

Abstract
Wilms’ Tumour 1 (WT1) is a zinc finger transcription factor that is over-expressed in acute myeloid leukaemia (AML). Its restricted expression in normal tissues makes it a promising target for novel immunotherapies aiming to accentuate the cytotoxic T lymphocyte (CTL) response against AML. Here we report a phase I/II clinical trial of subcutaneous peptide vaccination with two separate HLA-A2-binding peptide epitopes derived from WT1, together with a pan-DR binding peptide epitope (PADRE), in Montanide adjuvant. Eight HLA-A2-positive patients with poor risk AML received five vaccination cycles at 3-weekly intervals. The three cohorts received 0·3, 0·6 and 1 mg of each peptide, respectively. In six patients, WT1-specific CTL responses were detected using enzyme-linked immunosorbent spot assays and pWT126/HLA-A*0201 tetramer staining, after ex vivo stimulation with the relevant WT1 peptides. However, re-stimulation of these WT1-specific T cells failed to elicit secondary expansion in all four patients tested, suggesting that the WT1-specific CD8+ T cells generated following vaccination may be functionally impaired. No correlation was observed between peptide dose, cellular immune response, reduction in WT1mRNA expression and clinical response. Larger studies are indicated to confirm these findings.
Keywords: acute myeloid leukaemia, immunotherapy, tumour antigens, trials
The potential of T lymphocytes to eliminate leukaemia is demonstrated by the graft-versus-leukaemia effect following haematopoietic stem cell transplantation (Odom et al, 1978; Goldman et al, 1988; Horowitz et al, 1990). The identification of target antigens on leukaemic cells has renewed interest in the use of peptide vaccines to stimulate leukaemia-specific T cell responses (Oka et al, 2004; Rezvani et al, 2008; Keilholz et al, 2009; Maslak et al, 2010; Kuball et al, 2011). A target antigen of particular interest is the zinc finger transcription factor Wilms’ Tumour 1 (WT1), which is over expressed in several solid tumours as well as acute myeloid leukaemia (AML) and myelodysplasia (Inoue et al, 1997; Miyoshi et al, 2002; Oji et al, 2002). WT1 has several characteristics that render it a highly attractive target for immunotherapies: its expression in normal tissues is limited (Call et al, 1990); it is expressed by leukaemic stem cells in addition to more mature myeloid cells (Saito et al, 2010); and its expression increases as myeloid disease progresses (Cilloni et al, 2003). WT1 is required for proliferation of leukaemia cells (Algar et al, 1996), reducing the likelihood of tumour escape by down-modulation of the antigen. We have previously shown that WT1-specific cytotoxic T lymphocytes (CTLs) kill leukaemia cells and leukaemia stem cells without affecting normal stem cell function (Gao et al, 2000, 2003).
Although WT1 is expressed in normal tissues during embryogenesis, immunological tolerance to WT1 is not complete: WT1-specific CTLs have been detected and expanded following exposure to the peptide both in healthy donors (Rezvani et al, 2003) and in patients with AML (Scheibenbogen et al, 2002). Recent studies have shown immune and clinical responses with peptide vaccinations against single epitopes of WT1 (Oka et al, 2004; Keilholz et al, 2009); however, naturally occurring CD8+ T cell responses against myeloid leukaemias target multiple epitopes, potentially enhancing the strength and diversity of these responses (Gannagé et al, 2005; Rezvani et al, 2005). We have identified two distinct peptide epitopes of WT1 that are presented by HLA-A0201 (A2) and function as targets for leukaemia-reactive CD8+ CTL: pWT126 and pWT235 (Bellantuono et al, 2002). We hypothesized that the use of peptide vaccination with both epitopes would result in a robust immunological response with reduced risk of tumour escape. Here we report a phase I/II clinical trial of peptide vaccination in HLA-A2-positive patients with poor risk AML, using pWT126, pWT235 and the pan HLA-DR T helper cell epitope (PADRE) together with Montanide adjuvant.
Methods
Study design and patient population
This was a phase I/II multi-centre dose escalation trial in HLA-A*0201-positive patients with poor risk AML. The primary objectives of the study were to determine the safety and toxicity profile of peptide vaccination with a combination of WT1 peptides and PADRE in Montanide adjuvant, and to evaluate the induction of autologous WT1-specific CTL responses; the secondary objective was to document any disease response. Details of the clinical trial inclusion criteria are given in Table I.
Table I.
Inclusion criteria.
| HLA-A*0201-positive |
| 18–75 years |
| No fludarabine in previous 3 months |
| World Health Organization performance status 0–2 |
| Life expectancy 6 months or greater |
| Haemoglobin ≥70 g/l; neutrophil count ≥0·2 × 109/l; lymphocyte count >0·5 × 109/l; platelet count ≥40 × 109/l |
| Serum bilirubin, alanine aminotransferase and/or aspartate aminotransferase <3 times upper limit of normal reference range |
| Creatinine clearance ≥30 ml/min |
| Patients NOT eligible for haematopoietic stem cell transplantation, with: |
| AML in CR2 or greater |
| Good and standard risk AML in CR1 or stable PR (<20% blasts) in patients >60 years |
| Poor risk AML in CR1 or PR (slow remitters and/or adverse cytogenetics) |
| AML at first relapse post-HCST in CR or PR following re-induction and consolidation |
AML, acute myeloid leukaemia; CR, complete remission; CR1, first CR; CR2, second CR; PR, partial remission; HSCT, haematopoietic stem cell transplantation.
The trial was approved by the UK National Research Ethics Service and all participants gave written informed consent.
Vaccine preparation and administration
pWT126 (WT1 126–134: RMFPNAPYL), pWT235 (WT1 235–243: CMTWNQMNL) and PADRE [H-D-Ala-Cha-Val-Ala-Ala-Trp-Thr-Leu-Lys-Ala-Ala-D-Ala-εAhx-Cys-OH (Alexander et al, 1994)] peptides were synthesized to GMP grade by Bachem UK Ltd. (St Helens, Merseyside, UK). Peptides were reconstituted to 3 mg/ml in water with a maximum dimethyl sulfoxide (DMSO) concentration of 25% v/v and stored at −20°C. On the day of injection, peptides were thawed and a water-in-oil emulsion vaccine was prepared by mixing the peptides in 1:1 ratio (aqueous phase) with an equal volume of the adjuvant Montanide ISA-51 (oil phase; Seppic, Puteaux, France). Vaccines containing pWT126 with PADRE, and pWT235 with PADRE, were administered subcutaneously within 4 h of preparation at two separate sites, to avoid competitive binding of HLA-A*0201 by the WT1 peptides.
Dose escalation schedule
Three dose levels were used, comprising 0·3, 0·6 or 1·0 mg peptide. One vaccination cycle comprised separate subcutaneous injections of 0·3/0·6/1·0 mg pWT126 + 0·3/0·6/1·0 mg PADRE peptide, and of 0·3/0·6/1·0 mg pWT235 + 0·3/0·6/1·0 mg PADRE peptide, each admixed with Montanide ISA-51. Vaccination cycles were repeated every 3 weeks to a maximum of five cycles. After three of three patients had completed the initial 0·3 mg dose level with no evidence of grade 3–4 toxicity attributable to vaccination, a further three patients were treated at the 0·6 mg dose level. In the absence of grade 3–4 toxicity at this level, further patients received treatment at the 1·0 mg dose level.
Assessment of toxicity
At each outpatient visit, patients were evaluated for toxicities according to the National Cancer Institute, common toxicity criteria v2.0 (http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcmanual_v4_10-4-99.pdf). Dose limiting toxicity was defined as grade 3–4 non-haematological toxicity, or grade 4 haematological toxicity, that had been determined by defined criteria to be certainly or probably related to the vaccine.
Detection of antigen-specific CD8+ T cells
WT1-specific T cells were detected by staining with phycoerythrin-conjugated HLA-A*0201-pWT126 tetramer (Beckman Coulter, High Wycombe, Buckinghamshire, UK) or dextramer (Immudex, Copenhagen, Denmark), allophycocyanin-conjugated antibody to CD3 and fluorescein isothyocyanate-conjugated antibody to CD8 (BD Biosciences, Oxford, UK), as described previously (King et al, 2009). The multimers have mutations in the alpha-3 domain of the major histocompatibilty complex (MHC) molecule that reduces binding to CD8 molecules. The frequency of WT1-specific T cells was expressed as the percentage of gated CD3+ CD8+ T cells that were HLA-A*0201-pWT126-positive. The detection limit of this tetramer had previously been determined as 0·05% of viable CD3+ CD8+ T cells in patients with breast cancer and prostate cancer (Gillmore et al, 2006; King et al, 2009). Control cytomegalovirus (CMV)-specific T cells were detected by substituting the HLA-A*0201-pWT126 tetramer with HLA-A*0201-NLV tetramer (Beckman Coulter). Flow cytometry was performed on an LSRII flow cytometer (BD Biosciences) and analysed using FacsDiva software (BD Biosciences).
Heparinized peripheral blood samples were obtained prior to the first vaccination cycle, at weeks 3, 6, 9 and 12, and after the last vaccination at weeks 15 and 18–20. Serial cryopreserved mononuclear cell samples from each patient were analysed simultaneously. As primary assay, WT1-specific T cells were detected by tetramer staining following 8 d in culture with pWT126 peptide in the presence of interleukin (IL) 2 and IL7 at 20 units/ml and 5 ng/ml respectively (Roche, Welwyn Garden City, Herts, UK and R&D Systems, Abingdon, Oxfordshire, UK), as described previously (King et al, 2009). Induction of a T-cell response to WT1 vaccination was defined as an increase in frequency of tetramer-positive CD8+ T cells of >1·5 times that at baseline, as in previous studies (Oka et al, 2004).
Analysis of antigen-specific CD8+ T cell function in vitro
Functional analysis of WT1-specific T cell responses was carried out by detection of γ-interferon (IFN-γ) secretion using an enzyme-linked immunosorbent spot (ELISPOT) assay (BD Biosciences), as described previously (King et al, 2009). Autologous peripheral blood mononuclear cells (PBMC) were used to present peptides to the expanded T cells. ELISPOT assays were performed in triplicate, with each well containing 2 × 105 autologous PBMC, 5 μmol/l pWT126 or pWT235 peptide and 2 × 105 responder T cells. After incubation for 20 h at 37°C, the cells were discarded and plates were washed and developed with a second, biotinylated, antibody to human IFN-γ and streptavidin-alkaline phosphatase, according to the manufacturer's instructions. ELISPOT wells containing responder T cells and autologous PBMC with irrelevant peptides served as negative controls. Stimulation of 5 × 104 responder T cells with 200 ng/ml of staphylococcal enterotoxin B was used as a positive control. ELISPOT plates were read using an AxioCamMRc plate reader and KS Elispot software (Zeiss, Cambridge, UK). Results were expressed as the mean number of spot-forming units per 106 cells from triplicate wells, and a cytokine response was considered positive if the mean number of spot-forming units per 106 cells was greater than two standard deviations above that in the absence of cognate peptide.
The capacity of vaccination-induced WT1-specific CD8+ T cells to undergo antigen-specific expansion ex vivo was measured by HLA-A*0201-pWT126 dextramer staining and flow cytometric analysis following restimulation over 14 d: 3 × 106 responder T cells were incubated with 2 × 106 autologous PBMC and 5 μmol/l peptide in culture medium supplemented with 20 units/ml IL2 (Roche), 5 ng/ml IL7 (R&D Systems), together with additional 10 ng/ml IL15 (R&D Systems) and 30 ng/ml IL21 (Peprotech, London, UK), as previously shown (King et al, 2009) to maximize antigen-specific in vitro expansion. pWT126 peptide was used as test peptide, with CMV pp65 NLVPMVATV (NLV) peptide (ProImmune, Oxford, UK) as positive control in CMV-responsive patients.
Measurement of WT1 mRNA levels by real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR)
All samples for qRT-PCR were blinded. Total RNA was extracted from PBMC and reverse transcribed using the standardized protocol developed within the Europe Against Cancer (EAC) programme (Gabert et al, 2003). qRT-PCR assays were performed on an Applied Biosystems ABI7900 instrument (Applied Biosystems, Foster City, CA, USA) according to EAC conditions, with standardized WT1-specific primers (Van Dijk et al, 2002; Cilloni et al, 2009) and probes at final concentrations of 300 and 200 nmol/l respectively (Gabert et al, 2003). In patients with NPM1 mutations, the mutant transcript was monitored by qRT-PCR in parallel with WT1, according to the method of Gorello et al (2006). Reactions were performed in triplicate wells with appropriate water controls. Expression of the housekeeping gene ABL1 was used as the endogenous cDNA quantity control for all samples (Cilloni et al, 2009).
Statistics
The pre-specified primary immunological trial objective was to evaluate induction of autologous WT1-specific T cell responses. Baseline and maximum post-vaccination responses were compared using the paired two-tailed t-test. Spearman ρ was used to correlate antigen-specific responses to pWT126 and pWT235 peptides. GraphPad Prism version 5.0 for Windows (GraphPad Software, La Jolla, CA, USA) was used for statistical analyses. P values of 0·05 or less were considered statistically significant.
Results
Patient characteristics
Demographics of the patients are shown in Table II. The eight patients comprised three males and five females with AML. The median age was 65 years (range 56–75 years). All had received previous chemotherapy. At the start of vaccination, four were in morphological first complete remission (CR1) (one with persistent cytogenetic abnormalities), one in CR2 post-autograft, two in partial remission with 15% blasts and one with slowly progressive disease.
Table II.
Patient characteristics.
| Patient | Sex | Age (years) | Diagnosis (morphological subtype) | Cytogenetics at diagnosis | Previous treatment | Disease status pre-trial |
|---|---|---|---|---|---|---|
| RFH 001 | Male | 61 | AML (M0) | 46, XY, t(1;2)(p32;p13) | ADE 3 + 10 (no response); FlAG-Ida (to CR); FlAG; MACE | Morphological CR1; MRD positive |
| RFH 002 | Female | 68 | AML (M1) | Failed | DA + GO (to CR); DA | CR1; low platelet count |
| RFH 003 | Female | 56 | Secondary AML (M5) | Normal | MDS RAEB-1 treated with FlAG (to CR1); DA; MACE; MidAC Relapsed with AML after 3 years: treated with FlAG; FlAG-Ida (to CR2); Bu/Cy ASCT. | CR2 |
| UCH 001 | Female | 64 | AML (M1) | Normal | DA × 1; LD ara-C × 3 | Stable PR; 15% blasts |
| UCH 002 | Female | 65 | AML (M1) | Normal | DA × 1; LD ara-C × 8 | PR; 15% blasts |
| UCH 003 | Female | 75 | AML (M4) | Normal | LD ara-C + GO × 2 (to CR); LD ara-C × 2 | CR1 |
| BHM 001 | Male | 66 | AML | 46, XY, der(7)t(7;11) (q22;q13) | DA × 1; MidAC × 2 | PR; slowly progressive |
| BHM 002 | Male | 68 | AML (M0) | Normal | DA × 2; MidAC × 1 | CR1 |
RFH, Royal Free Hospital, London; UCH, University College Hospital, London; BHM, Queen Elizabeth Hospital, Birmingham; AML, acute myeloid leukaemia; CR, complete remission; CR1, first CR; CR2, second CR; PR, partial remission; MRD, minimal residual disease; MDS RAEB-1, myelodysplastic syndrome refractory anaemia with excess blasts-1; ADE, daunorubicin, cytarabine and etoposide; Bu/Cy ASCT, busulfan and cyclophosphamide-conditioned autologous stem cell transplantation; DA, daunorubicin and cytarabine; FlAG, fludarabine, cytarabine and granulocyte colony-stimulating factor; GO, gemtuzumabozogamicin; Ida, idarubicin; LD ara-C, low-dose cytarabine; MACE, amsacrine, cytarabine and etoposide; MidAC, mitoxantrone and cytarabine.
Protocol administration and toxicity
Three patients received vaccination at the 0·3 mg dose level, three at the 0·6 mg dose level and two at the 1·0 mg dose level. Five patients completed all five vaccination cycles; two received four cycles and one received two cycles, owing to disease progression. No toxicity greater than grade 3 was observed in any cohort; of note, no renal toxicity, no haematological toxicity and no autoimmune phenomena were attributable to vaccination. All patients developed local reactions comprising erythema, swelling and induration of up to grade 3 at vaccination sites, in one case requiring subsequent vaccination to be deferred by 3 weeks. These results indicate that vaccination with a combination of WT1 peptides and PADRE in Montanide adjuvant is well tolerated.
Vaccination induces WT1-specific immune responses
Seven of eight patients were evaluable for assessment of T cell response. PBMC samples were stained with HLA-A*0201-pWT126 tetramer and analysed for circulating CD8+ T cells specific for WT1 using flow cytometry. Overall, the frequency of WT1 tetramer-positive CD8+ T cells increased from a median of 0·1% at baseline to a median of 0·45% at maximum post-vaccination response (Fig1A and B). Six of seven patients showed an increase in frequency of WT1 tetramer-positive CD8+ T cells >1·5 times that at baseline, a criterion for response used in previous studies (Oka et al, 2004). However, the increase in WT1 tetramer-positive T cells was not significant when the pre- and post-vaccination data were analysed using a paired two-tailed t-test.
Fig 1.
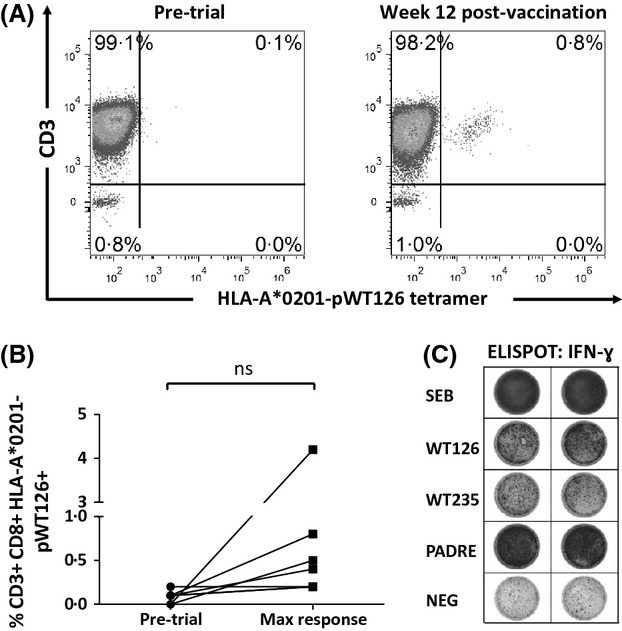
WT1-specific CD8+ T cell response to vaccination. Tetramer analysis of peripheral blood mononuclear cells was performed by flow cytometry. Plots shown are gated on viable CD8+ T cells. Representative data from Patient RFH003 (A) and maximal responses for all patients (B) are shown. (C) Functional responses were assessed by ELISPOT for γ-interferon (IFN-γ). Representative responses from Patient RFH003 are shown.
We next analysed functional CD8+ T cell responses to pWT126, pWT235 and PADRE peptides by IFN-γ ELISPOT. Six of seven patients showed antigen-specific IFN-γ responses to all three peptides following vaccination (Fig1C and Table III), the earliest response being detected after a single vaccination. Four of five patients in whom pWT126 and pWT235-specific responses were detected continued to demonstrate these responses at follow-up in weeks 15–18; however, one patient had lost pWT126- and pWT235-specific responses. Responses to pWT126 and pWT235 were highly correlated (P = 0·0008).
Table III.
Immunological and clinical response to vaccination.
| Patient | Vaccine dose (mg) | Cycles completed | Pre-trial WT1 tetramer (% of CD8+) | Maximum post-vaccination tetramer (% of CD8+) and time point | Tetramer response >1·5 × pre-trial? | IFN-γ response by ELISPOT | Time from start of study to disease progression | ||
|---|---|---|---|---|---|---|---|---|---|
| pWT126 | pWT235 | PADRE | |||||||
| RFH 001 | 0·3 | 5 | 0·1 | 0·2 maximum week 9 | + | + | + | + | 9 months |
| RFH 002 | 1·0 | 4 | 0·1 | 0·2 maximum week 9 | + | + | + | + | 39 months |
| RFH 003 | 1·0 | 5 | 0·1 | 0·8 maximum week 12 | + | + | + | + | >41 months |
| UCH 001 | 0·3 | 5 | N/A | 1·4 N/A | N/A | N/A | N/A | N/A | 18 months |
| UCH 002 | 0·3 | 4 | 0·2 | 0·2 N/A | − | + | + | + | 9 weeks |
| UCH 003 | 0·6 | 5 | 0·0 | 0·5 maximum week 15 | + | + | + | + | 7 months |
| BHM 001 | 0·6 | 2 | 0·1 | 0·4 maximum week 6 | + | − | − | − | 6 weeks |
| BHM 002 | 0·6 | 5 | 0·0 | 4·2 maximum week 6 | + | + | + | + | 10 months |
RFH, Royal Free Hospital, London; UCH, University College Hospital, London; BHM, Queen Elizabeth Hospital, Birmingham; ELISPOT, enzyme-linked immunosorbent spot; N/A, not applicable.
Vaccination-induced WT1-specific T cells do not expand in vitro
We then examined the potential for WT1-specific CD8+ T cells isolated from patients after vaccination to expand following re-challenge with the WT1 pWT126 peptide in vitro. PBMC samples from four patients with at least four-fold WT1 tetramer response were stimulated over 2 weeks in vitro with WT1 pWT126 peptide or CMV pp65 NLV peptide. Two of these patients were CMV IgG seropositive. Following stimulation, both of these patients showed a two-fold or greater increase in the frequency of CMV pp65-specific CD8+ T cells (Fig2). In contrast, WT1-specific CD8+ T cells did not respond to stimulation (Fig2 and data not shown), indicating that the vaccine-induced WT1-specific CD8+ T cells are unable to proliferate and expand upon re-stimulation with peptide antigen in vitro.
Fig 2.
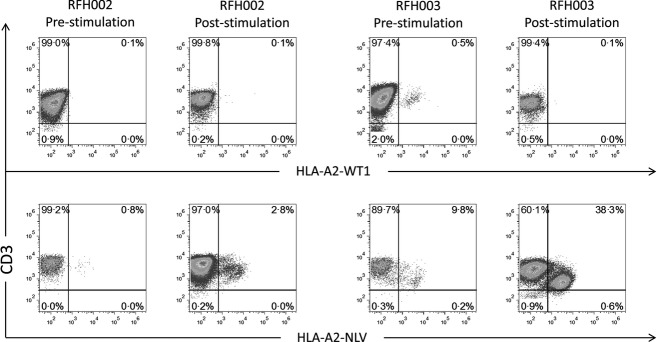
Vaccination-induced WT1-specific immune responses are short-lived. Peripheral blood mononuclear cells were isolated from two pWT126/pWT235/PADRE-vaccinated, cytomegalovirus (CMV) IgG seropositive patients and re-stimulated with WT126 (WT1) or NLV (CMV) peptides for 2 weeks. Frequencies of WT1- and CMV-specific CD8+ T cells were measured by dextramer staining and flow cytometric analysis. Plots shown are gated on viable CD8+ T cells.
Disease activity after vaccination
To assess the potential effect of vaccination on leukaemia, WT1 mRNA expression was used as a surrogate marker of minimal residual disease. In two patients, NPM1 mutations were present, allowing disease progression to be assessed independently of the targeted antigen.
In two of five evaluable patients (RFH001, RFH003), the emergence of functional WT1-specific T cells after vaccination at weeks 9 and 12 respectively, was followed by a decrease in WT1 mRNA levels to below the upper threshold limit observed in normal peripheral blood samples (Fig3). Patients in whom there was no increase in WT1-specific T cells, or in whom functional IFN-γ responses could not be detected, showed stable or increasing WT1 mRNA levels. However, there was no correlation between vaccine dose, emergence of WT1-specific T cells or disease response.
Fig 3.

Disease response to vaccination with pWT126, pWT235 and PADRE peptides as measured by WT1/ABL1 mRNA quantification. Results are shown for individual patients in complete remission (CR) (A) or partial remission (PR) (B) at the start of the trial. Expression of WT1 mRNA (solid, black squares) and, in patients with mutations in NPM1, NPM1 mutant mRNA (dashed, triangles) are shown as a ratio of the gene of interest to ABL1. Months from the start of trial are shown on the X axis. Bold arrows denote times of vaccination and the time at which maximal tetramer responses were detected are given in the results section. The horizontal dotted line corresponds to the upper limit of WT1 expression in normal peripheral blood, as previously defined (Cilloni et al, 2009). RFH, Royal Free Hospital, London; UCH, University College Hospital, London; BHM, Queen Elizabeth Hospital, Birmingham.
Discussion
Vaccination with a combination of WT1 and PADRE peptides in Montanide adjuvant was well tolerated and safe, with no evidence of renal toxicity nor the haematological toxicity that was previously observed in myelodysplastic syndrome patients in one study (Oka et al, 2003). The primary immunological objective of this trial was the assessment of WT1-specific CD8+ T cell responses following vaccination. In accordance with the recommendations of the Society for Biological Therapy (Keilholz et al, 2002), tetramer-based quantitative assays and ELISPOT assays of function were used to determine T cell responses. Both assays showed responses to vaccination in six/seven patients (86%), a response rate that compares favourably with that achieved in other trials of WT1 peptide vaccination in AML (Oka et al, 2004; Rezvani et al, 2008; Keilholz et al, 2009; Maslak et al, 2010; Kuball et al, 2011). Using WT1 peptides with Montanide adjuvant, several groups have reported immunological responses in 62·5–87·5% of patients with AML in CR (Oka et al, 2004; Rezvani et al, 2008; Maslak et al, 2010). In patients with residual disease, Keilholz et al (2009) used keyhole-limpet-haemocyanin (KLH) adjuvant with granulocyte-macrophage colony-stimulating factor to enhance immunogenicity, achieving WT1-specific tetramer responses in 44%: it was suggested that this lower response rate might be due either to an immunosuppressive effect of leukaemic blasts or to responses falling below the limits of detection of the assays used (Keilholz et al, 2009). In our study, immunological responses were seen within 2 weeks of a single vaccination: similarly rapid responses have been observed previously, suggesting that the vaccination-induced expansion of self-restricted WT1-specific cells originates from pre-existing memory CD8+ T cells (Oka et al, 2003; Rezvani et al, 2008). However, we did not find these early immunological responses to correlate with clinical outcome, possibly owing to the small sample size; only one group has previously been able to show a correlation between clinical and immunological responses (Oka et al, 2004).
Importantly, we were unable to elicit secondary expansion of WT1-specific CD8+ cells on re-stimulation with the cognate pWT126 antigen in vitro. By contrast, CMV-specific CD8+ T cells from the same patients exhibited robust potential for expansion, suggesting that the WT1-specific CD8+ T cells generated following vaccination were functionally impaired, or were of insufficient functional avidity. Such antigen-specific T cell expansion is a key feature of immune memory, and predicts tumour regression following cancer vaccination in mice (Janssen et al, 2005; Rosato et al, 2006). The failure of vaccination to establish functional immune memory may explain the transient nature of the immune responses typically observed in cancer vaccination trials. Rezvani et al (2011) recently found that initial WT1- and PR1-specific T cell responses were lost in all patients following repeated vaccination over 12 weeks; and although long-term follow-up of three patients from the first trial of WT1 peptide vaccination has demonstrated persistence of WT1-specific CD8+ T cells for over 8 years, in all cases these cells were detectable before vaccination, raising the possibility that their initial priming and expansion was a physiological response to tumour rather than to vaccination (Tsuboi et al, 2011).
Different approaches have been used to provide CD4+ T cell help following vaccination. In our study, PADRE peptide was used to stimulate CD4+ T cells through MHC class II. Both PADRE and WT1 peptides elicited an IFN-γ response following vaccination, suggesting that concurrent CD4+ and CD8+ T cell responses were achieved, but this did not correlate with establishment of a memory response. Similarly, Kuball et al (2011) were unable to demonstrate antigen-specific T cell responses in any of four patients with AML who were vaccinated with WT1, PR3 and PADRE peptides in Montanide and CpG adjuvant; PADRE-specific CD4+ T cells in their study were shown to lack the capacity to produce IL2, a cytokine that is required for potent CD8+ T cell responses to a vaccinated antigen.
The choice of adjuvant may also be important in determining the longevity of CD8+ T cell responses to vaccination. Following early murine experiments showing that oil-in-water adjuvants, such as Incomplete Freund's Adjuvant (IFA) and Montanide adjuvant, could enhance immune responses to peptide vaccination (Kast et al, 1993), Montanide adjuvant has been used by our group and others in clinical trials (Tsuboi et al, 2004; Rezvani et al, 2008; Maslak et al, 2010; Kuball et al, 2011). However, sustained systemic presentation of peptides from the oil-in-water depot in the absence of a continuous helper/danger signal may contribute to induction of CD8+ T cell tolerance, a hypothesis formed by Bijker et al (2007) and reinforced by their finding that substitution of IFA with phosphate-buffered saline shortened the duration of antigen presentation and resulted in durable CD8+ T cell responses to a minimal CD8+ epitope.
Although we found that, in two patients, the emergence of functional WT1-specific T cells after vaccination was followed by a decrease in WT1 mRNA levels to below the upper threshold limit observed in normal control samples, there was no significant correlation between the immunological and disease responses to vaccination. This may relate to the observation that secondary expansion of WT1-specific T cells in vitro failed despite IL2, IL7, IL15 and IL21 cytokine cocktail and good CMV-specific T cell expansion. It is well recognized that effective anti-tumour activity of cytotoxic T cells correlates with high avidity for tumour-associated antigens (Zeh et al, 1999; Dutoit et al, 2001; Yang et al, 2002); the lack of consistent clinical response in our study may reflect a failure of vaccination to result in sustained expansion of high-avidity WT1-specific T cell clones. Functional avidity using peptide titration experiments was not tested in our study. In common with other tumour-associated antigens, WT1 is expressed at low levels in some normal tissues (Mundlos et al, 1993; Ramani & Cowell, 1996; Baird & Simmons, 1997; Menssen et al, 1997), and high-avidity cytotoxic T cell clones may have been deleted or rendered unresponsive during the establishment of tolerance (Theobald et al, 1997; Nugent et al, 2000). In mice, repeated immunization with foreign antigen generates memory CD8+ T cells that have increased avidity for that antigen; in contrast, such avidity maturation is limited for self antigen, such as WT1 (Turner et al, 2008). Similarly, antigen-specific memory CD8+ T cells in patients vaccinated with a melanoma peptide failed to acquire the enhanced functional avidity usually associated with competent memory T cell maturation (Walker et al, 2008), while Rezvani et al (2011) demonstrated selective deletion of high-avidity leukaemia-specific CD8+ T cells following vaccination with WT1 and PR1 peptides.
Although early trials of peptide vaccination in cancer patients showed promising immunological responses, more recent reports have been predominantly negative (Kuball et al, 2011; Rezvani et al, 2011). Our results illustrate further the difficulties of expansion of high-avidity leukaemia-specific effector and memory CD8+ T cell clones through vaccination with short self-peptides, demonstrating that translational studies are still required to define the site, time period, dose and adjuvants that lead to optimal activation of both CD8+ and CD4+ T cell responses following delivery of peptide. The low toxicity and relative simplicity of administration of peptide vaccines remain attractive, with almost 100 open studies of peptide vaccination currently listed on clinicaltrials.gov; however, our data lend impetus to alternative strategies, such as adoptive cellular immunotherapy with T cell receptor gene-modified T cells, that have the potential to generate potent antigen-specific cytotoxic responses against cancer while circumventing the difficulties of peptide vaccination.
Acknowledgments
This study was funded by a grant from Leukaemia and Lymphoma Research and supported by the University College London Experimental Cancer Medicine Centre and the Medical Research Council. David Grimwade and Adam Ivey acknowledge support from the National Institute for Health Research for development and evaluation of minimal residual disease assays to predict outcome and guide treatment approach in AML. This paper presents independent research funded by the National Institute for Health Research (NIHR) under its Programme Grants for Applied Research Programme (Grant Reference Number RP- PG-0108-10093). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. In addition David Grimwade is grateful for research funding from the Guy's and St. Thomas’ Charity.
Author contributions
BU analysed the data and wrote the paper; IM-D performed the research and analysed the data; AI performed the research; CC performed the research; FC performed the research and analysed the data; AV performed the research; PK performed the research; DG performed the research, provided reagents and analysed the data; AK performed the research; HS designed the study and analysed the data; EM designed the study, performed the research, analysed the data and wrote the paper.
References
- Alexander J, Sidney J, Southwood S, Ruppert J, Oseroff C, Maewal A, Snoke K, Serra HM, Kubo RT. Sette A. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity. 1994;1:751–761. doi: 10.1016/s1074-7613(94)80017-0. [DOI] [PubMed] [Google Scholar]
- Algar EM, Khromykh T, Smith SI, Blackburn DM, Bryson GJ. Smith PJ. A WT1 antisense oligonucleotide inhibits proliferation and induces apoptosis in myeloid leukaemia cell lines. Oncogene. 1996;12:1005–1014. [PubMed] [Google Scholar]
- Baird PN. Simmons PJ. Expression of the Wilms’ tumor gene (WT1) in normal hemopoiesis. Experimental Hematology. 1997;25:312–320. [PubMed] [Google Scholar]
- Bellantuono I, Gao L, Parry S, Marley S, Dazzi F, Apperley J, Goldman JM. Stauss HJ. Two distinct HLA-A0201-presented epitopes of the Wilms tumor antigen 1 can function as targets for leukemia-reactive CTL. Blood. 2002;100:3835–3837. doi: 10.1182/blood.V100.10.3835. [DOI] [PubMed] [Google Scholar]
- Bijker MS, van den Eeden SJF, Franken KL, Melief CJM, Offringa R. van der Bur SH. CD8+ CTL priming by exact peptide epitopes in incomplete Freund's adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. Journal of Immunology. 2007;179:5033–5040. doi: 10.4049/jimmunol.179.8.5033. [DOI] [PubMed] [Google Scholar]
- Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H. Lewis WH. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- Cilloni D, Gottardi E, Messa F, Fava M, Scaravaglio P, Bertini M, Girotto M, Marinone C, Ferrero D, Gallamini A, Levis A. Saglio G. Significant correlation between the degree of WT1 expression and the International Prognostic Scoring System Score in patients with myelodysplastic syndromes. Journal of Clinical Oncology. 2003;21:1988–1995. doi: 10.1200/JCO.2003.10.503. [DOI] [PubMed] [Google Scholar]
- Cilloni D, Renneville A, Hermitte F, Hills RK, Daly S, Jovanovic JV, Gottardi E, Fava M, Schnittger S, Weiss T, Izzo B, Nomdedeu J, van der Heijden A, van der Reijden BA, Jansen JH, van der Velden VHJ, Ommen H, Preudhomme C, Saglio G. Grimwade D. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: a European Leukemia Net study. Journal of Clinical Oncology. 2009;27:5195–5201. doi: 10.1200/JCO.2009.22.4865. [DOI] [PubMed] [Google Scholar]
- Dutoit V, Rubio-Godoy V, Dietrich PY, Quiqueres AL, Schnuriger V, Rimoldi D, Liénard D, Speiser D, Guillaume P, Batard P, Cerottini JC, Romero P. Valmori D. Heterogeneous T-cell response to MAGE-A10 (254-262): high avidity-specific cytolytic T lymphocytes show superior antitumor activity. Cancer Research. 2001;61:5850–5856. [PubMed] [Google Scholar]
- Gabert J, Beillard E, van der Velden VHJ, Bi W, Grimwade D, Pallisgaard N, Barbany G, Cazzaniga G, Cayuela JM, Cavé H, Pane F, Aerts JLE, De Micheli D, Thirion X, Pradel V, González M, Viehmann S, Malec M, Saglio G. van Dongen JJM. Standardization and quality control studies of “real-time” quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia - a Europe Against Cancer program. Leukemia. 2003;17:2318–2357. doi: 10.1038/sj.leu.2403135. [DOI] [PubMed] [Google Scholar]
- Gannagé M, Abel M, Michallet A-S, Delluc S, Lambert M, Giraudier S, Kratzer R, Niedermann G, Saveanu L, Guilhot F, Camoin L, Varet B, Buzyn A. Caillat-Zucman S. Ex vivo characterization of multiepitopic tumor-specific CD8 T cells in patients with chronic myeloid leukemia: implications for vaccine development and adoptive cellular immunotherapy. Journal of Immunology. 2005;174:8210–8218. doi: 10.4049/jimmunol.174.12.8210. [DOI] [PubMed] [Google Scholar]
- Gao L, Bellantuono I, Elsasser A, Marley SB, Gordon MY, Goldman JM. Stauss HJ. Selective elimination of leukemic CD34+ progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood. 2000;95:2198–2203. [PubMed] [Google Scholar]
- Gao L, Xue S-A, Hasserjian R, Cotter F, Kaeda J, Goldman JM, Dazzi F. Stauss HJ. Human cytotoxic T lymphocytes specific for Wilms’ tumor antigen-1 inhibit engraftment of leukemia-initiating stem cells in non-obese diabetic-severe combined immunodeficient recipients. Transplantation. 2003;75:1429–1436. doi: 10.1097/01.TP.0000061516.57346.E8. [DOI] [PubMed] [Google Scholar]
- Gillmore R, Xue S-A, Holler A, Kaeda J, Hadjiminas D, Healy V, Dina R, Parry SC, Bellantuono I, Ghani Y, Coombes RC, Waxman J. Stauss HJ. Detection of Wilms’ tumor antigen–specific CTL in tumor-draining lymph nodes of patients with early breast cancer. Clinical Cancer Research. 2006;12:34–42. doi: 10.1158/1078-0432.CCR-05-1483. [DOI] [PubMed] [Google Scholar]
- Goldman JM, Gale RP, Horowitz MM, Biggs JC, Champlin RE, Gluckman E, Hoffmann RG, Jacobsen SJ, Marmont AM. McGlave PB. Bone marrow transplantation for chronic myelogenous leukemia in chronic phase. Increased risk for relapse associated with T-cell depletion. Annals of Internal Medicine. 1988;108:806–814. doi: 10.7326/0003-4819-108-6-806. [DOI] [PubMed] [Google Scholar]
- Gorello P, Cazzaniga G, Alberti F, Dell'Oro MG, Gottardi E, Specchia G, Roti G, Rosati R, Martelli MF, Diverio D, Lo Coco F, Biondi A, Saglio G, Mecucci C. Falini B. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia. 2006;20:1103–1108. doi: 10.1038/sj.leu.2404149. [DOI] [PubMed] [Google Scholar]
- Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, Rimm AA, Ringdén O, Rozman C. Speck B. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75:555–562. [PubMed] [Google Scholar]
- Inoue K, Ogawa H, Sonoda Y, Kimura T, Sakabe H, Oka Y, Miyake S, Tamaki H, Oji Y, Yamagami T, Tatekawa T, Soma T, Kishimoto T. Sugiyama H. Aberrant over expression of the Wilms Tumor gene (WT1) in human leukemia. Blood. 1997;89:1405–1412. [PubMed] [Google Scholar]
- Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR. Schoenberger SP. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- Kast WM, Brandt RM. Melief CJ. Strict peptide length is not required for the induction of cytotoxic T lymphocyte-mediated antiviral protection by peptide vaccination. European Journal of Immunology. 1993;23:1189–1192. doi: 10.1002/eji.1830230534. [DOI] [PubMed] [Google Scholar]
- Keilholz U, Weber J, Finke JH, Gabrilovich DI, Kast WM, Disis ML, Kirkwood JM, Scheibenbogen C, Schlom J, Maino VC, Lyerly HK, Lee PP, Storkus W, Marincola F, Worobec A. Atkins MB. Immunologic monitoring of cancer vaccine therapy: results of a workshop sponsored by the Society for Biological Therapy. Journal of Immunotherapy. 2002;25:97–138. doi: 10.1097/00002371-200203000-00001. (Hagerstown, Md.: 1997), [DOI] [PubMed] [Google Scholar]
- Keilholz U, Letsch A, Busse A, Asemissen AM, Bauer S, Blau IW, Hofmann W-K, Uharek L, Thiel E. Scheibenbogen C. A clinical and immunologic phase 2 trial of Wilms tumor gene product 1 (WT1) peptide vaccination in patients with AML and MDS. Blood. 2009;113:6541–6548. doi: 10.1182/blood-2009-02-202598. [DOI] [PubMed] [Google Scholar]
- King JW, Thomas S, Corsi F, Gao L, Dina R, Gillmore R, Pigott K, Kaisary A, Stauss HJ. Waxman J. IL15 can reverse the unresponsiveness of Wilms’ tumor antigen-specific CTL in patients with prostate cancer. Clinical Cancer Research. 2009;15:1145–1154. doi: 10.1158/1078-0432.CCR-08-1821. [DOI] [PubMed] [Google Scholar]
- Kuball J, de Boer K, Wagner E, Wattad M, Antunes E, Weeratna RD, Vicari AP, Lotz C, van Dorp S, Hol S, Greenberg PD, Heit W, Davis HL. Theobald M. Pitfalls of vaccinations with WT1-, Proteinase3- and MUC1-derived peptides in combination with Montanide ISA51 and CpG7909. Cancer Immunology, Immunotherapy: CII. 2011;60:161–171. doi: 10.1007/s00262-010-0929-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslak PG, Dao T, Krug LM, Chanel S, Korontsvit T, Zakhaleva V, Zhang R, Wolchok JD, Yuan J, Pinilla-Ibarz J, Berman E, Weiss M, Jurcic J, Frattini MG. Scheinberg DA. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses in patients with complete remission from acute myeloid leukemia. Blood. 2010;116:171–179. doi: 10.1182/blood-2009-10-250993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menssen HD, Renkl HJ, Entezami M. Thiel E. Wilms’ tumor gene expression in human CD34+ hematopoietic progenitors during fetal development and early clonogenic growth. Blood. 1997;89:3486–3487. [PubMed] [Google Scholar]
- Miyoshi Y, Ando A. Egawa C. High expression of Wilms ‘Tumor suppressor gene predicts poor prognosis in breast cancer patients. Clinical Cancer Research. 2002;8:1167–1171. [PubMed] [Google Scholar]
- Mundlos S, Pelletier J, Darveau A, Bachmann M, Winterpacht A. Zabel B. Nuclear localization of the protein encoded by the Wilms’ tumor gene WT1 in embryonic and adult tissues. Development (Cambridge, England) 1993;119:1329–1341. doi: 10.1242/dev.119.4.1329. [DOI] [PubMed] [Google Scholar]
- Nugent CT, Morgan DJ, Biggs JA, Ko A, Pilip IM, Pamer EG. Sherman LA. Characterization of CD8+ T lymphocytes that persist after peripheral tolerance to a self antigen expressed in the pancreas. Journal of Immunology. 2000;164:191–200. doi: 10.4049/jimmunol.164.1.191. (Baltimore, Md.: 1950), [DOI] [PubMed] [Google Scholar]
- Odom L, Githens J, Morse H, Sharma B, August C, Humbert J, Peakman D, Rusnak S. Johnson F. Remission of relapsed leukæmia during a graft-versus-host reaction a “graft-versus-leukæmia reaction” in man? The Lancet. 1978;312:537–540. doi: 10.1016/s0140-6736(78)92879-9. [DOI] [PubMed] [Google Scholar]
- Oji Y, Miyoshi S, Maeda H, Hayashi S, Tamaki H, Nakatsuka S-I, Yao M, Takahashi E, Nakano Y, Hirabayashi H, Shintani Y, Oka Y, Tsuboi A, Hosen N, Asada M, Fujioka T, Murakami M, Kanato K, Motomura M, Kim EH, Kawakami M, Ikegame K, Ogawa H, Aozasa K, Kawase I. Sugiyama H. Over expression of the Wilms’ tumor gene WT1 in de novo lung cancers. International Journal of Cancer. 2002;100:297–303. doi: 10.1002/ijc.10476. [DOI] [PubMed] [Google Scholar]
- Oka Y, Tsuboi A, Murakami M, Hirai M, Tominaga N, Nakajima H, Elisseeva OA, Masuda T, Nakano A, Kawakami M, Oji Y, Ikegame K, Hosen N, Udaka K, Yasukawa M, Ogawa H, Kawase I. Sugiyama H. Wilms tumor gene peptide-based immunotherapy for patients with overt leukemia from myelodysplastic syndrome (MDS) or MDS with myelofibrosis. International Journal of Hematology. 2003;78:56–61. doi: 10.1007/BF02983241. [DOI] [PubMed] [Google Scholar]
- Oka Y, Tsuboi A, Taguchi T, Osaki T, Kyo T, Nakajima H, Elisseeva OA, Oji Y, Kawakami M, Ikegame K, Hosen N, Yoshihara S, Wu F, Fujiki F, Murakami M, Masuda T, Nishida S, Shirakata T, Nakatsuka S-I, Sasaki A, Udaka K, Dohy H, Aozasa K, Noguchi S, Kawase I. Sugiyama H. Induction of WT1 (Wilms’ tumor gene)-specific cytotoxic T lymphocytes by WT1 peptide vaccine and the resultant cancer regression. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:13885–13890. doi: 10.1073/pnas.0405884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani P. Cowell JK. The expression pattern of Wilms’ tumour gene (WT1) product in normal tissues and paediatric renal tumours. The Journal of Pathology. 1996;179:162–168. doi: 10.1002/(SICI)1096-9896(199606)179:2<162::AID-PATH545>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Rezvani K, Grube M, Brenchley JM, Sconocchia G, Fujiwara H, Price DA, Gostick E, Yamada K, Melenhorst J, Childs R, Hensel N, Douek DC. Barrett AJ. Functional leukemia-associated antigen-specific memory CD8+ T cells exist in healthy individuals and in patients with chronic myelogenous leukemia before and after stem cell transplantation. Blood. 2003;102:2892–2900. doi: 10.1182/blood-2003-01-0150. [DOI] [PubMed] [Google Scholar]
- Rezvani K, Brenchley JM, Price DA, Kilical Y, Gostick E, Sewell AK, Li J, Mielke S, Douek DC. Barrett AJ. T-cell responses directed against multiple HLA-A*0201-restricted epitopes derived from Wilms’ tumor 1 protein in patients with leukemia and healthy donors: identification, quantification, and characterization. Clinical Cancer Research. 2005;11:8799–8807. doi: 10.1158/1078-0432.CCR-05-1314. [DOI] [PubMed] [Google Scholar]
- Rezvani K, Yong ASM, Mielke S, Savani BN, Musse L, Superata J, Jafarpour B, Boss C. Barrett AJ. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood. 2008;111:236–242. doi: 10.1182/blood-2007-08-108241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani K, Yong ASM, Mielke S, Jafarpour B, Savani BN, Le RQ, Eniafe R, Musse L, Boss C, Kurlander R. Barrett AJ. Repeated PR1 and WT1 peptide vaccination in Montanide-adjuvant fails to induce sustained high-avidity, epitope-specific CD8+ T cells in myeloid malignancies. Haematologica. 2011;96:432–440. doi: 10.3324/haematol.2010.031674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato A, Zoso A, Santa SD, Milan G, Del Bianco P, De Salvo GL. Zanovello P. Predicting tumor outcome following cancer vaccination by monitoring quantitative and qualitative CD8+ T cell parameters. Journal of Immunology. 2006;176:1999–2006. doi: 10.4049/jimmunol.176.3.1999. [DOI] [PubMed] [Google Scholar]
- Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, Uchida N, Suzuki N, Sone A, Najima Y, Ozawa H, Wake A, Taniguchi S, Shultz LD, Ohara O. Ishikawa F. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Science Translational Medicine. 2010;2:17ra9. doi: 10.1126/scitranslmed.3000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibenbogen C, Letsch A, Thiel E, Schmittel A, Mailaender V, Baerwolf S, Nagorsen D. Keilholz U. CD8 T-cell responses to Wilms tumor gene product WT1 and proteinase 3 in patients with acute myeloid leukemia. Blood. 2002;100:2132–2137. doi: 10.1182/blood-2002-01-0163. [DOI] [PubMed] [Google Scholar]
- Theobald M, Biggs J, Hernández J, Lustgarten J, Labadie C. Sherman LA. Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. The Journal of Experimental Medicine. 1997;185:833–841. doi: 10.1084/jem.185.5.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi A, Oka Y, Osaki T, Kumagai T, Tachibana I, Hayashi S, Murakami M, Nakajima H, Elisseeva OA, Fei W, Masuda T, Yasukawa M, Oji Y, Kawakami M, Hosen N, Ikegame K, Yoshihara S, Udaka K, Nakatsuka S-I, Aozasa K, Kawase I. Sugiyama H. WT1 peptide-based immunotherapy for patients with lung cancer: report of two cases. Microbiology and Immunology. 2004;48:175–184. doi: 10.1111/j.1348-0421.2004.tb03503.x. [DOI] [PubMed] [Google Scholar]
- Tsuboi A, Oka Y, Kyo T, Katayama Y, Elisseeva OA, Kawakami M, Nishida S, Morimoto S, Murao A, Nakajima H, Hosen N, Oji Y. Sugiyama H. Long-term WT1 peptide vaccination for patients with acute myeloid leukemia with minimal residual disease. Leukemia. 2011;26:1410–1413. doi: 10.1038/leu.2011.343. [DOI] [PubMed] [Google Scholar]
- Turner MJ, Jellison ER, Lingenheld EG, Puddington L. Lefrançois L. Avidity maturation of memory CD8 T cells is limited by self-antigen expression. The Journal of Experimental Medicine. 2008;205:1859–1868. doi: 10.1084/jem.20072390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk JP, Knops GHJN, Van De Locht LTF, Menke AL, Jansen JH, Mensink EJBM, Raymakers RAP. De Witte T. Abnormal WT1 expression in the CD34-negative compartment in myelodysplastic bone marrow. British Journal of Haematology. 2002;118:1027–1033. doi: 10.1046/j.1365-2141.2002.03728.x. [DOI] [PubMed] [Google Scholar]
- Walker EB, Haley D, Petrausch U, Floyd K, Miller W, Sanjuan N, Alvord G, Fox BA. Urba WJ. Phenotype and functional characterization of long-term gp100-specific memory CD8+ T cells in disease-free melanoma patients before and after boosting immunization. Clinical Cancer Research. 2008;14:5270–5283. doi: 10.1158/1078-0432.CCR-08-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Linette GP, Longerich S. Haluska FG. Antimelanoma activity of CTL generated from peripheral blood mononuclear cells after stimulation with autologous dendritic cells pulsed with melanoma gp100 peptide G209-2M is correlated to TCR avidity. Journal of Immunology. 2002;169:531–539. doi: 10.4049/jimmunol.169.1.531. Baltimore, Md.: 1950, [DOI] [PubMed] [Google Scholar]
- Zeh HJ, Perry-Lalley D, Dudley ME, Rosenberg SA. Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. Journal of Immunology. 1999;162:989–994. (Baltimore, Md.: 1950), [PubMed] [Google Scholar]