

Abstract
An important step in the malignant progression of HPV-associated lesions is the dysregulation of expression of the viral E6 and E7 oncogenes. This is often achieved through the loss of expression of E2, which represses the HPV LCR promoter and E6/E7 expression. Our previous studies confirmed a role for Brd4 in mediating the E2 transcriptional repression function, and identified JARID1C/SMCX and EP400 as contributors to E2-mediated repression. Here we show that TIP60, a component of the TIP60/TRRAP histone acetyltransferase complex, also contributes to the E2 repression function, and we extend our studies on SMCX. Di- and tri-methyl marks on histone H3K4 are reduced in the presence of E2 and SMCX, suggesting a mechanism by which SMCX contributes to E2-mediated repression of the HPV LCR. Together, these findings lead us to hypothesize that E2 recruits histone-modifying cellular proteins to the HPV LCR, resulting in transcriptional repression of E6 and E7.
Keywords: papillomavirus, E2, LCR, transcriptional repression, SMCX, methylation, chromatin, H3K4, EP400, TIP60
INTRODUCTION
Human papillomaviruses (HPV) infect squamous epithelial cells and cause a variety of epithelial lesions. ‘High risk’, alpha genus HPV types infect the mucosal epithelium and are associated with some cancers, most notably cervical cancer. A growing body of evidence also links HPV infection to other anogenital cancers and oropharyngeal cancers (zur Hausen, 2009). The viral E6 and E7 proteins account for the oncogenic potential of high-risk HPVs, in part through the ability of E6 to bind and degrade the tumor suppressor p53 and of E7 to bind and inactivate the retinoblastoma family of pocket proteins (Howley et al., 2013). Expression of the viral E6 and E7 genes is controlled through an upstream promoter and enhancer region referred to as the long control region (LCR). DNA viruses that usurp host cellular machinery for their replication, including papillomaviruses, tend to have chromatin structures much like cellular chromatin and to retain core histones when they are packaged into viral particles. Papillomavirus DNA is chromatinized much like cellular DNA, and it is thought to be subject to similar regulation by chromatin modifications. HPV genes impact chromatin modification in a variety of ways, for example HPV18 E6 protein interacts with and modulates the function of the histone acetyltransferase p300 (Thomas and Chiang, 2005) and HPV16 E7 protein has been reported to interact with the Mi2 ATPase-histone deacetylase complex (Brehm et al., 1999). In addition E7 induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming (McLaughlin-Drubin et al., 2011).
During the normal course of viral replication, the papillomavirus E2 protein regulates E6 and E7 oncogene expression. E2 binding to the four E2 binding sites within HPV LCRs results in diminished E6 and E7 expression (Bernard et al., 1989; Hwang et al., 1993; Thierry and Howley, 1991; Thierry and Yaniv, 1987). This could lead to repression of the HPV LCR in one or more of several ways. The binding of E2 to its cognate sites in the LCR may compete with binding of specific cellular transcription factors (Dong et al., 1994; Dostatni et al., 1991). Yet, the finding that single amino acid substitution mutants of E2 still competent for DNA binding are deficient in their ability to repress the E6/E7 promoter suggests that E2 recruits cellular factors to the LCR to mediate its repressive function (Dowhanick et al., 1995; Goodwin et al., 1998; Nishimura et al., 2000).
To identify the cellular proteins and pathways involved in E2-mediated transcriptional repression of the HPV18 LCR, we previously conducted a genome-wide siRNA screen (Smith et al., 2010). Our study confirmed the reported role for the E2-binding bromodomain protein Brd4 in E2-mediated silencing (Wu et al., 2006) and identified a number of genes that had not previously been implicated in E2 repression (Smith et al., 2010). We validated that both the demethylase JARID1C/SMCX and EP400, a component of the TIP60 histone acetyltransferase complex, contribute to E2-mediated repression and bind to E2 (Smith et al., 2010). Brd4, SMCX and EP400 contribute independently and additively to E2-mediated silencing, indicating that E2 functions through several distinct cellular complexes to repress E6 and E7 expression.
SMCX is a demethylase, also referred to as KDM5C or JARID1C. SMCX specifically demethylates tri- and di-methylated histone H3 lysine 4 (H3K4) (Iwase et al., 2007). EP400 is a component of the multi-subunit TIP60 histone acetyltransferase complex (Doyon and Cote, 2004) that was first identified through its ability to bind the adenovirus E1A protein (Fuchs et al., 2001). This complex also has ATP-dependent DNA helicase activities and is involved in transcriptional activation and repression functions (Ikura et al., 2000). In addition to EP400, this complex contains the TIP60 protein itself as well as other factors including include γ-actin, the actin-related-protein (ARP) BAF53 and EPC, a human homologue of the Drosophila Enhancer-of-Polycomb protein. TRRAP, a transcriptional regulatory protein found in the human STAGA/TFTC and PCAF complexes, is also a component of the TIP60 complex (Ikura et al., 2000; Vassilev et al., 1998). EP400 has been implicated in direct binding to the histone acetyltransferase domain of TIP60, blocking TIP60’s enzymatic activity and its co-activator function in regulation of p21 expression through the SWI3–ADA2–N-CoR–TFIIIB (SANT) domain of EP400 (Park et al., 2010).
We have continued these studies on E2-mediated transcriptional repression of the HPV18 LCR with the goal of elucidating the mechanisms by which SMCX and EP400 contribute to its downregulation. Here we show that E2, together with the cellular proteins SMCX and the TIP60 complex, contributes to the establishment of a specific pattern of chromatin modifications on and around the HPV LCR. This leads to repression of the papillomavirus oncogenes E6 and E7.
MATERIALS AND METHODS
Cell culture and cell lines
C33A and HeLa cells were maintained as monolayer cultures in high-glucose Dulbecco’s modified Eagle’s medium (hgDMEM) with 10% fetal bovine serum (FBS), 50 units/ml penicillin G and 50 μg/ml streptomycin sulfate (P/S). HeLa/sen2 RVY16E6 LXSN16E7 #21 cells (HeLa/16E6/16E7) were graciously provided by Dan DiMaio (Psyrri et al., 2004) and maintained as monolayer cultures in hgDMEM with 10% FBS, 10 mM HEPES (pH 7.2), 0.5 mg/ml G418, 125 μg/ml hygromycin B and P/S. To stably transduce HeLa/16E6/16E7 cells, pOZN or pOZN-HPV16E2 (p6072) were packaged following standard retrovirus production protocols and cells were infected with clarified supernatant of packaging cells (Ogawa et al., 2002; You et al., 2004; Zheng et al., 2009). The resulting cell lines are referred to as HeLa/16E6/16E7/pOZN and HeLa/16E6/16E7/16E2. C33A cells stably transduced with pOZN-HPV16E2 and HPV18LCR-luciferase (C33A/16E2/18LCR) have been previously described (Smith et al., 2010).
Expression plasmids
The following plasmids have been described: pcDVL (p1151); codon-optimized BPV1 E2 (C59 E2kz, p2450), pOZN-HA-BPV1E2 (p6094), pOZN-HA-HPV16E2 (p6093), pCMV-HPV16E2 (p3766), pDEST-HA-BPV1E2 (p6244), pDEST-HA-HPV16E2 (p6245), pOZ-N-HA-BPV-1 E2 (p6094), pOZ-N-FLAG-HA-BPV-1 E2 (p5066), pOZ-N-HA-BPV-1 E2R37A (p6752), pOZ-N-HA-BPV-1 E2I73A (p6753), and pOZ-N-HA-BPV-1 E2R37AI73A (p6754) (Schweiger et al., 2006; You et al., 2004; Zheng et al., 2009). pDEST-FLAG-SMCX and pDEST-HA-SMCX were graciously provided by Shigeki Iwase and Yang Shi (Iwase et al., 2007). pCMVb-FLAG-EP400 has been previously described (Fuchs et al., 2001). Plasmids derived from the C-βS mammalian expression vector, containing FLAG-tagged EP400 Fragments F1-F6 and the SANT domain as well as the FLAG-tagged EP400 full-length plasmid were kindly provided by Robert Roeder (Park et al., 2010). pcDNA-HA-TIP60 was graciously provided by Brendan Price (Sun et al., 2009). A retroviral pMSCV-V5-gateway vector was used in order to generate a pMSCV-V5-TIP60 plasmid (p6862).
siRNAs and transfections
siGENOME siRNA duplexes were purchased from Dharmacon/Thermo Fisher Scientific (Nontargeting siControl #1 (siC#1): D-001210-01; siGLO RED: D-001630-02; Brd4-05: D-004937-05; EP400-03: D021272-03; EPC1-01: D006376-01; TIP60 set of 4 duplexes: MQ-006301-01; SMCX set of 4 duplexes: MQ-010097-00; PLU1 set of 4 duplexes: MQ009899-00; RBP2 set of 4 duplexes: MQ003297-01; SMCY set of 4 duplexes: MQ-010820-00; and custom synthesis of siRNA duplex targeting HPV16E2 (16E2#2): GUUUAAAGAUGAUGCAGAA). Cells were transfected with siRNAs using DharmaFECT1 (HeLa cells) or DharmaFECT2 (C33A cells) following slight modifications to the manufacturer’s instructions (Dharmacon). Briefly, siRNAs were diluted in 1X siRNA buffer (Dharmacon), then complexed with DharmaFECT in OptiMEM in the wells/plates used for the experiment. Cells that had been depleted of antibiotics for one day were seeded on top of the siRNA/DharmaFECT mixture at densities to result in near confluent monolayers at the end of the experiment. After 24 h incubation at 37°C, media was removed and cells were incubated in hgDMEM plus 10% FCS for an additional 48 h.
Quantitation of luciferase activity
At the indicated times post-transfection, cells were washed, scraped and lysed in 1X Reporter Lysis Buffer (Promega). Clarified lysates were analyzed for luciferase activity using the Promega Luciferase Assay System and a luminometer. Lysate protein concentrations were determined using a protein assay kit (BCA protein assay kit, Thermo Fischer Scientific). All experiments were performed in triplicate.
Analysis of protein expression
Equivalent amounts of protein were suspended in protein sample buffer (50 mM Tris pH6.8, 5% SDS, 10% glycerol, 5% β-mercaptoethanol, 25mM NaF, 1mM Na3VO4, 5mM β-glycerophosphate, 1mM PMSF, 50 μM Leupeptin, 100 μM Pepstatin A), resolved by electrophoresis in Bis-Tris polyacrylamide gels and transferred to polyvinylidene fluoride membranes by electroblotting. Membranes were blocked in TNET (10 mM Tris pH 7.5, 50 mM NaCl, 2.5 mM EDTA pH 8.0, 0.1% tween) supplemented with 5% nonfat dry milk. Primary antibodies used in these experiments were against: HA (#12013819001, Roche), FLAG (F3165, Sigma), SMCX (NB100-55328, Novus Biologicals), EP400 (A300-541A, Bethyl Laboratories), Brd4 (Schweiger et al., 2006), TIP60 (sc-16623, Santa Cruz Biotechnology) and actin (MAB1501, Millipore). Bound antibodies were detected with horseradish peroxidase conjugated anti-rabbit or anti-mouse secondary antibodies. For detection of actin and GAPDH, membranes were incubated with an AlexaFluor 680 conjugated goat anti-mouse IgG secondary antibody (Molecular Probes/Invitrogen) and labeled proteins visualized using a LI-COR Odyssey Infrared Imaging System.
Quantitative RT-PCR
Total RNA was isolated from cell pellets using the NucleoSpin RNA II kit (Clontech) following manufacturer’s instructions. The concentration of each sample was determined by UV spectrophotometry (Nanodrop). To remove residual DNA contamination, purified RNAs were treated with DNase using the Turbo DNA-free kit (Ambion), which includes a DNase inactivation and removal step. Equal amounts of RNA were reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative real-time PCR was performed in an Applied Biosystems ABI 7500 Fast Sequence Detection System using TaqMan Fast Universal PCR Master Mix (Applied Biosystems), oligonucleotide primers and TaqMan dual-labeled probes (5′FAM, 3′Black hole quencher or 5′FAM, 3′Iowa Black Quencher; Integrated DNATechnologies). Real-time PCR assays to detect cellular genes were ordered from Applied Biosystems/Life Technologies and have assay ID numbers as follows: SMCX (KDM5C): Hs01011846_m1; PLU1 (KDM5B): Hs00981910_m1; actin: Hs01060665_g1.
Transient DNA transfections and immunoprecipitations
One day prior to transfection, cells were plated to result in near-confluent monolayers at the end of the experiment. Cells were transfected with the indicated plasmids using a 3:1 FuGENE (μl):DNA (μg) ratio according to the manufacturer’s protocol (Roche). At 48 h post-transfection, cells were washed with PBS, and then lysed in 1X lysis buffer (PBS, 0.5% NP40, 1X BD protease inhibitors, 1 mM DTT, 12.5 mM NaF, 1 mM Na3VO4, 12.5 mM β-glycerophosphate). Cell lysates were then sonicated for 8 sec at 35% intensity to shear nucleic acids and lysates were clarified by centrifugation. For immunoprecipitation experiments, anti-HA (Sigma A2095) or anti-FLAG beads (Sigma A2220) were added to lysates and incubated overnight at 4°C. Beads and bound proteins were washed 4x in 1X lysis buffer without DTT. Bound proteins were removed by briefly boiling beads in 1X sample buffer. Proteins were separated and analyzed as described above.
Chromatin Immunoprecipitations (ChIP)
At the time of harvest, cells were washed once with PBS, then incubated with 500 mM DTBP diluted in PBS for 30 min to crosslink DNA and protein. The reaction was quenched by adding 100 mM Tris/HCl (pH 8.0) and 150 mM NaCl, followed by 5 min incubation. Cells were washed with PBS, incubated for 10 min with 1% formaldehyde, followed by the addition of 100 mM glycine for an additional 10 min. Cells were lysed with cell lysis buffer (5 mM PIPES (pH 8.0), 85 mM KCl, 0.5% Nonidet P-40 and 1x BD protease inhibitors), pelleted and resuspended with buffer A (50 mM HEPES/KOH (pH 7.6), 500 mM NaCl, 1% Triton X100, 0.1% sodium deoxycholate, 1 mM EDTA). The mixture was then sonicated 5× 20 sec resulting in 200–500 base pair fragments, pelleted and resuspended in buffer A. The supernatant was pre-cleared with protein A/G-agarose (200 μL Protein A-agarose (PIERCE), 200 μL Protein G-agarose (PIERCE), 240 μL buffer A, 80 μL BSA (10 mg/mL), 80μL sheared salmon sperm DNA (10 mg/mL), 0.1% NaN3), diluted with buffer A and incubated with 5 μg of the appropriate antibody overnight at 4°C. Antibodies used were: Total H3 (ab1791, Abcam); H3K4me3 (ab1012, Abcam); H3K4me2 (07–030, Millipore); H3K4me1 (ab8895, Abcam); HA (ab9110, Abcam); rabbit IgG (ab46540, Abcam); and mouse IgG (ab37355, Abcam). Antibodies and bound protein were captured with protein A/G-agarose. The beads were washed with buffer A, buffer B (50 mM HEPES/KOH (pH 7.6), 0.3 M LiCl, 1 mM EDTA, 0.5% NP-40, 0.7% sodium deoxycholate) and TE (1 mM EDTA, 10 mM Tris (pH 8.0), 12.5 mM β-glycerophosphate, 12.5 mM NaF, 1 mM Na3VO4, 1X BD protease inhibitors) then the protein/DNA complexes were removed with elution buffer (50 mM NaHCO3, 1% SDS, 10 mM DTT, 0.3% sheared salmon sperm DNA, 10 mM EDTA and 0.2 M NaCl). Crosslinks were reversed by incubation at 65°C, followed by RNA and protein digestions with RNase A and proteinase K respectively. DNA was purified with the Qiagen PCR purification kit according to manufacturer’s instructions for qPCR quantification (Qiagen). Specific sequences present in equal volumes of input or immunoprecipitated DNA were quantified by real-time PCR performed in an Applied Biosystems ABI 7500 Fast Sequence Detection System using TaqMan Fast Universal PCR Master Mix (Applied Biosystems). Custom designed HPV18 and beta-actin promoter TaqMan assays (Integrated DNA Technologies) had sequences as follows: HPV18 L1 Forward primer 5′ AGCCTTTAGGTGTTGGCCTTAGT 3′, TaqMan probe 5′ 6-FAM-ACACTGAAAGTTCCCATGCCGCCA-Iowa Black quencher 3′, Reverse primer 5′ TTGTCCCTAACGTCCTCAGAAAC 3′. HPV18 LCR Forward primer 5′ GGTTGGGCAGCACATACTATACTTT 3′, TaqMan probe 5′ 6-FAM-CGGTCGGGACCGAAAACGGTG-Iowa Black quencher 3′, Reverse primer 5′ CATGGTATTGTGGTGTGTTTCTCACAT 3′. Beta-actin promoter Forward primer 5′ CGCACAGTGCAGCATTTTTT 3′, TaqMan probe 5′ 6-FAM-ACCCCCTCTCCCCTCCTTTTGCG 3′, Reverse primer 5′ CTCCCTCCTCCTCTTCCTCAA 3′. Unpaired t-tests were performed on all ChIP experiments to calculate two-tailed P values, with asterisks in the figures representing the level of significance (* ≤ 0.05 and ** ≤ 0.005).
RESULTS
Mutational analysis of the E2 transactivation domain in binding SMCX, Brd4 and EP400
The N-terminal transactivation domain of the papillomavirus E2 protein plays an essential role in E2-mediated repression due to its ability to complex with specific cellular factors such as Brd4 (You et al., 2004). Mutations in the E2 transactivation domain that obviate its transcriptional activation function affect the ability of E2 to repress the E6/E7 promoter (Dowhanick et al., 1995; Goodwin et al., 1998; Nishimura et al., 2000). Single amino acid substitution mutants of the BPV1 or HPV16 E2 transactivation domain at amino acids R37 or I73 are defective in binding Brd4 (Baxter and McBride, 2005; Schweiger et al., 2006), and R37 and I73 are exposed on the same face of the E2 transactivation domain (Antson et al., 2000; Schweiger et al., 2006). We have previously shown that both BPV1 E2 and HPV16 E2 coimmunoprecipitate with SMCX as well as EP400 (Smith et al., 2010).
To determine whether SMCX and/or EP400 also bind to the face of the E2 transactivation domain containing R37 and I73, we performed anti-HA immunoprecipitations of HA-tagged BPV1 E2, wild-type (wt) or alanine substitution mutants, in C33A cells co-transfected with expression plasmids encoding FLAG-tagged SMCX or EP400. BPV1 E2 was used in these studies because the binding of BPV1 E2 to Brd4, EP400 and SMCX is more robust and easier to detect (Smith et al., 2010). Expression plasmids were used to enhance their respective protein levels in the cells for detection in complex with E2. Cell lysates were harvested 48 h post-transfection and sonicated to reduce the size of nucleic acid fragments that might be involved in mediating E2 protein interactions with binding partners. Input and precipitated proteins were separated by SDS/PAGE and visualized by immunoblot analysis. Tagged BPV1 E2 (wt) as well as the R37A, I73A and R37A/I73A double mutants were expressed at comparable levels (Figure 1A) and could be efficiently immunoprecipitated with the HA antibody (Figure 1B). In agreement with previous studies, endogenous Brd4 co-immunoprecipitated with BPV1 E2 (wt), but not with the R37A, I73A or R37AI73A mutants (Figure 1B). SMCX co-immunoprecipitated with wt E2 as well as the R37A mutant, but co-immunoprecipitation with either the I73A or R37AI73A mutant was significantly reduced. These results suggest that the ability of SMCX to complex with E2 involves the same face of the E2 transactivation domain involved in Brd4 binding since the alanine point mutants have at least some effect on its co-immunoprecipitation with E2. In contrast, EP400 efficiently co-immunoprecipitated with the wt BPV1 E2 as well as each of the single mutants (R37A and I73A), but to a somewhat lesser extent with the double mutant R37A/I73A (compare the levels of EP400 in the E2 co-immunoprecipitation lanes with the levels in the input lanes). Neither BPV1 E2 R37 nor I73 is required for EP400 interaction with BPV1 E2, although the E2 double mutant is less efficient in binding.
Figure 1. N-terminal amino acids of BPV1 E2 contribute to interactions with Brd4, SMCX and EP400.

C33A cells were transfected with expression plasmids for HA-tagged wt or mutant (R37A, I73A, R37AI73A) BPV1 E2, FLAG-SMCX, FLAG-EP400 or the corresponding parental plasmids. 48 h post-transfection, cells were harvested and proteins immunoprecipitated with an antibody against HA. Total cellular proteins (A) and bound proteins (B) were separated by SDS-PAGE and visualized by immunoblot analysis using antibodies against HA (detection of BPV1 E2), Brd4, SMCX, EP400 and actin.
Mapping the regions of EP400 required for complexing with E2
To further characterize the interaction of E2 with EP400, we mapped the regions of EP400 involved in its ability to co-immunoprecipitate with E2 using six FLAG-tagged fragments of EP400 spanning the length of the protein (Park et al., 2010). A schematic representation of EP400 indicating its domains and the fragments used is illustrated in Figure 2A. We validated the expression of the various FLAG-tagged EP400 fragment constructs (Figure 2B) and used them to map the regions of EP400 involved in interacting with the papillomavirus E2 protein in C33A cells. In subsequent experiments, EP400 fragment levels were normalized based on the variation in EP400 fragment expression observed here by adjusting the amount of EP400 plasmid included in each condition. Fragment F3 was cytotoxic and could not be expressed at a higher level than shown. C33A cells were co-transfected with FLAG-tagged EP400 fragments and HA-tagged BPV1-E2, followed by HA-immunoprecipitation and western blotting with anti-HA and FLAG antibodies. Fragments F1, F2 and F4 of EP400, as well as the smaller SANT domain region of fragment F4 co-immunoprecipitated with E2 (Figure 2C and D). The regions of EP400 that complex with E2 are the same regions of EP400 that have been mapped for binding to TIP60 (Park et al., 2010).
Figure 2. The EP400 fragments F1, F2, F4 and the SANT domain co-immunoprecipitate with BPV1 E2.

(A) Schematic representation of the EP400 fragments relative to its identified domains (Park et al., 2010). (B) C33A cells were transfected with expression plasmids for the FLAG-tagged EP400 fragments (F1-F6 and SANT domain of F4). 48 h post-transfection, cells were harvested, total proteins separated by SDS-PAGE and visualized by immunoblot analysis using antibodies against FLAG (detection of EP400 fragments) and actin. (C) C33A cells were transfected with expression plasmids for HA-tagged BPV1 E2, FLAG-tagged EP400 fragments, or the corresponding parental plasmids. 48 h post-transfection, cells were harvested and proteins immunoprecipitated with an antibody against HA. Total cellular proteins (Input) and bound proteins (IP:HA) were separated by SDS-PAGE and visualized by immunoblot analysis using antibodies against HA (detection of BPV1 E2), FLAG (EP400 fragments) and actin. (D) The same experiment as described above in A was performed, except that only the FLAG-tagged F1, F4 or SANT domain of EP400 was co-transfected with HA-tagged BPV1 E2.
E2 interacts with TIP60
The finding that the regions of EP400 involved in complexing with E2 were the same as EP400 binding to TIP60, as well as the previous finding that other members of the TIP60 complex contributed to E2-mediated repression of the LCR (Smith et al., 2010), led us to investigate whether TIP60 is part of the cellular complex engaged by the papillomavirus E2 protein. We therefore tested whether TIP60 and BPV-1 E2 co-immunoprecipitate. C33A cells were co-transfected with the dual-tagged FLAG-HA-BPV1 E2 and HA-TIP60. As controls, cells were co-transfected with either the HA-TIP60 or the FLAG-HA-BPV1 E2 plasmid alone. FLAG-tagged EP400 fragment F4 was used as a positive control, since it has been documented to bind to TIP60 (Park et al., 2010). Anti-FLAG beads were used to immunoprecipitate BPV1 E2, and Western blot analysis for anti-FLAG revealed efficient immunoprecipitation for both FLAG-F4 and FLAG-HA-E2 (Figure 3A). Anti-HA immunoblotting revealed that either EP400 F4 or E2 pulled down HA-TIP60. The interaction between E2 and TIP60 was confirmed in a second cell line, HeLa cells (Figure 3B). HeLa is an HPV18 positive human cervical cancer cell line containing integrated HPV18 DNA; E6 and E7 are expressed from the viral endogenous LCR and HPV18 E2 is not expressed. The finding that E2 coimmunoprecipitated with both EP400 and TIP60 suggests that E2 interacts with the TIP60 complex, although we did not determine whether E2’s interactions with EP400 and TIP60 are direct or indirect, nor whether a single complex contains all three proteins.
Figure 3. BPV1 E2 co-immunoprecipitates with TIP60.

C33A (A) or HeLa (B) cells were transfected with expression plasmids for HA-tagged TIP60, FLAG-tagged F4 fragment of EP400 fragment, dual HA- and FLAG-tagged BPV1 E2 or the corresponding parental plasmids. 48 h post-transfection, cells were harvested and proteins immunoprecipitated with an antibody against FLAG. Total cellular proteins (Input) and bound proteins (IP:FLAG) were separated by SDS-PAGE and visualized by immunoblot analysis using antibodies against HA (detection of BPV1 E2 and TIP60), FLAG (F4 fragment of EP400 and BPV1 E2) and actin. Anti-Brd4 antibodies were used to detect Brd4 as a positive control for an E2 interacting protein in HeLa cells.
TIP60 contributes to the repression of the HPV18 LCR
Several components of the TIP60 complex (EP400, Brd8, EPC1 and GAS41) were hits in our previous siRNA screen that identified genes involved in E2-mediated repression of the HPV18 LCR (Smith et al., 2010). The TIP60 histone acetyltransferase protein was not identified as a repressor in the primary screen, which utilized the Dharmacon siGENOME SMARTpool library (primary screen data available via PubChem BioAssay AID: 624099). To test whether the TIP60 protein is a repressor of the HPV LCR that was not detected in the primary screen, we tested the TIP60 siRNA duplexes individually. Each duplex was transfected into C33A cells that stably express an HPV18 LCR luciferase reporter as well as HPV16E2 (C33A/16E2/18LCR), which effectively represses luciferase expression from the LCR. Non-targeting siRNA (siC#1) was included as a negative control and siRNA duplexes targeting HPV16 E2 and SMCX were included as positive controls. The impact of these siRNA duplexes on luciferase expression in these cells was established in our previous study (Smith et al., 2010). As expected, knockdown of HPV16 E2 resulted in a significant increase in luciferase, indicating an alleviation of repression of the HPV18 LCR. Luciferase levels also increased following SMCX knockdown. When compared to the siC#1-transfected cells, each of the four siRNA duplexes targeting TIP60 relieved E2-mediated repression of the LCR, although the extent varied (Figure 4A). Knockdown of HPV16 E2 and TIP60 protein levels were validated by western blot analysis (Figure 4B).
Figure 4. Depletion of TIP60 increases activity of the HPV-18 LCR.

C33A/16E2/18LCR cells were reverse transfected with non-targeting control siRNA (siC#1) or siRNA duplexes targeting HPV-16 E2, SMCX or TIP60. At 72 h post-transfection, lysates were harvested. Luciferase levels and total protein were quantitated. (A) Graph represents the relative luciferase units (RLU) normalized to protein extract concentration (RLU/[Protein]). (B) Western blots indicate depletion of HPV16 E2 and TIP60 proteins, as well as actin protein levels.
EP400 and TIP60 have similar effects on E2-mediated transcriptional repression of the HPV-18 LCR
Since EP400 and TIP60 are each components of the TIP60 histone acetyl transferase complex (Doyon and Cote, 2004) and each contributes to transcriptional repression of the HPV18 LCR, we next asked whether they contribute in concert or individually to E2 regulation of the LCR. To answer this question, C33A/16E2/18LCR cells were transfected with a non-targeting siRNA duplex (siC#1) or siRNAs directed at HPV16 E2, Brd4, EP400, TIP60 or EPC1 (Figure 5). EPC1 is another component of the TIP60 complex previously implicated in contributing to E2-mediated regulation of the LCR. As expected, the greatest increase in luciferase activity was observed when HPV16 E2 was depleted. In our previous study, we showed that depletion of both of these proteins resulted in an additive relief of E2-mediated repression (Smith et al., 2010). Co-transfection of the siRNA duplexes targeting TIP60 and EP400 resulted in an increase in luciferase activity comparable to that observed following the knockdown of TIP60 alone. Similar findings were obtained when EPC1 was depleted in combination with either EP400 or TIP60. These data indicate that depletion of the individual components of the TIP60 complex do not have additive or synergistic effects on the relief of repression of the HPV18 LCR, supporting the hypothesis that the TIP60 complex as a whole contributes to E2 regulation of E6 and E7 expression.
Figure 5. Combinatorial siRNA transfections reveal that members of the NuA4/TIP60 complex act together in contributing to E2-mediated repression.

C33A/16E2/18LCR cells were transfected with the indicated siRNA duplexes alone or in combination at a final concentration of 10 nM/duplex. At 72 h post-transfection, lysates were harvested. Luciferase levels and total protein were quantitated. The graph represents RLU/[protein]. Experiments were performed in triplicate, with each bar representing the average ± SD.
Other members of the JARID1 family of histone demethylases do not contribute to E2-mediated transcriptional repression of the HPV18 LCR in cervical cancer cells
SMCX is a member of the JARID1 family of histone demethylases, which includes three other members: SMCY/JARID1D/KDM5D, Rbp2/JARID1A/KDM5A and Plu1/JARID1B/KDM5B. None of these three genes scored as hits in our genome-wide siRNA screen (Smith et al., 2010). The gene encoding SMCY is located on the Y chromosome and its expression is limited to the testes. Therefore although SMCY is not expressed in C33A cells, the siRNAs targeting SMCY were included as negative controls. By qPCR we determined that Plu1, but not Rbp2 was expressed in the C33A cells, therefore Rbp2 could not be contributing to E2-mediated repression in C33A cells. To determine whether Plu1 was a false negative in our screen and might also contribute to E2-mediated repression of the HPV LCR, we tested the individual siRNA duplexes for each of the JARID1 family of histone demethylases in C33A/16E2/18LCR cells. As shown in Figure 6A, siRNA directed at either 16E2 or Brd4 significantly increased luciferase levels indicating a relief of E2-mediated repression of the HPV18 LCR. Similarly, siRNAs directed against SMCX relieved E2-mediated repression, most notably observed with the SMCX-02 and -04 siRNAs. In contrast, no change in luciferase levels was observed in cells transfected with any of the siRNAs against Plu1, SMCY or Rbp2. We therefore conclude that E2-mediated repression specifically involves SMCX and not other members of the JARID1 family of histone demethylases. Depletion of SMCX and PLU1 transcripts by the individual siRNAs were quantitated by qPCR and are shown in Figure 6B normalized to β-actin transcript levels.
Figure 6. E2-mediated transcriptional repression of the HPV18 LCR in cervical cancer cells does not require other JARID1 family proteins.

C33A/16E2/18LCR c1 cells were transfected with the indicated siRNAs at a final concentration of 20 nM. (A) Cell extracts were harvested 72 h post-transfection to determine RLU and total protein levels. Experiments were performed in triplicate, with each bar representing the average ± SD. (B) SMCX and PLU1 transcript levels were quantitated by qPCR in cells transfected with the indicated siRNAs. The graph displays the relative mRNA levels normalized to β-actin transcript levels.
Histone H3 trimethylation at lysine 4 is reduced at the HPV18 LCR in an E2- and SMCX-dependent manner
If SMCX contributes to E2-mediated repression of the LCR by altering the methylation status of histone H3 lysine 4 there, we would predict that the loss of E2 from HPV-positive cells would eliminate the recruitment of SMCX to the LCR and alter chromatin modifications at that site. To test this hypothesis, we first characterized E2 binding to the endogenous HPV18 LCR in HeLa cells by chromatin immunoprecipitation (ChIP). For these experiments, we utilized a HeLa cell line that had been engineered to additionally express HPV16 E6 and E7 from retroviral promoters, which are not subject to E2 repression (Psyrri et al., 2004). We further introduced into these cells either a vector encoding BPV1 HA-FLAG-E2 or the empty parental plasmid (Smith et al., 2010). By ChIP using anti-HA antibodies, we were able to detect E2 bound at the HPV18 LCR where four E2 binding sites are located but not over a region of the L1 gene devoid of any E2 binding sites (Figure 7).
Figure 7. BPV1 E2 is present at the HPV18 LCR in HeLa cells.
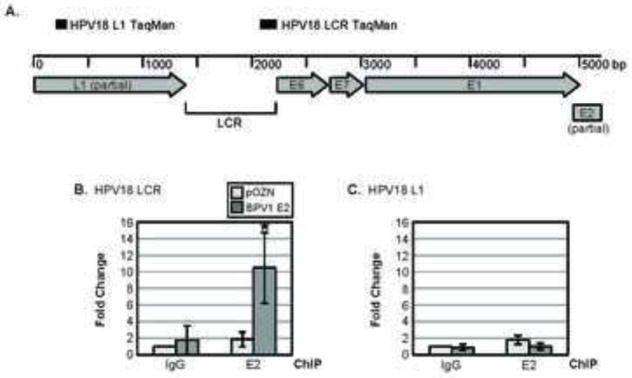
(A) Map of the HPV18 viral genome in HeLa cells, with the DNA sequences that were amplified via PCR indicated. DNA sequences encompassing the transcription start site in the HPV18 LCR (B) or L1 ORF (C) and associated with BPV1 E2 were quantitated in HeLa/16E6/16E7/BE2 or HeLa/16E6/16E7/pOZN cells after ChIP using antibodies against the HA-tag on BPV1 E2 via qPCR. The fold change displayed on the y-axis represents data normalized to the β-actin transcriptional start site, with each bar representing the average of three experiments ± SD. Asterisks represent the level of significance (* p-value ≤ 0.05 and ** p-value ≤ 0.005), as determined via unpaired t-tests.
To determine the potential effects of the E2-SMCX interaction, we next examined the methylation status of histone H3 lysine 4 at two regions of the HPV genome in the presence and absence of E2. Trimethylated histone H3 lysine 4 (H3K4me3) is one of the marks of transcriptionally active chromatin. In part through its ability to demethylate tri- and di-methylated lysine 4 of histone H3, SMCX functions as a transcriptional repressor (Iwase et al., 2007; Tahiliani et al., 2007). ChIPs were performed examining the LCR and the L1 region of the integrated HPV18 DNA sequences using antibodies specific to total histone H3, H3K4me3, H3K4me2 or H3K4me1 in HeLa/16E6/16E7/BE2 compared to HeLa/16E6/16E7/pOZN cells. The amount of DNA corresponding to the HPV18 LCR, L1 region or the β-actin promoter region was determined by qPCR. Results are expressed as the amount of the HPV18 LCR associated with a histone or modified histone relative to the amount of the β-actin promoter associated with the same histone and are the average of three independent experiments. Whereas the amount of total H3 in association with the HPV18 LCR E6/E7 promoter was similar in HeLa cells with or without E2, there was a significant, greater than 10-fold decrease in the amount of H3K4me3 associated with the HPV18 LCR when E2 was present (Figure 8A). There was also a 3-fold decrease in H3K4me2 bound to the HPV18 LCR promoter in the E2 expressing cells compared to the control HeLa/16E6/16E7/pOZN cells. H3K4me1 was not affected by E2. In contrast, the analysis of H3K4 modifications in the L1 ORF showed minimal difference in the presence or absence of E2 (Figure 8B), indicating that the E2-dependent decrease in H3K4me3 and H3K4me2 at the HPV18 LCR was specific to the LCR E6/E7 promoter. We note that due to the lack of antibodies suitable to ChIP SMCX, we were unable to examine whether or not SMCX itself was at the LCR.
Figure 8. E2 and SMCX contribute to a reduction in H3K4me3 at the HPV18 LCR.

The amount of total H3, H34K4me3, H3K4me2 and H3K4me1 associated with the transcription start site present in the HPV18 LCR (A) or HPV18 L1 ORF (B) was determined in HeLa/16E6/16E7/BE2 or HeLa/16E6/16E7/pOZN cells by ChIP. The fold change displayed on the y-axis represents data normalized to the β-actin transcriptional start site, with each bar representing the average of three experiments ± SD. HeLa/16E6/16E7/BE2 (C, E) or HeLa/16E6/16E7/pOZN cells (D, F) were transfected with the indicated siRNAs (legend in C). 72 h post-transfection, the amount of total H3, H34K4me3, H3K4me2 and H3K4me1 associated with the HPV18 LCR (C, D) or L1 ORF (E, F) was determined by ChIP. The fold change displayed on the y-axis represents data normalized to the β-actin transcriptional start site, with each bar representing the average of two experiments ± the difference between them. Asterisks represent the level of significance (* p-value ≤ 0.05 and ** p-value ≤ 0.005), as determined via unpaired t-tests.
To determine whether the decreased H3K4me3 and H3K4me2 at the HPV18 E6/E7 LCR was dependent upon SMCX, we performed similar ChIP experiments in cells depleted of SMCX by siRNA knockdown. Compared to non-targeting siRNA-treated cells (siC#1), the knockdown of SMCX in the E2-expressing HeLa/16E6/16E7 cells led to increased H3K4me3, H3K4me2 and H3K4me1 at the HPV18 E6/E7 promoter (Figure 8C, white bars versus light grey bars). Similar increases in H3K4me3 and H3K4me2 were observed in the cells transfected with BPV1 E2 siRNA (Figure 8C, dark grey bars). In contrast, the methylation levels of H3K4 at the HPV18 E6/E7 promoter in either siC#1 siRNA- or in SMCX siRNA-transfected HeLa/16E6/16E7/pOZN cells did not affect H3K4me3, H3K4me2 or H3K4me1 levels (Figure 8D, white bars versus grey bars). Together, these results indicate that SMCX is responsible for the decreased methylation of H3K4 at the HPV18 LCR in an E2-dependent manner.
To determine if the SMCX- and E2-dependent changes in the methylation status of H3K4 described above are specific to the HPV18 LCR, we performed the same siRNA knock down/ChIP experiments examining the L1 ORF since E2 does not affect transcription from this region of the HPV18 genome. Knockdown of BPV1 E2 did not impact the methylation states of H3K4 in the L1 ORF (Figure 8E, white bars versus dark grey bars). While the depletion of SMCX did increase L1-associated H3K4me3, H3K4me2 and H3K4me1 compared to the siC#1-transfected cells (Figure 8E, white bars versus light grey bars), the change was modest (2.5-fold) compared to the over 10-fold increase of HPV18 E6/E7 promoter-associated H3K4 in HeLa/16E6/16E7/BE2 cells (Figure 8C). The knockdown of SMCX also increased H3K4 methylation at the HPV18 L1 ORF in HeLa/16E6/16E7/pOZN cells (Figure 8F), further supporting the interpretation that any SMCX-mediated effect of H3K4 methylation at the L1 ORF was not mediated by E2.
These data indicate that there was a specific E2- and SMCX-dependent decrease in the methylation of H3K4me3 and H3K4me2 at the HPV18 LCR (Figure 8). Based on this observation and evidence indicating that specific nucleosome structures along the HPV16 and HPV18 LCRs contribute to E6 and E7 expression (Stunkel and Bernard, 1999), we hypothesize that E2 recruits SMCX to the HPV18 LCR to maintain a transcriptionally inactive promoter, thus decreasing E6 and E7 expression. Using siRNA knockdown and ChIP experiments, we demonstrate a specific E2- and SMCX-dependent decrease in the methylation of H3K4me3 and H3K4me2 at the HPV18 LCR.
DISCUSSION
In this study, we have extended our analysis of the cellular genes and pathways that are engaged by the papillomavirus E2 regulatory protein in mediating its transcriptional repression function. This E2 mediated repression function is important during papillomavirus replication in the negative regulation of the long control region (LCR) promoter that drives E6 and E7 expression. For the high risk HPVs associated with human cancer, E6 and E7 are responsible for cellular transformation. Thus, through its repression of E6 and E7, E2 effectively functions as a tumor suppressor gene in HPV-positive cancers. In HPV-associated cancers, the expression of E2 is frequently lost following integration of the HPV genome into cellular DNA in a manner that disrupts the integrity of the E2 ORF (Howley et al., 2013). The re-expression of E2 in HPV positive cancers causes cellular senescence due to the silencing of the E6 and E7 oncoproteins (Goodwin and DiMaio, 2000; Wells et al., 2000).
Our previous unbiased, genome-wide siRNA screen demonstrated that 96 cellular genes are involved in repression of the HPV LCR promoter (Smith et al., 2010). Although some of these genes could repress the HPV LCR in the absence of E2, most could not. From this study came the realization that E6/E7 repression occurs through multiple E2-dependent and -independent mechanisms. Three of the strongest hits in the screen were Brd4, EP400 and SMCX, and each of these proteins contributes to E2-mediated repression in an independent and additive fashion. Brd4, a cellular protein previously implicated in E2-mediated repression of the LCR (Wu et al., 2006), was the focus of our initial follow-up studies. In addition, we showed that E2 could bind SMCX and EP400. These binding partners of E2 are notable since both have the potential to modify chromatin – either directly, in the case of SMCX, or in association with the TIP60 complex, as for EP400. The goal of the current study was to further investigate how chromatin modifying enzymes or complexes contribute to E2-mediated repression of the HPV LCR.
Virus-mediated control of chromatin modification has been established as an important regulatory mechanism. Many viruses target TIP60, often through a protein-protein interaction. Indeed TIP60 is named Tat interacting protein, 60 kDa based on its initial identification as a binding partner of HIV Tat (Kamine et al., 1996). In general, TIP60 functions as a repressor of transcription, and some virus interactions with TIP60 result in TIP60 inactivation, often through degradation. In addition to HIV Tat, HCMV pUL27 (Reitsma et al., 2011), Adenovirus E1B55K and E4orf6 (Gupta et al., 2013) and HPV16 and HPV18 E6 (Jha et al., 2010) have been reported to target TIP60 for proteasome-mediated degradation. Consistent with its inactivation by a broad range of viruses, TIP60 and other members of its complex have recently been shown to have anti-flaviviral effects in mammalian cells (Yasunaga et al., 2014). Thus TIP60 serves as a regulator of antiviral activity for many diverse viruses. In contrast, TIP60 promotes progression of some virus infections. KSHV LANA binds but does not degrade TIP60 (Shamay et al., 2012), and it appears that a variety of herpsesviruses exhibit impaired replication when TIP60 is depleted (Li et al., 2011). It seems that TIP60 could have context-dependent positive or negative effects on viral functions and that viruses could redirect, not just inactivate, its functions to promote virus replication.
Although our previous study focused on EP400, one component of the TIP60 histone complex, other components including Brd8 and EPC1, also scored in our assay and suggested a role for the complex as a whole (Smith et al., 2010). We therefore wished to determine whether other components of the TRRAP/TIP60 complex also contributed to repression or whether EP400 contributes uniquely. Subsequent to our publication, the TIP60 protein was shown to function in the negative regulation of the high risk HPV LCR E6/E7 promoter, although in a manner that was independent of E2 (Jha et al., 2010). Since TIP60 had not scored as a hit in our whole genome siRNA screen, we examined each of the four siRNA duplexes in the TIP60 SMARTpool individually to determine their effects on E2-mediated repression. Although the TIP60 SMARTpool itself did not relieve E2-mediated repression of the HPV18 LCR in the initial screen, each of the individual siRNAs did when tested individually (Figure 4). Combination experiments with siRNAs targeting TIP60, EP400 and EPC1 did not provide additive relief of E2 repression compared to the knockdown of individual TIP60 complex components. Thus, depletion of any one subunit of the TIP60 histone acetyl transferase complex may be sufficient to override E2-driven repression.
In addition to E2’s ability to bind EP400, we demonstrate co-immunoprecipitation between E2 and TIP60, and mapped the binding regions on EP400 for E2 to the same regions that have been previously mapped for the binding of TIP60 to EP400 (Park et al., 2010). Although our data indicate that E2 engages the TIP60 complex, we have not identified which specific protein (or proteins) E2 binds within the complex. Nonetheless, our results validate a role for the TIP60 complex in E2-mediated repression of the high risk HPV LCR. Our previous study showed an E2-dependence for both EP400 and EPC in E2-mediated repression of E6/E7 expression (Smith et al., 2010). We note however that an E2-independent role for TIP60 repression of the LCR has been established and shown to occur in a Brd4-dependent manner (Jha et al., 2010).
To our knowledge, our study describes the first example of a virus-encoded protein that binds to and/or alters the target of SMCX. Several viruses modulate the function of other lysine demethylases in the KDM family. For example herpes simplex virus and varicella zoster virus recruit the lysine-specific demethylase 1 (LSD1/KDM1A) to the viral immediate-early promoter, where it enables viral lytic gene expression (Liang et al., 2009), and consistent with this an inhibition of LSD1 blocks HSV gene expression (Liang et al., 2013). The latency-associated nuclear antigen (LANA) encoded by the Kaposi’s Sarcoma herpesvirus (KSHV) binds to KDM3A, and the KSHV K-bZIP binds to KDM4A (Chang et al., 2011; Kim et al., 2013), and one report suggests that KDM6B overexpression results from Epstein-Barr virus infection (Anderton et al., 2011).
Our data examining SMCX and other members of the JARID1 family of histone demethylases (Rbp2, Plu1 and SMCY) indicate that SMCX is the sole member that contributes to E2-mediated transcriptional repression. As demonstrated in the ChIP experiments, there is a clear decrease in the level of trimethylated H3K4 at the HPV18 LCR in the presence of E2. We go on to show that this decrease in H3K4me3 is specific to the HPV18 LCR and requires both SMCX and the papillomavirus E2 protein. We hypothesize that E2, through its ability to bind to the E2 binding sites within the HPV18 LCR, recruits additional cellular proteins, including SMCX. SMCX then demethylates H3K4, thus contributing to decreased transcription of the papillomavirus E6 and E7 oncogenes through this chromatin modification.
The ability of the papillomavirus E2 protein to regulate expression of E6 and E7 from the LCR is complex, involving multiple cellular proteins. These proteins, including Brd4, SMCX and the TRAAP/TIP60 complex, contribute to repression through distinct mechanisms and their activities are required in order to maximize/to ensure maximal E2-mediated repression. The chromatin microenvironment surrounding the LCR is key, as all of these proteins modulate chromatin modifications. E2 plays a central role, as it is able to recruit cellular proteins to the E6 and E7 promoter. Thus, the elimination of E2 during carcinogenesis disrupts the ability of Brd4, SMCX and members of the TIP60 complex to influence expression from the LCR, resulting in increased expression of the papillomavirus oncogenes and HPV-associated cancer.
Research Highlights (for review).
Chromatin modifying enzymes contribute to E2-medaied transcriptional repression.
Papillomavirus E2 proteins bind TIP60.
TIP60/TRRAP histone acetylase complex contributes to E2-mediated repression.
SMCX is the sole JARID1 histone demethylase involved in E2-mediated repression.
Acknowledgments
We thank S. Iwase and Y. Shi (Harvard Medical School) for providing SMCX expression constructs, D. DiMaio (Yale University) for the HeLa/16E6/16E7 cells, B. Price (Dana Farber Cancer Institute) for TIP60 expression construct, and R. Roeder (Rockefeller University) for EP400 fragment expression constructs. We are grateful to members of the Howley lab for helpful discussions and feedback on various aspects on this project, in particular L. Zhao for constructing the mutant BPV1 E2 plasmids. This work was supported by NIH grants T32CA009361 to J.A.S., individual NRSA F32AI080075 to E.A.W., R01CA116720 to P.M.H. and P01CA050661 to D.M.L., J.A.D. and P.M.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderton JA, Bose S, Vockerodt M, Vrzalikova K, Wei W, Kuo M, Helin K, Christensen J, Rowe M, Murray PG, et al. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin’s Lymphoma. Oncogene. 2011;30:2037–2043. doi: 10.1038/onc.2010.579. [DOI] [PubMed] [Google Scholar]
- Antson AA, Burns JE, Moroz OV, Scott DJ, Sanders CM, Bronstein IB, Dodson GG, Wilson KS, Maitland NJ. Structure of the intact transactivation domain of the human papillomavirus E2 protein. Nature. 2000;403:805–809. doi: 10.1038/35001638. [DOI] [PubMed] [Google Scholar]
- Baxter MK, McBride AA. An acidic amphipathic helix in the Bovine Papillomavirus E2 protein is critical for DNA replication and interaction with the E1 protein. Virology. 2005;332:78–88. doi: 10.1016/j.virol.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Bernard BA, Bailly C, Lenoir MC, Darmon M, Thierry F, Yaniv M. The human papillomavirus type 18 (HPV18) E2 gene product is a repressor of the HPV18 regulatory region in human keratinocytes. J Virol. 1989;63:4317–4324. doi: 10.1128/jvi.63.10.4317-4324.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm A, Nielsen SJ, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999;18:2449–2458. doi: 10.1093/emboj/18.9.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PC, Fitzgerald LD, Hsia DA, Izumiya Y, Wu CY, Hsieh WP, Lin SF, Campbell M, Lam KS, Luciw PA, et al. Histone demethylase JMJD2A regulates Kaposi’s sarcoma-associated herpesvirus replication and is targeted by a viral transcriptional factor. J Virol. 2011;85:3283–3293. doi: 10.1128/JVI.02485-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G, Broker TR, Chow LT. Human papillomavirus type 11 E2 proteins repress the homologous E6 promoter by interfering with the binding of host transcription factors to adjacent elements. J Virol. 1994;68:1115–1127. doi: 10.1128/jvi.68.2.1115-1127.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostatni N, Lambert PF, Sousa R, Ham J, Howley PM, Yaniv M. The functional BPV-1 E2 transactiving protein can act as a repressor by preventing formulation of the initiation complex. Genes & Development. 1991;5:1657–1671. doi: 10.1101/gad.5.9.1657. [DOI] [PubMed] [Google Scholar]
- Dowhanick JJ, McBride AA, Howley PM. Suppression of cellular proliferation by the papillomavirus E2 protein. J Virol. 1995;69:7791–7799. doi: 10.1128/jvi.69.12.7791-7799.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y, Cote J. The highly conserved and multifunctional NuA4 HAT complex. Curr Opin Genet Dev. 2004;14:147–154. doi: 10.1016/j.gde.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Fuchs M, Gerber J, Drapkin R, Sif S, Ikura T, Ogryzko V, Lane WS, Nakatani Y, Livingston DM. The p400 complex is an essential E1A transformation target. Cell. 2001;106:297–307. doi: 10.1016/s0092-8674(01)00450-0. [DOI] [PubMed] [Google Scholar]
- Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci U S A. 2000;97:12513–12518. doi: 10.1073/pnas.97.23.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin EC, Naeger LK, Breiding DE, Androphy EJ, DiMaio D. Transactivation-competent bovine papillomavirus E2 protein is specifically required for efficient repression of human papillomavirus oncogene expression and for acute growth inhibition of cervical carcinoma cell lines. J Virol. 1998;72:3925–3934. doi: 10.1128/jvi.72.5.3925-3934.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Jha S, Engel DA, Ornelles DA, Dutta A. Tip60 degradation by adenovirus relieves transcriptional repression of viral transcriptional activator EIA. Oncogene. 2013;32:5017–5025. doi: 10.1038/onc.2012.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howley PM, Schiller JT, Lowy DR. Papillomaviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, Lippincott: Williams and Wilkins; 2013. pp. 1662–1703. [Google Scholar]
- Hwang ES, Riese DJ, Settleman J, Nilson LA, Honig J, Flynn S, DiMaio D. Inhibition of cervical carcinoma cell line proliferation by the introduction of a bovine papillomavirus regulatory gene. J Virol. 1993;67:3720–3729. doi: 10.1128/jvi.67.7.3720-3729.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–473. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Jha S, Vande Pol S, Banerjee NS, Dutta AB, Chow LT, Dutta A. Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol Cell. 2010;38:700–711. doi: 10.1016/j.molcel.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamine J, Elangovan B, Subramanian T, Coleman D, Chinnadurai G. Identification of a cellular protein that specifically interacts with the essential cysteine region of the HIV-1 Tat transactivator. Virology. 1996;216:357–366. doi: 10.1006/viro.1996.0071. [DOI] [PubMed] [Google Scholar]
- Kim KY, Huerta SB, Izumiya C, Wang DH, Martinez A, Shevchenko B, Kung HJ, Campbell M, Izumiya Y. Kaposi’s sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen regulates the KSHV epigenome by association with the histone demethylase KDM3A. J Virol. 2013;87:6782–6793. doi: 10.1128/JVI.00011-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhu J, Xie Z, Liao G, Liu J, Chen MR, Hu S, Woodard C, Lin J, Taverna SD, et al. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe. 2011;10:390–400. doi: 10.1016/j.chom.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Quenelle D, Vogel JL, Mascaro C, Ortega A, Kristie TM. A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. MBio. 2013;4:e00558–00512. doi: 10.1128/mBio.00558-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med. 2009;15:1312–1317. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Crum CP, Munger K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc Natl Acad Sci U S A. 2011;108:2130–2135. doi: 10.1073/pnas.1009933108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura A, Ono T, Ishimoto A, Dowhanick JJ, Frizzell MA, Howley PM, Sakai H. Mechanisms of human papillomavirus E2-mediated repression of viral oncogene expression and cervical cancer cell growth inhibition. J Virol. 2000;74:3752–3760. doi: 10.1128/jvi.74.8.3752-3760.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]
- Park JH, Sun XJ, Roeder RG. The SANT domain of p400 ATPase represses acetyltransferase activity and coactivator function of TIP60 in basal p21 gene expression. Mol Cell Biol. 2010;30:2750–2761. doi: 10.1128/MCB.00804-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psyrri A, DeFilippis RA, Edwards AP, Yates KE, Manuelidis L, DiMaio D. Role of the retinoblastoma pathway in senescence triggered by repression of the human papillomavirus E7 protein in cervical carcinoma cells. Cancer Res. 2004;64:3079–3086. doi: 10.1158/0008-5472.can-03-3739. [DOI] [PubMed] [Google Scholar]
- Reitsma JM, Savaryn JP, Faust K, Sato H, Halligan BD, Terhune SS. Antiviral inhibition targeting the HCMV kinase pUL97 requires pUL27-dependent degradation of Tip60 acetyltransferase and cell-cycle arrest. Cell Host Microbe. 2011;9:103–114. doi: 10.1016/j.chom.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Schweiger MR, You J, Howley PM. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J Virol. 2006;80:4276–4285. doi: 10.1128/JVI.80.9.4276-4285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamay M, Liu J, Li R, Liao G, Shen L, Greenway M, Hu S, Zhu J, Xie Z, Ambinder RF, et al. A protein array screen for Kaposi’s sarcoma-associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity, and telomere shortening. J Virol. 2012;86:5179–5191. doi: 10.1128/JVI.00169-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, White EA, Sowa ME, Powell ML, Ottinger M, Harper JW, Howley PM. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc Natl Acad Sci U S A. 2010;107:3752–3757. doi: 10.1073/pnas.0914818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stunkel W, Bernard HU. The chromatin structure of the long control region of human papillomavirus type 16 represses viral oncoprotein expression. J Virol. 1999;73:1918–1930. doi: 10.1128/jvi.73.3.1918-1930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11:1376–1382. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- Thierry F, Howley PM. Functional analysis of E2 mediated repression of the HPV-18 P105 promoter. New Biologist. 1991;3:90–100. [PubMed] [Google Scholar]
- Thierry F, Yaniv M. The BPV1 E2 trans-acting protein can be either an activator or a repressor of the HPV18 regulatory region. EMBO J. 1987;6:3391–3397. doi: 10.1002/j.1460-2075.1987.tb02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MC, Chiang CM. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol Cell. 2005;17:251–264. doi: 10.1016/j.molcel.2004.12.016. [DOI] [PubMed] [Google Scholar]
- Vassilev A, Yamauchi J, Kotani T, Prives C, Avantaggiati ML, Qin J, Nakatani Y. The 400 kDa subunit of the PCAF histone acetylase complex belongs to the ATM superfamily. Mol Cell. 1998;2:869–875. doi: 10.1016/s1097-2765(00)80301-9. [DOI] [PubMed] [Google Scholar]
- Wells SI, Francis DA, Karpova AY, Dowhanick JJ, Benson JD, Howley PM. Papillomavirus E2 induces senescence in HPV positive cells via pRB- and p21CIP-dependent pathways. EMBO J. 2000;19:5762–5771. doi: 10.1093/emboj/19.21.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang CM. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006;20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga A, Hanna SL, Li J, Cho H, Rose PP, Spiridigliozzi A, Gold B, Diamond MS, Cherry S. Genome-wide RNAi screen identifies broadly-acting host factors that inhibit arbovirus infection. PLoS Pathog. 2014;10:e1003914. doi: 10.1371/journal.ppat.1003914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J, Croyle JL, Nishimura A, Ozato K, Howley PM. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell. 2004;117:349–360. doi: 10.1016/s0092-8674(04)00402-7. [DOI] [PubMed] [Google Scholar]
- Zheng G, Schweiger MR, Martinez-Noel G, Zheng L, Smith JA, Harper JW, Howley PM. Brd4 Regulation of Papillomavirus E2 Protein Stability. J Virol. 2009;83:8683–8692. doi: 10.1128/JVI.00674-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses in the causation of human cancers - a brief historical account. Virology. 2009;384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]