

Abstract
Objective:
Mutations in exon 1 of ARHGAP29, a RhoA specific GTPase have been identified in North American and Filipino subjects with nonsyndromic cleft palate and cleft lip with or without cleft palate. Since the genetic status of ARHGAP29 in Indian subjects with nonsyndromic oral clefts is not known, we designed the present study to investigate the occurrence of the above mutations in them.
Materials and Methods:
Total genomic DNA extracted from peripheral blood of 60 subjects with nonsyndromic cleft palate and cleft lip with or without cleft palate, and equal number of control healthy subjects were amplified with primers flanking exon 1 of ARHGAP29 gene and subjected to direct sequencing.
Results:
Sequencing analysis identified a nonsense mutation in exon 1 of ARHGAP29 that caused substitution of lysine to stop codon at codon position 32 in a subject with nonsyndromic cleft lip with cleft palate. The mutation, however, occurred in heterozygous condition. None of the other subjects carried mutation in this region.
Conclusion:
The study has thus identified a rare but novel truncation mutation in ARHGAP29 gene for the first time in nonsyndromic oral clefts.
Keywords: ARHGAP29, gene mutation, Indian population, cleft lip, cleft palate
INTRODUCTION
The development of the maxillary division of oro-facial region is regulated by a cascade of signaling molecules, the expression and/or activation of which is tightly regulated. Disturbances in the signaling cascade due to function impairing defects in developmental genes may cause failure of meeting and fusion of the developing primordium. The extent of manifestation though may vary among the affected children and may include either cleft lip (CL) or cleft palate (CP) (isolated CL or CP) or cleft lip with cleft palate (CLP),[1] that may involve one or both halves of the facial region and are classified as unilateral or bilateral clefts.[2] Oro-facial cleft may either occur as an isolated event or as a part of other clinical symptoms and are designated as nonsyndromic or syndromic cases, respectively. Nonsyndromic clefts make about 70% of all reported oro-facial clefts.[1,2] Syndrome associated clefts occur in several hundreds of mendelian (autosomal dominant, autosomal recessive, or X-linked) chromosomal conditions,[3] that may at times include mental retardation, myopia and cardiac anomalies[4,5,6,7,8] as additional clinical manifestation. Of the above two, nonsyndromic clefts of the lip and palate (CL, CP, CLP) occurs in every 1/500-1/2500 of live births[9,10] and hence is considered as the most common of all congenital defects.
The cause of development of CL, CP and CLP is believed to be multi-factorial in nature and may include both environmental factors and genetic defects.[11] Two major environmental factors that the developing embryos get exposed to are byproducts of tobacco smoke and alcohol binging by expectant mothers especially during the early trimester. Both of these agents have been found to be associated with the development of oro-facial clefts in infants.[12,13,14,15,16,17,18,19,20,21,22,23] Besides the above, oro-facial cleft has also been observed in the fetus of mothers undergoing phenytoin drug therapy for epileptic conditions or who were consuming dietary supplement, retinoic acid.[21,22] Though the exact the mechanism of development of oro-facial clefts in embryos exposed to these environmental and dietary factors remains yet to be understood, association between functional impairment of molecules responsible for detoxification of environmental agents and drugs and oro-facial clefts have been observed in infants. For example, significant over transmission of G590A variant of NAT2 gene coding N-acetyltransferase 2 enzyme that activates and deactivates arylamine and hydrazine drugs and carcinogens have been observed in subjects with oro-facial clefts relative to controls.[23,24] On the other hand, genetic alteration in developmental regulators of palatal shelf such as TGFA, TGFβ,[25,26] MSX1,[27] IRF6,[28,29] PVR, PVRL1, PVRL2,[30,31] TBX22,[32] FGFR1,[33] ABCA4,[34,35] MAFB1,[34,35] and ARHGAP29 have also been observed in several subjects with oro-facial clefts.
Of the above developmental regulators, ARHGAP29 gene encodes for a GTPase-activating protein (GAP) that has a strong affinity to the small GTP-binding protein RhoA than Rac1 or CDC42.[36] By binding to the active form of RhoA, the GTPase function of ARHGAP29 converts RhoA-GTP to RhoA-GDP.[37] Expression of ARHGAP29 is seen in the mandibular and maxillary processes of developing mouse embryo at E10.5 and the shelves of the secondary palate at E13.5.[37] Nonsense mutation with consequent premature truncation of the protein has been identified within the coding region of exon 1 of ARHGAP29 gene in oro-facial cleft subjects from other races.[37] Studies from the Indian Genome Variation Consortium have suggested that most of the populations in the Indian subcontinent are distinct from HapMap populations, and that the genetic basis of diseases in the Indian population may be different due to differences in the risk allele frequency and pattern of linkage disequilibrium.[38] This prompted us to design the present study, in which, the exon 1 of ARHGAP29 gene was investigated in a small group of Indian subjects with CL, CP or CLP, who belonged to a defined South Indian Dravidian race.
MATERIALS AND METHODS
Study design and subjects
A cross-sectional study was designed and 60 subjects with either NS-CL or CP or CLP, but otherwise medically able were included in the study after obtaining informed consent. As controls, an equal number of race matched healthy volunteers were included. Subjects with syndromic CL or CP or CLP, and those who were not willing to be a part of the study were excluded.
DNA extraction and polymerase chain reaction
About 200 μl of peripheral blood was processed to extract chromosomal DNA with Gen-Elute Blood DNA Extraction Kit (Sigma-Aldrich, USA) according to the manufacturer's protocol. The exon 1 region of ARHGAP29 was amplified with intronic primers (sequence information available upon request) flanking splice donor and acceptor sites. The gene region was amplified under following conditions: Initial denaturation at 94°C for 5 min, followed by 35 cycles of denaturation at 94°C for 45 s, primer annealing at 55°C for 45 s, primer extension at 72°C for 1 min, with a final extension at 72°C for 5 min. The PCR amplicons were confirmed in comparison to DNA size markers and were subsequently eluted with Genelute DNA gel elution kit (Sigma Aldrich, cat# NA1111) and subjected to direct sequencing with same set of primers that were used for PCR amplification.
RESULTS
In order to determine the occurrence of mutation in exon 1 of ARHGAP29 gene in Indian subjects with either NS-CL or CP or CLP, and thus to understand the risk potential of mutant ARHGAP29 in the development of CL/CP/CLP, we screened the chromosomal DNA from 60 CL/CP/CLP subjects along with equal number of healthy volunteers. DNA from peripheral blood was amplified with exon 1 specific primers, and the PCR amplicons were subjected to direct sequencing after gel purification. Analysis of the sequencing data showed a nonsense mutation, c. 94A>T, in a subject with NS-CLP that resulted in the substitution of lysine amino acid with stop codon at codon position 32 in exon 1 (AAG32TAG). However, the mutation was identified to be heterozygous as both wild type AAG, and mutant TAG codons were present at codon position 32 [Figure 1].
Figure 1.
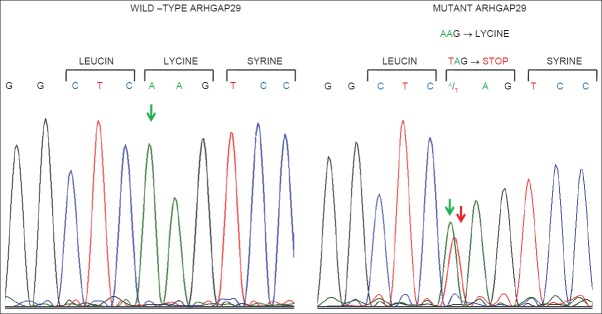
Chromatogram of wild type and mutant sequences within exon 1 of ARHGAP29 gene. Note that the mutant sample carries both wild type nucleotide “A” (indicated by a green arrow) and mutant nucleotide “T” (indicated by a red arrow) at first position of codon 32
DISCUSSION
A total of 60 subjects with NS-CL, CP or CLP and 60 normal subjects were analyzed for the presence of the mutation in ARHGAP29 gene. Both groups of subjects were selected from Tamil speaking Dravidians from the Southern region of India, in order to minimize race dependent genetic variations as genetic diversity has been observed in the population from different parts of India.[38] Though the development of NS-CP/CL/CLP is believed to be due to gene–environment interaction, the present study was focused only on the genetic aspect as no records of the subject's prenatal period or that of respective mother's were available. Direct sequencing analysis of PCR amplicons of exon 1 coding region of ARHGAP29 identified a single mutation in a subject with NS-CLP. The mutation caused substitution of lysine amino acid at codon 32 with a premature stop codon in one copy of ARHGAP29, as the mutant codon occurred along with wild-type lysine codon (indicated by two arrows in mutant ARHGAP29 panel in Figure 1). The identification of K32STOP mutation in only one of 60 CL, CP or CLP subjects who were investigated in the present study indicates that the mutation may be a rare event in CL, CP or CLP from this region. By analyzing 1440 and 360 subjects with CL, CP or CLP from Philippines and US respectively, Leslie et al. identified a nonsense (c. 62_63delCT) and a missense mutation (c. 76A>G) in two independent subjects from Philippines, and a missense mutation (c. 137A>G) in a subject from the US.[37] Considering the occurrence of c. 94A>T mutation in a single NS-CLP subject from a sample size of 60 analyzed in the present study, it may be inferred that the relative prevalence of this mutation may be higher than those observed by Leslie et al. This claim, however, requires to be confirmed by analyzing a higher sample size by including subjects from similar racial background.
Both nonsense mutations, c. 94A>T identified by us, and c. 62_63delCT causes premature truncation of the ARHGAP29 protein. While c. 94A>T creates an in-frame stop codon, c. 62_63delCT causes a frame-shift that consequently results in the premature occurrence of stop codon after twenty misread codons. Both mutations, however, are expected to cause loss of function of ARHGAP29 protein. In contrast, the two missense mutations, c. 76A>G and c. 137A>G that causes substitution of threonine with alanine at codon 26 (T26A) and lysine with arginine at codon 46 (K46R) respectively are expected to have a benign effect on the function of ARHGAP29.[37]
The identification of c. 94A>T mutant allele of ARHGAP29 in heterozygous condition along with wild-type allele suggests that the mutation event causes haplo-insufficiency. It is important to note that the deletion mutation (c. 62_63delCT) reported by Leslie et al. was homozygous in nature.[37] Although biallelic inactivation of genes has often been described as a cause for development and progression of diseases, haplo-insufficiency of genes has also been observed in several disease conditions. For example, haplo-insufficiency has been observed in cancerous lesions,[39,40,41] neurodegenerative disorders,[42] myopathies,[43] inflammatory conditions,[44] and developing tissues leading to birth defects including syndromes such as Frias syndrome (haplo-insufficiency of BMP4),[45] and CNS malformation syndrome (haplo-insufficiency of NFIA).[46] These findings clearly suggest that the heterozygous c. 94A>T mutation identified in the present study may present haplo-insufficiency effect during the development of oro-facial cleft region. As expression of ARHGAP29 transcript is detected strongly in medial and lateral nasal processes and to a lesser extent in the mandibular and maxillary processes at E10.5 and shelves of the secondary palate at E13.5,[37] the haplo-insufficiency effect of c. 94A>T mutant ARHGAP29 may have acted as one of the causative factor in the development of NS-CLP as the etiology of NS-CL/CP/CLP is believed to be polygenic. However, a knock-in experiment to recreate the above heterozygous condition along with other reported mutations in transgenic mice is essential to validate the above observation.
CONCLUSION
The present study has identified for the first time a novel nonsense mutation c. 94A>T in the gene ARHGAP29 in a South Indian Dravidian subject with NS-CLP. The occurrence of a mutation in one of 60 subjects with NS-CL/CP/CLP suggests that the mutation event may be rare.
ACKNOWLEDGMENT
The authors would like to thank the Central Research Funding Authority of Sree Balaji Medical and Dental College and Hospital, Bharath University, Chennai - 600 100, for providing financial support to conduct the research work.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared
REFERENCES
- 1.Lidral AC, Moreno LM, Bullard SA. Genetic factors and orofacial clefting. Semin Orthod. 2008;14:103–14. doi: 10.1053/j.sodo.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jugessur A, Murray JC. Orofacial clefting: recent insights into a complex trait. Curr Opin Genet Dev. 2005;15:270–8. doi: 10.1016/j.gde.2005.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carinci F, Scapoli L, Palmieri A, Zollino I, Pezzetti F. Human genetic factors in nonsyndromic cleft lip and palate: an update. Int J Pediatr Otorhinolaryngol. 2007;71:1509–19. doi: 10.1016/j.ijporl.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 4.Hu DN, Li JH, Chen HY, Chang HS, Wu BX, Lu ZK, et al. Genetics of cleft lip and cleft palate in China. Am J Hum Genet. 1982;34:999–1002. [PMC free article] [PubMed] [Google Scholar]
- 5.Venkatesh R. Syndromes and anomalies associated with cleft. Indian J Plast Surg. 2009;42(Suppl):S51–5. doi: 10.4103/0970-0358.57187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murray JC. Face facts: genes, environment, and clefts. Am J Hum Genet. 1995;57:227–32. [PMC free article] [PubMed] [Google Scholar]
- 7.Lacombe D, Pedespan JM, Fontan D, Chateil JF, Verloes A. Phenotypic variability in van der Woude syndrome. Genet Couns. 1995;6:221–6. [PubMed] [Google Scholar]
- 8.Stottmann RW, Bjork BC, Doyle JB, Beier DR. Identification of a Van der Woude syndrome mutation in the cleft palate 1 mutant mouse. Genesis. 2010;48:303–8. doi: 10.1002/dvg.20618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaturvedi P, Banerjee KS. Spectrum of congenital malformations in the newborns from rural Maharashtra. Indian J Pediatr. 1989;56:501–7. doi: 10.1007/BF02722424. [DOI] [PubMed] [Google Scholar]
- 10.Grosen D, Chevrier C, Skytthe A, Bille C, Mølsted K, Sivertsen A, et al. A cohort study of recurrence patterns among more than 54,000 relatives of oral cleft cases in Denmark: support for the multifactorial threshold model of inheritance. J Med Genet. 2010;47:162–8. doi: 10.1136/jmg.2009.069385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC. Cleft lip and palate. Lancet. 2009;374:1773–85. doi: 10.1016/S0140-6736(09)60695-4. [DOI] [PubMed] [Google Scholar]
- 12.Chung KC, Kowalski CP, Kim HM, Buchman SR. Maternal cigarette smoking during pregnancy and the risk of having a child with cleft lip/palate. Plast Reconstr Surg. 2000;105:485–91. doi: 10.1097/00006534-200002000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Munger RG, Romitti PA, Daack-Hirsch S, Burns TL, Murray JC, Hanson J. Maternal alcohol use and risk of orofacial cleft birth defects. Teratology. 1996;54:27–33. doi: 10.1002/(SICI)1096-9926(199607)54:1<27::AID-TERA4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 14.Romitti PA, Lidral AC, Munger RG, Daack-Hirsch S, Burns TL, Murray JC. Candidate genes for nonsyndromic cleft lip and palate and maternal cigarette smoking and alcohol consumption: evaluation of genotype-environment interactions from a population-based case-control study of orofacial clefts. Teratology. 1999;59:39–50. doi: 10.1002/(SICI)1096-9926(199901)59:1<39::AID-TERA9>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 15.Romitti PA, Sun L, Honein MA, Reefhuis J, Correa A, Rasmussen SA. Maternal periconceptional alcohol consumption and risk of orofacial clefts. Am J Epidemiol. 2007;166:775–85. doi: 10.1093/aje/kwm146. [DOI] [PubMed] [Google Scholar]
- 16.Saad AY, Gartner LP, Hiatt JL. Teratogenic effects of nicotine on palate formation in mice. Biol Struct Morphog. 1990;3:31–5. [PubMed] [Google Scholar]
- 17.Saito T, Cui XM, Yamamoto T, Shiomi N, Bringas P, Jr, Shuler CF. Effect of N’-nitrosonornicotine (NNN) on murine palatal fusion in vitro. Toxicology. 2005;207:475–85. doi: 10.1016/j.tox.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 18.Seller MJ, Bnait KS. Effects of tobacco smoke inhalation on the developing mouse embryo and fetus. Reprod Toxicol. 1995;9:449–59. doi: 10.1016/0890-6238(95)00037-b. [DOI] [PubMed] [Google Scholar]
- 19.Shi M, Wehby GL, Murray JC. Review on genetic variants and maternal smoking in the etiology of oral clefts and other birth defects. Birth Defects Res C Embryo Today. 2008;84:16–29. doi: 10.1002/bdrc.20117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang P, Svoboda KK. Nicotine inhibits palatal fusion and modulates nicotinic receptors and the PI-3 kinase pathway in medial edge epithelia. Orthod Craniofac Res. 2003;6:129–42. doi: 10.1034/j.1600-0544.2003.02236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunt JR. Teratogenicity of high vitamin A intake. N Engl J Med. 1996;334:1197. doi: 10.1056/NEJM199605023341814. [DOI] [PubMed] [Google Scholar]
- 22.Cobourne MT. The complex genetics of cleft lip and palate. Eur J Orthod. 2004;26:7–16. doi: 10.1093/ejo/26.1.7. [DOI] [PubMed] [Google Scholar]
- 23.Ramirez D, Lammer EJ, Iovannisci DM, Laurent C, Finnell RH, Shaw GM. Maternal smoking during early pregnancy, GSTP1 and EPHX1 variants, and risk of isolated orofacial clefts. Cleft Palate Craniofac J. 2007;44:366–73. doi: 10.1597/06-011.1. [DOI] [PubMed] [Google Scholar]
- 24.Shi M, Christensen K, Weinberg CR, Romitti P, Bathum L, Lozada A, et al. Orofacial cleft risk is increased with maternal smoking and specific detoxification-gene variants. Am J Hum Genet. 2007;80:76–90. doi: 10.1086/510518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lidral AC, Murray JC, Buetow KH, Basart AM, Schearer H, Shiang R, et al. Studies of the candidate genes TGFB2, MSX1, TGFA, and TGFB3 in the etiology of cleft lip and palate in the Philippines. Cleft Palate Craniofac J. 1997;34:1–6. doi: 10.1597/1545-1569_1997_034_0001_sotcgt_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 26.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–81. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 27.van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HK. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet. 2000;24:342–3. doi: 10.1038/74155. [DOI] [PubMed] [Google Scholar]
- 28.Vieira AR, Cooper ME, Marazita ML, Orioli IM, Castilla EE. Interferon regulatory factor 6 (IRF6) is associated with oral-facial cleft in individuals that originate in South America. Am J Med Genet A. 2007;143A:2075–8. doi: 10.1002/ajmg.a.31884. [DOI] [PubMed] [Google Scholar]
- 29.Pan Y, Ma J, Zhang W, Du Y, Niu Y, Wang M, et al. IRF6 polymorphisms are associated with nonsyndromic orofacial clefts in a Chinese Han population. Am J Med Genet A. 2010;152A:2505–11. doi: 10.1002/ajmg.a.33624. [DOI] [PubMed] [Google Scholar]
- 30.Sözen MA, Hecht JT, Spritz RA. Mutation and association analysis of the PVR and PVRL2 genes in patients with non-syndromic cleft lip and palate. Genet Mol Biol. 2009;32:466–9. doi: 10.1590/S1415-47572009000300007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sözen MA, Hecht JT, Spritz RA. Mutation analysis of the PVRL1 gene in caucasians with nonsyndromic cleft lip/palate. Genet Test Mol Biomarkers. 2009;13:617–21. doi: 10.1089/gtmb.2009.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaewkhampa A, Jotikasthira D, Malaivijitnond S, Kantaputra P. TBX22 Mutation Associated With Cleft Lip/Palate, Hypodontia, and Limb Anomaly. Cleft Palate Craniofac J. 2012;49:240–4. doi: 10.1597/10-208. [DOI] [PubMed] [Google Scholar]
- 33.Wang H, Zhang T, Wu T, Hetmanski JB, Ruczinski I, Schwender H, et al. The FGF and FGFR Gene Family and Risk of Cleft Lip With or Without Cleft Palate. Cleft Palate Craniofac J. 2013;50:96–103. doi: 10.1597/11-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beaty TH, Murray JC, Marazita ML, Munger RG, Ruczinski I, Hetmanski JB, et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet. 2010;42:525–9. doi: 10.1038/ng.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan Q, Blanton SH, Hecht JT. Association of ABCA4 and MAFB with non-syndromic cleft lip with or without cleft palate. Am J Med Genet A. 2011;155A:1469–71. doi: 10.1002/ajmg.a.33940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birnbaum S, Ludwig KU, Reutter H, Herms S, Steffens M, Rubini M, et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat Genet. 2009;41:473–7. doi: 10.1038/ng.333. [DOI] [PubMed] [Google Scholar]
- 37.Leslie EJ, Mansilla MA, Biggs LC, Schuette K, Bullard S, Cooper M, et al. Expression and mutation analyses implicate ARHGAP29 as the etiologic gene for the cleft lip with or without cleft palate locus identified by genome-wide association on chromosome 1p22. Birth Defects Res A Clin Mol Teratol. 2012;94:934–42. doi: 10.1002/bdra.23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Indian Genome Variation Consortium. Genetic landscape of the people of India: a canvas for disease gene exploration. J Genet. 2008;87:3–20. doi: 10.1007/s12041-008-0002-x. [DOI] [PubMed] [Google Scholar]
- 39.Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Philipp-Staheli J, Payne SR, Kemp CJ. p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res. 2001;264:148–68. doi: 10.1006/excr.2000.5143. [DOI] [PubMed] [Google Scholar]
- 41.Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 2009;23:2700–4. doi: 10.1101/gad.1848209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ganji H, Nouri N, Salehi M, Aryani O, Houshmand M, Basiri K, et al. Detection of intragenic SMN1 mutations in spinal muscular atrophy patients with a single copy of SMN1. J Child Neurol. 2014 doi: 10.1177/0883073814521297. [DOI] [PubMed] [Google Scholar]
- 43.Lamandé SR, Bateman JF, Hutchison W, McKinlay Gardner RJ, Bower SP, Byrne E, et al. Reduced collagen VI causes Bethlem myopathy: a heterozygous COL6A1 nonsense mutation results in mRNA decay and functional haploinsufficiency. Hum Mol Genet. 1998;7:981–9. doi: 10.1093/hmg/7.6.981. [DOI] [PubMed] [Google Scholar]
- 44.Szigeti R, Pangas SA, Nagy-Szakal D, Dowd SE, Shulman RJ, Olive AP, et al. SMAD4 haploinsufficiency associates with augmented colonic inflammation in select humans and mice. Ann Clin Lab Sci. 2012;42:401–8. [PMC free article] [PubMed] [Google Scholar]
- 45.Martínez-Fernández ML, Bermejo-Sánchez E, Fernández B, MacDonald A, Fernández-Toral J, Martínez-Frías ML. Haploinsufficiency of BMP4 gene may be the underlying cause of Frías syndrome. Am J Med Genet A. 2014;164A:338–45. doi: 10.1002/ajmg.a.36224. [DOI] [PubMed] [Google Scholar]
- 46.Lu W, Quintero-Rivera SF, Fan Y, Alkuraya FS, Donovan DJ, Xi Q, et al. NFIA haploinsufficiency is associated with a CNS malformation syndrome and urinary tract defects. PLoS Genet. 2007;25:e80. doi: 10.1371/journal.pgen.0030080. [DOI] [PMC free article] [PubMed] [Google Scholar]