

Abstract
Mitochondrial myopathy, lactic acidosis and sideroblastic anemia (MLASA) is a rare mitochondrial disorder that has previously been associated with mutations in PUS1 and YARS2. In the present report, we describe a 6 year old male with an MLASA plus phenotype. This patient had features of MLASA in the setting of developmental delay, sensorineural hearing loss, epilepsy, agenesis of the corpus callosum, failure to thrive, and stroke-like episodes. Sequencing of the mitochondrial genome identified a novel de novo, heteroplasmic mutation in the mitochondrial DNA (mtDNA) encoded ATP6 gene (m.8969G>A, p.S148N). Whole exome sequencing did not identify mutations or variants in PUS1 or YARS2 or any known nuclear genes that could affect mitochondrial function and explain this phenotype. Studies of fibroblasts derived from the patient revealed a decrease in oligomycin-sensitive respiration, a finding which is consistent with a complex V defect. Thus, this mutation in MT-ATP6 may represent the first mtDNA point mutation associated with the MLASA phenotype.
Keywords: MLASA, lactic acidosis, mitochondria, ATP6, mitochondrial myopathy
1. Introduction
MLASA1 (OMIM # 600462, OMIM #613561) is a rare mitochondrial disorder characterized by myopathy, lactic acidosis, and sideroblastic anemia. Variable features of this condition include failure to thrive [1–6], and developmental delay or intellectual disability [3–5, 7]. In addition, one patient with MLASA has had pigmentary retinopathy [3], one patient has been reported with renal involvement and multiple metabolic decompensations [8] and several patients have been described with cardiac involvement, typically hypertrophic cardiomyopathy [4, 6, 8–10]. The age of onset of symptoms of patients with MLASA varies with some patients diagnosed in infancy with sideroblastic anemia and/or failure to thrive whereas other patients present in childhood or later with exercise intolerance secondary to myopathy. Biochemical investigations in all patients described have demonstrated lactic acidemia, and although respiratory chain studies in fibroblasts may be normal, skeletal muscle biopsies typically display decreased complex I and IV activities with reduced complex III activity in a subset of patients [3–5, 7, 10–13]. The lack of pancreatic dysfunction distinguishes MLASA from Pearson syndrome, an mtDNA single deletion disorder that is also associated with sideroblastic anemia.
Previously, MLASA has been associated with autosomal recessive inheritance. Two nuclear encoded genes (PUS1 and YARS2) have been associated with the phenotype [1, 4]. Mutations in PUS1 have been associated with the MLASA phenotype in patients of Persian Jewish and Italian descent [1, 3]. PUS1 encodes the nuclear pseudouridine synthase 1 which pseudouridinylates tRNA, and patients with mutations in PUS1 have tRNAs which lack pseudouridine [14]. The mechanism by which mutations in PUS1 result in mitochondrial dysfunction remains unclear although an effect on mitochondrial protein synthesis is hypothesized [1]. Mutations in YARS2, which encodes the human mitochondrial tyrosyl-tRNA, have been associated with the MLASA phenotype in patients of Lebanese descent [4, 6, 10, 11], in one French patient [10], and in Turkish siblings [8]. As with PUS1 mutations, a reduction in mitochondrial protein synthesis is the proposed mechanism by which YARS2 mutations result in the MLASA phenotype, and overexpression of YARS2 rescued the mitochondrial translation defect observed in the myotubes of one subject with mutations in this gene [4, 10, 11]. To date, mutations in no other autosomal or mitochondrial genes have been associated with this phenotype.
In the present report, we describe a 6 year old boy with a severe form of MLASA (eg. MLASA plus) with an infantile presentation of sideroblastic anemia, severe developmental delay, failure to thrive, epilepsy, agenesis of the corpus callosum, sensorineural hearing loss, and stroke-like episodes. Sequencing of his mitochondrial genome revealed a novel de novo, heteroplasmic mutation in the mitochondrial encoded ATP6 gene (m.8969G>A, p.S148N) [15] and whole exome sequencing did not identify any mutations or variants in known nuclear genes associated with mitochondrial dysfunction or disease. Thus, this is the first report of an MLASA phenotype associated with a point mutation in an mtDNA-encoded gene.
2. Materials and Methods
2.1 Patients and DNA
For research testing, tissue samples were collected according to IRB approved research protocols. DNA from blood, muscle, urine sediments, and hair follicles was extracted according to published and manufacturer procedures (Gentra Systems Inc., Minneapolis, MN).
2.2 Mitochondrial whole genome sequencing analysis
The entire mitochondrial genome was amplified in 24 overlapping fragments followed by Sanger sequencing [16]. In addition, mitochondrial whole-genome amplification by single-amplicon LR-PCR and construction of Illumina indexed libraries were performed as previously described [17, 18]. Twelve indexed DNA libraries were pooled together at equal molar ratio and sequenced in a single lane of one flow cell on HiSeq2000 (Illumina, San Diego, CA) with 76-bp single-end reads. The average coverage depth for the next-generation sequencing studies was >20,000X per base.
2.3 Whole exome sequencing and targeted confirmation
Exome sequencing and data interpretation were performed at the Whole Genome Laboratory and Medical Genetics Laboratory at Baylor College of Medicine. Briefly, the DNA extracted from a blood sample of this patient was sonicated and the fragmented genomic DNA was ligated to the Illumina multiplexing PE adapters. The adapter-ligated DNA is then PCR amplified using primers with sequencing barcodes. For target enrichment/exome capture procedure, the pre-capture library is enriched by hybridizing to biotin labeled VCRome 2.1. Subsequently, the post capture library DNA is subjected to sequence analysis on Illumina HiSeq platform for 100 bp paired-end reads. Whole exome sequencing data were then analyzed and annotated using the HGSC-Mercury pipeline (www.tinyurl.com/HGSC-Mercury) [19]. Variants that were regarded clinically significant were confirmed by targeted Sanger sequencing.
2.4 Biochemical analysis
The electron transport chain enzyme activities assayed on skeletal muscle or cultured fibroblasts derived from the proband were assayed at 30°C using a temperature-controlled spectrometer. Each assay was performed in duplicate. The activities of complex I (NADH: Ferricyanide dehydrogenase), complex II (succinate dehydrogenase), complex I+III (NADH: cytochrome c oxidoreductase), complex II+III (succinate: cytochrome c oxidase) were measured using different electron donors/acceptors. The increase or decrease of cytochrome c at 550 nm was measured for complex I+III, II+III, or complex IV (cytochrome c oxidase). The activity of complex I was measured by following the oxidation of NADH at 340 nm. The activity of complex II (succinate dehydrogenase) was analyzed by tracking the secondary reduction of 2,6 –dichlorophenolindophenol (DCIP) by ubiquinone-2 at 600 nm. Citrate synthase was used as a marker for mitochondrial content and its activity was determined by the reduction of 5,5′-dithiobis-2-nitrobenzoic acid at 412 nm in the presence of acetyl-CoA and oxaloacetate.
2.5 Cellular respiration assay
XF24 extracellular flux analyzer from Seahorse Biosciences was used to measure the rates of oxygen consumption in fibroblasts derived from the patient as well as from 5 deidentified, unrelated and unaffected individuals as previously described [20]. All cell lines were cultured at similar low passage number and harvested at similar confluence. Cells were plated the day prior to the experiment on the XF24 cell culture microplates at a density of 60,000 cells per well. XF24 cartridge was equilibrated with the calibration solution overnight at 37 °C. XF assay media (5mM glucose/5mM galactose, 2mM pyruvate, in DMEM (Seahorse Biosciences)) was prepared and pH adjusted to 7.0 on the day of the experiment. XF assay media was used to prepare cellular stress reagents, 500nM oligomycin, 500nM FCCP, 100nM antimycin A and 100nM rotenone (final concentration). All the reagents were loaded in the ports as suggested by Seahorse Biosciences. Oxygen consumption rates (OCR) were measured for 3 minutes with 3 minutes of mixing and 2 minutes of waiting period. After the assay was completed, viable cells in each well were counted using the ViCell cell counter, and the counts were used to normalize the OCR. OCR is expressed as pmoles of oxygen/min/1000cells. Basal respiration (BR) was calculated as the difference between the pre-oligomycin OCR and the Rotenone/Antimycin A resistant OCR. Oligomycin sensitive respiration (OSR) was calculated as the difference between the pre-oligomycin OCR and the oligomycin resistant OCR. Respiratory capacity (RC) was calculated as the difference between the FCCP-stimulated OCR and the Rotenone/Antimycin A resistant OCR. Spare respiratory capacity (SRC) was calculated as the difference between RC and BR.
3. Case Report
The proband was born via repeat Caesarean section at 32 weeks to non-consanguineous parents with a birth weight of 2.01 kg (50–70th %tile). Pregnancy was complicated by oligohydramnios. Mother denied smoking, alcohol or drug use during the pregnancy. The infant was admitted to the neonatal intensive care unit (NICU) for oxygen and feeding difficulties for five weeks. During his admission, head imaging (ultrasound and MRI) revealed evidence of a Grade II intraventricular hemorrhage and agenesis of the corpus callosum. In addition, he had an abnormal EEG that showed evidence of bicentral and bitemporal sharp spikes of low to moderate amplitude. He also had an abnormal brainstem auditory evoked potential study and was eventually diagnosed with sensorineural hearing loss.
At approximately 2 months of age, the infant was re-admitted for anemia requiring packed red blood cell transfusions. He was subsequently diagnosed with sideroblastic anemia and has required regular packed red blood cell transfusions to maintain his hemoglobin in the normal range. At 5 months of age, he developed epilepsy requiring anti-epileptic medication. In addition, he has had failure to thrive requiring gastrostomy tube feeds and severe global developmental delay. At four years of age, he was admitted for abnormal movements and head CT and MRI revealed evidence for a metabolic stroke (Figure 1). This imaging also confirmed the persistence of agenesis of the corpus callosum. Following this event, parents noted residual left sided weakness. By history, parents have since reported a second right sided stroke-like episode (at six years of age) for which he was hospitalized, although imaging was not available for our review. A coagulopathy workup was completed, and no clinically significant abnormalities were noted. By the time of his most recent visit to genetics, he had also been diagnosed with Wolff-Parkinson-White (WPW) by EKG, was followed by cardiology and was taking a beta-blocking medication.
Figure 1.
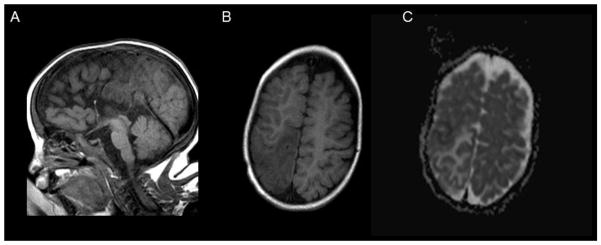
Brain MR imaging performed during first stroke-like episode. A. A midline sagittal T1-weighted image demonstrates agenesis of the corpus callosum. B. An axial image demonstrates T1 hypointensity in a non-vascular pattern of distribution in the right posterior parietal region. C. The ADC (apparent diffusion coefficient) image of the same demonstrates a mixed pattern of restricted (black) and unrestricted (white) diffusion most consistent with metabolic failure
At his most recent visit to the genetics clinic at 6 years of age, he was non-ambulatory and non-verbal. Parents reported that he could turn himself and that he was attempting to hold his head up and grasp for objects. He had no regression. On physical examination, his height and weight were below 3rd centile. His head circumference was at the 8th centile for age. His examination was notable for an inability to track, high arched palate, contractures at knees and ankles, axial hypotonia, increased tone of lower extremities, muscle weakness and atrophy. He had a history of bilateral post-axial toe duplication. The reported left sided weakness was not evident on our examination.
4. Results
Lactate (2.4–6.1 mmol/L) was repeatedly elevated in the proband’s blood. In addition, large quantities of fumarate, malate, β-hydroxybutyrate, and lactate were detected in urine organic acid analysis, and plasma amino acids showed elevated alanine. An acylcarnitine profile in plasma was normal. Pyruvate carboxylase and pyruvate dehydrogenase complex activities in cultured fibroblasts were normal.
Exon targeted MitoMet® array CGH (44,000 oligo probes) did not detect any intragenic deletions [21, 22]. PDHA1 and PUS1 sequencing analysis did not reveal deleterious mutations in the coding regions of those genes. Sanger sequencing of the mitochondrial genome revealed an apparently homoplasmic novel variant, m.8969G>A (p.S148N, ATP6), and two apparently homoplasmic rare variants, m.8702C>T (p.T59I, ATP6) and m.14757T>C (p.M4T, CYTB), in the proband’s muscle and blood specimens. The novel variant, m.8969G>A (p.S148N, ATP6), results in the change of serine to asparagine in the ATP6 protein. The serine at position 148 of ATP6 is highly conserved from yeast to human (Figure 2). The computer-based algorithms, PolyPhen2 and SIFT, predict the m.8969G>A (p.S148N in ATP6) to be deleterious [23, 24]. The m.8702C>T (p.T59I in ATP6) and m.14757T>C (p.M4T in CYTB) variants have been reported in mtDB at the frequencies of 0.04 % and 0.11 %, respectively (www.genpat.uu.se/mtDB/). The threonine at position 59 of the ATP6 protein and methionine at position 4 of CYTB protein are not evolutionarily conserved. Additional variants were identified from the proband (Supplemental Table 1), and they were either synonymous changes or previously reported polymorphisms most likely to be benign. Both m.8702C>T (p.T59I, ATP6) and m.14757T>C (p.M4T, CYTB) variant are homoplasmic in the proband and in his mother by next-generation sequencing (NGS). Because both homoplasmic m.8702C>T (p.T59I, ATP6) and m.14757T>C (p.M4T, CYTB) variants were identified in the asymptomatic mother, these two variants are unlikely to be the primary cause of the clinical symptoms in the proband. Subsequent amplification refractory mutation system (ARMS) quantitative qPCR and allele specific oligonucleotide hybridization (ASO) analyses detected heteroplasmic m.8969G>A (p.S148N, ATP6) at 89%, 80% and 85% of mutation load, respectively, in the blood, muscle and skin fibroblast cell specimens from the proband, but this variant was absent in the blood, urine and hair samples from the mother. The more accurate and sensitive NGS method confirmed the presence of the m.8969G>A (p.S148N, ATP6) novel variant at 96% and 88% heteroplasmy in the proband’s blood and muscle specimens respectively, but this variant was not detected in his mother’s blood sample using this method (Figure 3) [17, 18]. The failure to detect the m.8969G>A variant in the blood, urine and hair specimens of the mother suggested that it may have arisen de novo in the proband. However, maternal gonadal mosaicism cannot be ruled out. Clinical whole exome sequencing was also performed to determine if an autosomal locus might be contributing to his phenotype, but mutations or likely pathogenic variants in known nuclear genes associated with mitochondrial disease were not detected. Furthermore, complete coverage of YARS2 was achieved with Sanger sequencing in combination with whole exome sequencing and no mutations were identified.
Figure 2.
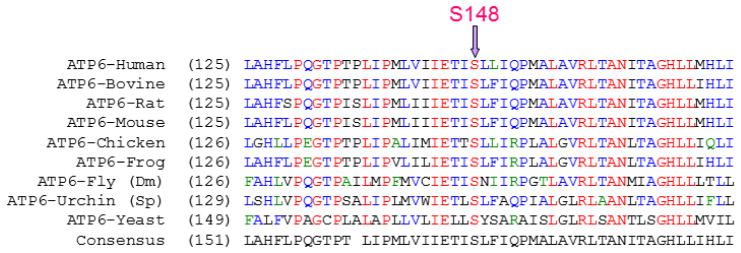
Conservation of serine residue at position 148 in ATP6. The serine at position 148 in mtATP6 is conserved from yeast to human.
Figure 3.

Heteroplasmic m.8969G>A (p.S148N, ATP6) detected in the proband.
A muscle biopsy was performed, and enzymatic activity assays of the electron transport chain (ETC) showed increased citrate synthase (CS) activity, indicating mitochondrial proliferation. After normalization to CS, no major defects were observed in the ETC complex I–IV activities (all >70% of control means). Because the ETC studies only evaluate for defects in complexes I to IV, patient fibroblasts along with control cell lines (5 independent lines) were also analyzed for respiration defects using XF24 analyzer (Seahorse). A fibroblast cell line derived from the patient and 5 independent control fibroblast cell lines were analyzed for cellular respiration using an XF24 analyzer (Seahorse Biosciences). Basal respiration (BR), oligomycin-sensitive respiration (OSR), and respiratory capacity (RC) were determined with the sequential addition of the complex V inhibitor, oligomycin, the uncoupler FCCP (carbonyl cyanide-p-trifluromethoxyphenylhydrazone), and ETC inhibitors rotenone (R) and antimycin A (A). Consistent with a complex V defect, the patient cells exhibited a dramatic decrease in OSR compared to controls. In addition, the patient cells demonstrated significant reductions in BR, RC, and spare respiratory capacity (SRC, difference between RC and BR) (Table 1, Figure 4).
Table 1.
Cellular respiration studies in fibroblast cell line derived from the proband (P) and unrelated normal controls (C). Respiration rates are presented as mean ± standard deviation. Values represent data from two replicate studies in the same cells.
| 5mm Glucose | 5mm Galactose | |||
|---|---|---|---|---|
| C1 | P2 | C1 | P2 | |
| Basal respiration3 | 2.09±0.3 | 1.6±0.23# | 2.41±0.23 | 1.37±0.14* |
| Oligomycin sensitive respiration3 | 0.8±0.07 | 0.19±0.09* | 1.16±0.08 | 0.02±0.006* |
| Respiratory capacity3 | 4.03±0.16 | 2.19±0.12* | 5.58±0.22 | 1.9±0.13* |
| Spare respiratory capacity3 | 2.9±0.09 | 1.01±0.1* | 3.78±0.09 | 1.28±0.08* |
N = 5 independent control fibroblasts cell lines
N = 5 independent wells
pMoles oxygen/min/1×103 cells
p-value = 0.02, unpaired Student’s T-test
p-value = 0.0001, unpaired Student’s T-test
Figure 4.
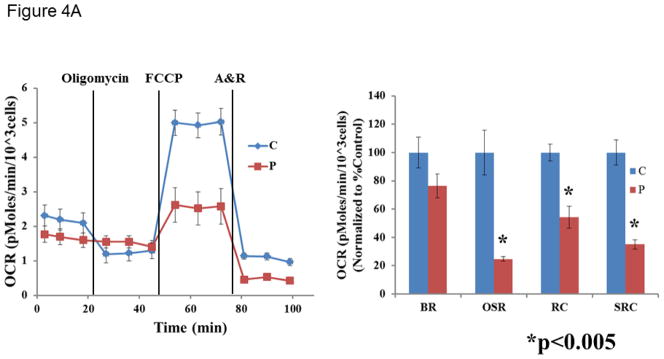

Cellular respiration measured in fibroblast cell line derived from patient #1 and controls in the presence of 5 mM glucose (A) or 5 mM galactose (B). Data is representative of two replicate analyses. The control cells are shown in blue and the patient cells are shown in red. OCR = oxygen consumption rate. A&R = antimycin A and rotenone. BR = basal respiration. OSR = oligomycin-sensitive respiration. RC = respiratory capacity. SRC = spare respiratory capacity. An unpaired Student’s t-test was used to compare the patient fibroblast cell line with the control fibroblast cell lines.
5. Discussion
Although MLASA has not been previously associated with mutations in the mitochondrial genome, multiple lines of evidence support the conclusion that the m.8969G>A (p.S148N, ATP6) mutation identified in MT-ATP6 is associated with the MLASA plus phenotype in our patient. First, the amino acid change is de novo, affects a highly conserved residue and is predicted to be deleterious by two different software algorithms (SIFT and Polyphen2). In addition, whole exome sequencing did not identify deleterious mutations in relevant genes. Furthermore, functional studies performed using a skin fibroblast cell line derived from the patient showed a profound reduction in oligomycin sensitive respiration. A reduction in oligomycin sensitive respiration provides strong evidence for a defect in complex V, which would be expected with a deleterious mutation in MT-ATP6. Interestingly, the cellular respiration studies performed in fibroblasts also revealed a decrease in basal respiration and a decrease in respiratory capacity. This severe defect in cellular respiration likely explains the phenotype in our patient. It is possible that a complex V defect may result in instability of ETC supercomplexes without compromising the integrity of monomeric ETC complexes.
Missense mutations in MT-ATP6 have been associated with a wide variety of phenotypes including Leigh Syndrome, late onset Leigh Syndrome, neuropathy, ataxia and retinitis pigmentosa (NARP), bilateral striatal necrosis, ataxia, and developmental delay, which have recently been summarized elsewhere [25]. This report expands the clinical spectrum associated with MT-ATP6 mutations as this case is the first to associate the MLASA phenotype with MT-ATP6.
The phenotypes of previously reported patients with MLASA have ranged in severity from early infantile presentation with failure to thrive or sideroblastic anemia to presentation in later childhood or early adulthood with myopathy and/or exercise tolerance [1–5, 7–9, 11, 12]. Our patient is on the severe end of the MLASA phenotypic spectrum with early infantile presentation of transfusion-dependent sideroblastic anemia in the setting of failure to thrive, hearing loss, epilepsy, stroke-like episodes, and severe developmental delay. To our knowledge, no other patients with MLASA have been reported with sensorineural hearing loss, stroke-like episodes, or WPW syndrome. Given that no known genes associated with either hearing loss, early-onset stroke-like episodes, or WPW were identified with whole exome sequencing, these phenotypes may be associated with the MLASA phenotype rather than a separate disorder. The biochemical phenotype of our patient is also unique when compared to other previously reported patients with MLASA because activities of complex I, III, and IV were normal as would be expected with a defect on complex V. However, a severe respiratory phenotype observed in skin fibroblast cell studies reflects the defect in complex V and may explain why his clinical phenotype is so severe. Thus, his clinical and biochemical presentation is unique compared to previously described patients with MLASA.
To our knowledge, the present case is the first case of MLASA associated with a mutation in MT-ATP6 or any other mitochondrial DNA-encoded gene and thus, extends the etiologies of the MLASA phenotype and the phenotypic spectrum of mutations in MT-ATP6. Therefore, next generation sequencing of the mitochondrial genome should be considered in patients with MLASA or MLASA plus, especially if mutations in PUS1 and YARS2 are not detected.
Supplementary Material
Highlights.
A novel, de novo, heteroplasmic mutation in MT-ATP6 has been identified in a patient with MLASA.
A decrease in oligomycin-sensitive respiration was identified in fibroblasts from this patient.
This mutation in MT-ATP6 may represent the first mtDNA point mutation associated with MLASA.
Acknowledgments
L.C.B. was supported by the Genzyme/ACMG Foundation for Genetic and Genomic Medicine Medical Genetics Training Award in Clinical Biochemical Genetics and the Medical Genetics Research Fellowship Program NIH/NIGMS NIH T32 GM07526. T.D. and B.H.G. were supported by NIH/NIGMS R01 GM098387 (B.H.G.). This project was also supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (NIAID P30AI036211, NCI P30CA125123, and NCRR S10RR024574) and the assistance of Joel M. Sederstrom.
Footnotes
Abbreviations: MLASA – Mitochondrial myopathy, lactic acidosis, and sideroblastic anemia; ETC-electron transport chain; CS – citrate synthase; OCR – oxygen consumption rate; OSR – oligomycin sensitive respiration; RC-respiratory capacity; SRC – spare respiratory capacity; WPW - Wolff-Parkinson-White; NGS – Next generation sequencing;
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Lindsay C. Burrage, Email: burrage@bcm.edu.
Sha Tang, Email: stang@ambrygen.com.
Jing Wang, Email: jwang7@bcm.edu.
Taraka R. Donti, Email: donti@bcm.edu.
Magdalena Walkiewicz, Email: Magdalena.Walkiewicz@bcm.edu.
J. Michael Luchak, Email: jluchak@bcm.edu.
Li-Chieh Chen, Email: ziv28@hotmail.com.
Eric S. Schmitt, Email: eschmitt@bcm.edu.
Zhiyv Niu, Email: zniu@bcm.edu.
Rodrigo Erana, Email: rxerana@txch.org.
Jill V. Hunter, Email: jvhunter@texaschildrens.org.
Brett H. Graham, Email: bgraham@bcm.edu.
Lee-Jun Wong, Email: ljwong@bcm.edu.
Fernando Scaglia, Email: fscaglia@bcm.edu.
References
- 1.Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA) American journal of human genetics. 2004;74:1303–1308. doi: 10.1086/421530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casas K, Bykhovskaya Y, Mengesha E, Wang D, Yang H, Taylor K, Inbal A, Fischel-Ghodsian N. Gene responsible for mitochondrial myopathy and sideroblastic anemia (MSA) maps to chromosome 12q24.33. American journal of medical genetics Part A. 2004;127A:44–49. doi: 10.1002/ajmg.a.20652. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez-Vizarra E, Berardinelli A, Valente L, Tiranti V, Zeviani M. Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA) Journal of medical genetics. 2007;44:173–180. doi: 10.1136/jmg.2006.045252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riley LG, Cooper S, Hickey P, Rudinger-Thirion J, McKenzie M, Compton A, Lim SC, Thorburn D, Ryan MT, Giege R, Bahlo M, Christodoulou J. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia--MLASA syndrome. American journal of human genetics. 2010;87:52–59. doi: 10.1016/j.ajhg.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeharia A, Fischel-Ghodsian N, Casas K, Bykhocskaya Y, Tamari H, Lev D, Mimouni M, Lerman-Sagie T. Mitochondrial myopathy, sideroblastic anemia, and lactic acidosis: an autosomal recessive syndrome in Persian Jews caused by a mutation in the PUS1 gene. Journal of child neurology. 2005;20:449–452. doi: 10.1177/08830738050200051301. [DOI] [PubMed] [Google Scholar]
- 6.Shahni R, Wedatilake Y, Cleary MA, Lindley KJ, Sibson KR, Rahman S. A distinct mitochondrial myopathy, lactic acidosis and sideroblastic anemia (MLASA) phenotype associates with YARS2 mutations. American journal of medical genetics Part A. 2013;161:2334–2338. doi: 10.1002/ajmg.a.36065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inbal A, Avissar N, Shaklai M, Kuritzky A, Schejter A, Ben-David E, Shanske S, Garty BZ. Myopathy, lactic acidosis, and sideroblastic anemia: a new syndrome. American journal of medical genetics. 1995;55:372–378. doi: 10.1002/ajmg.1320550325. [DOI] [PubMed] [Google Scholar]
- 8.Nakajima J, Eminoglu TF, Vatansever G, Nakashima M, Tsurusaki Y, Saitsu H, Kawashima H, Matsumoto N, Miyake N. A novel homozygous YARS2 mutation causes severe myopathy, lactic acidosis, and sideroblastic anemia 2. Journal of human genetics. 2014 doi: 10.1038/jhg.2013.143. [DOI] [PubMed] [Google Scholar]
- 9.Rawles JM, Weller RO. Familial association of metabolic myopathy, lactic acidosis and sideroblastic anemia. The American journal of medicine. 1974;56:891–897. doi: 10.1016/0002-9343(74)90820-1. [DOI] [PubMed] [Google Scholar]
- 10.Riley LG, Menezes MJ, Rudinger-Thirion J, Duff R, de Lonlay P, Rotig A, Tchan MC, Davis M, Cooper ST, Christodoulou J. Phenotypic variability and identification of novel YARS2 mutations in YARS2 mitochondrial myopathy, lactic acidosis and sideroblastic anaemia. Orphanet journal of rare diseases. 2013;8:193. doi: 10.1186/1750-1172-8-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sasarman F, Nishimura T, Thiffault I, Shoubridge EA. A novel mutation in YARS2 causes myopathy with lactic acidosis and sideroblastic anemia. Human mutation. 2012;33:1201–1206. doi: 10.1002/humu.22098. [DOI] [PubMed] [Google Scholar]
- 12.Bachmeyer C, Ferroir JP, Eymard B, Maier-Redelsperger M, Lebre AS, Girot R. Coenzyme Q is effective on anemia in a patient with sideroblastic anemia and mitochondrial myopathy. Blood. 2010;116:3681–3682. doi: 10.1182/blood-2010-07-299453. [DOI] [PubMed] [Google Scholar]
- 13.Sasarman F, Karpati G, Shoubridge EA. Nuclear genetic control of mitochondrial translation in skeletal muscle revealed in patients with mitochondrial myopathy. Human molecular genetics. 2002;11:1669–1681. doi: 10.1093/hmg/11.14.1669. [DOI] [PubMed] [Google Scholar]
- 14.Patton JR, Bykhovskaya Y, Mengesha E, Bertolotto C, Fischel-Ghodsian N. Mitochondrial myopathy and sideroblastic anemia (MLASA): missense mutation in the pseudouridine synthase 1 (PUS1) gene is associated with the loss of tRNA pseudouridylation. The Journal of biological chemistry. 2005;280:19823–19828. doi: 10.1074/jbc.M500216200. [DOI] [PubMed] [Google Scholar]
- 15.Tang S, Wang J, Zhang VW, Li FY, Landsverk M, Cui H, Truong CK, Wang G, Chen LC, Graham B, Scaglia F, Schmitt ES, Craigen WJ, Wong LJ. Transition to next generation analysis of the whole mitochondrial genome: a summary of molecular defects. Human mutation. 2013;34:882–893. doi: 10.1002/humu.22307. [DOI] [PubMed] [Google Scholar]
- 16.Shanske S, Wong LJ. Molecular analysis for mitochondrial DNA disorders. Mitochondrion. 2004;4:403–415. doi: 10.1016/j.mito.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 17.Zhang W, Cui H, Wong LJ. Comprehensive one-step molecular analyses of mitochondrial genome by massively parallel sequencing. Clinical chemistry. 2012;58:1322–1331. doi: 10.1373/clinchem.2011.181438. [DOI] [PubMed] [Google Scholar]
- 18.Cui H, Li F, Chen D, Wang G, Truong CK, Enns GM, Graham B, Milone M, Landsverk ML, Wang J, Zhang W, Wong LJ. Comprehensive next-generation sequence analyses of the entire mitochondrial genome reveal new insights into the molecular diagnosis of mitochondrial DNA disorders. Genetics in medicine: official journal of the American College of Medical Genetics. 2013;15:388–394. doi: 10.1038/gim.2012.144. [DOI] [PubMed] [Google Scholar]
- 19.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, Bekheirnia MR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Eng CM. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. The New England journal of medicine. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donti TR, Stromberger C, Ge M, Eldin KW, Craigen WJ, Graham BH. Screen for abnormal mitochondrial phenotypes in mouse embryonic stem cells identifies a model for succinyl-CoA ligase deficiency and mtDNA depletion. Disease models & mechanisms. 2014;7:271–280. doi: 10.1242/dmm.013466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong LJ, Dimmock D, Geraghty MT, Quan R, Lichter-Konecki U, Wang J, Brundage EK, Scaglia F, Chinault AC. Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions. Clinical chemistry. 2008;54:1141–1148. doi: 10.1373/clinchem.2008.103721. [DOI] [PubMed] [Google Scholar]
- 22.Chinault AC, Shaw CA, Brundage EK, Tang LY, Wong LJ. Application of dual-genome oligonucleotide array-based comparative genomic hybridization to the molecular diagnosis of mitochondrial DNA deletion and depletion syndromes. Genetics in medicine: official journal of the American College of Medical Genetics. 2009;11:518–526. doi: 10.1097/GIM.0b013e3181abd83c. [DOI] [PubMed] [Google Scholar]
- 23.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome research. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial ATP synthase: architecture, function and pathology. Journal of inherited metabolic disease. 2012;35:211–225. doi: 10.1007/s10545-011-9382-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.