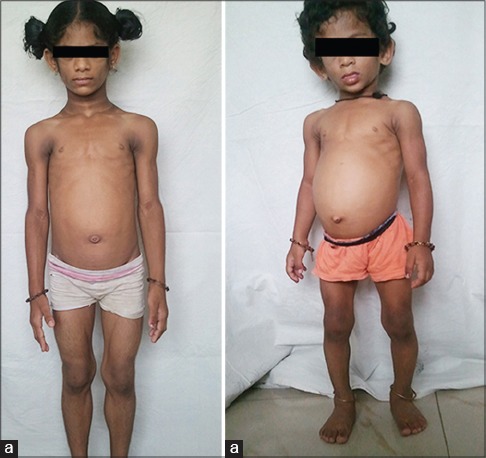
Six year (a) and 3 year (b) old siblings with loss of subcutaneous fat, protuberant abdomen, and prominent musculature
On examination, both the siblings had gener alized loss of subcutaneous fat, muscular appearance with prominent pectoral, deltoid and thigh muscles, acanthosis nigricans of the neck and all flexures, increased lanugo hair, prominent subcutaneous veins, protuberant abdomen with hepatomegaly, [Figure 2a-c] prominent and enlarged wrists, knees and ankle joints.
Figure 2.
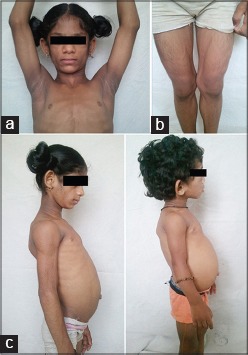
(a) Acanthosis nigricans of both axillae and neck (b) Increased lanugo hair, prominent subcutaneous veins over both lower extremities (c) Protuberant abdomen in both siblings due to hepatomegaly
Hematological investigations of the elder sister revealed microcytic hypochromic anemia with a raised erythrocyte sedimentation rate. Peripheral smear showed mild anisocytosis and hypochromasia of red blood cells. She had subclinical hypothyroidism with mild elevation of thyroid stimulating hormone. Ultrasonography abdomen revealed mild hepatomegaly. The serum triglycerides were elevated, but other parameters in her lipid profile were normal. Biochemical parameters such as liver function tests, renal function tests, blood sugar, and serum cortisol were within normal ranges. Radiological analysis of wrists done at the age of 23 months revealed normal soft tissue, normal bone density, normal joint space, and a bone age of three years as per Greulich and Pyle classification. Chest X-ray revealed pneumonitis of the middle lobe of the right lung and mild cardiomegaly. However, further cardiac evaluation with two-dimensional echocardiography revealed no cardiac involvement. Laboratory investigations of the younger sibling are within normal limits. Human immunodeficiency virus infection was ruled out in both the siblings. In view of presence of the above features, they were diagnosed to have Berardinelli-Seip syndrome.
DISCUSSION
Generalized congenital lipoatrophy or BSCL was first described in 1954 by Berardinelli in a 2-year-old Brazilian boy and later by Seip in three patients in 1959.[1] BSCL1 is the milder variant presenting mostly in the 2nd or 3rd decade of life, commonly prevalent in African-American population. The gene for BSCL1 was first identified by Garg et al.,[5] on chromosome 9q34. Agarwal et al.,[6] detected different mutations in the gene encoding AGPAT2 involved in the biosynthesis of triacylglycerol and glycerophospholipids, in individuals showing linkage to chromosome 9q34.
BSCL2 is more common and severe form than BSCL1, with onset in the neonatal period or in early infancy, being more prevalent in Portugal, Lebanon, Norway, and the Middle East. The locus for BSCL2 has been identified on chromosome 11q13 by Magré et al.[7] Various mutations were identified in the seipin gene, which encodes a 398-amino acid protein of unknown function. Windpassinger et al.,[8] in 2004 showed that seipin is an integral membrane protein of the endoplasmic reticulum. BSCLX families are very rare; they show evidence against cosegregation with either 9q34 or 11q13.
There are approximately 250 cases registered all over the world, with greater frequency reported in some ethnic groups such as Latin Americans and Arabs (individuals of Portuguese and Norwegian ancestry).[9]
The major diagnostic features of BSCL, as described by Maldergem[10] are lipoatrophy affecting trunk, limbs and face, acromegaloid features, hepatomegaly, elevated serum triglycerides, and insulin resistance (manifested as acanthosis nigricans); all of these were found in our patients. Minor features include hypertrophic cardiomyopathy, mental retardation, hypertrichosis, bone cysts, precocious puberty (in females), and phlebomegaly. Both the siblings had hypertrichosis and phlebomegaly. Diagnosis requires the presence of three major criteria or two major and two minor criteria, which are satisfied by our patients. In type 2 disease, patients lose both subcutaneous and visceral types of adipose tissue.[11] Muscular hypertrophy is present from birth, with an increase in the formation of muscle glycogen and creatinine. Skeletal maturation occurs quickly during the first 6−8 years of life[12] and there may be enlarged joints of hands and feet.[13] Association with bone cysts has been reported, which was previously described as Brunzell syndrome.
In Bjornstad et al.,'s study,[14] myocardial hypertrophy and ventricular diastolic dysfunction were found in a greater frequency in patients with lipodystrophy, risk increasing with age. In patients who are in an insulin-resistant state and therefore, hyperinsulinemia, the linkage of insulin to insulin-like growth factor-1[15] receptors present in the myocardial tissues results in their hypertrophy.[15,16,17] The histological differentiation from the classic hypertrophic cardiomyopathy is made by the absence of rearrangement of fibres in cases of myocardial hypertrophy associated with lipodystrophy.
These children may have associated immune deficiency as described by Kher et al.[18] in 1990. This could be the cause of recurrent upper respiratory tract infections and skin infections in our patient. However, no polymorphonuclear cell function studies were performed in our patients.
BSCL should be differentiated from other congenital generalized lipodystrophy syndromes like mandibuloacral dysplasia, Hutchinson-Gilford progeria syndrome, and Werner syndrome (pangeria), which are all premature ageing syndromes.
Evaluation includes assessment of pubertal states according to Tanner charts, and neurological, endocrinological, cardiovascular, orthopedic, and opthalmological consultations. Recently published studies show that leptin-replacement therapy over a period of 4 months appears as a promising tool in the correction of the metabolic complications in BSCL patients. BSCL patients should consume a low-fat diet (<20%-30% of total dietary energy) with low saturated transfats and cholesterol. This should be combined with increase in physical activity.[12] Other therapeutic agents include metformin, fibric acid derivatives, n-3 polyunsaturated fatty acids. Our patients are currently on lifestyle regulation to postpone the onset of metabolic syndrome and may require growth hormone, leptin or other specific agents in the future.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Seip M, Trygstad O. Generalized lipodystrophy, congenital and acquired (lipoatrophy) Acta Paediatr Suppl. 1996;413:2–28. doi: 10.1111/j.1651-2227.1996.tb14262.x. [DOI] [PubMed] [Google Scholar]
- 2.Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350:1220–34. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- 3.Van Maldergem L, Magre J, Khallouf TE, Gedde-Dahl T, Jr, Delepine M, Trygstad O, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet. 2002;39:722–33. doi: 10.1136/jmg.39.10.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hegele RA, Joy TR, Al-Attar SA, Rutt BK. Thematic review series: Adipocyte Biology. Lipodystrophies: Windows on adipose biology and metabolism. J Lipid Res. 2007;48:1433–44. doi: 10.1194/jlr.R700004-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Garg A, Wilson R, Barnes R, Arioglu E, Zaidi Z, Gurakan F, et al. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab. 1999;84:3390–4. doi: 10.1210/jcem.84.9.6103. [DOI] [PubMed] [Google Scholar]
- 6.Agarwal AK, Arioglu E, De Almeida S, Akkoc N, Taylor SI, Bowcock AM, et al. AGPAT2 is mutated in congenital generalized lipo-dystrophy linked to chromosome 9q34. Nat Genet. 2002;31:21–3. doi: 10.1038/ng880. [DOI] [PubMed] [Google Scholar]
- 7.Magre J, Delepine M, Khallouf E, Gedde-Dahl T, Jr, Van Maldergem L, Sobel E, et al. BSCL Working Group. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Gen. 2001;28:365–70. doi: 10.1038/ng585. [DOI] [PubMed] [Google Scholar]
- 8.Windpassinger C, Auer-Grumbach M, Irobi J, Patel H, Petek E, Horl G, et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Gen. 2004;36:271–6. doi: 10.1038/ng1313. [DOI] [PubMed] [Google Scholar]
- 9.Daher E, Silva G, Júnior, Benevides V, Mendonça P, Bezerra H, Silva A, et al. Berardinelli syndrome. A case report with fatal outcome. Invest Clin. 2008;49:251–5. [PubMed] [Google Scholar]
- 10.Van Maldergem L. Berardinelli-Seip congenital lipodystrophy. Orphanetencyclopedia November. 2001. [Last accessed on 2005 May 12]. Available from: http://www.orpha.net/data/patho/GB/uk-berad.pdf .
- 11.Garg A, Fleckenstein JL, Peshock RM, Grundy SM. Peculiar distribution of adipose tissue in patients with congenital generalized lipodystrophy. J Clin Endocrinol Metab. 1992;75:358–61. doi: 10.1210/jcem.75.2.1639935. [DOI] [PubMed] [Google Scholar]
- 12.Gomes KB, Pardini VC, Fernandes AP. Clinical and molecular aspects of Berardinelli-Seip Congenital Lipodystrophy (BSCL) Clin Chim Acta. 2009;402:1–6. doi: 10.1016/j.cca.2008.12.032. [DOI] [PubMed] [Google Scholar]
- 13.Darmasdt GL, Sidbury R. The Skin. In: Behrman RE, Kliegman RM, Jenson HB, editors. Nelson Textbook of Paediatrics. 17th ed. Philadelphia: W B Saunders; 2003. pp. 2153–250. [Google Scholar]
- 14.Bjornstad PG, Foerster A, Ìhlen H. Cardiac findings in generalized lipodystrophy. Acta Paediatr Suppl. 1996;413:39–43. doi: 10.1111/j.1651-2227.1996.tb14264.x. [DOI] [PubMed] [Google Scholar]
- 15.Geffner ME, Golde DW. Selective insulin action on skin, ovary, and heart in insulin-resistant states. Diabetes Care. 1988;11:500–5. doi: 10.2337/diacare.11.6.500. [DOI] [PubMed] [Google Scholar]
- 16.Rheuban KS, Blizzard RM, Parker MA, Carter T, Wilson T, Gutgesell HP. Hypertrophic cardiomyopathy in total lipodystrophy. J Pediatr. 1986;109:301–2. doi: 10.1016/s0022-3476(86)80389-4. [DOI] [PubMed] [Google Scholar]
- 17.Geffner ME, Santulli TV, Jr, Kaplan AS. Hypertrophic cardiomyopathy in total lipodystrophy: Insulin action in the face of insulin resistance? J Pediatr. 1987;110:161. doi: 10.1016/s0022-3476(87)80317-7. [DOI] [PubMed] [Google Scholar]
- 18.Kher AS, Lahiri KR, Jain MK, Shah MD. Congenital lipodystrophy with defective leucocyte function (a case report) J Postgrad Med. 1990;36:48–50. [PubMed] [Google Scholar]