

Abstract
Background
Recessive mutations in the PTEN-induced putative kinase 1 (PINK1) gene cause early-onset Parkinson's disease (EOPD). The clinical phenotype of families that have this PINK1-associated disease may present with different symptoms, including typical PD. The loss of the PINK1 protein may lead to mitochondrial dysfunction, which causes dopaminergic neuron death.
Methods
The clinical phenotypes of a large Polish family with EOPD and an identified PINK1 homozygous nonsense mutation were assessed. Ubiquitination and degradation of mitochondrial parkin substrates as well as mitochondrial bioenergetics were investigated as direct functional readouts for PINK1's kinase activity in biopsied dermal fibroblasts.
Results
A four-generation family was genealogically evaluated. Genetic screening identified two affected subjects who were both homozygous carriers of the pathogenic PINK1 p.Q456X substitution. Both patients presented with dystonia and gait disorders at symptom onset. Seven heterozygous mutation carriers remained unaffected. Functional studies revealed that the PINK1 p.Q456X protein is non-functional in activating the downstream ubiquitin ligase parkin and priming the ubiquitination of its substrates, and that the RNA levels of PINK1 were significantly reduced.
Conclusions
The PINK1 p.Q456X mutation leads to a decrease in mRNA and a loss of protein function. The foot dystonia and gait disorders seen at disease onset in affected members of our family, which were accompanied by parkinsonism had a similar clinical presentation to what has been described in previous reports of PINK1 mutation carriers.
Keywords: PINK1 p.Q456X mutation, familial Parkinson's disease, autosomal recessive parkinsonism, clinical characteristic, mitochondrial dysfunction
Introduction
Early-onset Parkinson's disease (EOPD) is a genetic condition closely resembling idiopathic Parkinson's disease (PD). Mutations in the PARKIN (PARK2), PINK1 (PARK6), and DJ-1 (PARK7) genes are causative for autosomal recessive EOPD. Approximately 50 pathogenic mutations in PINK1 have been reported. In 2005, Bonifati et al. first reported a nonsense mutation in exon 7 of the PINK1 gene (c.1366C>T) in three Italian patients with sporadic EOPD, which resulted in a premature stop codon (p.Q456X) [1]. Subsequently several additional reports describing PINK1 p.Q456X mutation carriers have been published [2, 3, 4, 5, 6]. PINK1 encodes a putative serine/threonine kinase that is widely expressed in the human brain and is involved in the mitochondrial response to cellular and oxidative stress. Recent studies have demonstrated that mitochondrial dysfunction plays a major role in neurodegeneration, especially in the pathogenesis of PD [7]. The PINK1 response is mediated by regulation of the calcium efflux process, which includes mitochondrial trafficking, reactive oxygen species (ROS) formation, and mitochondrial respiration efficacy. PINK1 also plays a role in regulating mitochondrial morphology and quality control through the PINK1/parkin-directed pathway. In brief, upon the loss of mitochondrial membrane potential, the PINK1 protein is stabilized on the outer mitochondrial membrane and thereby activates and recruits the E3 ubiquitin ligase parkin. PINK1 not only phosphorylates parkin, but it also primes its mitochondrial ubiquitination targets through phosphorylation [8]. Upon decoration of damaged mitochondria with Ubiquitin, individual substrate proteins are degraded through the proteasome, while eventually whole organelles are cleared via the autophagy/lysosome system (mitophagy). Through this presumably neuroprotective pathway, dysfunctional mitochondria (or their components) are eliminated to prevent destruction of the remaining (functional) mitochondrial network. However, the biochemical consequences are variable between different mutations in PINK1 [7]. The homozygous PINK1 p.Q456X nonsense mutation leads to a premature stop codon and a reduction in PINK1 mRNA levels, which is probably caused by nonsense-mediated mRNA decay [7]. In a study of the mitochondrial function of skin fibroblasts derived from four PD patients with the homozygous p.Q456X mutation, Grunewald et al. showed that the PINK1 p.Q456X was found to cause a significant depletion of steady-state ATP levels, but it had no effect on ATP synthesis or enzyme activity of the respiratory chain [7]. These patients also exhibited reduced mitochondrial membrane potentials [8]. Here, we present a clinical vignette and the results of functional studies completed on a Polish family who have EOPD and a PINK1 p.Q456X mutation.
Material and Methods
The family was evaluated during three field trips. All twelve family members enrolled in the study provided written informed consent for clinical examinations, molecular genetic studies, and skin biopsies. The study protocol was approved by the IRB committees of the Mayo Clinic, Johns Hopkins University, and Silesian Medical University. Standard genealogical studies were performed. Clinical examinations were performed by movement disorders specialists using the Unified Parkinson's Disease Rating Scale (UPDRS), the Hoehn and Yahr Scale (H&Y), and the Schwab and England activities of daily living scale (ADL). The Mini Mental State Examination (MMSE) and the Clock Drawing Test (CDT) were used to assess cognitive functions. Major depressive symptoms were excluded by applying the Beck Depression Inventory (BDI). The clinicians who assessed the subjects were blinded to the genetic test results. The diagnosis of definite PD was established according to the United Kingdom Parkinson's disease Brain Bank diagnostic criteria, with the exception that a positive family history was not regarded as a criterion for exclusion. Structural brain imaging was performed in symptomatic subjects with computerized axial tomography (CT) and/or 1.5T (T1, T2, FLAIR) magnetic resonance imaging (MRI). Genomic DNA was extracted from frozen peripheral blood samples by using standard procedures. The eight exons and exon-intron junctions of PINK1 were amplified by using a polymerase chain reaction (PCR). The products were then directly sequenced in at the 5′ and 3′ ends. Dermal fibroblasts were obtained from skin biopsies taken from the arm under local anesthesia. The Supplementary Material contains detailed information about primers, fibroblast cultures [9], and conditions.
Results
Clinical and molecular studies
The resulting pedigree, without any known consanguineous loops, consisted of 33 individuals and spanned four generations. We identified six clinically-affected subjects in the pedigree based on the detailed histories, chart reviews, and interviews with family members (Suppl. Figure 1). For the present study, we were able to examine two affected subjects who had EOPD (III.5 and III.8) and were homozygous for the PINK1 p.Q456X mutation (Table 1, contains detailed descriptions of these cases' phenotypes and are available in the Supplementary Material). We also have examined seven family members (4 men and 3 women) with a mean age at examination 40 years (range 19 to 72 years). These individuals were heterozygous PINK1 p.Q456X carriers (Suppl. Table 1). Their examinations were normal without any features of parkinsonism. Additionally, three family members (2 men and 1 woman) were clinically unaffected and did not carry the PINK1 p.Q456X mutation. The remaining clinically-affected family members (I.1, II.2s, III.1, III.2) identified on genealogical studies were reported to have parkinsonism. Their blood samples were not available for genetic studies. The spouse of II.1 (II.2s) also had a dementia of an unknown cause. The proband's maternal grandfather (I.1) was already deceased at the time of our research.
Table 1. Phenotype data of p.Q456X homozygous PINK1 mutation carriers.
| Present study | Bonifati et al. 2005 [1] |
Hedrich et al. 2006 [4], | Zadikoff et al. 2006 [2] |
Ibanez et al. 2006 [3] |
Scornaienchi et al. 2012 [6] |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Country of origin | Poland (Upper Silesia) | Italy | Germany | Canada/Italy | France/Italy | Italy (Sicily) | |||||
| Ethnicity* | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian |
| Gender | male | female | female | male | female | female | female | male | female | Female | male |
| PD | familial | sporadic | familial | familial | familial | ||||||
| AAO | 16 | 27 | 35 | 39 | 61 | 53 | 47 | 37 | 24 | 36 | 38 |
| First symptoms | B, R, D | D | D | R | B, R, PI | B, R, PI | B, PI | T | A, D | A | NA |
| Asymetric onset | + | - | - | + | + | + | + | + | + | - | NA |
| AAE | 36 | 46 | 37 | 69 | 67 | 60 | 71 | 57 | 42 | 47 | NA |
| Disease duration | 20 | 19 | 2 | 30 | 6 | 7 | 24 | 20 | 18 | 11 | 3 |
| Progression | slow | slow | slow | slow | slow | slow | slow | slow | slow | slow | NA |
| Parkinsonism | B, R, T | B, R | B, R, T | B, R, PI | B, R, PI | B, R, PI | B, R, T, PI | B, T | B, R, T | B, R | NA |
| Gait disorders | + | + | NA | - | - | - | + | + | + | + | NA |
| ON/OFF UPDRSIII | NA/26 | NA/18 | 15/NA | 18/34 | 20/27 | 21/27 | 24/35 | NA | 2/NA | 14/NA | NA/30 |
| Hoehn&Yahr | 2.0 | 2.0 | NA | 3.0 | 2.0 | 2.5 | 3.0 | NA | NA | NA | 2.0 |
| Levodopa daily dose | 800 | 150 | not treated | 500 | 300 | 150 | 500 | NA | 600 | 300 | NA |
| Levodopa response | very good | very good | NA | very good | very good | very good | very good | good | very good | very good | excellent |
| Levodopa induced motor sings | fluctuations, foot dystonia, dyskinesias | fluctuations, peak-dose dyskinesias | NA | mild choreiform dyskinesias | - | - | mild choreiform dyskinesias | leg dystonia, generalized dyskinesias | painful torticollis, dyskinesias | foot dystonia, dyskinesias, fluctuations | NA |
| Schwab&England | 100% | 100% | NA | 80% | 90% | 90% | 90% | NA | NA | NA | NA |
| MMSE | 30 | 28 | NA | 27 | 29 | 27 | 28 | NA | NA | NA | 29 |
| BDI | 2 | 1 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Depression | - | - | + | - | + | + | + | - | + | + | NA |
| Psychotic signs | - | - | severe anxiety | - | OCPD | anxiety | - | anxiety | anxiety | - | NA |
| Dysautonomia | night sweating | - | - | - | - | - | - | - | - | - | NA |
| Other signs and symptoms | sleep fragmentation | lower limbs paresthesia | - | tics, myalgia, hyposmia, | hyposmia | RLS, hyposmia | myalgia, hyposmia | back pain, extremity pain | increase of motor symptoms with hormonal changes | NA | |
| MRI | normal | normal | NA | normal | normal | normal | normal | normal | NA | NA | NA |
legend: AAO age at onset, AAE age at examination, PD Parkinson's disease, B bradykinesia, R rigidity, D dystonia, T tremor, PI postural instability, A akinesia, NA, not assessed or not applicable, UPDRS III Unified Parkinson's Disease Rating Scale part III, MMSE Mini Mental State Examination, BDI Beck Depression Inventory, OCPD obsessive-compulsive personality disorder, RLS restless legs syndrome, MRI magnetic resonance imaging,
This table does not include data from Ref. [5], which contained cases of Arab-Berber ethnicity due to the fact that the information given was not retrieved in the manner that would enable it to be entered into this table.
Functional studies
Skin biopsies were obtained from 12 family members: two affected (PINK1 p.Q456X homozygotes) and 10 unaffected (7 PINK1 p.Q456X heterozygotes and 3 wild-type homozygotes). An analysis of the levels of PINK1 RNA in the fibroblasts from the two p.Q456X homozygote patients showed significantly reduced PINK1 RNA levels. This was approximately 4-6% of the control levels (Figure 1A). Supporting this, biochemical analysis of PINK1 and mitochondrial parkin substrates from fibroblast cultures revealed that the p.Q456X protein variant was weakly expressed and/or unstable, and it was only minimally stabilized upon mitochondrial depolarization compared to the wild-type protein (Figure 1B). Due to this and to potentially reduced kinase activity of the truncated PINK1 mutant, parkin was not properly activated to ubiquitinate and degrade its mitochondrial substrates, e.g., Mitofusins (Mfn1/2) or Hexokinase I (HKI) in a mitophagy paradigm. Results from both homozygous carriers showed that the PINK1 p.Q456X protein was non-functional in the paradigm of activating parkin and priming the ubiquitination of its substrates (Figure 1B). Because previous reports linked the loss of PINK1 to mitochondrial dysfunction, the Seahorse XF analyzer was used to measure oxygen consumption rate (OCR) in these patient-derived fibroblasts to evaluate electron transport chain function. PINK1 p.Q456X fibroblasts showed no obvious defects in respiration compared to control fibroblasts (Suppl. Figure 2A-B). Next, tetramethylrhodamine, methyl ester (TMRM) staining was used to measure mitochondrial membrane potential, while mitotracker green was used as a control for mitochondrial mass (Suppl. Figure 2C-D). PINK1 p.Q456X fibroblasts had a similar membrane potential compared to wild type control cells. The loss of TMRM staining upon depolarization of mitochondria by FCCP shows the specificity of TMRM staining in both PINK1 p.Q456X and control cells (Suppl. Figure 2D-E).
Figure 1.
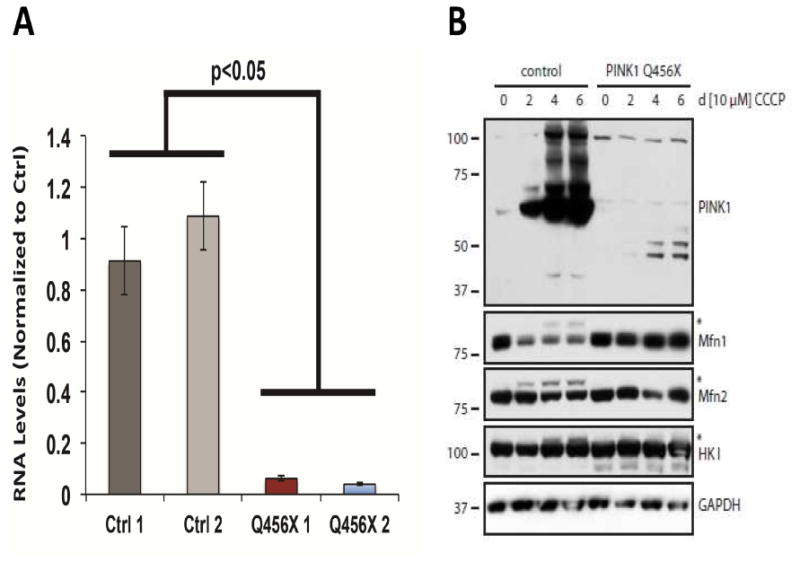
PINK1 p.Q456X fibroblasts showed reduced mRNA levels and a loss of protein expression. A) Quantitative reverse transcription PCR shows that PINK1 RNA levels were reduced in p.Q456X cells. Displayed are PINK1 RNA levels normalized to GAPDH and to average control PINK1 levels. Error bars displayed represent standard error of the mean. p < 0.05 by ANOVA with Tukey's post-hoc test. B) Control human dermal fibroblasts and fibroblasts from homozygous PINK1 p.Q456X mutation carriers were treated with CCCP at the indicated time points and analyzed by Western blot. A representative example is shown. Endogenous full-length PINK1 WT protein (63kDa) is greatly stabilized over time upon dissipation of the mitochondrial membrane potential. Moreover, PINK1 accumulation and kinase activity results in Parkin-dependent ubiquitination and degradation of mitochondrial substrates including the Mitofusins (Mfn1/2) and Hexokinase I (HKI). In contrast, the truncated PINK1 p.Q456X mutant (48.8 kDa) is not efficiently stabilized on de-energized mitochondria, likely due to the generally low expression levels caused by nonsense-mediated mRNA decay and/or general instability of the truncated protein. As a result of this and potentially because of reduced PINK1 kinase activity, Parkin is not properly activated and thus is not able to ubiquitinate its mitochondrial substrate proteins.
Discussion
PINK1 mutations cause recessively inherited EOPD. In a recent systematic review, Kilarski et al. estimated that the frequency of PINK1 homozygous mutations in EOPD patients was 3.7%, with a very low proportion in Caucasians, reaching 0.6% [10]. In a study of 150 early-onset PD patients of Polish origin, the PINK1 mutations were also rare; the overall frequency was 2.4% [11]. In general, parkinsonian patients with PINK1 mutations show typical features of resting tremor, rigidity, and bradykinesia as the most common initial symptoms; they typically have good levodopa responsiveness [1]. However, several clinical presentations, such as younger age at onset, slower disease progression, and early levodopa-induced dyskinesias may distinguish PINK1-disease from patients with late-onset PD [12]. PINK1 p.Q456X has been described in 16 homozygous mutation carriers from Italy, Germany, and Tunisia (Table 1) [2-6]. Both homozygous mutation carriers in our family developed bradykinesia, rigidity, and foot dystonia at a very young age, 16 and 27 years, which is clinically indicative of EOPD. Whereas, in the first reported case of PINK1 p.Q456X [1] and also in other cases, the age of disease onset ranged from 24-61 years [4-8]. Besides having typical PD features, mutation carriers with PINK1-associated phenotypes have been reported to have presented with levodopa-responsive dystonia, restless leg syndrome (RLS) with parkinsonism, and parkinsonism with cognitive and psychiatric problems [12]. Atypical clinical features were also described in PINK1 p.Q456X mutation carriers in addition to the typical clinical presentation of parkinsonism (Table 1). Our family confirms that the PINK1-associated phenotype may be variable. In both our homozygous PINK1 p.Q456X carriers, the first disease symptom was foot dystonia, and this was followed by progressive gait difficulties and sensory symptoms in the lower limbs, but this could have possibly been undiagnosed RLS, which is suggestive of atypical parkinsonism rather than feature of typical PD. However, during the disease course there were no other atypical features or signs of pyramidal system dysfunction, cerebellar syndrome, supranuclear gaze palsy, cognitive impairment, or orthostatic hypotension that could fulfill the atypical parkinsonism diagnostic criteria.
Although mild dystonia mainly in the feet was previously described as a clinical feature of EOPD, mild foot dystonia was rarely seen at the onset of PINK1-associated disease [1,3,12]. Predominant disease onset in the lower limbs and early gait impairment has been previously described in patients with homozygous p.Q456X and other PINK1 mutations [2,3,12]. It has even been suggested that it could be recognized as a feature specific of PINK1-associated parkinsonism. However, these phenotypic characteristics are not sufficient to clinically distinguish PINK1 from PARKIN mutation carriers. We did not find any studies that provided a direct comparison of gait disturbances in PARKIN and PINK1 mutation carriers in our literature review. Therefore, we can only speculate that the gait dysfunction associated with mutation carriers is related to other comorbidities, an earlier age of onset, or some other genetic and environmental factors.
Levodopa treatment was started in our affected subjects late in the disease course, but the subjects had a very good and sustained response to levodopa therapy. Besides mild motor fluctuations with peak dose dyskinesias and foot dystonia, no other complications were observed (Table 1). This is in concordance with the literature; EOPD is known to have an excellent and lasting response to levodopa therapy [1].
Both of our patients were able to continue their daily tasks while maintaining a high quality of life (ADL 100%). Despite a long disease duration, the UPDRS scores and H&Y stages were low in both cases, which confirms that PINK1-associated PD has a more benign disease course and milder symptoms than typical sporadic late-onset parkinsonism [2-6].
Similar to previous observations in leukocytes [9], our data suggests that the PINK1 p.Q456X mutation leads to an almost complete loss of PINK1 at the RNA level in patient-derived fibroblasts. This leads to impairment in the ability of parkin to translocate to the mitochondria and ubiquitinate mitochondrial substrates, such as mitofusins and hexokinase, following treatment with the mitochondrial uncoupler CCCP. However, fibroblasts from these patients showed no major defects in mitochondrial bioenergetics or membrane potential. In accordance with these findings, another group found that there were no defects in complex activity from patients with the same mutation. Abramov et al. described decreased mitochondrial membrane potential in three of the four p.Q456X patient fibroblast populations they studied [8], while no cells from the two patients studied here had decreased mitochondrial membrane potential. This suggests that the loss of PINK1 alone may not be sufficient to cause decreased membrane potential in human fibroblasts, but it may make cells more sensitive to other stressors. However, the loss of the PINK1 protein and its function, as seen with p.Q456X, abrogates the mitochondrial quality control mediated by the E3 ubiquitin ligase parkin. We can speculate that the loss of PINK1 may affect mitochondrial bioenergetics in a cell type-specific manner. Supporting this, it has recently been demonstrated that the PINK1 p.Q456X mutation may affect basal mitochondrial respiration as well as respiratory capacity in human dopaminergic neurons that were differentiated from induced pluripotent cells derived from patient fibroblasts [9]. Therefore, more studies of the effects of the PINK1 p.Q456X mutation in human dopaminergic neurons are warranted.
Supplementary Material
Supplementary Table 1. Phenotype data of p.Q456X heterozygous PINK1 mutation carriers
Supplementary Figure.1. Pedigree. Square, male. Circle, female. Diamond, gender masked to reduce the pedigree structure. Diagonal line, deceased. Arrow, proband. Symbols with black marks, lower right quarter, parkinsonism; lower left quarter, dementia; circle in the middle, dystonia. Numbers inside symbols, numbers of siblings. Numbers on the right lower side of the symbol, successive subject's number in each generation. Roman number, generation. s, spouse.
Supplementary Figure.2. PINK1 p.Q456X fibroblasts showed no significant mitochondrial bioenergetics defects. A) Mitochondrial respiration was measured at baseline and then after the administration of of oligomycin (to inhibit the ATP synthase and abolish ATP synthesis-coupled respiration), FCCP (to increase respiration to maximal respiratory capacity) and rotenone (to inhibit mitochondrial respiration). Oxygen consumption rate (OCR) is displayed as a percentage of baseline respiration. Cells from each patient were analyzed in side-by-side comparison with control cells. While cells from the first PINK1 p.Q456X patient showed no differences in respiration compared to control cells (left), the cells from the second patient had slightly increased maximal respiration (right). B) PINK1 p.Q456X cells showed no difference in absolute baseline respiration normalized to protein content. C) Cells were stained with TMRM and mitotracker green for 30 minutes and then visualized by confocal microscopy. D) No differences in membrane potential normalized to mitochondrial mass (TMRM/ mitotracker green) were observed in p.Q456X cells. E) Application of FCCP to cells drastically decreased TMRM staining, showing that staining is specific to polarized mitochondria. Error bars displayed represent standard error of the mean. *p < 0.05 by ANOVA with Tukey post-hoc test.
Highlights.
A PINK1 mutation was identified in a Polish family with EOPD.
Two affected members were homozygous for the PINK1 p.Q456X substitution mutation.
These two carriers had parkinsonism, dystonia, and foot drop.
The PINK1 p.Q456X mutant leads to loss of mRNA and protein function.
Our report supports the presence of PINK1 p.Q456X-related EOPD in Eastern Europe.
Acknowledgments
We would like to thank all those who have contributed to our research, particularly the patients and family members who participated in the study. We would like also to thank Audrey J. Strongosky for a pedigree preparation and for logistic and administrative support in collecting material for this study, and Kelly E. Viola, ELS for editorial assistance.
JS is supported by the Stowarzyszenie na Rzecz Rozwoju Neurologii Wieku Podeszlego grant. LAS is the recipient of a Canadian Institutes of Health Research Doctoral Foreign Study Award. VLD is supported by grants from the NIH/NINDS NS38377, MDSCRF 2007-MSCRFI-0420-00, 2009-MSCRFII-0125-00, MDSCRF 2012-MSCRFII-0268-00, MDSCRF 2013-MSCRFII-0105-00. OAR is partially supported by NIH/NINDS P50 NS072187 and NINDS R01 NS078086. WS is supported by the Mayo Clinic Center for Individualized Medicine, the GHR Foundation, the Marriott Family Foundation and a Gerstner Family Career Development Award. TMD is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases, and is supported by grants from the NIH/NINDS NS38377, MDSCRF 2007-MSCRFI-0420-00, 2009-MSCRFII-0125-00, MDSCRF 2012-MSCRFII-0268-00, MDSCRF 2013-MSCRFII-0105-00, and the JPB Foundation. TMD and VLD acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through their direct engagement in the continuous active conduct of medical research in conjunction with the Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation's Parkinson's Disease Program. ZKW is partially supported by NIH/NINDS P50 NS072187, and a gift from Carl Edward Bolch, Jr., and Susan Bass Bolch.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bonifati V, Rohe CF, Breedveld GJ, Fabrizio E, De Mari M, Tassorelli C, et al. Early-onset parkinsonism associated with PINK1 mutations. Frequency, genotypes and phenotypes. Neurology. 2005;65:87–95. doi: 10.1212/01.wnl.0000167546.39375.82. [DOI] [PubMed] [Google Scholar]
- 2.Zadikoff C, Rogaeva E, Djarmati A, Sato C, Salehi-Rad S, George-Hyslop P, et al. Homozygous and heterozygous PINK1 mutations: considerations for diagnosis and care of Parkinson's disease patients. Mov Disord. 2006;21(6):875–9. doi: 10.1002/mds.20854. [DOI] [PubMed] [Google Scholar]
- 3.Ibanez P, Lesage S, Lohmann E, Thobois S, De Michele G, Borg M, et al. Mutational analysis of the PINK1 gene in ealry-onset parkinsonism in Europe and North Africa. Brain. 2006;129:686–94. doi: 10.1093/brain/awl005. [DOI] [PubMed] [Google Scholar]
- 4.Hedrich KH, Hagenah J, Djarmati A, Hiller A, Lohnau T, Lasek K, et al. Clinical spectrum of homozygous and heterozygous PINK1 mutations in a large German family with Parkinson disease. Arch Neurol. 2006;63:833–8. doi: 10.1001/archneur.63.6.833. [DOI] [PubMed] [Google Scholar]
- 5.Ishihara-Paul L, Hulihan MM, Kachergus J, Upmanyu R, Warren L, Amouri R, et al. PINK1 mutations and parkinsonism. Neurology. 2008;71:896–902. doi: 10.1212/01.wnl.0000323812.40708.1f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scornaienchi V, Civitelli D, De Marco EV, Annesi G, Tarantino P, Rocca FE, et al. Mutation analysis of the PINK1 gene in Southern Italian patients with early-onset and late-onset parkinsonism. Parkinsonism Relat Disord. 2012;18:651–3. doi: 10.1016/j.parkreldis.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 7.Grunewald A, Gegg ME, Taanman JW, King RH, Kock N, Klein C, et al. Differential effects of PINK1 nonsense and missense mutations on mitochondrial function and morphology. Experimental neurology. 2009;219:266–73. doi: 10.1016/j.expneurol.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 8.Abramov AY, Gegg M, Grunewald A, Wood NW, Klein C, Schapira AH. Bioenergetic consequences of PINK1 mutations in Parkinson disease. PLoS ONE. 2011;6(10):e25622. doi: 10.1371/journal.pone.0025622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, et al. Familial Parkinson's disease iPSCs show cellular deficits in mitochondrial responses that can be pharmacologically rescued. Sci Transl Med. 2012;4(141):141ra90. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kilarski LL, Pearson JP, Newsway V, Majounie E, Knipe MD, Misbahuddin A, et al. Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson's disease. Mov Disord. 2012;27(12):1522–9. doi: 10.1002/mds.25132. [DOI] [PubMed] [Google Scholar]
- 11.Koziorowski D, Hoffman-Zacharska D, Slawek J, Jamrozik Z, Janik P, Potulska-Chromik A, et al. Incidence of mutations in the PARK2, PINK1, PARK7 genes in Polish early-onset Parkinson disease patients. Neurol Neurochir Pol. 2013;47(4):319–24. doi: 10.5114/ninp.2013.36756. [DOI] [PubMed] [Google Scholar]
- 12.Nishioka K, Kefi M, Jasinska-Myga B, Wider C, Vilarino-Guell C, Ross OA, et al. A comparative study of LRRK2, PINK1 and genetically undefined familial Parkinson's disease. J Neurol Neurosurg Psychiatry. 2010;81:391–5. doi: 10.1136/jnnp.2009.185231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Phenotype data of p.Q456X heterozygous PINK1 mutation carriers
Supplementary Figure.1. Pedigree. Square, male. Circle, female. Diamond, gender masked to reduce the pedigree structure. Diagonal line, deceased. Arrow, proband. Symbols with black marks, lower right quarter, parkinsonism; lower left quarter, dementia; circle in the middle, dystonia. Numbers inside symbols, numbers of siblings. Numbers on the right lower side of the symbol, successive subject's number in each generation. Roman number, generation. s, spouse.
Supplementary Figure.2. PINK1 p.Q456X fibroblasts showed no significant mitochondrial bioenergetics defects. A) Mitochondrial respiration was measured at baseline and then after the administration of of oligomycin (to inhibit the ATP synthase and abolish ATP synthesis-coupled respiration), FCCP (to increase respiration to maximal respiratory capacity) and rotenone (to inhibit mitochondrial respiration). Oxygen consumption rate (OCR) is displayed as a percentage of baseline respiration. Cells from each patient were analyzed in side-by-side comparison with control cells. While cells from the first PINK1 p.Q456X patient showed no differences in respiration compared to control cells (left), the cells from the second patient had slightly increased maximal respiration (right). B) PINK1 p.Q456X cells showed no difference in absolute baseline respiration normalized to protein content. C) Cells were stained with TMRM and mitotracker green for 30 minutes and then visualized by confocal microscopy. D) No differences in membrane potential normalized to mitochondrial mass (TMRM/ mitotracker green) were observed in p.Q456X cells. E) Application of FCCP to cells drastically decreased TMRM staining, showing that staining is specific to polarized mitochondria. Error bars displayed represent standard error of the mean. *p < 0.05 by ANOVA with Tukey post-hoc test.