

Abstract
Background
Less is known about the genetic basis of Alzheimer disease (AD) in African Americans (AAs) than in non-Hispanic whites.
Methods
Whole exome sequencing (WES) was performed on seven AA AD cases. Disease association with potentially AD-related variants from WES was assessed in an AA discovery cohort of 422 cases and 394 controls. Replication was sought in an AA sample of 1,037 cases and 1,869 controls from the Alzheimer Disease Genetics Consortium (ADGC).
Results
Forty-four SNPs from WES passed filtering criteria and were successfully genotyped, Nominally significant (p<0.05) association to AD was observed with two African-descent specific AKAP9 SNPs in tight linkage disequilibrium: rs144662445 (p=0.014) and rs149979685 (p=0.037). These associations were replicated in the ADGC sample (rs144662445: p=0.0022, odds ratio [OR]=2.75; rs149979685: p=0.0022, OR=3.61).
Conclusions
Because AKAP9 was not previously linked to AD risk, this study indicates a potential new disease mechanism.
Keywords: whole-exome sequencing, late-onset Alzheimer disease, rare variant, genetic association, African American, AKAP9
1. Background
Late onset Alzheimer disease (AD) is the most common form of dementia in the elderly. It has a substantial genetic component, with heritability as high as 75%[1]. The apolipoprotein E (APOE) gene ε4 variant has been shown to be a major component of this risk[2]; among non-Hispanics whites (NHW), ε4/ε4 homozygotes are estimated to have up to 15-fold increased odds of AD compared to those with the most common ε3/ε3 genotype[3]. Genome-wide association studies (GWAS) including very large NHW samples have identified genes harboring additional modest-effect AD risk susceptibility loci (odds ratios between 1.1 and 1.3) [4–8].
Interestingly, even though the proportion of risk due to genetic influences is similar in African Americans (AA) and NHWs[9], there is evidence that the genetic architecture of the disease differs between these two populations. For example, the effect of APOE ε4 on AD risk is considerably weaker in AAs than NHWs, [3, 10, 11] although this difference may be accounted for in part by the age-dependent effect of ε4[11]. Several genes associated with AD and AD-related hippocampal atrophy measured by magnetic resonance imaging in NHWs also show evidence of association in AAs, however most SNPs associated with NHWs have not been replicated in AAs[12–14]. While it may be that the AA studies are underpowered compared to studies with NHWs, it also may be due to differences in the linkage disequilibrium (LD) structure between AAs and NHWs or population-specific causal variants underlying the associations. Interestingly, two GWAS replicated the association with ABCA7 in AAs[12, 14] and the risk associated with ABCA7 (odds ratio [OR]=1.79) approaches that of APOE ε4 in AAs (OR=2.31)[14]. Nevertheless, a large proportion of heritability in AAs remains unexplained. Much of the heritability may be ascribed to uncommon (frequency < 5%) or rare (frequency < 1%) variants which would be undetectable in GWAS except in very large samples[15]. Causal high-penetrant rare variants in APP, PSEN1 and PSEN2 have been identified primarily in the early-onset familial form of the disease [16]. However, to date no causal variants in these gene have been reported in AAs.
Next-generation sequencing methodologies have the potential to identify rare or private risk variants many of which would be expected to exert moderate to high effects on disease risk compared to most common disease-associated variants[15, 17]. One efficient study design to detect highly penetrant disease variants entails sequencing individuals from families with multiple affected members who are more likely than sporadic cases to possess such variants. Potentially disease-causing variants identified in familial cases can then be genotyped in other family members or in a larger sample of unrelated individuals to evaluate association with disease. This study design was recently used in Caucasian samples to identify rare variants in the phospholipase D3 (PLD3) gene[18]. Resequencing PLD3 exons in a larger number of cases and controls identified additional AD-associated PLD3 variants including Ala442Ala, which was subsequently shown to be nominally-significantly associated (p=0.03) with AD in a modest AA sample (130 cases, 172 controls). In this study, we performed whole-exome sequencing (WES) of 7 familial AA late-onset AD cases and subsequently tested association of AD risk with 44 rare variants selected on the basis of frequency, sequencing quality, and relationship to previously implicated AD genes in 422 AA cases and 394 cognitively normal AA controls. We identified two African-descent specific genetic variants in the A kinase anchor protein 9 (AKAP9) gene which are significantly enriched in AA AD cases versus controls, and subsequently replicated this association in a large independent AA cohort. The study design is summarized in Figure 1.
Figure 1.

The overall study design including a whole-exome sequencing (WES) of 7 AA late-onset AD cases, followed by variant prioritization and filtering, genotyping in a larger AA discovery sample, and replication in a large AA ADGC cohort.
2. Methods
2.1 Study samples
The discovery sample included AA participants of the Multi-Institutional Research on Alzheimer Genetic Epidemiology (MIRAGE) Study and the Genetic and Environmental Risk Factors for Alzheimer Disease among African Americans Study (GenerAAtions). Ascertainment methods, inclusion criteria, and genome-wide genotyping in the MIRAGE and GenerAAtions cohorts are described in detail elsewhere[9, 12]. The MIRAGE cohort consists of nuclear families most of which are discordant sib pairs. There are also several affected sib pairs and larger sibships. Seven MIRAGE-Study AD cases with multiple affected family members, and thus more likely to be enriched with rare AD-related variants, were selected for whole-exome sequencing. Descriptive statistics for these subjects are summarized in Supplementary Table 1. Four of these subjects were affected sib-pairs. The genotypes of single nucleotide polymorphisms (SNPs) identified as potentially AD related in WES were genotyped in 218 cases (including the seven sequenced subjects) and 237 controls from the MIRAGE cohort and 223 cases and 190 controls from the GenerAAtions cohort. Analyses were performed on a discovery cohort made up of the genotyped subjects excluding the seven sequenced AD cases and subjects less than 60 years at the time of assessment. The discovery cohort includes 199 cases and 209 controls from the MIRAGE cohort and 223 cases and 185 controls from the GenerAAtions cohort. Characteristics of the discovery cohort are summarized in Supplementary Table 1. The most significant variants were genotyped in replication cohort comprising a subset of the Alzheimer Disease Genetics Consortium (ADGC) AA sample, excluding the MIRAGE and GenerAAtions cohorts, which contains 1,037 cases and 1,869 controls (Supplementary Table 2). The ADGC AA cohort is described in detail elsewhere[14]. Studies were performed with appropriate institutional review board approval and informed consent was obtained from all study participants.
2.2. Sequencing and Genotyping
A detailed description of whole-exome sequencing, SNP genotyping methods and quality control, and Sanger-sequencing used to confirm genotypes of the most significant SNPs from the analysis of the discovery data is provided in Supplementary Methods.
2.3. Variant Filtering
Variants identified by whole exome sequencing and passing quality control (QC) were filtered for novelty (absence in the dbSNP database v. 132, using time since discovery as a proxy for low frequency because rare SNPs are more likely to be included in later dbSNP revisions) and potential for deleterious effect (must be a non-synonymous base pair change). To filter out possible sequencing artifacts, we additionally required that variants be observed in more than one case, whether or not the variants occurred in siblings or unrelated subjects. The novel non-synonymous variants were selected for genotyping if they were in genes which had been either previously-identified as associated with AD—that is, genes that are listed in the AlzGene database—or in genes belonging to networks with established AD genes. We constructed gene networks using the INGENUITY Pathway Analysis (IPA) software. IPA analyses were seeded with genes emerging from WES and established risk genes for early or late onset AD (APOE, BIN1, CLU, CR1, PICALM, MS4A6E, CD2AP, CD33, ABCA7, EPHA1, SORL1, ACE, PSEN1, PSEN2, and APP) [2, 4–8, 19–25] in order to identify genes and the variants contained therein that might be related physiologically to AD-related processes or are related to genes with a known connection to AD. These networks do not exclusively consist of genes involved in a single AD-related biological function, but instead include genes that interact, either directly or indirectly, with known AD genes and hence may implicate new disease-related pathways.
2.4. Statistical Analysis
Tests of association between genotyped SNPs and AD were performed using R[26]. Significance was determined using a one-sided Fisher’s exact test because rare SNPs can cause numerical stability issues in logistic models, and because we specified a priori that our search was focused on deleterious rare variants. The test of association within the discovery cohort may underestimate the strength of a true association in the MIRAGE dataset because many of the controls in this sample are unaffected siblings of AD cases who would presumably be carriers of risk alleles at a higher rate than unrelated controls. Although the MIRAGE study ascertained primarily discordant sib-pairs, the sample contained three affected sib-pairs including the two affected sib-pairs in the WES sample. Both members of the sib-pair not included in the WES sample were included in the discovery cohort. The possibility of inflated significance due to non-independence of the samples is unlikely (see the generalized estimating equation [GEE] analysis in the Supplementary Results). Principal components (PC) analysis with EIGENSTRAT[27] based on all minor allele frequency (MAF) >5% markers from a whole-genome genotyping panel (described in [12]) found no evidence of AD-associated population substructure within the discovery sample (all p>0.05 on tests of association between AD and each of the first 10 PCs). In the replication sample, SNPs were analyzed for association with AD using a logistic regression model adjusting for sex and study site. Methods for the haplotype, phylogenetic dendogram, and bioinformatic analyses (using PolyPhen-2[28], SIFT[29], and MutPred[30] software) are described in Supplementary Methods. Haplotype analyses were performed with PLINK[31] using both whole-exome sequence variant data and tag SNPs from whole-genome genotyping.
3. Results
A total of 88,867 variants were identified by WES. No coding APP, PSEN1, PSEN2 or PLD3 variants were identified in the 7 AA AD cases. After filtering, 63 SNPs from 58 genes were selected for further study and 44 of these SNPs were successfully genotyped in the entire sample. Detailed results of sequencing and variant filtering are presented in the supplementary materials, including a complete list of variants that were genotyped (Supplementary Table 4). The majority of genotyped SNPs were rare (29 SNPs had a MAF<1%). Nominally significant results were obtained with AKAP9 SNPs rs144662445 (p = 0.014, OR = 8.4) and rs149979685 (p = 0.037; the OR for rs149979685 could not be estimated because the rare allele was not observed in any controls) (Table 1, Supplementary Table 5). The genotypes for these AKAP9 SNPs were confirmed by Sanger sequencing. Five AD cases, in addition to the two cases included in WES, were observed with rare alleles for both rs144662445 and rs149979685 (Supplementary Table 6). Five other subjects (four AD cases and one control) had the minor allele for rs144662445 but not rs149979685. Subjects having at least one of these two rare variants are designated AKAP9+. No subjects were observed having the minor allele for rs149979685 but not rs144662445. No rare-allele homozygotes were observed for either SNP. One control subject who was genotyped but not included in the discovery sample due to age—a 54 year-old unaffected sibling of an AD proband (whose age at onset was 64)—had rare alleles for both rs144662445 and rs149979685. The AKAP9+ subjects are all unrelated according to their self-reported data and identity by descent (IBD) estimates calculated by PLINK[31] using genome-wide SNP data. A PC analysis in EIGENSTRAT[27] indicated that these subjects were not members of a clearly defined population subgroup within AAs (Supplementary Figure 1). AKAP9 genotypes were available for relatives of four AKAP9+ AD cases. Two of these cases each had an unaffected sibling who was AKAP9-. Each of the other two AKAP9+ cases who were included in the WES discovery sample had an AKAP9− affected sibling.
Table 1.
The most significant associations between AD and genotyped SNPs in the discovery cohort.
| Gene | Position | SNP ID | Freq Cases | Freq Controls |
P |
|---|---|---|---|---|---|
| AKAP9* | 7:91709085 | rs144662445 | 0.011 | 0.0013 | 0.014* |
| AKAP9* | 7:91732110 | rs149979685 | 0.0060 | 0.000 | 0.037* |
| ATP2B4 | 1:203672867 | rs145963279 | 0.023 | 0.013 | 0.092 |
| LRP4 | 11:46911956 | rs138878258 | 0.0072 | 0.0026 | 0.17 |
| ITGA9 | 3: 37774225 | rs142726080 | 0.032 | 0.024 | 0.20 |
Abbreviations: AD, Alzheimer disease; Freq, frequency; SNP, single-nucleotide polymorphism.
Genotypes for AKAP9 SNPs confirmed via Sanger sequencing.
Next, we sought replication of the association only with the two AKAP9 SNPs in the ADGC AA sample. Valid genotypes for both SNPs were obtained in 1,037 AD cases and 1,869 controls. In this sample, the average age at onset among AD cases was 80.1 and the average exam age for controls was 74.5. Average ages were similar among AKAP9+ (79.7) and AKAP-(80.1) cases (p=0.83) and among AKAP9+ (75.9) and AKAP− (74.5) controls (p=0.47). The APOE ε4 allele frequency was 33.7% in cases and 18.5% in controls in the replication sample. This does not differ between AKAP9+ (38.8%) and AKAP9− (33.6%) cases (p=0.59) or between AKAP9+ (12.5%) and AKAP9− (18.6%) controls (p=0.50). A logistic regression model adjusting for age and study site demonstrated significant association of AD risk with the minor alleles of both rs144662445 (p=0.0022, OR=2.75) and rs149979685 (p=0.0022, OR=3.61) (Table 2). Hence, the association observed in the discovery data was replicated in a larger independent sample. A logistic regression analysis of the subset of ADGC AA replication subjects who had both AKAP9 SNP genotypes and genome-wide genotype data (877 cases and 1641 controls) yielded nearly identical results for both SNPs when PCs were included (rs144662445 p=0.0034, OR=2.72 versus p=0.0035, OR=2.72 with PCs; 149979685 p=0.0064, OR=3.17 versus p=0.0063, OR=3.19 with PCs), indicating that population substructure did not contribute to the confirmation of the AKAP9 association in the replication sample.
Table 2.
The association between AKAP9 variants and AD using a logistic regression adjusted for sex and study site in the replication cohort.
| SNP ID | Frequency (Proportion) of Minor Alleles |
Odds Ratio * |
95% CI | P | |
|---|---|---|---|---|---|
| Cases | Controls | ||||
| rs144662445 | 0.011 (22†/2052) | 0.0043 (16/3722) | 2.75 | 1.38 –5.61 | 0.0022 |
| rs149979685 | 0.0072 (15/1059) | 0.0027 (10/3728) | 3.61 | 1.51–9.00 | 0.0022 |
Abbreviations: AD, Alzheimer disease; CI, confidence interval; OR, odds ratio; SNP, single nucleotide polymorphism.
Effect size estimates, CI, and P values were derived from a logistic regression model adjusted for sex and study site.
Includes one AD case who was homozygous for the minor allele
To examine the possibility that an untested AD causal variant in AKAP9 or surrounding region could account for the observed association, we compared haplotypes of AKAP9+ and AKAP9− individuals in the discovery sample. The 5’ end of AKAP9 overlaps a large LD block which includes the neighboring genes CYP51A1, KRIT1, ANKIB1, and LRRD1 (see supplementary Figure 2). Haplotype analysis of the 10 common (MAF>0.05) AKAP9 SNPs in the GWAS-genotyping array revealed that all of the AKAP9+ subjects harbored the same haplotype (designated HAP0). That is, the minor alleles (either as a singleton or together) are present on the haplotype GGAAGGAAGC as defined by rs1859037, rs2299233, rs2282973, rs6465347, rs2158138, rs733957, rs2079082, rs13239875, rs4265, and rs1063243 (eTable 7). The frequency of this haplotype is greater in AD cases (6.1%) compared to unrelated controls (4.1%), but this difference is not quite significant (p=0.071). No other coding variants in this region were observed in the two AKAP9+ subjects with WES data through whom this association was ascertained.
Next, we performed phylogenetic dendogram analysis of the SNP information in the haplotype region among 11 of the 246 African-descent subjects in 1000 Genomes for whom genotypes are available (including 88 Yorubans from Nigeria, 97 Luhyans from Kenya, and 61 AAs from the American Southwest) and who had the HAP0 haplotype as defined by the common SNPs. Three of these 11 individuals (NA18499, NA19448, and NA20126) had minor alleles at both rs144662445 and rs149979685, and one individual (NA20298) had only the rs144662445 variant. These AKAP9 variants were observed in each of the three African-descent groups suggesting that the AKAP9 variants are present in multiple African ancestral populations. Cluster analysis of the phased alleles for the 22 haplotypes further discriminated the HAP0+ haplotypes from the HAP0− haplotypes (Figure 2A). There is also a branch point separating the HAP0+ haplotype containing one AKAP9 variant (designated HAP0+1) from the haplotype containing both AKAP9 variants (HAP0+2).
Figure 2.
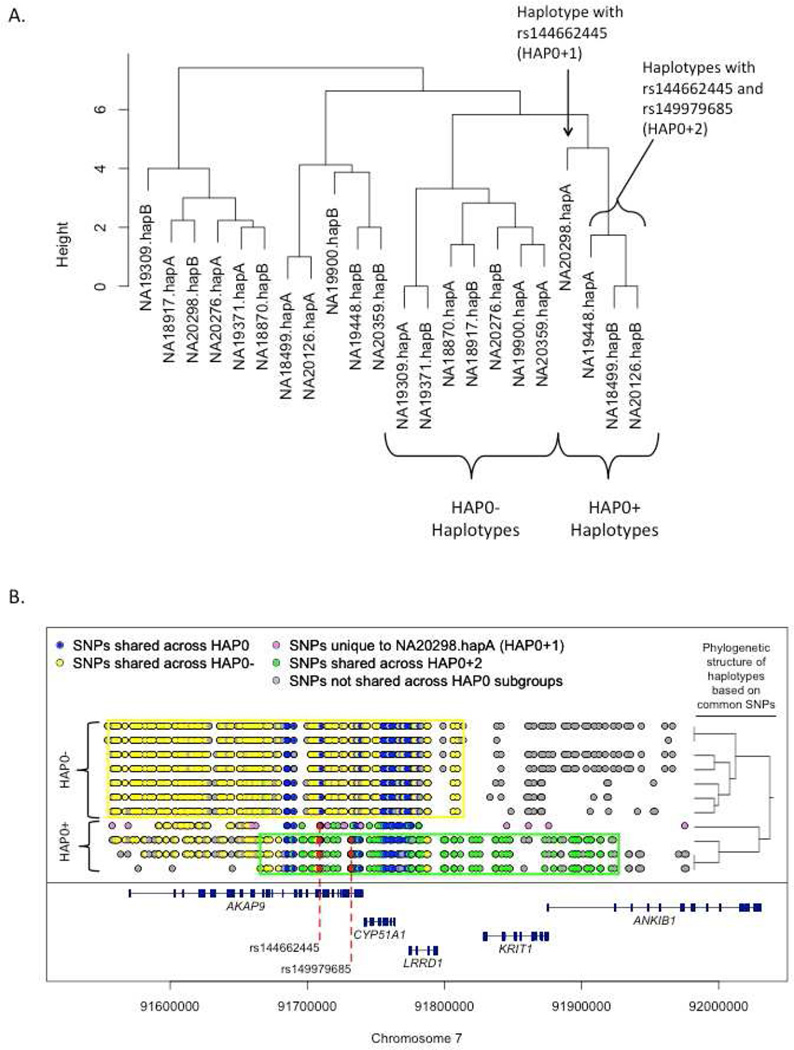
Examination of the 1000 Genomes subjects with the haplotype containing the two identified AKAP9 variants including (A) a phylogenetic dendogram of haplotypes and (B) positions of infrequent SNPs (<5% MAF) shared by haplotypes with 0, 1, and 2 variants. Key: HAP0: The background haplotype which contains the two identified AKAP9 variants. HAP0+ the background variant with one or more of the two putatively AD-associated variants; HAP0− the background haplotype without either AD-associated variants; HAP0+1 the background haplotype with one of the AD-associated AKAP9 variants; HAP0+2 the background haplotype with both AD-associated AKAP9 variants.
Based on these results, we examined the 1000 Genomes phased haplotypes for plausible exonic candidate AD-risk SNPs on the same haplotypes as rs144662445 and rs149979685 by comparing the high-risk haplotypes (HAP0+2 and HAP0+1) to the low-risk haplotype (HAP0−). Within the region from 91.3 Mb to 92.1 Mb—which extends well beyond the regions shared across those harboring AKAP9 mutations (Figure 2B)—the three subjects with HAP0+2 haplotypes shared 69 variants with MAF< 10% that were not observed in subjects with the HAP0− haplotype. Rs144662445 and rs149979685 are the only coding variants in this group of SNPs. Next, we compared the single copy of HAP0+1 from 1000 Genomes (NA20298A) to HAP0−. In the same region, NA20298A contains 49 variants with MAF< 10%, which were not observed on the (low-risk) HAP0− haplotypes, including three coding variants: rs144662445 and two additional non-synonymous coding AKAP9 SNPs rs186619641 and rs201858518. Rs186619641 and rs201858518 are unique to individual NA20298 in the 1000 Genomes database. Using only 1000 Genomes data we cannot determine whether or not these two additional non-synonymous variants are shared by all subjects in our study with HAP0+1, present in only a portion of subjects with HAP0+1 haplotypes, or unique to 1000 Genomes individual NA20298 who is the only AA subject in 1000 Genomes with one HAP0+1 haplotype. We sought clarification in the ESP database which, based on allele counts of the AD-associated SNPs rs144662445 and rs149979685, contains 3 copies of HAP0+1. These two exonic SNPs (rs186619641 and rs201858518) were absent from the ESP database suggesting that the two AD-associated variants defining HAP0+1 and HAP0+2 are not surrogate markers for rs186619641 and rs201858518. Therefore, based on our examination of HAP0+1 and HAP0+2 in public databases, we conclude that rs144662445 and rs149979685 are the only plausible AD-related coding variants on these background haplotypes within AKAP9 or the surrounding region.
Bioinformatic analysis using several methods suggests that rs149979685 (S3771L transcript Q99996-1) is likely to be functional whereas rs144662445 (I2558M transcript Q99996-1) is not (Table 3). The MutPred analysis also indicated that the most likely impact of the rs149979685 variant is loss of a helix (p=0.0033), but gain of a loop (p=0.024) or loss of phosphorylation (p=0.029) are also possible. However, since MutPred makes these predictions based on homology to known functional variants and the 3D structure of AKAP9 is yet unknown, it is unclear whether these structures are present near rs149979685 or critical to the function of AKAP9. The two NA20298-specific non-synonymous SNPs (rs186619641 and rs201858518) were also predicted to be benign according to Polyphen-2.
Table 3.
Bioinformatic Examination of the Alzheimer disease-associated non-synonymous AKAP9 variants.
| Polyphen-2 | Sift | MutPred | ||||
|---|---|---|---|---|---|---|
| SNP | Score* | Prediction | Score† | Prediction | Probability Deleterious‡ |
Actionable Hypotheses‡ |
| rs144662445 | 0.008 | Benign | 0.11 | Tolerated | 21% | NA |
| rs149979685 | 0.99 | Probably | 0.01 | Damaging | 57% | Loss of helix (P = 0.0033) |
| Damaging | Gain of loop (P = 0.024 Loss of phosphorylation (P = 0.029) |
|||||
Abbreviations: NA, not applicable.
In Polyphen-2 higher scores indicate that a SNP is more likely to be damaging.
In SIFT, lower scores indicate that a SNP is more likely to be deleterious.
In MutPred, a higher probability indicates that a is more likely to be damaging.
Actionable hypotheses are predicted functional changes for which the probability deleterious is <50% and for which the structural change P<.05.
4. Discussion
In a small WES study of seven familial AD cases, we identified two AKAP9 SNPs which co-occur on a single uncommon background haplotype (HAP0) spanning AKAP9 and the surrounding area. These SNPs were significantly enriched in AD cases compared to cognitively normal controls in a large African-American sample, and the associations were replicated in a larger independent AA cohort. These missense variants are rare in African-descent populations (ESP AA sample: MAFrs144662445=0.43% and MAFrs149979685=0.36%; 1000 Genomes AFR sample: MAFrs144662445=0.81% and MAFrs149979685=0.61%). They are not present in approximately 4,000 Caucasians in the ESP database or in currently available data for 379 European and 286 East-Asian subjects in the 1000 Genomes database. Although the estimated OR’s are lower in the replication sample compared to the discovery sample (as expected due to ‘winner’s curse’), the magnitude of effect of these variants on AD risk in the replication sample (estimated ORs of 2.75 and 3.61) is slightly larger than that reported for APOE ε4 in AAs (OR=2.31)[14]. Hence, as expected given their frequency, the identified AKAP9 risk variants not likely to explain a great proportion of AD risk in AAs. However, those inheriting at least one risk allele have a substantially increased risk of AD. Bioinformatic analysis indicated that one of these SNPs (rs149979685) likely has an adverse impact on the structure or post-translational modification of the AKAP protein.
We observed many differences in the 1000 Genomes database between the HAP0 haplotype containing the AD-associated AKAP9 SNPs and HAP0 haplotypes lacking both mutations, suggesting that the instances of the rare rs149979685 and rs144662445 variants are likely identical by descent, although derived from a very old mutational event. This conclusion is supported by the observation of these variants in subjects of diverse African ancestry and by evidence in our study that subjects sharing these alleles do not appear to be related or from a particular population subgroup based on genome-wide measures of IBD sharing and PC analysis.
AKAP9, located on chromosome 7q21–22, encodes a member of the A kinase anchoring protein (AKAP) family. AKAPs bind or tether protein kinase A (PKA) and other signaling molecules to relevant targets[32]. AKAP9 is differentially spliced into isoforms which localize to different cellular compartments[32] and are involved in distinct biological processes. AKAP9 gene products include a short isoform called Yotiao and long isoforms (AKAP350, AKAP450, and CG-NAP) often referred to collectively as AKAP450 based on their molecular weight.
AKAP9 was identified in a cDNA library of human brain where the Yotiao isoform is expressed in the hippocampus, cerebellum, and cerebral cortex[33]. A SNP in Yotiao has been identified as a cause of long-QT syndrome[34], a congenital heart abnormality characterized by long-QT intervals and arrhythmias. Disruption of the binding of PKA and AKAPs (including AKAP5 and Yotiao) at nerve terminals in the hippocampus has been shown to interfere with cellular mechanisms associated with spatial memory[35]. The locations of rs144662445 and rs149979685 are approximately 38 kb 3’ and 61 kb 3’, respectively, from the boundaries of the Yotiao coding region. Therefore, these variants probably do not affect the Yotiao isoform, unless they are regulatory or simply in LD with unidentified non-coding risk variants. Hence it is more likely that the AD-associated AKAP9 variants affect the structure or function of AKAP450. AKAP450 localizes to centrosomes of cells[36]. It is expressed in most tissues including brain. Disrupted binding of AKAP450 to the centrosome interferes with centriole duplication and cell cycle progression[37]. Studies have also shown that AKAP450 is involved in microtubule anchoring and organization at the centrosome[38] and necessary for the initiation of new microtubule formation on the cis-side of the golgi[39]. The non-synonymous change induced by rs144662445 (I2558M transcript Q99996-1) is 5 amino acids from the R2 binding site of AKAP450 (Figure 3). Rs149979685 (S3771L transcript Q99996-1) is located within the pericentrin/AKAP450 centrosomal targeting (PACT) domain which is a highly conserved region near the 3’ end of AKAP450 and is the region responsible for the localization of AKAP450 to the centrosome[40] (Figure 3). The role of AKAP450 in AD is unclear, however, it has functional similarity with tau protein which is involved in microtubule stability and assembly and is a key constituent of neurofibrillary tangles which accumulate in AD brain[41–44]. Reports also indicate that phosphorylated tau does not account for all of the microtubule loss and shortening observed in neurons of those with AD[45]. Thus, it is possible that functional defects in AKAP450 may contribute to AD pathogenesis.
Figure 3.

Annotated protein domain of AKAP9 canonical transcript Q99996-1 including Yotiao; AKAP450; the PKA binding sites; the PACT domain; Long QT syndrome amino-acid change; and the conservation of the two AD-associated variants rs144662445 (I2558M) and rs149979685 (S3771L).
These results should be interpreted cautiously. The bioinformatics programs have predicted that rs149979685 has a possible function while rs144662445 does not. However, without functional data and supporting molecular work, we cannot definitively demonstrate that rs149979685 is the causal locus or whether or not rs144662445 is potentially causal due to some other mechanism not accounted for in the software used, or even whether another non-coding regulatory variant in LD with both rs144662445 and rs149979685 is responsible for the observed association with AD. However, examination of the region in 1000 Genomes and the ESP database indicates that such a variant is not likely to be a non-synonymous coding change. Resolution of this question will require functional experimentation. Finally, we acknowledge that our findings do not represent the breadth of possible AD-related rare variation in African Americans because the majority of such variants would not be present in the exons of seven individuals. Thus, for example, it is not surprising that we did not observe previously reported rare AD-risk variants in PLD3 [18]. It is also possible that several of the non-AKAP9 variants identified by WES and selected for genotyping are actually true risk loci which the discovery sample was underpowered to confirm by association. Targeted resequencing in both African- and European-descent populations will be necessary to identify the full complement of genetic variants which will provide greater understanding of the role of AKAP9 in AD pathogenesis.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge the assistance of Madison Steele and Alison Peltz, participants in Boston University’s Research Internship in Science & Engineering (RISE) Program.
Additional Members of the Alzheimer Disease Genetics Consortium Who Contributed to This Study: Lisa L. Barnes, PhD, Guiqing Cai, PhD, Laura B. Cantwell, MPH, Philip L. De Jager, MD, PhD, Rodney C. P. Go, PhD, Patrick Griffith, MD, Rosalyn Lang, PhD, Oscar L. Lopez, MD, Thomas O. Obisesan, MD, Towfique Raj, PhD
Funding Support:
This work supported by NIA/NIH grants R01-AG09029, R01-AG025259, P30-AG13846 (Dr. Farrer); U01-AG016976 (Dr. Kukull); U24-AG026395, U24-AG026390; R01-AG037212, R37-AG015473 (Dr. Mayeux); U24-AG021886 (Dr. Foroud); R01-AG20688 (Dr. Fallin); P50-AG005133, AG041718, AG030653 (Dr. Kamboh); R01-AG019085 (Dr. Haines); R01-AG1101, R01-AG030146, RC2-AG036650 (Dr. Evans); P30-AG10161, R01-AG15819, R01-AG17917 (Dr. Bennett); R01AG028786 (Dr. Manly); R01-AG22018, P30-AG10161 (Dr. Barnes); P50AG-16574, R01-032990, KL2-RR024151 (Dr. Ertekin-Taner); R01-AG027944, R01-AG028786 (Dr. Pericak-Vance); P20-MD000546, R01-AG28786 (Dr. Byrd); AG005138 (Dr. Buxbaum); P50-AG05681, P01-AG03991, P01-AG026276 (Dr. Goate); and U01-AG032984, RC2-AG036528 (Dr. Schellenberg).
Abbreviations
- AA
African Americans
- AD
Alzheimer disease
- ADGC
Alzheimer Disease Genetics Consortium
- GEE
generalized estimating equation analysis
- GenerAAtions
Genetic and Environmental Risk Factors for Alzheimer Disease among African Americans Study
- GWAS
Genome-wide association study
- IBD
identity by descent
- IPA
INGENUITY Pathway Analysis software
- LD
linkage disequilibrium
- MAF
minor allele frequency
- MIRAGE
Multi-Institutional Research on Alzheimer Genetic Epidemiology Study
- NHW
non-Hispanics whites
- OR
odds ratio
- PC
principal components
- SNP
single nucleotide polymorphism
- WES
whole-exome sequencing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest:
The authors of this paper have no conflicts of interest to report.
References
- 1.Bienvenu OJ, Davydow DS, Kendler KS. Psychiatric ‘diseases’ versus behavioral disorders and degree of genetic influence. Psychol Med. 2011;41:33–40. doi: 10.1017/S003329171000084X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 3.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype, Alzheimer disease. A meta-analysis APOE and Alzheimer Disease Meta Analysis Consortium. Jama. 1997;278:1349–1356. [PubMed] [Google Scholar]
- 4.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 5.Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green RC, Cupples LA, Go R, Benke KS, Edeki T, Griffith PA, et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. Jama. 2002;287:329–336. doi: 10.1001/jama.287.3.329. [DOI] [PubMed] [Google Scholar]
- 10.Tang MX, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, et al. The APOE-epsilon4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. Jama. 1998;279:751–755. doi: 10.1001/jama.279.10.751. [DOI] [PubMed] [Google Scholar]
- 11.Graff-Radford NR, Green RC, Go RC, Hutton ML, Edeki T, Bachman D, et al. Association between apolipoprotein E genotype and Alzheimer disease in African American subjects. Arch Neurol. 2002;59:594–600. doi: 10.1001/archneur.59.4.594. [DOI] [PubMed] [Google Scholar]
- 12.Logue MW, Schu M, Vardarajan BN, Buros J, Green RC, Go RC, et al. A comprehensive genetic association study of Alzheimer disease in African Americans. Arch Neurol. 2011;68:1569–1579. doi: 10.1001/archneurol.2011.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melville SA, Buros J, Parrado AR, Vardarajan B, Logue MW, Shen L, et al. Multiple loci influencing hippocampal degeneration identified by genome scan. Ann Neurol. 2012;72:65–75. doi: 10.1002/ana.23644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4, and the risk of late-onset Alzheimer disease in African Americans. Jama. 2013;309:1483–1492. doi: 10.1001/jama.2013.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 16.Bird TD. Genetic aspects of Alzheimer disease. Genetics in medicine : official journal of the American College of Medical Genetics. 2008;10:231–239. doi: 10.1097/GIM.0b013e31816b64dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schork NJ, Murray SS, Frazer KA, Topol EJCommon vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014;505:550–554. doi: 10.1038/nature12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reitz C, Cheng R, Rogaeva E, Lee JH, Tokuhiro S, Zou F, et al. Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch Neurol. 2011;68:99–106. doi: 10.1001/archneurol.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehmann DJ, Cortina-Borja M, Warden DR, Smith AD, Sleegers K, Prince JA, et al. Large meta-analysis establishes the ACE insertion-deletion polymorphism as a marker of Alzheimer’s disease. Am J Epidemiol. 2005;162:305–317. doi: 10.1093/aje/kwi202. [DOI] [PubMed] [Google Scholar]
- 22.Meng Y, Baldwin CT, Bowirrat A, Waraska K, Inzelberg R, Friedland RP, et al. Association of polymorphisms in the Angiotensin-converting enzyme gene with Alzheimer disease in an Israeli Arab community. Am J Hum Genet. 2006;78:871–877. doi: 10.1086/503687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 24.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 25.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 26.R Development Core Team. R: A language and environment for statistical computing. 2008 [Google Scholar]
- 27.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. Epub 2006 Jul 23. [DOI] [PubMed] [Google Scholar]
- 28.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25:2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. Epub 2007 Jul 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 33.Lin JW, Wyszynski M, Madhavan R, Sealock R, Kim JU, Sheng M. Yotiao, a novel protein of neuromuscular junction and brain that interacts with specific splice variants of NMDA receptor subunit NR1. J Neurosci. 1998;18:2017–2027. doi: 10.1523/JNEUROSCI.18-06-02017.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A. 2007;104:20990–20995. doi: 10.1073/pnas.0710527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nie T, McDonough CB, Huang T, Nguyen PV, Abel T. Genetic disruption of protein kinase A anchoring reveals a role for compartmentalized kinase signaling in theta-burst long-term potentiation and spatial memory. J Neurosci. 2007;27:10278–10288. doi: 10.1523/JNEUROSCI.1602-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Witczak O, Skalhegg BS, Keryer G, Bornens M, Tasken K, Jahnsen T, et al. Cloning and characterization of a cDNA encoding an A-kinase anchoring protein located in the centrosome, AKAP450. Embo J. 1999;18:1858–1868. doi: 10.1093/emboj/18.7.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keryer G, Witczak O, Delouvee A, Kemmner WA, Rouillard D, Tasken K, et al. Dissociating the centrosomal matrix protein AKAP450 from centrioles impairs centriole duplication and cell cycle progression. Mol Biol Cell. 2003;14:2436–2446. doi: 10.1091/mbc.E02-09-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keryer G, Di Fiore B, Celati C, Lechtreck KF, Mogensen M, Delouvee A, et al. Part of Ran is associated with AKAP450 at the centrosome: involvement in microtubule-organizing activity. Mol Biol Cell. 2003;14:4260–4271. doi: 10.1091/mbc.E02-11-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rivero S, Cardenas J, Bornens M, Rios RM. Microtubule nucleation at the cis-side of the Golgi apparatus requires AKAP450 and GM130. Embo J. 2009;28:1016–1028. doi: 10.1038/emboj.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gillingham AK, Munro S. The PACT domain, a conserved centrosomal targeting motif in the coiled-coil proteins AKAP450 and pericentrin. EMBO Rep. 2000;1:524–529. doi: 10.1093/embo-reports/kvd105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- 42.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, et al. Defective brain microtubule assembly in Alzheimer’s disease. Lancet. 1986;2:421–426. doi: 10.1016/s0140-6736(86)92134-3. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida H, Ihara Y. Tau in paired helical filaments is functionally distinct from fetal tau: assembly incompetence of paired helical filament-tau. J Neurochem. 1993;61:1183–1186. doi: 10.1111/j.1471-4159.1993.tb03642.x. [DOI] [PubMed] [Google Scholar]
- 45.Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, et al. Microtubule reduction in Alzheimer’s disease and aging is independent of tau filament formation. Am J Pathol. 2003;162:1623–1627. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.