

Abstract
DNA has become a prime material for assembling complex three-dimensional objects that promise utility in various areas of application. However, achieving user-defined goals with DNA objects has been hampered by the difficulty to prepare them at arbitrary concentrations and in user-defined solution conditions. Here, we describe a method that solves this problem. The method is based on poly(ethylene glycol)-induced depletion of species with high molecular weight. We demonstrate that our method is applicable to a wide spectrum of DNA shapes and that it achieves excellent recovery yields of target objects up to 97 %, while providing efficient separation from non-integrated DNA strands. DNA objects may be prepared at concentrations up to the limit of solubility, including the possibility for bringing DNA objects into a solid phase. Due to the fidelity and simplicity of our method we anticipate that it will help to catalyze the development of new types of applications that use self-assembled DNA objects.
Keywords: DNA nanotechnology, DNA origami, molecular crowding, poly(ethylene glycol), self-assembly
In the quest for achieving precise control over the positioning of matter from the bottom-up, various strategies for creating feature-rich DNA objects of arbitrary shapes with molecular weights in the multiple-megadalton regime have been developed.1–3 Objects made using a particular approach known as DNA origami,2a in which many short DNA single-strands are designed to form double-helical DNA domains with a long single-strand template, have already found use in applications such as single-molecule sensing,4 plasmonics,5 and structural biology.6 Other objects are being explored as potential drug delivery vehicles.7
However, more often than not application requirements are at odds with the restricted set of conditions at which productive synthesis of DNA objects can be performed.8 For instance, the concentration of objects is typically limited to the dilute nanomole-per-liter regime when using current methods of assembly and purification.8b,c Yet, many potential applications—whether in solution, in cell culture, or in organisms—do require much more concentrated solutions to achieve measurable effects. Furthermore, co-factors such as the calibrated amounts of cations that are required for the self-assembly reactions2b, 9 may interfere in prospective applications. Hence, in order to reach utility, the restricted conditions at which productive self-assembly of DNA objects can be carried out must be uncoupled from those required for intended applications. Here, we solve this problem and describe a scalable method for purifying and dissolving repeatedly and efficiently megadalton-scale DNA objects in user-defined buffers at user-defined concentrations. We demonstrate the utility of our method with 19 different objects made using the DNA origami approach, but the method is likely to be generally applicable to any high-molecular-weight DNA object, including objects made using either the DNA origami2a or template-free DNA tile approach.3
Our method exploits the phenomenon of depletion of high-molecular-weight species by excluded volume effects that occur when adding poly(ethylene glycol) (PEG) polymers as a crowding agent to a solution. Precipitation by PEG crowding has been used for decades to purify and concentrate DNA plasmids10 and phage particles.11 In previous work with one particular type of DNA objects that resembled filamentous phage particles,12 PEG precipitation was employed to reduce large elution volumes after an ion-exchange-based purification of the folded objects from non-integrated DNA strands—a task for which the step involving PEG-induced sedimentation apparently lacked in fidelity. The procedure that we describe here builds on these important previous successes, but we have tested and refined the method significantly with respect to recovery yields, separation capability from non-integrated DNA strands, reproducibility, scalability, and applicability to DNA objects with arbitrary shapes. The procedure that we arrived at consists in simply adding one volume of a calibrated precipitation buffer, followed by centrifugation to pellet precipitates, and exchanging the buffer (Scheme S1). Tests in which we varied the composition of the precipitation buffer (type and concentration of depletant, type and concentration of cations), the time of incubation in the precipitation buffer, and details of the pelleting procedure helped us converge on conditions that give simultaneously excellent recovery of the high-molecular-weight target species and good separation from non-integrated DNA oligonucleotides (Note S3).
First, we demonstrate that our method can be used for purifying and concentrating a broad range of DNA objects with various aspect ratios and mechanical properties. To this end, we have self-assembled and purified a library of different DNA origami objects (Figure 1). Unpurified and purified samples were analyzed using agarose gel electrophoresis and direct imaging by transmission electron microscopy (TEM; Figure 1 b,c). Using our method, all of the objects that we tested could be purified satisfactorily from non-integrated low-molecular-weight DNA oligonucleotides (Figure 1 b). Some of the objects that we tested contained by design polythymidine tails at the helical interfaces (10- and 100-helix bundle, plate-like objects, and pointer object), whereas all other objects had single-stranded scaffold DNA loops with various sequences at the helical interfaces. The loops and the tails both served the purpose of preventing aggregation by blunt-end association. After PEG purification, the samples with polythymidine tails exhibited the same electrophoretic mobility as before the treatment. By contrast, the samples with single-stranded scaffold loops had greater mobility after PEG purification than before. We attribute the enhanced mobility to the removal of excess DNA oligonucleotides that stick to the single-stranded loops in the unpurified samples. When we added excess DNA oligonucleotides to a PEG-purified object with single-stranded DNA loops, it migrated again with the reduced mobility as observed before PEG purification (Figure S6b).
Figure 1.

PEG purification of a library of DNA origami objects. a) Illustration (RR to 100 hb) and CanDo-computed20 models (plate1 to 42 hbb) of a panel of DNA objects comprising a variant of Rothemund’s single-layer DNA rectangle (RR)2a and five multilayer objects (four-, ten-, 18-, or 42-helix bundle in honeycomb lattice design; 100-helix bundle in square lattice design),9 a plate-like object with aperture and double-stranded loop (plate1),4c a plate-like object (plate2),4c an asymmetric 82-helix bundle in square lattice design (pointer),2h a gear-like object (gear),18 a flexible hinged-bar object (hinge),18 a letter-A-like object (“A”),20b a straight and bent version of a robot-like object (robot, robotb),20 a letter-S-like object (“S”),20b a bow-like object (bow),20b and bent versions of an 18-helix bundle and a 42-helix bundle (18hbb, 42hbb).2c, 18 The library of objects thus samples a wide spectrum of shapes, aspect ratios, and mechanical properties. b) Images of agarose gels on which unpurified self-assembly reaction mixtures (U) and PEG purified samples (P) of the objects listed above in (a) were electrophoretically separated. Labels: po, gel loading pocket; m, properly folded monomers; ex, non-integrated excess staple strands. c) Exemplary TEM micrographs of single particles in unpurified reaction mixtures (U) and PEG-purified samples (P). Scale bars: 50 nm.
To test the efficacy of our method with respect to recovery yield and purity of the target species, we performed ten successive cycles of precipitating and redissolving one particular multilayer DNA origami object (the 42-helix bundle, Figure 1, lane 5). A gel-electrophoretic analysis of samples taken after each cycle (Figure 2 a–c) indicated an average recovery efficiency of 97±5 % per cycle (N=30). One cycle of PEG purification removed 96 % of non-integrated staple strands. Two cycles removed 99.4 % of all excess staples, and three cycles gave samples that were essentially free of non-integrated staple strands (Figure 2 b,c).
Figure 2.
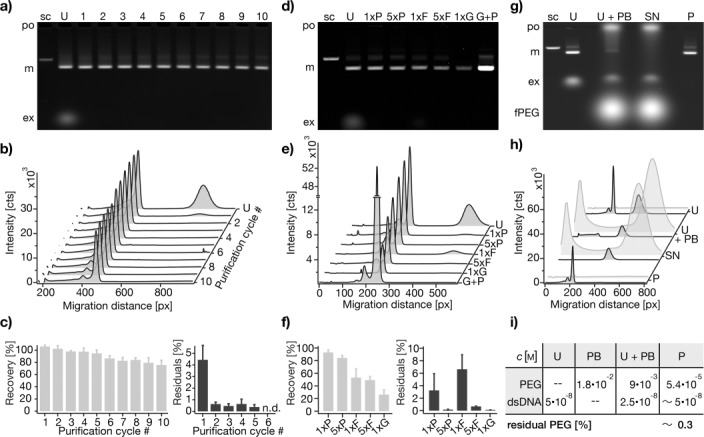
Agarose-gel-electrophoretic characterization of PEG purification using exemplarily a 42-helix bundle object. a) Image of a gel on which samples extracted after each of ten consecutive PEG purification cycles (1–10) were separated. Labels: sc, reference sample containing only scaffold strands; U, unpurified self-assembly reaction mixture; po, gel loading pocket; m, folded objects; ex, non-integrated excess staple strands. b) Cross-sectional lane profiles determined from (a). c) Recovery of folded objects (left) and residuals of excess staple strands (right) relative to unpurified reaction mixture, as determined by integrating and comparing the areas of the peaks reflecting folded objects and excess strands, respectively. The experiment was run in triplicate, each experiment gave data as in (a). Error bars in (c) indicate the standard deviation in the recovery and residuals, respectively. d) Image of a gel containing samples extracted after one and five cycles of PEG purification (1xP, 5xP), after one and five cycles of molecular-weight cut-off filtration (1xF, 5xF), after AGE extraction (1xG), and after PEG purification of a previously AGE-extracted sample (G+P). U refers to the unpurified self-assembly reaction mixture. Other labels as in (a). e) Cross-sectional lane profiles determined from (d). f) Recovery of folded objects and residual excess strands as in (c) but for samples as in (d). Values were obtained from three independent experiments, each giving data as in (d). Error bars indicate the standard deviation in the recovery and residuals, respectively. g–i) Estimation of residual PEG content in purified samples using fluorescein-labeled PEG (fPEG). g) Overlay image of two scans of the same gel, recorded separately for the ethidium bromide and fluorescein emission channels. Samples were taken before (U) and after (U+PB) addition of precipitation buffer (PB) and compared to the supernatant (SN) and the redissolved pellet (P) of the precipitation. h) Cross-sectional lane profiles from ethidium bromide channel (dark gray) and fluorescein channel (light gray). i) Estimated concentrations of folded DNA objects (dsDNA) and PEG at all steps of a PEG purification cycle. Labels: fPEG, fluorescein-labeled PEG; other labels as in (a).
Similarly to PEG-based purification, molecular weight cut-off filtration also allows for separating non-integrated DNA staple strands from high-molecular-weight self-assembly products.13 To compare the performance of PEG purification versus filtration, we subjected test reaction mixtures to one and five rounds of PEG-based purification and filtration, respectively (Figure 2 d–f). According to a gel-electrophoretic analysis of the samples after the procedure, we recovered 53 % of the sample after one cycle of filtration, and 49 % after five cycles. We attribute the significant losses incurred in particular during the first cycle of filtration to material that adhered irreversibly to the filtration membrane. By contrast, after one cycle of PEG purification we recovered 93 % of the sample in this experiment, and 84 % after five cycles. Filtration was also less efficient than PEG purification with respect to staple strand removal: twice as many residual staple strands were present in the once-filtered sample as compared to a once-PEG-precipitated sample. Five cycles of filtration were necessary to remove more than 99.3 % of residual staple strands (Figure 2 e,f).
Agarose gel electrophoresis (AGE) is a popular method for purifying selected species with a particular electrophoretic mobility by band excision and physical extraction from the gel matrix.14 AGE extraction can thus be employed to remove non-integrated DNA oligonucleotides as well as species of greater molecular weight such as aggregates that might have been produced during molecular self-assembly reactions or postprocessing steps. Two important drawbacks of AGE extraction, however, are notoriously low recovery yields (Figure 2 d–f)8b,c and the fact that the procedure results in dilute solutions containing the purified species at concentrations in the low nanomolar range. Combining AGE extraction with our PEG purification method, however, provides a convenient route for preparing pure target species at essentially any desired concentration, which we illustrate here exemplarily by first AGE-purifying a multilayer DNA origami object from all other species in the self-assembly reaction mixture, and then concentrating this sample tenfold (Figure 2 d–f).
Our method relies on adding PEG polymers as a precipitating agent. To quantify exemplarily the extent of residual PEG polymers in a purified sample with a particular scale, we prepared a precipitation buffer that contained a fraction of PEG polymers labeled with fluorescein isothiocyanate (FITC) and used it to precipitate a multilayer DNA origami object (the 42-helix bundle). The pelleted precipitate was redissolved in PEG-free buffer in the starting volume. Agarose gel electrophoresis of the thus treated sample and laser scanning of the gel allowed us to quantify the remainder of FITC-labeled PEG polymers in the purified sample (Figure 2 g,h). Our analysis shows that 99.7 % of the FITC-labeled PEG molecules were removed. We consider this as an estimate for the overall rate of removal of PEG molecules (Figure 2 i). However, if desired, residual PEG can be removed completely from the PEG-purified and concentration-enhanced sample using low-molecular-weight filtration membranes (Note S4).
Our refined PEG purification method is scalable and free from restrictions with respect to the volumes and compositions of the starting sample and the target sample. To illustrate these properties, we have self-assembled one single-layer and two multilayer DNA origami test objects at a large 20 mL scale (as opposed to the typical 100 μL scale reaction mixtures). The single-layer object was a variant of Rothemund’s rectangle (Figure 1, lane 1). One multilayer object was the 42-helix bundle introduced above, but modified with a single fluorescent Atto655 dye. The other multilayer object was a 24-helix bundle in honeycomb-packing that was labeled with, on the average, ten fluorescent cyanine-3 dye molecules distributed over 30 single-stranded sites per object. We precipitated the high-molecular-weight reaction products, removed the supernatant that contained non-integrated DNA oligonucleotides and then dried the precipitates to obtain several milligrams of each object within solid material (Figure 3 a–c). We attribute about 50 % of the measured dry weight to PEG molecules, salts, and residual water (Note S5).
Figure 3.

Preparation of solid material and dense solutions containing intact DNA objects. a) Approximately 6.8 mg of solid material containing a variant of Rothemund′s rectangle (unlabeled). b) Approximately 5.5 mg of solid material containing 42-helix bundle objects, labeled each with one ATTO655 dye. c) Approximately 8.4 mg of solid material containing 24-helix bundle objects, labeled each with ten cyanine-3 dyes on average. d) Left: unpurified self-assembly reaction mixture containing approximately 50 nm of folded 24-helix bundle objects and 150 nm excess staple strands; center: dense solution containing approximately 5.6 μm of folded 24-helix bundle objects, prepared by redissolving the materials from (c); right: reference sample containing 70 μm of a cyanine-3 modified DNA oligonucleotide. e–g) TEM micrographs of dried and redissolved single-layer rectangle (e), 42-helix bundle (f), and 24-helix bundle (g); insets: average single-particle micrographs obtained from non-dried (left) and dried and redissolved samples (right). Scale bars: 50 nm (field-of-view micrographs), 10 nm (insets).
To test whether the objects survive drying, we redissolved the solid material in buffer and analyzed the thus obtained solutions with TEM and gel electrophoresis. TEM micrographs revealed well-folded particles that were indistinguishable both from unpurified and once precipitated, but not dried, particles (Figure 3 e–g). To evaluate the structural integrity in greater detail, we computed average single-particle micrographs but could not discern significant differences in the average micrographs obtained from dried versus non-dried objects (Figure 3 e–g, insets). Furthermore, a gel-electrophoretic analysis showed that the electrophoretic mobility of Rothemund’s rectangle, 42-helix bundle, and 24-helix bundle particles remained unaffected by the precipitation, drying, and redissolving treatment (Figures S8 and S9). To test whether the treatment caused a greater amount of unpaired DNA defects in the objects, we labeled dried and non-dried samples of 24-helix bundle using a fluorescent de Bruijn “defect” probe.15 The labeling intensity was comparable for dried and non-dried samples which suggests that no additional unpaired DNA emerged by drying the objects (Figure S9). Taken together, the data indicates that single-layer and multilayer DNA origami objects do survive the precipitation and drying treatment without taking damage.
One key advantage of the ability to prepare DNA objects in a solid phase is that the material can be used to create solutions with arbitrary object concentrations, at least within the limits of solubility of the object. We redissolved the dried material to prepare concentrated solutions containing up to ca. 5.6 μm of the DNA objects (as measured by absorption spectroscopy), as opposed to the low 0.05 μm concentration at which the objects were self-assembled. The average interparticle distance at 5 μm concentration is on the order of 70 nm, which is comparable to the absolute dimensions of the objects. The solutions were slightly turbid suggesting that the limit of solubility was reached.
The experiment performed here with the three objects illustrates how our method uncouples the highly restricted conditions at which productive self-assembly of DNA origami objects can be carried out from those required for downstream applications. For example, at the elevated concentrations that we prepared, the main functionality of the 24-helix bundle object, which is to carry fluorescent labels, can now be readily observed in solution by eye due to the intense color of the solution (Figure 3 d, center). By contrast, at the initial self-assembly reaction conditions, the solutions were colorless (Figure 3 d, left). The perception of color is due to the wavelength-specific absorption of transmitted and reflected light and depends on the concentration of absorbing particles in solution. Here, color may thus serve as a placeholder for other desirable effects that depend on particle concentration. Preparing sufficiently dense solutions so that macroscopic properties result from the features of designed nanoparticles is standard in other areas of nanotechnology,16 but has so far been quite difficult to achieve when dealing with self-assembled DNA objects.
To conclude, we showed that PEG purification is a versatile method for efficiently purifying and concentrating a broad range of DNA origami objects with various aspect ratios and mechanical properties. Combining AGE extraction with our PEG purification method enables preparing pure objects at essentially any desired concentration, albeit with material losses that are caused by AGE. In our experience, PEG purification by itself does not enhance (and also does not reduce) the tendency for aggregation of a given object at a given solution condition (see Figure 1). Recent advances in design strategies9, 17 and synthesis methods18 support the high-yield self-assembly of desired objects with a significantly reduced formation of byproducts, which diminishes greatly the need for laborious and inefficient molecular-weight-specific purification such as AGE. For example, we designed the 42-helix bundle and the 100-helix bundle using improved strand-breaking and self-assembly procedures. These objects, and also the variant of Rothemund’s single-layer DNA rectangle, the plates, and the pointer self-assembled with close to 100 % yield (Figure 1). In such cases, our refined PEG purification method is an ideal choice for the high-yield preparation of pure samples that can be directly used in downstream applications.
Since our method allows preparing samples routinely at arbitrary concentrations, we anticipate that it will help to catalyze the development of new types of applications that use self-assembled DNA objects. For example, crystallization assays, protein–DNA interaction assays, and cell-culture-based screens for immune response or cytotoxicity typically all require micromolar concentrations of the active agent, which we can now prepare. Because PEG purification also allows exchanging the solvent repeatedly without incurring significant sample losses, it opens a route for using DNA objects as solid support platforms for chemical synthesis.
Experimental Section
PEG precipitation: Self-assembly reaction mixtures at 20 mm MgCl2 were mixed 1:1 (v/v) with precipitation buffer containing 15 % PEG 8000 (w/v) (Ph.Eur.), 5 mm Tris, 1 mm EDTA, and 505 mm NaCl (all chemicals from Carl Roth, Karlsruhe, Germany). The solution was mixed by tube inversion and spinned at 16 000 g, at room temperature (RT) for 25 min using a microcentrifuge (Eppendorf 5420, Hamburg, Germany). The supernatant was removed using a pipette. The pellet was dissolved in target buffer as indicated for each set of experiments and incubated for approximately 20 h at RT or 30 °C.
DNA object self-assembly: Structures were designed using caDNAno v0.2.19 DNA scaffold strands of 7249, 7560, 7704, and 8064 bases length derived from the genome of bacteriophage M13 were used for assembly reactions.2b Staple oligonucleotide strands were prepared by solid-phase chemical synthesis (Eurofins MWG, Ebersberg, Germany, HPSF grade). Production of DNA objects was accomplished in one-pot reaction mixtures containing scaffold DNA at a concentration of 20 nm (default) or 50 nm (pointer in Figure S1, RR in Figures 3 and S8, 24 hb, 42 hb, 100 hb), staple DNA oligonucleotides at 200 nm each, and 5 mm TRIS, 1 mm EDTA, 20 mm MgCl2, and 5 mm NaCl (pH 8). The reaction mixtures were subjected to a thermal annealing protocol using TETRAD (Biorad) thermal cycling devices. The mixtures were first incubated at 65 °C for 15 min and then annealed from 60 to 40 °C in steps of 1 °C per 2–3 h. The reaction products were stored at RT.
Agarose gel electrophoresis: Electrophoresis of the folded DNA objects was carried out in 2 % agarose gels containing electrophoresis buffer (1 mm EDTA, 44.5 mm Tris base, 44.5 mm boric acid, and 11 mm MgCl2, pH 8.4). The samples were electrophoresed for two hours at 70–90 V in a water-cooled gel box filled with electrophoresis buffer. The gels typically contained ethidium bromide at a concentration of 1 μm. The agarose gels were scanned using a Typhoon 9500 FLA laser scanner (GE Healthcare) at a resolution of 50 μm/px (ethidium bromide: excitation at 535 nm, emission> 575 nm; fluorescein: excitation at 473 nm, emission 520–540 nm) to give 16-bit tif image files, which we analyzed using ImageJ64 V1.47 (U.S. National Institutes of Health). Cross-sectional lane intensity profiles were computed by averaging over grayscale values within a 20–75 pixel wide box drawn over the lane of interest. After linear background correction, the regions of interest were quantified by integrating the area under the peaks. Yields were estimated by comparing the intensity of bands of interest for treated versus untreated samples.
Negative-staining TEM: Samples were adsorbed on glow-discharged formvar-supported carbon-coated Cu400 TEM grids (Science Services, Munich) and stained using a 2 % aqueous uranyl formate solution containing 25 mm sodium hydroxide. Imaging was performed using a Philips CM100 EM operated at 100 kV. Images were acquired using an AMT 4 Megapixel CCD camera. Micrograph scale bars were calibrated by imaging 2D catalase crystals and using the lattice constants as length reference. Imaging was performed at ×28 500 magnification.
Supporting Information
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201405991.
miscellaneous_information
References
- 1a).Seeman NC. Annu. Rev. Biochem. 2010;79:65–87. doi: 10.1146/annurev-biochem-060308-102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b).Linko V, Dietz H. Curr. Opin. Biotechnol. 2013;24:555–561. doi: 10.1016/j.copbio.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 2a).Rothemund PW. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 2b).Douglas SM, Dietz H, Liedl T, Hogberg B, Graf F, Shih WM. Nature. 2009;459:414–418. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c).Dietz H, Douglas SM, Shih WM. Science. 2009;325:725–730. doi: 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d).Ke Y, Douglas SM, Liu M, Sharma J, Cheng A, Leung A, Liu Y, Shih WM, Yan H. J. Am. Chem. Soc. 2009;131:15903–15908. doi: 10.1021/ja906381y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e).Han D, Pal S, Nangreave J, Deng Z, Liu Y, Yan H. Science. 2011;332:342–346. doi: 10.1126/science.1202998. [DOI] [PubMed] [Google Scholar]
- 2f).Andersen ES, Dong M, Nielsen MM, Jahn K, Subramani R, Mamdouh W, Golas MM, Sander B, Stark H, Oliveira CL, Pedersen JS, Birkedal V, Besenbacher F, Gothelf KV, Kjems J. Nature. 2009;459:73–76. doi: 10.1038/nature07971. [DOI] [PubMed] [Google Scholar]
- 2g).Ke Y, Voigt NV, Gothelf KV, Shih WM. J. Am. Chem. Soc. 2012;134:1770–1774. doi: 10.1021/ja209719k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2h).Bai XC, Martin TG, Scheres SH, Dietz H. Proc. Natl. Acad. Sci. USA. 2012;109:20012–20017. doi: 10.1073/pnas.1215713109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3a).Wei B, Dai M, Yin P. Nature. 2012;485:623–626. doi: 10.1038/nature11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b).Ke Y, Ong LL, Shih WM, Yin P. Science. 2012;338 doi: 10.1126/science.1227268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a).Derr ND, Goodman BS, Jungmann R, Leschziner AE, Shih WM, Reck-Peterson SL. Science. 2012;338:662–665. doi: 10.1126/science.1226734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b).Pfitzner E, Wachauf C, Kilchherr F, Pelz B, Shih WM, Rief M, Dietz H. Angew. Chem. Int. Ed. 2013;52:7766–7771. doi: 10.1002/anie.201302727. Angew. Chem2013125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c).Wei R, Martin TG, Rant U, Dietz H. Angew. Chem. Int. Ed. 2012;51:4864–4867. doi: 10.1002/anie.201200688. Angew. Chem2012, 124, 4948–4951; [DOI] [PubMed] [Google Scholar]
- 4d).Langecker M, Arnaut V, Martin TG, List J, Renner S, Mayer M, Dietz H, Simmel FC. Science. 2012;338:932–936. doi: 10.1126/science.1225624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuzyk A, Schreiber R, Fan Z, Pardatscher G, Roller EM, Hogele A, Simmel FC, Govorov AO, Liedl T. Nature. 2012;483:311–314. doi: 10.1038/nature10889. [DOI] [PubMed] [Google Scholar]
- 6a).Douglas SM, Chou JJ, Shih WM. Proc. Natl. Acad. Sci. USA. 2007;104:6644–6648. doi: 10.1073/pnas.0700930104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b).Berardi MJ, Shih WM, Harrison SC, Chou JJ. Nature. 2011;476:476–113. doi: 10.1038/nature10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c).Selmi DN, Adamson RJ, Attrill H, Goddard AD, Gilbert RJ, Watts A, Turberfield AJ. Nano Lett. 2011;11:657–660. doi: 10.1021/nl1037769. [DOI] [PubMed] [Google Scholar]
- 7a).Douglas SM, Bachelet I, Church GM. Science. 2012;335:831–834. doi: 10.1126/science.1214081. [DOI] [PubMed] [Google Scholar]
- 7b).Perrault SD, Shih WM. ACS Nano. 2014;8:5132–5140. doi: 10.1021/nn5011914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8a).Pinheiro AV, Han D, Shih WM, Yan H. Nat. Nanotechnol. 2011;6:763–772. doi: 10.1038/nnano.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b).Lin C, Perrault SD, Kwak M, Graf F, Shih WM. Nucleic Acids Res. 2013;41:e40. doi: 10.1093/nar/gks1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c).Bellot G, McClintock MA, Lin C, Shih WM. Nat. Methods. 2011;8:5132–5140. doi: 10.1038/nmeth0311-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin TG, Dietz H. Nat. Commun. 2012;3:1103. doi: 10.1038/ncomms2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humphreys GO, Willshaw GA, Anderson ES. Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1975;383:457–463. doi: 10.1016/0005-2787(75)90318-4. [DOI] [PubMed] [Google Scholar]
- 11a).Yamamoto KR, Alberts BM, Benzinger R, Lawhorne L, Treiber G. Virology. 1970;40:734–744. doi: 10.1016/0042-6822(70)90218-7. [DOI] [PubMed] [Google Scholar]
- 11b).Gibaud T, Barry E, Zakhary MJ, Henglin M, Ward A, Yang Y, Berciu C, Oldenbourg R, Hagan MF, Nicastro D, Meyer RB, Dogic Z. Nature. 2012;481 doi: 10.1038/nature10769. [DOI] [PubMed] [Google Scholar]
- 12.Bellot G, McClintock MA, Chou JJ, Shih WM. Nat. Protoc. 2013;8:755–770. doi: 10.1038/nprot.2013.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinhauer C, Jungmann R, Sobey TL, Simmel FC, Tinnefeld P. Angew. Chem. Int. Ed. 2009;48:8870–8873. doi: 10.1002/anie.200903308. Angew. Chem20091219030–9034. [DOI] [PubMed] [Google Scholar]
- 14.Thuring RW, Sanders JP, Borst P. Anal. Biochem. 1975;66:213–220. doi: 10.1016/0003-2697(75)90739-3. [DOI] [PubMed] [Google Scholar]
- 15.Wagenbauer KF, Wachauf CH, Dietz H. Nat. Commun. 2014;5:3691. doi: 10.1038/ncomms4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boisselier E, Astruc D. Chem. Soc. Rev. 2009;38:1759–1782. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 17.Ke Y, Bellot G, Voigt NV, Fradkov E, Shih WM. Chem. Sci. 2012;3:2587–2597. doi: 10.1039/C2SC20446K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobczak JP, Martin TG, Gerling T, Dietz H. Science. 2012;338:1458–1461. doi: 10.1126/science.1229919. [DOI] [PubMed] [Google Scholar]
- 19.Douglas SM, Marblestone AH, Teerapittayanon S, Vazquez A, Church GM, Shih WM. Nucleic Acids Res. 2009;37:5001–5006. doi: 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20a).Castro C, Kilchherr F, Kim D, Shiao E, Wauer T, Wortmann P, Bathe M, Dietz H. Nat. Methods. 2011;8:221. doi: 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- 20b).Kim D, Kilchherr F, Dietz H, Bathe M. Nucleic Acids Res. 2012;40:2862. doi: 10.1093/nar/gkr1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information