

Abstract
Tight junctions create a paracellular barrier that is essential for survival of complex organisms. In many cases tight junctions define separate, generally sterile, tissue compartments. In the skin and gut, tight junctions must also seal the paracellular space to prevent microbiota from accessing the internal milieu. This is a relatively simple task in the integument, where an absolute barrier is effective. However, intestinal epithelial tight junctions are charged with the far more complex task of supporting paracellular transport of water, ions, and nutrients while providing a barrier to microbial translocation. The delicate nature of this balance, which is disrupted in disease, makes the intestine a unique organ in which to explore the complexities of tight junction permeability and barrier regulation. Here we review recent progress in understanding the molecular determinants of barrier function and events responsible for regulation, and dysregulation, of tight junction permeability.
Keywords: claudin, inflammatory bowel disease, leak pathway, myosin light chain kinase, occludin, pore pathway, ZO-1
1. Introduction to tight junction physiology
Epithelial and endothelial tight junctions can be simply characterized on the basis of their permeability. For example tight junctions within epithelia of the skin and urinary bladder are relatively impermeable, with electrical resistances exceeding 5,000 Ω•cm2. In contrast, resistance of small intestinal, colonic, and proximal renal tubular tight junctions is typically below 100 Ω•cm2. These differences reflect tissue function, as a robust epidermal barrier is essential to survival and the urinary bladder must prevent dissipation of ion gradients and urine concentration or dilution achieved within the renal tubules.
In contrast to skin and bladder, transporting epithelia take advantage of selectively-permeable tight junctions to drive passive, paracellular absorption and secretion. Because tight junctions are incapable of active transport of the sort accomplished by transcellular transporters, it is critical that the latter generate favorable gradients that drive passive, trans-tight junction transport in the appropriate direction. For example, claudin-16-mediated paracellular Ca2+ and Mg2+ reabsorption in the renal tubule depends on the lumen-positive potential generated by transepithelial transporters [1].
Paracellular permeability is also essential for recycling of ions, such as Na+, that drive absorption [2]. This is highlighted by the fact that mice lacking claudins 2 and 15, both of which can form paracellular Na+ channels, die of malnutrition as a result of decreased paracellular cation flux and limited recycling of Na+ absorbed by transcellular pathways back to the lumen. The resulting reduced luminal Na+ is insufficient to serve as the driving force for transcellular Na+- dependent nutrient absorption (Fig 1) [2]. Thus, in transporting epithelia, the tight junction allows passive paracellular flux that contributes significantly to overall transepithelial absorption and secretion.
Figure 1. Paracellular flux is required for Na+ recycling.

Claudin-2 and -15 form paracellular channels that facilitate Na+ flux. When claudin-2 and claudin-15 are absent, the tight junction is relatively impermeant to Na+. Thus, transcellular Na+ absorption, e.g. by apical Na+-nutrient cotransport and the basolateral Na+/K+-ATPase, depletes luminal Na+. The residual luminal Na+ is insufficient to support further Na+-nutrient cotransport.
2. Intestinal epithelial tight junction barriers are selectively-permeable and can be modulated by diverse stimuli
Leaky epithelia, such as those found in the small intestine and colon, are not simply less effective barriers than tight epithelia. Instead, different leaky epithelia have increased permeability to specific types of solutes and water. For example, paracellular permeability within the small intestinal crypt epithelium is greater than that of villous epithelium [3]. This allows the crypt to be a primarily secretory compartment, which helps limit microbial colonization within the crypt space.
The most well-defined example of tight junction regulation in response to physiological stimuli is that following activation of SGLT1-mediated Na+-nutrient cotransport [4, 5]. In this process, brush border Na+-nutrient cotransport activates NHE3-mediated Na+-H+ exchange [6] and myosin light chain kinase (MLCK) - dependent actomyosin contraction [5]. These events augment the transcellular Na+ gradient and increase tight junction permeability [5], respectively, to enhance paracellular water absorption. This physiology is, in part, responsible for the great success of oral rehydration solutions containing glucose and carbohydrates. Notably, the increase in tight junction permeability is limited to small molecules, e.g. mannitol, with radii ≤3.6 Å; larger molecules such as inulin, with a radius of 11.5 Å, are excluded [5, 7]. This allows small nutrients, such as glucose, to be carried along with water and results in paracellular amplification of transcellular glucose transport when luminal glucose concentrations are high [8]. At the same time, the size-selectivity of permeability regulation limits paracellular flux of proteins and microbial products between the lamina propria and gut lumen (Fig. 2A).
Figure 2. Physiological and pathophysiological regulation of tight junction permeability and passive water transport s by diverse stimuli.
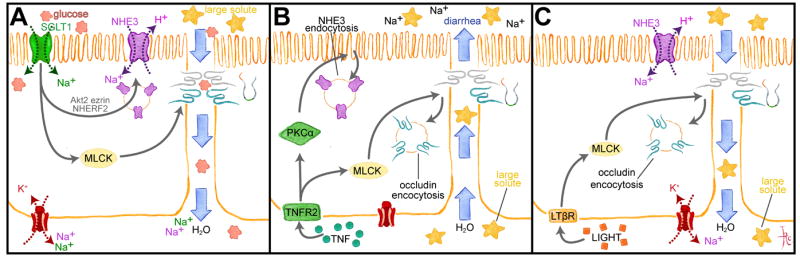
A. SGLT1-dependent Na+ and glucose cotransport triggers a signaling cascade in which Akt2 and MLCK are activated. Akt2 and ezrin promote NHE3 trafficking to the apical membrane to increase transcellular Na+ transport, while MLCK enhances tight junction permeability to small, nutrient-sized molecules, e.g. glucose. Transcellular deposition of Na+ and glucose in the basolateral space creates an osmotic gradient that draws water and glucose across the more permeable paracellular path. B. TNF binds to TNFR2 and activates PKCα and MLCK. NHE3 endocytosis is triggered by PKCα, which reduces transcellular Na+ absorption, thereby reducing the transepithelial Na+ gradient. MLCK activation causes occludin endocytosis that increases tight junction permeability to large solutes, including proteins. These changes result in passive water and solute flow into the lumen. C. Like TNF, LIGHT (lymphotoxin-like inducible protein that competes with glycoprotein D for herpes virus entry on T cells) activates MLCK to cause occludin endocytosis and increased tight junction permeability to large solutes. However, LIGHT does not inhibit NHE3, which continues to generate a transepithelial Na+ gradient that enhances passive water absorption.
Processes similar to those that enhance paracellular water and nutrient absorption following initiation of Na+-glucose cotransport also contribute to cytokine-mediated diarrheal disease. For example tumor necrosis factor-α (TNF) and the TNF core family member LIGHT both enhance tight junction permeability by a myosin light chain kinase-dependent process similar to that associated with Na+-glucose cotransport [9-12]. However, while TNF induces net water secretion into the gut lumen, LIGHT modestly augments paracellular water absorption [12]. These disparate effects do not reflect differences in extent or mechanism of tight junction regulation triggered by the two cytokines. Instead, the distinct effects of TNF and LIGHT reflect inhibition of NHE3-mediated Na+ absorption by TNF but not LIGHT [12]. TNF-induced, PKCα-mediated NHE3 inhibition markedly reduces transcellular Na+ absorption, thereby reducing the transepithelial Na+ gradient that normally drives water absorption (Fig. 2B). In contrast, NHE3 is not inhibited by LIGHT [12]. Thus, the enhanced absorption observed in mice treated with LIGHT [12] occurs by a mechanism similar to the increased absorption following initiation of Na+-glucose cotransport (Fig. 2C). However, the increased tight junction permeability triggered by TNF and LIGHT differs in one important respect from that induced by Na+-glucose cotransport; TNF- and LIGHT-induced barrier loss is not size-selective, and molecules as large as albumin easily leak into the lumen [12]. This important distinction is discussed in greater detail below.
2.1 Distinct pathways of paracellular flux
The existence of multiple routes across the tight junction has been recognized for many years [13]. Study of paracellular size-selectivity using small intestinal tissue autoradiographs after perfusion with 51Cr-EDTA and 14C-mannitol identified progressive increases in silver grains, corresponding to more isotope, within deeper portions of the mucosa [7]. Together with analysis of 51Cr-EDTA, 14C-mannitol, and 3H-inulin clearance, the data suggest that paracellular conductance channels have radii <6 Å in upper portion of the villus, 10 -15 Å in the lower part of the villus, and 50 - 60 Å in the crypts [7]. Activation of Na+- glucose cotransport increased 14C-mannitol flux, i.e. that across the small channels in the upper villus, by up to 15-fold. In contrast, 51Cr-EDTA and 3H-inulin flux were unaffected [7]. These data indicate that the small intestine contains at least three distinct paracellular flux routes that can be differentiated on the basis of their size-selectivity, location, and regulation by Na+-glucose cotransport (Fig. 3). It is important to recognize that commonly used electrical conductance and transepithelial electrical (TER) resistance measures do not assess size selectivity and, primarily, reflect flux of small ions.
Figure 3. Paracellular flux routes vary along the crypt-villus.
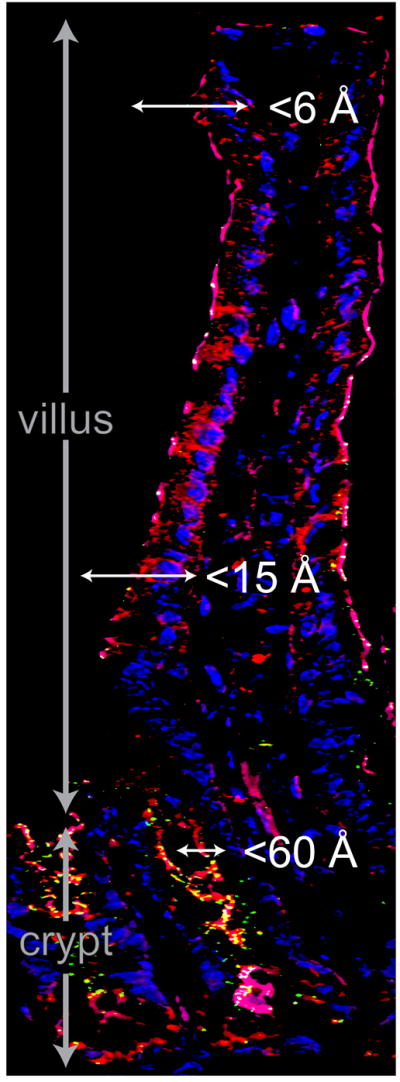
Fluorescence microscopy showing F-actin (red), claudin-2 (green), and nuclei (blue) in mouse jejunum. Note the increased claudin-2 expression in the crypt region. Tight junctions in the upper villus are permeable to small molecules (<6 Å), such as glucose, while channels with radii estimated to be 10-15 Å have been described in the lower part of the villus. Crypt tight junctions are permeable to macromolecules with radii up to 60 Å. Along with enhanced expression of claudin-2, which mediates paracellular Na+ and, possibly, water flux, this may promote net water secretion into the crypt space.
Similar to size-selectivity, tight junctions can be selectively permeable on the basis of solute charge [14]. This differs greatly between tissues, and, as detailed below, can be acutely regulated. Dilution potentials, the electrical potential generated when some ionic components, e.g. Na+ and Cl-, of either the apical or basolateral bathing solution are iso-osmotically replaced by a non-electrolyte, e.g. mannitol, can be used to determine charge selectivity [14].
In recent years several additional methods have become available to measure tight junction size-selectivity with high resolution. The bi-ionic potential technique is similar to dilution potentials, except that Na+ in the apical or basolateral bathing solution is iso-osmotically replaced by larger monovalent cations [15]. Alternatively, it is possible to assess transepithelial, i.e. paracellular, flux of a series of polyethylene glycol (PEG) oligomers with radii from 3.5 to 7.4 Å [16]. When flux of PEGs across Caco-2 and T84 cell monolayers was plotted as a function of PEG radius it demonstrated a biphasic relationship, with a sharp decrease in permeability as PEG radius increased from 3.5 to ~4 Å [16]. Although permeability of larger PEGs was limited, this part of the curve asymptotically approached, but never reached, zero [16]. In Caco-2 monolayers this relationship could be modeled by ‘restrictive,’ i.e. size-selective, pores with radii of ~4.5 Å and a second population of ‘nonrestrictive,’ i.e. relatively nonselective pores with radii of at least 7.5 Å [16].
Tight junction disruption with EGTA increased permeability of all PEGs and eliminated size-selectivity [16]. In contrast, sodium caprate, which reduces epithelial barrier function by incompletely defined mechanisms [17], increases flux of all PEGs while retaining relative size-selectivity and the biphasic relationship between PEG radius and permeability [16]. A subsequent study showed that interferon-γ (IFN-γ) treatment of intestinal epithelial cell monolayers had an effect similar to sodium caprate, i.e. paracellular flux of all PEGs was increased, but size-selectivity and the biphasic relationship between PEG radius and permeability was maintained [18]. The effects of sodium caprate and IFN-γ could be modeled as increases in flux across the nonrestrictive class of pores [18].
More recently, the same PEG-based approach was used to assess permeability of high and low resistance MDCK monolayers [19]. Both demonstrated a biphasic relationship between PEG radius and paracellular permeability that was generally similar to that observed in Caco-2 and T84 monolayers [16]. When the two types of MDCK monolayers were compared, it was apparent that the primary difference was reduced permeability of small PEGs in high resistance MDCK monolayers [19]. Claudin-2 expression increased permeability of small PEGs, with radii less than 4 Å, in both high and low resistance MDCK monolayers, but did not affect paracellular flux of larger PEGs [19]. Thus, in contrast to IFN-γ, it appears that claudin-2 enhances flux across the small, or restrictive, class of pores [18, 19]. Overall, these studies using PEGs to assess size-selectivity indicate that paracellular permeability cannot be simply thought of as one function, but must be regarded as the sum of at least two distinct flux pathways.
2.2 Immune regulation of tight junction permeability
While IFN-γ may be a pathophysiologically relevant regulator of intestinal epithelial tight junction permeability, TNF, a critical mediator of Crohn’s disease, has been studied far more extensively [9, 11, 20]. TNF-induced barrier loss could reflect epithelial apoptosis, as described in an analysis using a scanning electrode to detect local conductance variations [21]. That study found, however, that only 56% of barrier loss induced by TNF could be explained by apoptosis, with the remainder occurring in non-apoptotic areas [21]. This was confirmed by in vivo and in vitro analyses showing that TNF-induced barrier loss was not due to epithelial apoptosis [9, 11, 22]. TNF reduced TER and increased paracellular flux of 3kD dextran in vitro [20, 23] and increased paracellular flux of albumin in vivo [11, 12]. Thus, a major component of TNF-induced increases in paracellular permeability is due to enhanced flux across the nonrestrictive class of pores [20, 23-25]. Because an upper size limit has not been defined for this route, it is best referred to as the leak pathway [26, 27], rather than one containing large pores. In general, the leak pathway is a low capacity, size- and charge-nonselective paracellular route (Fig. 4).
Figure 4. Differential activation of pore and leak pathways by cytokine signaling. The relationship between tight junction proteins and the pore and leak pathways.
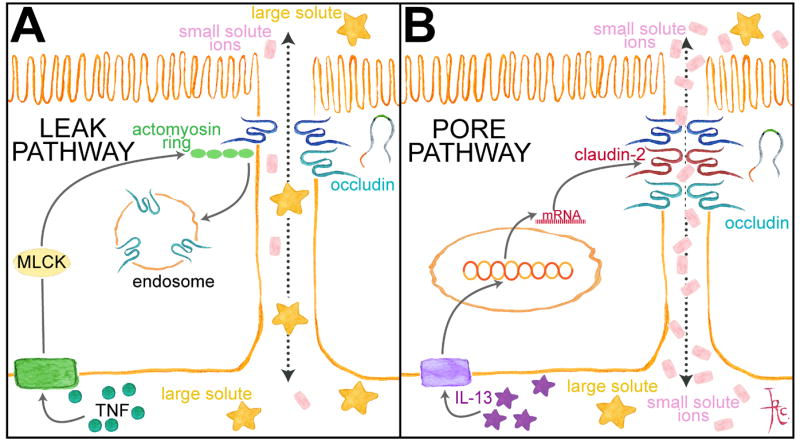
A. TNF increases leak pathway paracellular permeability via MLCK activation and occludin endocytosis. B. IL-13 increases pore pathway permeability by upregulating claudin-2 expression. See text for details.
Separate studies showed that interleukin-13 (IL-13), a cytokine involved in ulcerative colitis is also able to increase paracellular permeability of cultured intestinal epithelial monolayers [25, 28]. Detailed analyses showed that IL-13-induced barrier loss was quite different from that induced by TNF; IL-13 increased paracellular flux of Na+, but not Cl- or 4kD dextran [25]. This is consistent with flux via restrictive small pores and also indicates that this pathway can be charge selective. This high capacity paracellular flux route, the pore pathway, is size- and charge-selective (Fig. 4) [26, 27].
2.3 Pore pathway effectors
In the gastrointestinal tract, tight junction permeability is exquisitely sensitive to immune cell signaling. IL-13 can enhance pore pathway permeability of intestinal epithelial monolayers in vitro and mouse colon in vivo [25]. In vitro studies showed that claudin-2-targeted siRNA could limit IL-13-induced claudin-2 expression and completely prevent IL-13-induced increases in PNa+/PCl- [25]. However, claudin-2-targeted siRNA only partially prevented IL-13-induced TER loss [25]. This difference could reflect the incomplete inhibition of claudin-2 expression by siRNA or, alternatively, that factors other than claudin-2 upregulation can contribute to increased pore pathway permeability. Thus, while claudin-2 expression can clearly enhance pore pathway permeability (Fig. 4), other effectors cannot be entirely excluded.
Activation of physiological Na+-glucose cotransport in intestinal epithelia enhances pore pathway permeability without altering charge-selectivity or claudin-protein expression [5]. This form of tight junction regulation depends on myosin light chain kinase (MLCK) activation [5] similar to that induced by Rock II activation in other epithelia [29]. One key distinction between Na+-glucose cotransport and TNF-induced tight junction regulation that may explain pore vs. leak pathway upregulation is the involvement of occludin endocytosis only in TNF-induced barrier regulation [10, 24, 30].
2.4 Leak pathway regulation
TNF has been recognized as a regulator of tight junction permeability for over 20 years [31]. However, the requisite role of myosin II regulatory light chain (MLC) phosphorylation in this process was not discovered until a highly specific MLCK inhibitor was developed [9]. The roles of myosin II and MLCK in tight junction regulation were subsequently reproduced in vivo [11]. MLCK was also shown to be required for barrier loss induced by the TNF family member LIGHT and IL-1β [10-12, 32]. This tight junction regulation was accompanied by ultrastructural condensation of the perijunctional actomyosin ring [11] that was remarkably similar to that previously associated with Na+-glucose cotransport [5, 33, 34]. Although Rho effectors have been implicated in some forms of tight junction regulation, particularly during wound repair, they do not appear to be involved in TNF-induced permeability increases [11].
When analyzed in vivo, occludin internalization was the most prominent tight junction alteration following TNF-induced barrier loss (Fig. 4) [11, 12]. This occludin internalization did not occur in vivo following treatment with a specific MLCK inhibitor or in knockout mice lacking long MLCK [11]. Further analyses showed that LIGHT and TNF triggered occludin internalization via a caveolar process, both in vitro and in vivo [10, 30]. Morphometric analyses of mouse enterocytes showed an abrupt increase in caveolin-1-positive, occluding-containing vesicles, beginning 90 minutes after TNF administration, that coincided with appearance of 80 nm diameter vesicles [30]. These vesicles became EEA1-positive and grew to 125 nm diameter over the next 15 minutes, suggesting maturation of occludin-containing endosomes [30]. Further, pharmacological inhibition of caveolar endocytosis prevented LIGHT and TNF-induced increases in leak pathway permeability in vitro and in vivo [10, 30], and TNF did not induce occludin internalization, barrier loss, or diarrhea in caevolin-1 knockout mice [30]. Thus, genetic and pharmacological inhibitor studies, colocalization analyses, and EM morphometry all indicate that MLCK-dependent caveolar endocytosis is required for TNF-induced barrier loss.
While the data above confirm the essential role of endocytosis in TNF-induced leak pathway regulation, they do not unambiguously demonstrate that occludin plays a functional role. In support of the idea that occludin may merely be a useful marker of endocytosis associated with tight junction regulation, occludin knockout mice have been reported to have normal intestinal barrier function [35]. To assess the role of occludin endocytosis in TNF-induced barrier loss, recombinant TNF was administered to wild type mice and transgenic mice that express an excess of EGFP-occludin within the intestinal epithelium [30]. While occludin endocytosis occurred in both groups of mice, a continuous ring of occludin remained at the tight junction of transgenic mice, while the tight junction-associated occludin pool was markedly diminished in wild type mice. EGFP-occludin transgenic mice were also partially protected from TNF-induced increases in leak pathway permeability and completely protected from TNF-induced diarrhea [30]. It can therefore be concluded that MLCK-dependent, caveolar endocytosis of occludin is required for TNF-induced leak pathway regulation. Consistent with this, occludin knockdown Caco-2 or MDCK cells display increased leak pathway permeability at baseline, but no further barrier loss following TNF treatment, despite increased MLC phosphorylation [24, 36, 37]. A detailed analysis in Caco-2 cell monolayers showed that occludin specifically enhanced paracellular flux by a pathway that could be modeled as having a ~60 Å radius [24]. While this may not be identical to the leak pathway activated by TNF, it is remarkable that this is consistent with the 50 - 60 Å paracellular ‘pores’ described previously in small intestinal crypts [7]. Importantly, this increased paracellular permeability to large solutes should be distinguished from in vitro knockdown studies showing that cells deficient in myosin II isoforms fail to properly establish apical junctions.
3. Tight junction function is regulated by continuous remodeling of protein-protein interactions
An enormous number of protein-protein interactions occur at the tight junction. Together with the observation that fluorescence recovery after photobleaching (FRAP) of claudin-1 expressed in fibroblasts was limited, this led to the conclusion that tight junction complexes are stable at steady state [38]. This prevailing model was, however, upended when it was shown that claudin-1, occludin, ZO-1, and β-actin all have distinct mechanisms and kinetics of exchange at the junction [39]. Claudin-1 demonstrated extremely limited FRAP, indicating a large stably anchored pool at the tight junction. In contrast, occludin had a mobile fraction of ~70%, and recovery was limited by interventions that disrupted membrane fluidity, such as methyl-β-cyclodextrin or reduced temperature, but not by ATP depletion [39]. Moreover, when large regions of the tight junction were bleached, occludin recovery could be seen to begin at the edges of the bleached zone, suggesting that recovery occurred by diffusion (Fig. 5). In silico modeling based on of intramembranous diffusion of tight junction and lateral membrane occludin pools recapitulated recovery in FRAP and fluorescence loss in photobleaching (FLIP) experiments [39]. In contrast, ZO-1 recovery was energy-dependent, unaffected by methyl-β-cyclodextrin or reduced temperature, and distributed uniformly along the full length of an extended bleach region [39, 40]. ZO-1 FRAP and FLIP behavior could be modeled as exchange between a large cytosolic pool and a smaller tight junction-associated pool [39]. Thus, occludin, ZO-1, claudin-1, and β-actin interactions must be highly dynamic, even at steady state (Fig. 5). This led to the hypothesis that modification of the kinetics and stability of these and other interprotein interactions could be a mechanism of barrier regulation. As discussed below, that has proven to be the case for both pore and leak pathways.
Figure 5. Tight junction proteins display distinct dynamic behaviors.
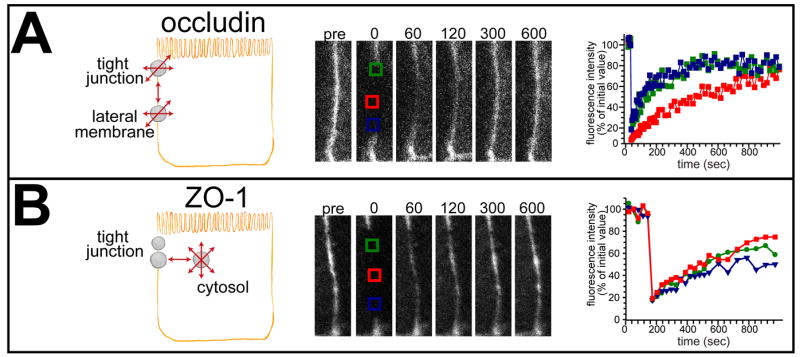
A. Occludin diffuses passively within tight junction (TJ) and lateral membrane pools. Consistent with this, fluorescence recovery begins peripherally when a large area of the tight junction is bleached. B. Part of tight junction-associated ZO-1 exchanges by an active, energy-dependent process with a large cytosolic pool. Consistent with this, fluorescence recovery occurs uniformly across a large bleached tight junction region. From Shen et al. J Cell Biol. 2008.
3.1 Analysis of tight junction remodeling provides insights into mechanisms of pore pathway regulation and potential for therapeutic modulation
Pore pathway flux is physiologically increased following initiation of Na+-glucose cotransport, and this regulation can be reversed by MLCK inhibition [5, 40]. To determine if MLCK inhibition also affected dynamic behaviors of tight junction proteins, occludin, ZO-1, claudin-1, and β-actin FRAP kinetics were assessed in monolayers with active Na+-glucose cotransport before and after MLCK inhibition [40]. ZO-1 recovery was reduced in vitro and was similarly reduced in vivo following MLCK inhibitor treatment of wild type, but not long MLCK knockout, mice [40]. The reduction in fluorescence recovery indicates that, despite no apparent change in localization, ZO-1 anchoring at the tight junction is reduced by Na+-glucose cotransport and enhanced by MLCK inhibition [40]. Stabilization following MLCK inhibition was mediated by the ZO-1 actin binding region (ABR), as exchange of ZO-1 lacking the ABR was increased at baseline and unaffected by MLCK inhibition. Further, when the ABR was expressed as a dominant negative domain it blocked the effects of MLCK inhibition on both ZO-1 FRAP behavior and pore pathway permeability. These data show that ABR-dependent ZO-1 anchoring at the tight junction is one mechanisms of reducing flux across the pore pathway [41].
The carboxy terminal tail of occludin is extensively phosphorylated, particularly when occludin is at the tight junction [42, 43]. Casein kinase 2 (CK2) is one of the many kinases responsible for this phosphorylation [44, 45]. To determine whether CK2-mediated phosphorylation might regulate occludin function, Caco-2 monolayers were treated with specific CK2 inhibitors, which reduced pore pathway permeability specifically [46]. siRNA knockdown studies showed that this effect required expression of both CK2 and occludin [46]. Moreover, CK2 inhibition resulted in occludin stabilization, i.e. reduced exchange, at the tight junction [46]. Both occludin stabilization and reduced pore pathway permeability were mapped to dephosphorylation of serine 408, a known CK2 substrate [46]. These data suggest that S408 is normally phosphorylated by CK2, and that CK2 inhibition leads to S408 dephosphorylation and enhanced occludin anchoring at the tight junction. To identify the binding partners responsible for this increased anchoring, S408A and S408D nonphosphorylatable and pseudophosphorylated occludin tails were used as bait to capture proteins from cell lysates [46]. The S408A occludin tail captured more claudin-1 and claudin-2, but less occludin, relative to the S408D tail. In contrast, both tails bound similar quantities of claudin-4 and the occludin-related protein marvelD3. As direct binding of occludin to claudins has not been described, it was postulated that ZO-1, which binds both occludin and claudins, might be an intermediate in this interaction. Consistent with this, the S408A occludin tail bound more of the ZO-1 U5-GuK occludin binding domain that did the S408D tail, and neither occludin tail was able to recover claudins from lysates of ZO-1-deficient cells [46].
In contrast to occludin, claudin-1 and claudin-2 diffusion within the tight junction were increased following CK2 inhibition [46]. This could be recapitulated by expression of S408A, but not S408D, mutant occludin in occludin-deficient monolayers [46]. In contrast, claudin-4 displayed limited anchoring, i.e. a large mobile fraction, that was unaffected by occludin S408 phosphorylation, consistent with the similar capture of claudin-4 by S408A and S408D occludin tails. CK2 inhibition did not affect claudin-2 FRAP behavior in ZO-1-deficient monolayers or after expression of EGFP-ZO-1 lacking either the PDZ1 domain required for claudin binding or the U5-GuK domain required for occludin binding [46]. In contrast, claudin-2 mobility was increased by CK2 inhibition in ZO-1-deficient monolayers expressing full length EGFP-ZO-1 [46]. Thus, all of the data support a model where S408 phosphorylated occludin forms dimers with high mobility, and little anchoring, within the tight junction while S408 dephosphorylated occludin bind to ZO-1 and, in turn, to claudin-2 (Fig. 6A,B). The assembly of a complex containing occludin, ZO-1, and claudin-2 is further supported by the observation that CK2 inhibition leads to the development of a diffuse component of ZO-1 recovery (Fig. 6C) as well as the convergence of occludin, ZO-1, and claudin-2 mobile fractions [46]. This increased claudin-2 mobility disrupts claudin-2 function and reduces pore pathway permeability [46]. If this model is correct, CK2 inhibitors should be able to inhibit the claudin-2- dependent effects of IL-13 on pore pathway permeability [46]. Consistent with this, CK2 inhibition reversed IL-13-induced pore pathway permeability increases [46]. Thus, CK2 inhibition may be one means of preventing disease-associated increases in pore pathway permeability due to increased claudin-2 expression.
Figure 6. TJ dynamic behavior and barrier function are regulated through occludin S408 phosphorylation.
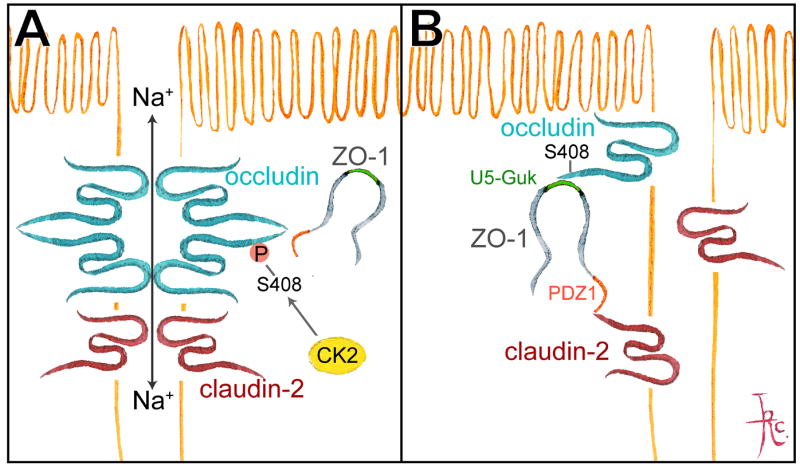
A. CK2-mediated occludin S408 phosphorylation promotes assembly of occludin dimers that are highly mobile within the tight junction. Claudin-2 forms complexes with claudin-2 on adjacent cells to create paracellular cation pores. B. CK2 inhibition and occludin S408 dephosphorylation promotes occludin binding to ZO-1 which, in turn, binds to claudin-2. This increases claudin-2 mobility, which disrupts claudin-2 interactions and cation pores. C. Stable association of ZO-1 with occludin (after CK2 inhibition) converts part of ZO-1 mobility to a diffusive process, with fluorescence recovery beginning at the edges of the bleached region. From Raleigh et al. J Cell Biol. 2011.
3.2 TNF-induced barrier regulation requires altered tight junction protein interactions and dynamic behavior
As noted above, occludin endocytosis is required for TNF-induced, MLCK-dependent leak pathway regulation [24, 30, 36]. While the mechanisms that trigger occludin internalization are not well-defined, it is reasonable to hypothesize that this must involve some release of occludin from protein binding partners at the tight junction. To test this idea, FRAP behavior of EGFP-occludin was assessed before and after TNF treatment [24]. Notably, monolayers were treated for ≤4 hr incubation with a low concentration of TNF [23, 24]. Although the occludin mobile fraction was unaffected by TNF, the rate of occludin exchange was significantly increased, as indicated by a reduction in the time required for half-maximal fluorescent recovery (t1/2). This accelerated occludin exchange reflected loss of C-terminal occludin/ELL (OCEL) domain interactions, as EGFP-occludin lacking the OCEL domain (occludinΔOCEL) recovered rapidly in the absence of TNF and was unaffected by TNF treatment [24]. Consistent with a critical role for OCEL-mediated interactions in leak pathway maintenance, EGFP-occludin, but not EGFP occludinΔOCEL, sealed the leak pathway in occludin knockdown Caco-2 cell monolayers [24]. Further, EGFP-occludin, but not EGFP occludinΔOCEL, expression restored TNF-induced leak pathway flux increases in occludin knockdown Caco-2 monolayers [24].
EGFP-occludinΔOCEL was not internalized following TNF treatment. In addition, expression of the OCEL domain as a free protein blocked TNF effects on occludin FRAP, endocytosis, and barrier loss [24]. Thus, in addition to a critical role in stabilizing occludin at the tight junction, the OCEL domain is essential for activating TNF-induced occludin endocytosis [24]. This OCEL domain function is mediated by K433, as a K433D OCEL mutant failed to block TNF-induced effects on occludin FRAP and endocytosis [24]. K433 has not generally been considered to be part of the OCEL binding site for ZO-1 [47]. However, one study showed that while K433D OCEL bound a ZO-1 PDZ3-SH3-GuK construct, it did not effectively bind to full-length ZO-1 [48]. This suggests that, while the ZO-1 GuK domain is essential for occludin binding, other sites must also be involved. Overall, these data demonstrate that K433-dependent, OCEL-mediated interactions are critical for occludin anchoring and exchange at the tight junction as well as TNF-induced occludin endocytosis and leak pathway regulation (Fig. 7).
Figure 7. mCherry-OCEL expression prevents TNF-induced occludin mobilization and internalization.
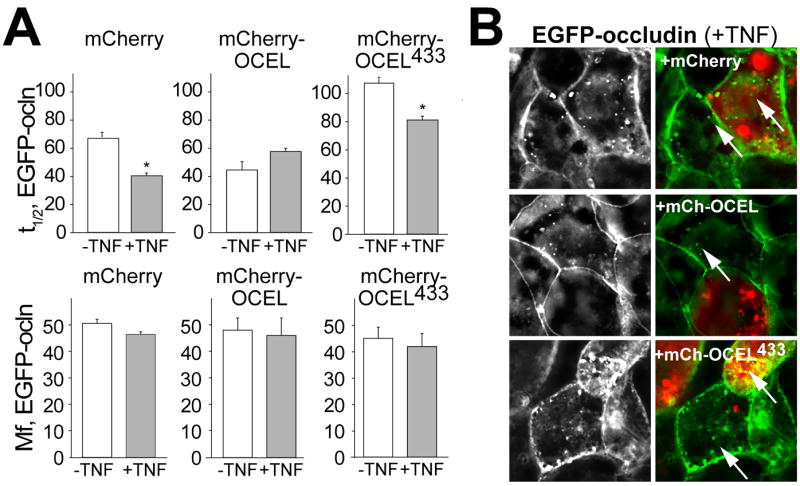
A. mCherry-OCEL, but not mCherry-OCELK433D, prevented TNF-induced acceleration of EGFP-occludin fluorescence recovery. B. mCherry-OCEL, but not mCherry-OCELK433D, blocked TNF-induced EGFP-occludin (green) endocytosis (arrows). Cells expressing mCherry-OCEL or mCherry-OCELK433D, can be identified by their red fluorescence. Adjacent cells not expressing mCherry-OCEL or mCherry-OCELK433D, all show TNF-induced EGFP-occludin internalization. From Buschmann et al. Mol Biol Cell. 2013.
The studies above focused on acute, TNF-induced leak pathway regulation. However, it has been reported that longer TNF treatments, e.g. ≥ 24 hrs, are able to increase claudin-2 expression [49]. While not examined in detail, it would be reasonable to conclude that this claudin-2 upregulation enhances pore pathway permeability. The role of claudin-2 in TNF-induced barrier loss was recently examined in the context of claudin-4 FRAP behavior [50]. This study found that the claudin-4 mobile fraction, but not t1/2, was increased following 48 hours of treatment with an IFN-γ/TNF cocktail [50]. This suggests that claudin-4 function at the tight junction may be affected by IFN-γ/TNF treatment. Consistent with this, expression of GFP-tagged claudin-4 increased the TER of cytokine-treated monolayers [50]. However, GFP-claudin-4 expression also increased TER of monolayers not exposed to cytokines, and cytokines significantly reduced TER of GFP-claudin-4 expressing monolayers [50]. Unfortunately, it is not clear whether the TER effects analyzed reflected pore or leak pathway permeability changes. Nevertheless, these data do lend further support to the idea that tight junction protein dynamic behavior is a critical regulator of paracellular barrier function.
4. Future Directions
A multitude of tight junction proteins have been identified over the more than 25 years since the discovery of ZO-1. Although it is likely that additional tight junction-associated proteins will be found, the challenge that now faces the field is to build our understanding of how these proteins interact with one another and other cellular components to create semi-permeable barriers. Some examples of such work have been described here. Among important remaining challenges will be the critical task of defining the differences between barrier function and paracellular permeability, by either pathway, in molecular terms. For example, although claudin proteins must be involved, the molecular structure of the barrier is not understood. Similarly, it is not known how occludin regulates the leak pathway or why occludin knockout mice are free of apparent intestinal barrier defects. Thus, despite great strides in identifying signaling events that can regulate barrier function, detailed characterization of the molecular underpinnings and development of means to modulate these processes for therapeutic purposes is limited. The golden age of tight junction protein discovery may therefore be ending, but a new era in tight junction biology and pathobiology is only beginning.
Acknowledgments
We thank past and present members of our laboratory for their insight and helpful suggestions of our work. We also thank Dr. Judith Turner for her critical reading of this manuscript. We apologize to all colleagues whose work could not be discussed or cited because of lack of space. Work in our laboratory is supported by the National Institutes of Health (R01DK61931, R01DK68271, F32DK094550, K01DK092381, R24DK099803), the Broad Medical Research Foundation (IBD-022), and the Crohn’s and Colitis Foundation of America.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science. 1999;285:103–6. doi: 10.1126/science.285.5424.103. [DOI] [PubMed] [Google Scholar]
- 2.Wada M, Tamura A, Takahashi N, Tsukita S. Loss of Claudins 2 and 15 From Mice Causes Defects in Paracellular Na(+) Flow and Nutrient Transport in Gut and Leads to Death from Malnutrition. Gastroenterol. 2013;144:369–80. doi: 10.1053/j.gastro.2012.10.035. [DOI] [PubMed] [Google Scholar]
- 3.Marcial MA, Carlson SL, Madara JL. Partitioning of paracellular conductance along the ileal crypt-villus axis: a hypothesis based on structural analysis with detailed consideration of tight junction structure-function relationships. J Membr Biol. 1984;80:59–70. doi: 10.1007/BF01868690. [DOI] [PubMed] [Google Scholar]
- 4.Pappenheimer JR, Reiss KZ. Contribution of Solvent Drag through Intercellular-Junctions to Absorption of Nutrients by the Small-Intestine of the Rat. J Membrane Biol. 1987;100:123–36. doi: 10.1007/BF02209145. [DOI] [PubMed] [Google Scholar]
- 5.Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273:C1378–85. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- 6.Zhao H, Shiue H, Palkon S, Wang Y, Cullinan P, Burkhardt JK, et al. Ezrin regulates NHE3 translocation and activation after Na+-glucose cotransport. Proc Natl Acad Sci USA. 2004;101:9485–90. doi: 10.1073/pnas.0308400101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fihn BM, Sjoqvist A, Jodal M. Permeability of the rat small intestinal epithelium along the villus- crypt axis: effects of glucose transport. Gastroenterol. 2000;119:1029–36. doi: 10.1053/gast.2000.18148. [DOI] [PubMed] [Google Scholar]
- 8.Meddings JB, Westergaard H. Intestinal glucose transport using perfused rat jejunum in vivo: model analysis and derivation of corrected kinetic constants. Clin Sci (Lond) 1989;76:403–13. doi: 10.1042/cs0760403. [DOI] [PubMed] [Google Scholar]
- 9.Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, et al. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterol. 2002;123:163–72. doi: 10.1053/gast.2002.34235. [DOI] [PubMed] [Google Scholar]
- 10.Schwarz BT, Wang F, Shen L, Clayburgh DR, Su L, Wang Y, et al. LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterol. 2007;132:2383–94. doi: 10.1053/j.gastro.2007.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, et al. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–15. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clayburgh DR, Musch MW, Leitges M, Fu YX, Turner JR. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest. 2006;116:2682–94. doi: 10.1172/JCI29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moreno JH, Diamond JM. Discrimination of monovalent inorganic cations by “tight” junctions of gallbladder epithelium. J Membr Biol. 1974;15:277–318. doi: 10.1007/BF01870092. [DOI] [PubMed] [Google Scholar]
- 14.Reuss L, Finn AL. Electrical properties of the cellular transepithelial pathway in Necturus gallbladder. I. Circuit analysis and steady-state effects of mucosal solution ionic substitutions. J Membr Biol. 1975;25:115–39. doi: 10.1007/BF01868571. [DOI] [PubMed] [Google Scholar]
- 15.Angelow S, Yu AS. Structure-function studies of claudin extracellular domains by cysteine-scanning mutagenesis. J Biol Chem. 2009;284:29205–17. doi: 10.1074/jbc.M109.043752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watson CJ, Rowland M, Warhurst G. Functional modeling of tight junctions in intestinal cell monolayers using polyethylene glycol oligomers. Am J Physiol - Cell Physiol. 2001;281:C388–97. doi: 10.1152/ajpcell.2001.281.2.C388. [DOI] [PubMed] [Google Scholar]
- 17.Krug SM, Amasheh M, Dittmann I, Christoffel I, Fromm M, Amasheh S. Sodium caprate as an enhancer of macromolecule permeation across tricellular tight junctions of intestinal cells. Biomaterials. 2013;34:275–82. doi: 10.1016/j.biomaterials.2012.09.051. [DOI] [PubMed] [Google Scholar]
- 18.Watson CJ, Hoare CJ, Garrod DR, Carlson GL, Warhurst G. Interferon-gamma selectively increases epithelial permeability to large molecules by activating different populations of paracellular pores. J Cell Sci. 2005;118:5221–30. doi: 10.1242/jcs.02630. [DOI] [PubMed] [Google Scholar]
- 19.Van Itallie CM, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, et al. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci. 2008;121:298–305. doi: 10.1242/jcs.021485. [DOI] [PubMed] [Google Scholar]
- 20.Taylor CT, Dzus AL, Colgan SP. Autocrine regulation of epithelial permeability by hypoxia: role for polarized release of tumor necrosis factor alpha. Gastroenterol. 1998;114:657–68. doi: 10.1016/s0016-5085(98)70579-7. [DOI] [PubMed] [Google Scholar]
- 21.Gitter AH, Bendfeldt K, Schulzke JD, Fromm M. Leaks in the epithelial barrier caused by spontaneous and TNF-alpha-induced single-cell apoptosis. FASEB J. 2000;14:1749–53. doi: 10.1096/fj.99-0898com. [DOI] [PubMed] [Google Scholar]
- 22.Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788:864–71. doi: 10.1016/j.bbamem.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–19. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buschmann MM, Shen L, Rajapakse H, Raleigh DR, Wang Y, Wang Y, et al. Occludin OCEL-domain interactions are required for maintenance and regulation of the tight junction barrier to macromolecular flux. Mol Biol Cell. 2013;24:3056–68. doi: 10.1091/mbc.E12-09-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weber CR, Raleigh DR, Su L, Shen L, Sullivan EA, Wang Y, et al. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J Biol Chem. 2010;285:12037–46. doi: 10.1074/jbc.M109.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 27.Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterol. 2005;129:550–64. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Terry SJ, Zihni C, Elbediwy A, Vitiello E, Leefa Chong San IV, Balda MS, et al. Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nat Cell Biol. 2011;13:159–66. doi: 10.1038/ncb2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, 2nd, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189:111–26. doi: 10.1083/jcb.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mullin JM, Snock KV. Effect of tumor necrosis factor on epithelial tight junctions and transepithelial permeability. Cancer Res. 1990;50:2172–6. [PubMed] [Google Scholar]
- 32.Al-Sadi R, Ye D, Dokladny K, Ma TY. Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. J Immunol. 2008;180:5653–61. doi: 10.4049/jimmunol.180.8.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madara JL, Pappenheimer JR. Structural basis for physiological regulation of paracellular pathways in intestinal epithelia. J Membr Biol. 1987;100:149–64. doi: 10.1007/BF02209147. [DOI] [PubMed] [Google Scholar]
- 34.Atisook K, Carlson S, Madara JL. Effects of phlorizin and sodium on glucose-elicited alterations of cell junctions in intestinal epithelia. Am J Physiol. 1990;258:C77–C85. doi: 10.1152/ajpcell.1990.258.1.C77. [DOI] [PubMed] [Google Scholar]
- 35.Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11:4131–42. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Itallie CM, Fanning AS, Holmes J, Anderson JM. Occludin is required for cytokine-induced regulation of tight junction barriers. J Cell Sci. 2010:2844–52. doi: 10.1242/jcs.065581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu AS, McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, et al. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am J Physiol - Cell Physiol. 2005;288:C1231–41. doi: 10.1152/ajpcell.00581.2004. [DOI] [PubMed] [Google Scholar]
- 38.Sasaki H, Matsui C, Furuse K, Mimori-Kiyosue Y, Furuse M, Tsukita S. Dynamic behavior of paired claudin strands within apposing plasma membranes. Proc Natl Acad Sci USA. 2003;100:3971–6. doi: 10.1073/pnas.0630649100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen L, Weber CR, Turner JR. The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J Cell Biol. 2008;181:683–95. doi: 10.1083/jcb.200711165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu D, Marchiando AM, Weber CR, Raleigh DR, Wang Y, Shen L, et al. MLCK-dependent exchange and actin binding region-dependent anchoring of ZO-1 regulate tight junction barrier function. Proc Natl Acad Sci USA. 2010;107:8237–41. doi: 10.1073/pnas.0908869107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol. 2011;73:283–309. doi: 10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cordenonsi M, Mazzon E, De Rigo L, Baraldo S, Meggio F, Citi S. Occludin dephosphorylation in early development of Xenopus laevis. J Cell Sci. 1997;110:3131–9. doi: 10.1242/jcs.110.24.3131. [DOI] [PubMed] [Google Scholar]
- 43.Sakakibara A, Furuse M, Saitou M, Ando-Akatsuka Y, Tsukita S. Possible involvement of phosphorylation of occludin in tight junction formation. J Cell Biol. 1997;137:1393–401. doi: 10.1083/jcb.137.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cordenonsi M, Turco F, D’Atri F, Hammar E, Martinucci G, Meggio F, et al. Xenopus laevis occludin. Identification of in vitro phosphorylation sites by protein kinase CK2 and association with cingulin. Eur J Biochem. 1999;264:374–84. doi: 10.1046/j.1432-1327.1999.00616.x. [DOI] [PubMed] [Google Scholar]
- 45.Smales C, Ellis M, Baumber R, Hussain N, Desmond H, Staddon JM. Occludin phosphorylation: identification of an occludin kinase in brain and cell extracts as CK2. FEBS Lett. 2003;545:161–6. doi: 10.1016/s0014-5793(03)00525-8. [DOI] [PubMed] [Google Scholar]
- 46.Raleigh DR, Boe DM, Yu D, Weber CR, Marchiando AM, Bradford EM, et al. Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J Cell Biol. 2011;193:565–82. doi: 10.1083/jcb.201010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, Fanning AS, Anderson JM, Lavie A. Structure of the conserved cytoplasmic C-terminal domain of occludin: identification of the ZO-1 binding surface. J Mol Biol. 2005;352:151–64. doi: 10.1016/j.jmb.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 48.Tash BR, Bewley MC, Russo M, Keil JM, Griffin KA, Sundstrom JM, et al. The occludin and ZO-1 complex, defined by small angle X-ray scattering and NMR, has implications for modulating tight junction permeability. Proc Natl Acad Sci USA. 2012;109:10855–60. doi: 10.1073/pnas.1121390109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mankertz J, Amasheh M, Krug SM, Fromm A, Amasheh S, Hillenbrand B, et al. TNFalpha up-regulates claudin-2 expression in epithelial HT-29/B6 cells via phosphatidylinositol-3-kinase signaling. Cell Tissue Res. 2009;336:67–77. doi: 10.1007/s00441-009-0751-8. [DOI] [PubMed] [Google Scholar]
- 50.Capaldo CT, Beeman N, Hilgarth RS, Nava P, Louis NA, Naschberger E, et al. IFN-gamma and TNF-alpha-induced GBP-1 inhibits epithelial cell proliferation through suppression of beta-catenin/TCF signaling. Mucosal Immunol. 2012;5:681–90. doi: 10.1038/mi.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]