

Abstract
Background and Purpose
Chronic hepatic inflammation results in liver fibrosis. As effective anti-fibrogenic agents are lacking, we investigated ursodeoxycholyl lysophosphatidylethanolamide (UDCA-LPE), a synthetic bile acid–phospholipid conjugate with anti-inflammatory and anti-apoptotic properties for tis effects on hepatic fibrogenesis.
Experimental Approach
To stimulate fibrogenesis, LX2 hepatic stellate cells were cultured with conditioned medium from CL48 liver cells after exposure to stress-inducing conditions – methionine–choline-deficient (MCD) medium or TNFα/cycloheximide (CHX) – with or without UDCA-LPE preincubation. Anti-fibrogenic effects of UDCA-LPE were further studied in CL48 and LX2 cells and in primary human hepatic stellate cells (HHStec) directly exposed to TGF-β1. To test UDCA-LPE in vivo, C57BL/6 mice were fed a MCD diet for 11 weeks followed by 30 mg·kg−1 UDCA-LPE 3× per week for 2.5 weeks.
Key Results
Expression of α-smooth muscle actin (α-SMA), α1-collagen, vimentin and TGF-β1 was down-regulated by up to 93% by UDCA-LPE in LX-2 cells cultured with conditioned medium. Also, UDCA-LPE inhibited Smad3 phosphorylation in CL48 cells incubated with MCD medium or TNFα/CHX and in LX2 cells exposed to conditioned medium. UDCA-LPE also decreased phosphorylated Smad3 and Smad2 directly induced by TGF-β1. Inhibition of TGF-β1/Smad2/3 signalling with down-regulation of target genes was confirmed in HHStec. In vivo, UDCA-LPE decreased hepatic α-SMA, α1-collagen and TGF-β1 expression and markedly lowered α-SMA protein and collagen deposition in MCD mice.
Conclusions and Implications
By blocking TGF-β1/Smad2/3 signalling, UDCA-LPE suppressed key mediators of hepatic fibrogenesis. Thus, UDCA-LPE could be suitable for prevention of fibrotic progression of chronic liver disease.
Table of Links
| TARGETS | LIGANDS | |
|---|---|---|
| Collagen | CHX, cycloheximide | TGF-β1 |
| ERK | TNFα | UDCA, ursodeoxycholic acid |
This Table lists key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Liver fibrosis generally evolves from persisting inflammatory conditions of the liver independent of their underlying causes. Chronic liver injury leads to an activation of hepatic stellate cells with subsequent extracellular matrix (ECM) production resulting in a replacement of liver parenchymal cells by scar tissue, which finally ends in irreversible organ dysfunction (Bataller and Brenner, 2005). With increasing incidence of chronic inflammatory liver diseases such as non-alcoholic fatty disease (NAFLD) (Angulo, 2002; Clark and Diehl, 2003), new therapeutic approaches to prevent fibrotic progression are urgently needed. As inflammation is the fuel perpetuating fibrogenesis, potential anti-fibrogenic compounds capable of dampening the inflammatory response within the liver in addition to the ability to inhibit ECM deposition and organ remodelling would be desirable.
Acknowledging the strong anti-apoptotic and anti-inflammatory properties of certain phospholipids such as phosphatidylcholine (PC) (Oneta et al., 1999; Treede et al., 2007), and its earlier-described anti-fibrogenic functions (Li et al., 1992; Lieber et al., 1994; Ma et al., 1996), we designed the bile acid–phospholipid conjugate ursodeoxycholyl lysophosphatidylethanolamide (UDCA-LPE) consisting of ursodeoxycholic acid (UDCA) coupled to lysophosphatidylethanolamine (LPE) (Chamulitrat et al., 2009). The bile acid UDCA has been used for hepatic drug targeting and improvement of bioavailability in earlier studies (Fiorucci et al., 2001; Goto et al., 2001). We have already shown that UDCA-LPE exerts potent anti-apoptotic and anti-inflammatory effects against TNFα-induced cytotoxicity in vitro and further confirmed its hepatoprotective functions in mouse models of endotoxin-mediated fulminant hepatitis and NAFLD in vivo (Chamulitrat et al., 2009; Pathil et al., 2011; 2012).
In our present work, we aimed to study the effects of UDCA-LPE on hepatofibrogenesis. We analysed anti-fibrogenic functions of the conjugate in experimental models of stellate cell activation in vitro. We used LX2 hepatic stellate cells cultured with conditioned medium from liver cells exposed to agents such as TNFα/cycloheximide (CHX) causing acute hepatotoxicity or chronic injurious conditions such as methionine–choline-deficient (MCD) medium. The MCD medium induces chronic hepatocellular inflammation by an impairment of hepatic lipid metabolism with a disturbed β-oxidation of fatty acids and an inhibition of the normal secretion of very low density lipoprotein (George et al., 2003; Rinella et al., 2008). In order to assess immediate anti-fibrogenic properties of UDCA-LPE, primary human hepatic stellate cells (HHStec) were analysed after exposure to the pro-fibrogenic master cytokine TGF-β1. Finally, for evaluation of the in vivo efficacy of the conjugate, we further studied the anti-fibrogenic effects of UDCA-LPE in a mouse model of advanced non-alcoholic steatohepatitis (NASH) with fibrosis induced by long-term MCD diet.
Methods
Cell culture
Human embryonic liver cell line CL48 (WRL-68) was from American Type Culture Collection (Manassas, VA, USA) and was cultured in DMEM containing 10% FCS. Human hepatic stellate cell line LX2 (a gift from S. Friedman) was maintained in DMEM containing 2% FCS. Media and supplements were from PAA Laboratories GmbH (Cölbe, Germany). Customized MCD DMEM was from PAA. UDCA-LPE as well as the other compounds UDCA, LPE and PC were solubilized in PBS containing 5% ethanol. Control cells were treated with PBS with ethanol correspondingly. For generation of conditioned medium CL48 liver cells were either cultured in control medium (DMEM without FCS), MCD medium and MCD medium + 90 μM UDCA-LPE for 48 h or in control medium, TNFα/CHX (15 ng mL−1; 40 μM respectively) and TNFα/CHX + 90 μM UDCA-LPE for 4 h or in control medium, ethanol (50 mM) and ethanol + 90 μM UDCA-LPE for 24 h. LX-2 cells were incubated with medium supernatant from CL48 cells (conditioned medium) for fibrogenic activation. HHStec (Innoprot, Biscay, Spain; Cat.-Ref.: P10653) were maintained in HHStec medium (Innoprot; Cat.-Ref.: P60126) and cultured in poly-L-lysine-coated flasks according to the manufacturer's protocol.
Animal model
All animal care and experimental procedures were in compliance with ethical guidelines of the institution and were approved by the Animal Care and Use Committee of the University of Heidelberg. Furthermore, all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
A total of 24 male C57BL/6 mice (Charles River Laboratories, Sulzfeld, Germany) were used, at 8 weeks of age. The animals were housed in the animal facility for 2 weeks before beginning the experimental procedures and the median body weight of the mice was approximately 23 g when starting the experiment. Food and water were provided ad libitum. For induction of fibrosis, mice were fed an MCD diet (Research Diets, Inc., New Brunswick, NJ, USA) for 11 weeks, control mice received a standard diet containing 10% fat. All diets were γ-irradiated. Development of liver injury was followed by i.p. injections of 30 mg·kg−1 UDCA-LPE, which was solubilized in PBS with 0.5% carboxymethylcellulose (CMC), three times a week for the last 2.5 weeks on diet in MCD mice. Control mice and MCD mice without UDCA-LPE treatment received CMC and PBS. At the end of the feeding period mice were anesthetized and killed by cardiac puncture. Livers were harvested, a portion of fresh tissue was fixed in 10% buffered formalin, the remaining liver tissue was snap frozen in liquid nitrogen and stored at −80°C. Blood samples were allowed to clot and subsequently centrifuged at 1000× g for 15 min. Serum was collected and stored at −80°C.
Sirus red staining and immunohistochemistry
Paraffin-embedded liver sections were stained with 0.1% Sirius red for evaluation of hepatic collagen deposition. Immunohistochemistry staining was performed with anti-α-smooth muscle actin (α-SMA) antibody (Epitomics, Inc., Burlingame, CA, USA). Antigens were visualized with HRP-conjugated secondary antibody (Abcam, Cambridge, UK) and Diaminobenzidine solution (42 Life Sciences GmbH & Co. KG, Bremerhaven, Germany).
Western blotting
Cell lysates (20 μg protein) were separated by gel electrophoresis and transferred onto a PVDF membrane. Blots were stained with dilutions of primary antibodies overnight at 4°C, washed and stained with secondary antibodies for 1 h at room temperature. Protein bands were visualized using Luminata Forte ECL system (Merck Millipore, Darmstadt, Germany). As recommended by the manufacturer, the HRP substrate was added to the blot and incubated for 5 min at room temperature. After draining excess substrate the blot was exposed to an X-ray film for different exposure times. The following antibodies were used at specified concentrations: monoclonal rabbit antibody to α-SMA at 1:15000 (Epitomics, Inc.); monoclonal rabbit antibody to phospho-Smad3 (pSmad3) at 1:10000 (Epitomics, Inc.); monoclonal rabbit antibody to Smad3 1:1000 (Epitomics, Inc.); polyclonal rabbit antibody to pSmad2 (Ser245/250/255) at 1:1000 (Cell Signaling, Danvers, MA, USA); polyclonal rabbit antibody to pSmad2 (Ser455/465) at 1:10000 (Cell Signaling); monoclonal rabbit antibody to Smad2 at 1:1000 (Epitomics, Inc.); polyclonal rabbit antibody to ERK1/2 (Thr202/Tyr204) at 1:1000 (Cell Signaling); monoclonal rabbit antibody to Smad ubiquitination regulatory factor 2 (Smurf2) at 1:1000 (Epitomics, Inc.) and monoclonal rabbit antibody to GAPDH 1:50000 (Cell Signaling). Secondary goat anti-rabbit antibody was used at 1:10000 (BioRad, Munich, Germany) Density of protein bands was quantified using Image J software (W. Rasband, NIH; http://rsb.info.nih.gov/ij/).
Gene expression analysis by quantitative real-time PCR
TaqMan Gene Expression Assays™ (Applied Biosystems, Darmstadt, Germany) were used as recommended by the manufacturer and were employed to relatively quantify the expression of following genes in LX2 hepatic stellate cells, CL48 liver cells and mouse liver samples. We used α-SMA (Hs00426835_g1), α1-collagen (Hs00164004_m1), vimentin (Hs00185584_m1), TGF-β1 (Hs00998133_m1), connective tissue growth factor(CTGF; Hs01026927_g1), Smad7 (Hs00998193_m1), GAPDH (Hs99999905_m1), α-SMA (Mm01204962_gH), α1-collagen (Mm00801666_g1), TGF-β1 (Mm01178820_m1) and GAPDH (Mm99999915_g1) assays. Total RNA from liver tissue samples was extracted employing the RNAqueous® Kit (Ambion, Darmstadt, Germany). RNA concentration and purity was assessed spectophotometrically at 260 nm in relation to the absorbance at 280 nm. We removed genomic DNA contamination in the RNA with the Turbo DNA-free™ kit (Ambion). cDNA was synthesized from 2 μg of total RNA using the SuperScript Double-Stranded cDNA Synthesis Kit (Life Technologies, Carlsbad, CA, USA) and Oligo(dT)(12–18) as suggested by the manufacturer. Real-time RT-PCR was carried out using a 7500 Fast Sequence Detector System (Applied Biosystems) and TaqMan Universal PCR Master Mix 2X (Applied Biosystems). Universal PCR conditions recommended by the manufacturer were followed. All samples were processed in quadruplicate. We analysed TaqMan gene expression data based on normalized expression values. GAPDH was used as housekeeping gene to normalize mRNA levels. The relative change of expression was calculated using the 2-ΔΔCT method.
Data analysis
Results are presented as mean ± SEM unless stated otherwise. Data were analysed with Prism Software version 4.0 (GraphPad, La Jolla, CA, USA). The significance of differences between two groups was determined by unpaired two-tailed Student's t-test. For comparison of multiple groups, we applied one-way anova with Dunnett's post test. A P value < 0.05 was considered significant.
Materials
Custom synthesis of UDCA-LPE was performed by ChemCon (Freiburg, Germany). All other chemicals were obtained from Sigma (Munich, Germany) unless stated otherwise.
Results
UDCA-LPE reduces MCD- and TNFα-induced pro-fibrogenic gene expression in the hepatic stellate cell line LX2
In order to stimulate fibrogenic activation, LX2 hepatic stellate cells were exposed to different stress-inducing conditions. Incubation of LX2 cells for 48 h with conditioned medium from MCD-cultured CL48 liver cells induced the expression of crucial pro-fibrogenic genes such as those for α-SMA, α1-collagen, vimentin and TGF-β1 (Figure 1A–D) with an almost 50-fold increase in the mRNA for TGF-β1 (Figure 1D).
Figure 1.
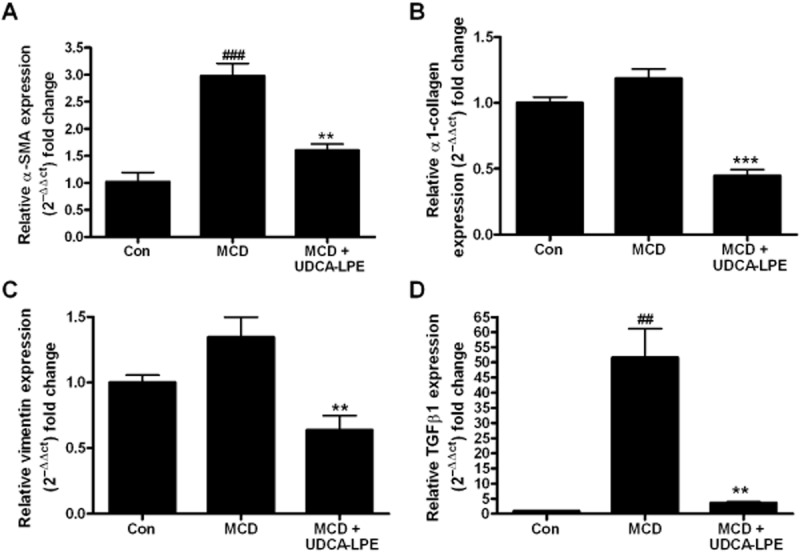
UDCA-LPE reduces MCD-induced pro-fibrogenic gene expression in hepatic stellate cells. (A–D) LX2 cells were cultured for 48 h with conditioned medium from CL48 liver cells exposed to control medium, MCD medium or MCD medium + 90 μM UDCA-LPE for 48 h. Expression levels of (A) α-SMA, (B) α1-collagen, (C) vimentin and (D) TGF-β1 in LX2 cells were quantified by qRT-PCR (N = 4). ##P < 0.01, ###P < 0.001 versus Con; **P < 0.01, ***P < 0.001 versus MCD.
In contrast, preincubation with UDCA-LPE down-regulated expression of these genes by 47–93% with the most pronounced effect on TGF-β1 (Figure 1D). We previously showed that treatment of CL48 liver cells with TNFα and CHX induced apoptosis (Pathil et al., 2011). As hepatocellular apoptosis is an important trigger for stellate cell activation, we next analysed the effect of conditioned medium from TNFα/CHX-treated CL48 liver cells on fibrogenic gene expression in hepatic stellate cells. The results showed that incubation with conditioned medium from apoptotic liver cells led to a marked fibrogenic activation of LX2 cells followed by a strong up-regulation of markers of ECM deposition (Figure 2A–D) with a more than 200-fold increase of α-SMA expression (Figure 2A) and a 7.5-fold rise of α1-collagen mRNA transcripts (Figure 2B). As observed earlier, pretreatment with UDCA-LPE significantly reduced the expression of pro-fibrotic genes by up to 86–89% (Figure 2A–D).
Figure 2.
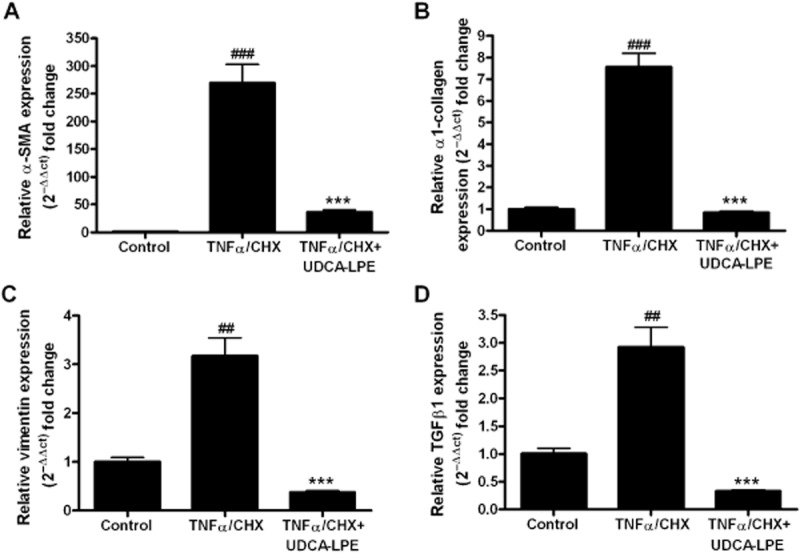
Reduction of TNFα-induced pro-fibrogenic genes in hepatic stellate cells by UDCA-LPE. (A–D) LX2 cells were cultured for 48 h with conditioned medium from CL48 liver cells exposed to control medium, TNFα/CHX (15 ng·mL−1·per 40 μM) or TNFα/CHX + 90 μM UDCA-LPE for 4 h. Expression levels of (A) α-SMA, (B) α1-collagen, (C) vimentin and (D) TGF-β1 in LX2 cells were quantified by qRT-PCR (N = 4). ##P < 0.01, ###P < 0.001 versus Con; ***P < 0.001 versus TNFα/CHX.
Stress-induced and TGF-β1-mediated phosphorylation of Smad3 is suppressed by UDCA-LPE
Because of the fundamental role of TGF-β1/Smad signalling for fibrogenesis in the liver, we evaluated the influence of UDCA-LPE on components of the signalling pathway downstream of TGF-β1. Exposure of hepatic stellate cells to conditioned medium from MCD-cultured CL48 liver cells increased phosphorylated Smad3 (pSmad) protein, without changing levels of unphosphorylated Smad3 (Figure 3A,C). Accumulation of pSmad3 was even more pronounced in LX2 cells cultured with conditioned medium from TNFα/CHX-treated CL48 cells (Figure 3B,D). Treatment with UDCA-LPE reduced protein levels of pSmad3 under both conditions (Figure 3A–D), the amount of phosphorylated Smad3 was even below levels of controls in LX2 cells cultured with conditioned medium from TNFα/CHX-exposed liver cells. Additionally, reduction of pSmad3 following UDCA-LPE was also observed in LX2 cells exposed to conditioned medium from ethanol-treated CL48 liver cells (Supporting Information Fig. S1). Notably, CL48 cells cultured with stress-inducing agents, such as MCD medium or TNFα/CHX, displayed a similar activation of Smad3, which was almost totally blocked by UDCA-LPE treatment (Figure 3E–H). Thus, the results showed that UDCA-LPE markedly decreased activation of Smad3, as shown by the extent of phosphorylation, in hepatic stellate cells and liver cells. To confirm that the impaired activation of Smad3 by the conjugate was not only a consequence of reduced TGF-β1 expression, but was caused by the inhibition of downstream-signalling events, liver cells and hepatic stellate cells were directly exposed to TGF-β1. Incubation with the pro-fibrogenic master cytokine resulted in an activation of Smad3 in LX2 hepatic stellate cells (Figure 4A,C) and CL48 liver cells (Figure 4B,D). As noted previously, UDCA-LPE reduced pSmad3 in both cell lines below baseline levels of control cells (Figure 4A–D) with the most pronounced effect in CL48 liver cells (Figure 4B,D). Moreover, UDCA-LPE suppressed Smad3 phosphorylation more than its separate components (UDCA or LPE) or other phospholipids such as PC, which had either no or rather limited inhibitory effect on Smad3 phosphorylation (Supporting Information Fig. S2).
Figure 3.
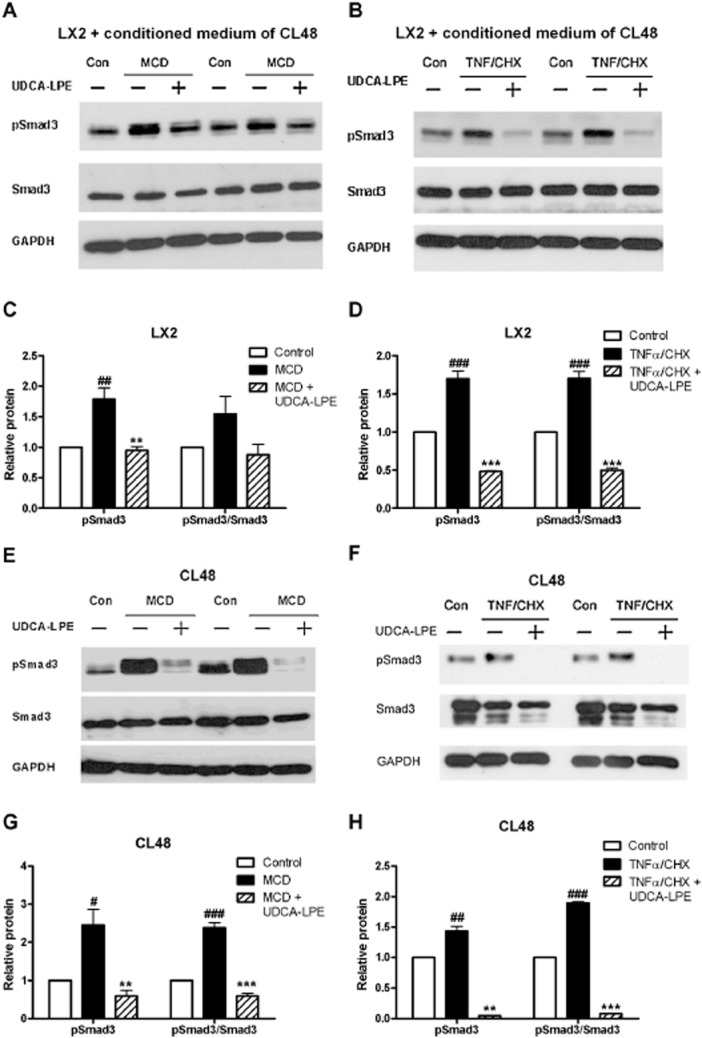
Inhibition of stress-induced pSmad3 by UDCA-LPE. (A,B) Representative Western blot analysis of pSmad3 and Smad3 in LX2 hepatic stellate cells (A) cultured for 12 h with conditioned medium from CL48 liver cells exposed to control medium, MCD medium or MCD medium + 90 μM UDCA-LPE for 12 h or (B) incubated for 1 h with conditioned medium from CL48 liver cells exposed to control medium, TNFα/CHX (15 ng·mL−1 per 40 μM) or TNFα/CHX + 90 μM UDCA-LPE for 4 h. (E,F) Representative Western blot analysis of pSmad3 and Smad3 in CL48 cells incubated with (E) control medium, MCD medium or MCD medium + 90 μM UDCA-LPE for 12 h or (F) control medium, TNFα/CHX (15 ng·mL−1 per 40 μM) or TNFα/CHX + 90 μM UDCA-LPE for 4 h. GAPDH was used as control for equal protein loading. (C,D,G,H) Graphical presentation of relative protein levels of pSmad3 and pSmad3/Smad3 ratio as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3. #P < 0.05, ##P < 0.01, ###P < 0.001 versus Con; **P < 0.01, ***P < 0.001 versus MCD; **P < 0.01, ***P < 0.001 versus TNFα/CHX.
Figure 4.
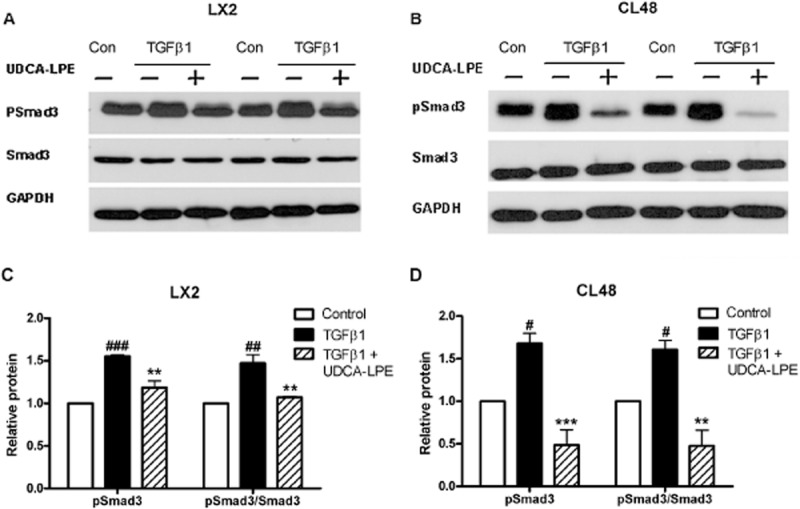
UDCA-LPE reduces TGF-β1-mediated phosphorylation of Smad3. (A,B) Representative Western blot analysis of pSmad3 and Smad3 in (A) LX2 hepatic stellate cells or (B) CL48 liver cells incubated with control medium, TGF-β1 (4 ng·mL−1) or TGF-β1 + 90 μM UDCA-LPE for 1 h. GAPDH was used as control for equal protein loading. (C,D) Graphical presentation of relative protein levels of pSmad3 and pSmad3/Smad3 ratio as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3. #P < 0.05, ##P < 0.01, ###P < 0.001 versus Con; **P < 0.01, ***P < 0.001 versus TGF-β1.
Reduction of TGF-β1-induced pSmad2 by UDCA-LPE
In addition to the effects on Smad3 activation, we next studied the effect of UDCA-LPE on Smad2 in hepatic stellate cells and liver cells. Fibrogenic activation following incubation with TGF-β1 resulted in a marked C-terminal phosphorylation of Smad2(at Ser465) in both cell types (Figure 5A–D). As observed with pSmad3, Western blot analysis revealed that UDCA-LPE decreased the amount of pSmad2 protein in LX2 hepatic stellate cells and CL48 liver cells (Figure 5A–D). Interestingly, in contrast to pSmad3, these lower levels of pSmad2 were accompanied by decreased levels of unphosphorylated Smad2 protein (Figure 5A–F). Therefore, Western blot analysis of Smurf2 was performed, which showed that the lower Smad2 levels were not related to changes in Smurf2 (Supporting Information Fig. S3A,B). Analysis of the phosphorylation status of the linker region in LX2 cells showed a similar increase of pSmad2(Ser245) after TGF-β1, which was again subsequently reduced together with the level of unphosphorylated Smad2 on treatment with UDCA-LPE (Figure 5E,F). TGF-β1-dependent ERK activation was reported to be mediated by linker-phosphorylated Smad2 (Li et al., 2009). However, impaired phosphorylation of Smad2 at the linker region after UDCA-LPE was not accompanied by decreased ERK1/2 activation, on the contrary, the conjugate slightly increased pERK1/2 protein levels in LX2 cells (Figure 5E,F).
Figure 5.

Reduction of TGF-β1-induced pSmad2 by UDCA-LPE. (A–C) Representative Western blot analysis of pSmad2 (Ser465) and Smad2 in (A) LX2 hepatic stellate cells or (C) CL48 liver cells incubated with control medium, TGF-β1 (4 ng·mL−1) or TGF-β1 + 90 μM UDCA-LPE for 1 h. (E) Representative Western blot analysis of pSmad2 (Ser245), Smad2 and pERK1/2 in LX2 hepatic stellate cells incubated with control medium, TGF-β1 (4 ng·mL−1) or TGF-β1 + 90 μM UDCA-LPE for 1 h. GAPDH was used as control for equal protein loading. (B,D,F) Graphical presentation of relative protein levels of pSmad2, Smad2, pSmad2/Smad2 ratio and pERK1/2 as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3–4. #P < 0.05, ##P < 0.01, ###P < 0.001 versus Con; *P < 0.05, **P < 0.01, ***P < 0.001 versus TGF-β1.
Impairment of TGF-β1-induced Smad signalling and repression of TGF-β1 target genes in HHStec
In order to analyse further the direct effects of UDCA-LPE on TGF-β1 signalling in hepatic stellate cells, culture-activated primary HHStec were exposed to TGF-β1 after varying days of culture on plastic. Incubation with TGF-β1 led to an increased expression of α-SMA and α1-collagen with a stronger up-regulation of these genes in earlier days of culture activation (Figure 6A,B). In contrast, UDCA-LPE was able to significantly down-regulate the expression of these ECM markers in HHStec irrespective of their activation stage (Figure 6A,B). TGF-β1 also induced a pronounced accumulation of pSmad3 at different stages of HHStec culture activation, whereas UDCA-LPE almost abolished TGF-β1-mediated phosphorylation of Smad3 at any time (Figure 6C,D). Furthermore, a strong activation of Smad2 was found in HHStec upon TGF-β1 exposure (Figure 6E–G). Accordingly, incubation with UDCA-LPE totally blocked C-terminal phosphorylation of Smad2 and reduced the amount of linker-phosphorylated Smad2, whereas levels of unphosphorylated Smad2 protein appeared to be unchanged (Figure 6E–G). Suppression of TGF-β1 signal transduction was additionally reflected in an impaired expression of TGF-β1 target genes such as Smad7 and CTGF in UDCA-LPE-treated HHStec (Figure 6H,I). Thus, the results showed that the anti-fibrogenic effects of UDCA-LPE were not only the outcome of its anti-inflammatory and anti-apoptotic actions on liver cells, but reflected an immediate inhibitory effect of UDCA-LPE on TGF-β1 signalling in hepatic stellate cells.
Figure 6.
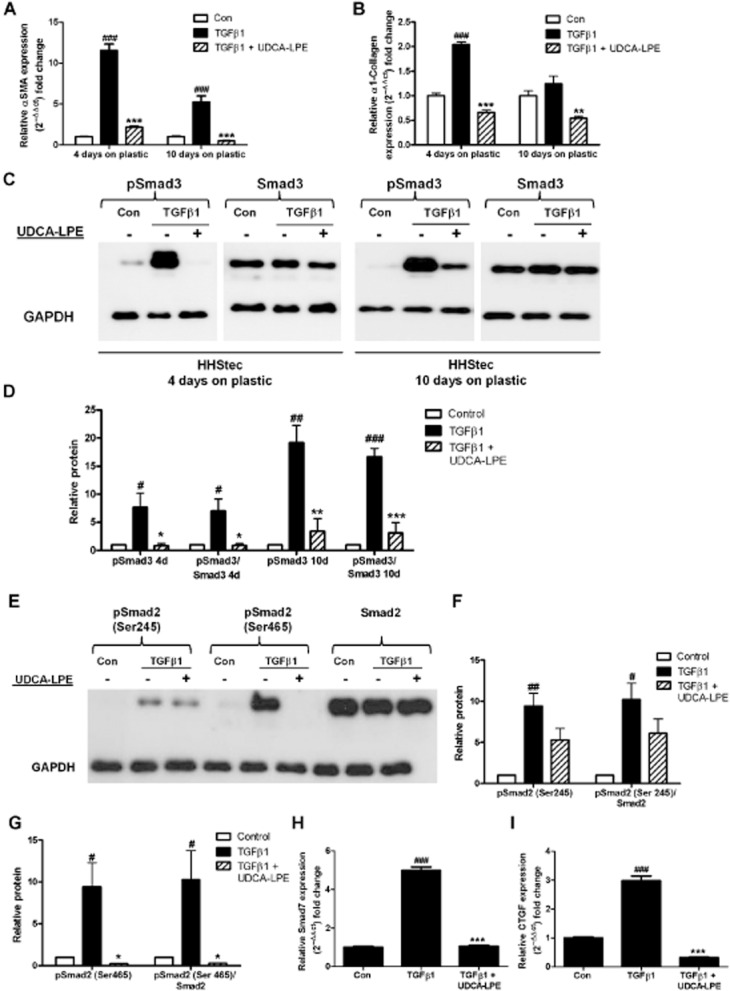
UDCA-LPE inhibits expression of TGF-β1 target genes and Smad phosphorylation in HHStec. (A,B) HHStec were incubated with control medium, TGF-β1 (4 ng·mL−1) or TGF-β1 + 90 μM UDCA-LPE for 24 h after 4 days and 10 days culture on plastic. Expression of (A) α-SMA and (B) α1-collagen was quantified by qRT-PCR (N = 4). (C) Representative Western blot analysis of pSmad3 and Smad3 in HHStec incubated with control medium, TGF-β1 (4 ng·mL−1) or TGF-β1 + 90 μM UDCA-LPE for 1 h after 4 days and 10 days culture on plastic. (E) Representative Western blot analysis of pSmad2 (Ser245), pSmad2 (Ser465) and Smad2 in HHStec incubated with control medium, TGF-β1 (4 ng·mL−1) or TGF-β1 + 90 μM UDCA-LPE for 1 h after 4 days culture on plastic. GAPDH was used as control for equal protein loading. (D,F,G) Graphical presentation of relative protein levels of (D) pSmad3 and pSmad3/Smad3 ratio and (F,G) pSmad2 and pSmad2/Smad2 ratio as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3. #P < 0.05, ##P < 0.01, ###P < 0.001 versus Con; *P < 0.05, **P < 0.01, ***P < 0.001 versus TGF-β1. (H,I) HHStec were incubated with control medium, TGF-β1 (4 ng·mL−1) or TGF-β1 + 90 μM UDCA-LPE after 4 days culture on plastic. Expression of (H) Smad7 after 1 h TGF-β1 exposure and expression of (I) CTGF after 4 h TGF-β1 exposure quantified by qRT-PCR (N = 4). GAPDH was used as control for equal protein loading. ###P < 0.001 versus Con; **P < 0.01, ***P < 0.001 versus TGF-β1.
UDCA-LPE ameliorates fibrotic response in a murine model of MCD-induced liver fibrosis
To confirm anti-fibrogenic functions of UDCA-LPE in vivo, mice received a MCD diet for 11 weeks, which induced advanced NASH with up to fivefold increase of transaminase values. Establishment of liver injury was followed by treatment with UDCA-LPE at 30 mg·kg−1 three times a week for 2.5 weeks. Decrease of transaminase levels to near normal levels and improvement of liver histology following UDCA-LPE treatment have already been shown (Pathil et al., 2012). In our present experiments, chronic liver injury induced by the MCD diet was accompanied by an up-regulation of crucial pro-fibrogenic genes with a 25-fold increase of α-SMA, a 47-fold induction of α1-collagen and a fourfold increase of TGF-β1 (Figure 7A–C). Nevertheless, administration of UDCA-LPE was able to significantly down-regulate these genes by 75% for α-SMA, by 78% for α1-collagen and by 59% for TGF-β1 (Figure 7A–C). Furthermore, in terms of protein, treatment with the conjugate achieved a marked reduction of α-SMA in both liver tissue lysates and in the α-SMA staining of liver sections (Figure 7D–F). Additionally, liver sections of MCD mice treated with UDCA-LPE displayed less collagen deposition as visualized by Sirius red staining (Figure 7G). The amelioration of fibrotic response because of UDCA-LPE was further accompanied by a reduction of pSmad3 levels in liver tissue lysates of MCD mice (Supporting Information Fig. S4).
Figure 7.

Reduction of fibrotic response in a murine model of MCD-induced liver fibrosis by UDCA-LPE administration. (A–F) C57BL6 mice were fed an MCD diet for 11 weeks followed by 30 mg·kg−1 UDCA-LPE 3× per week for the last 2.5 weeks on diet (N = 8). (A–C) Expression levels of (A) α-SMA, (B) α1-collagen and (C) TGF-β1 in liver tissue were quantified by qRT-PCR. ###P < 0.001 versus Con; ***P < 0.001 versus MCD. (D) Representative Western blot analysis of α-SMA in liver tissue lysates. GAPDH was used as control for equal protein loading. (E) Graphical presentation of relative α-SMA protein levels as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3. ###P < 0.001 versus Con; **P < 0.01 versus MCD; ***P < 0.001 versus MCD. (F) α-SMA staining of liver sections of two different mice and (G) representative Sirus red staining of one mouse fed a control diet, MCD diet or MCD diet + UDCA-LPE respectively.
Discussion
Liver fibrosis is the frequent result of chronic inflammatory liver diseases and the prerequisite for end-stage liver cirrhosis. As effective therapeutic options are lacking to date, novel compounds suitable for the treatment of hepatofibrogenesis are urgently needed. In our current work, we report that the bile acid–phospholipid conjugate UDCA-LPE showed potent anti-fibrogenic functions in different in vitro models of stellate cell activation with the ability to down-regulate genes crucial to ECM production. Moreover, UDCA-LPE inhibited the normal activation of the pro-fibrogenic TGF-β1/Smad signalling pathway by reducing phosphorylation of Smad3 and Smad2. Administration of UDCA-LPE in vivo further resulted in an impairment of fibrotic response in a MCD diet-mediated mouse model of advanced NASH with fibrosis. Taken together, UDCA-LPE exhibited characteristics very relevant to the prevention of hepatofibrogenesis.
Chronic inflammation and hepatocyte injury are the major causes for transdifferentiation of hepatic stellate cells resulting in increased deposition of ECM. Our previous work demonstrated that UDCA-LPE lowered the susceptibility of hepatocytes towards apoptosis and inflammation. Culture of liver cells with MCD medium results in an inflammatory condition with steatosis because of impaired hepatic lipid metabolism (Kohli et al., 2007). In addition to chronic inflammation, phagocytosis of hepatocellular apoptotic bodies by hepatic stellate cells was described to be another important stimulus for fibrogenic activation with subsequent matrix production (Canbay et al., 2003; Malhi and Gores, 2008). Accordingly, we were able to show that conditioned medium of liver cells exposed to stress-inducing agents provoked a significantly weaker fibrotic response in hepatic stellate cells when liver cells were additionally treated with UDCA-LPE. Thus, the anti-apoptotic and anti-inflammatory effects of UDCA-LPE on hepatocytes seem to be important to prevent fibrogenic activation of hepatic stellate cells and to reduce ECM production in the liver.
Among the different factors able to promote hepatofibrogenesis, TGF-β1 is the most potent cytokine for the initiation of fibrogenic signalling in the liver. Binding of TGF-β1 to its type II receptor is followed by recruitment of the type I receptor, which subsequently phosphorylates Smad3 and Smad2, the so-called receptor-activated Smad (R-Smad) proteins. This step is crucial for the intracellular delivery of TGF-β1-mediated signals to the nucleus and the induction of pro-fibrogenic gene expression (Wrana et al., 1994; Moustakas et al., 2001; Derynck and Zhang, 2003; Shi and Massague, 2003). Accordingly, in chronic fibrotic liver disease, Smad3 was constitutively phosphorylated and translocated to the nucleus (Inagaki et al., 2001). The results of our current work demonstrated the ability of UDCA-LPE to interfere with this signalling cascade, at the stage of Smad2/3 phosphorylation. Earlier studies employing genetic approaches suggested that strategies aiming at decreasing Smad2/3 activation might ameliorate TGF-β1-mediated hepatofibrogenesis. Knock-down of Smad3 resulted in less α1-collagen production in mice exposed to CCl4 compared with wild-type mice (Schnabl et al., 2001). Adenoviral expression of Smad7, an inhibitory Smad protein, which interferes with Smad2/3 phosphorylation (Nakao et al., 1997) decreased ECM components in liver fibrosis induced by bile-duct ligation (Dooley et al., 2003). Moreover, reduction of Smad2 levels by the methylxanthine. Caffeine. inhibited the expression of CTGF (Gressner et al., 2008), a pro-fibrogenic amplifier of TGF-β1 signalling (Leask and Abraham, 2006), presumably by enforcing proteosomal degradation of Smad2 depending on the E3 ubiquitin ligase Smurf2 (Lo and Massague, 1999; Zhang et al., 2001). Our results showed that UDCA-LPE markedly reduced levels of pSmad2 and unphosphorylated Smad2 protein in LX2 cells in a similar fashion. However, analysis of Smurf2 protein levels revealed no changes following UDCA-LPE treatment. It is not known at this time whether UDCA-LPE could accelerate Smad2 ubiquitination and/or activate proteosomal degradation of ubiquitinated Smad2.
In addition to the phosphorylation status at the C-terminus, recent studies have also focused at the linker region of Smad proteins (Sapkota et al., 2006; Wrighton et al., 2009). Among others MAPK family members ERK, JNK and p38 catalyse regulatory phosphorylation of the linker domain of R-Smads (Kretzschmar et al., 1997; Furukawa et al., 2003; Mori et al., 2004; Li et al., 2009). TGF-β1-dependent ERK activation followed by subsequent collagen synthesis was reported to be mediated by phosphorylation of the linker region of Smad2 (Li et al., 2009). In contrast, activated ERK signalling has also been implicated in the inhibition of type I collagen expression in fibroblasts (Reunanen et al., 2000). In our study, UDCA-LPE significantly decreased linker-phosphorylated Smad2 in LX2 cells, accompanied by slightly elevated pERK1/2 protein levels. Furthermore, extensive ERK1/2 activation was detected in CL48 liver cells after incubation with the conjugate (data not shown). Thus, further experimental studies are needed to elucidate the potential influence of UDCA-LPE on the crosstalk between TGF-β1/Smad2/3 and ERK signalling pathways.
Earlier studies proposed that the hepatic stellate cell line LX2 displays characteristics of fully transdifferentiated myofibroblasts (Xu et al., 2005), which have been reported to be less sensitive towards TGF-β1 (Dooley et al., 2001). However, our results showed an appropriate response of LX2 cells to different profibrogenic stimuli within our experimental setting, most probably because of the early passages of LX2 cells employed in this study. Analysis of HHStec at different stages of culture activation confirmed a marked suppression of TGF-β1 signalling by UDCA-LPE with impaired phosphorylation of R-Smads and subsequent down-regulation of TGF-β1 target genes. Taken together, our results strongly imply that the anti-fibrogenic effects of UDCA-LPE in hepatic stellate cells are caused by an immediate inhibition of TGF-β1 signal transduction, which add to, but are completely independent of, anti-inflammatory and anti-apoptotic actions of the conjugate in liver cells. Recently, we showed that UDCA-LPE protected hepatocytes from lipoapoptosis by altering cellular fatty acid pools, which may influence cell membrane remodelling and stabilization (Chamulitrat et al., 2013). TGF-β1 signalling can be regulated negatively by enhanced TGF-β receptor degradation depending on receptor localization within certain membrane microdomains such as lipid rafts (Di Guglielmo et al., 2003; Chen, 2009). As UDCA-LPE may influence membrane composition, ongoing studies will have to address whether the conjugate thereby interferes with the intracellular delivery of TGF-β1-mediated signals.
Apart from the anti-fibrogenic effects of UDCA-LPE on non-parenchymal cells, our results further indicate beneficial effects of the conjugate on hepatocytes during the epithelial-to-mesenchymal transition (EMT). This cellular process, which is characterized by the loss of epithelial functions and the acquisition of mesenchymal properties in epithelial cells (Thiery and Sleeman, 2006), plays an important physiological role during embryonic development, but is also tightly linked to fibrotic disorders and carcinogenesis (Kalluri and Neilson, 2003). EMT can contribute to the pool of matrix-producing cells in the liver and TGF-β1 was shown to induce EMT in hepatocytes (Blobe et al., 2000; Kaimori et al., 2007). Analysis of pSmad3 and pSmad2 in CL48 liver cells exposed to stress-inducing conditions or directly cultured with TGF-β1 revealed an almost complete suppression of Smad3 phosphorylation as well as reduced pSmad2 content because of UDCA-LPE. Thus, it seems reasonable that UDCA-LPE may block TGF-β1-mediated EMT during fibrogenesis and tumour progression. Based upon ongoing experimental work employing primary liver cells the potential effects of UDCA-LPE on EMT have to be addressed in the future.
In summary, the results of the current study have yielded evidence that the bile acid–phospholipid conjugate UDCA-LPE exhibits excellent anti-fibrogenic properties with the ability to interfere with the TGF-β1/Smad2/3 signalling pathway. By modifying key mediators of fibrotic response in the liver, UDCA-LPE qualifies as a promising compound suitable for the prevention of fibrogenic progression of chronic liver disease.
Acknowledgments
The authors thank Sabine Tuma, Nenad Katava and Fortunata Jung for excellent technical assistance.
Glossary
- CHX
cycloheximide
- CTGF
connective tissue growth factor
- ECM
extracellular matrix
- EMT
epithelial-to-mesenchymal transition
- HHStec
primary human hepatic stellate cells
- MCD
methionine–choline-deficient
- NAFLD
non-alcoholic fatty disease
- UDCA
ursodeoxycholic acid
- UDCA-LPE
ursodeoxycholyl lysophosphatidylethanolamide
- NASH
non-alcoholic steatohepatitis
- PC
phosphatidylcholine
- α-SMA
α-smooth muscle actin
- Smurf2
Smad ubiquitination regulatory factor 2
Author contributions
A.P. – study concept and design, data interpretation, article drafting, and final revision; J. M. – data acquisition, analysis and interpretation; J. M. L. – data acquisition, analysis and interpretation; J. W. – data acquisition, analysis and interpretation; A. W. – data analysis and interpretation; C. W. – data interpretation and article revision; and W. S. – overall supervision of the study concept and final revision.
Funding
This study was supported by the German Research Foundation (PA 2365/1-1). Anita Pathil was further funded by the Olympia Morata Postdoctoral Fellowship of the Medical Faculty of University of Heidelberg.
Conflict of interest
The authors who have taken part in this study declare that they do not have anything to disclose regarding funding or conflict of interest with respect to this paper. For Wolfgang Stremmel, there is a patent application pending for UDCA-LPE as a drug for treatment of liver diseases (no industrial funding).
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12837
Figure S1 UDCA-LPE decreases ethanol-induced pSmad3 in hepatic stellate cells. (A) Representative Western blot analysis of pSmad3 and Smad3 in LX2 hepatic stellate cells cultured for 4 h with conditioned medium from CL48 liver cells exposed to control medium, ethanol 50 mM (Eth) or ethanol + 90 μM UDCA-LPE for 24 h. (B) Graphical presentation of relative protein levels of pSmad3 and pSmad3/Smad3 ratio as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3. ###P < 0.001 versus Con; ***P < 0.001 versus Eth.
Figure S2 Reduction of pSmad3 by UDCA-LPE was greated than that after UDCA, LPE or PC separately. (A) Representative Western blot analysis of pSmad3 and Smad3 in CL48 liver cells incubated with control medium, TGF-β1 (4 ng·mL−1), TGF-β1 + 90 μM UDCA, TGF-β1 + 90 μM LPE, TGF-β1 + 90 μM PC or TGF-β1 + 90 μM UDCA-LPE for 1 h. GAPDH was used as control for equal protein loading. (B) Graphical presentation of relative protein levels of pSmad3 and pSmad3/Smad3 ratio as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3. ###P < 0.001 versus Con; ***P < 0.001 versus TGF-β1.
Figure S3 Smurf2 protein levels are not changed by UDCA-LPE treatment. (A,B) Representative Western blot analysis of Smurf2 in (A) LX2 hepatic stellate cells and (B) CL48 liver cells incubated with control medium, TGF-β1 (4 ng·mL−1) or TGF-β1 + 90 μM UDCA-LPE for 1 h. GAPDH was used as control for equal protein loading. (C,D) Graphical presentation of relative Smurf2 protein levels in (C) CL48 cells and (D) LX2 cells as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3.
Figure S4 Reduction of pSmad3 in livers of MCD mice by UDCA-LPE. (A) Representative Western blot analysis of pSmad3 in liver tissue lysates of C57BL6 mice fed a MCD diet for 11 weeks followed by 30 mg·kg−1 UDCA-LPE 3× per week for the last 2.5 weeks on diet. GAPDH was used as control for equal protein loading. (B) Graphical presentation of relative protein levels of pSmad3 and pSmad3/Smad3 ratio as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3. ###P < 0.001 versus Con; ***P < 0.001 versus MCD.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003;83:655–663. doi: 10.1097/01.lab.0000069036.63405.5c. [DOI] [PubMed] [Google Scholar]
- Chamulitrat W, Burhenne J, Rehlen T, Pathil A, Stremmel W. Bile salt-phospholipid conjugate ursodeoxycholyl lysophosphatidylethanolamide as a hepatoprotective agent. Hepatology. 2009;50:143–154. doi: 10.1002/hep.22955. [DOI] [PubMed] [Google Scholar]
- Chamulitrat W, Liebisch G, Xu W, Gan-Schreier H, Pathil A, Schmitz G, et al. Ursodeoxycholyl lysophosphatidylethanolamide inhibits lipoapoptosis by shifting fatty acid pools toward monosaturated and polyunsaturated fatty acids in mouse hepatocytes. Mol Pharmacol. 2013;84:696–709. doi: 10.1124/mol.113.088039. [DOI] [PubMed] [Google Scholar]
- Chen YG. Endocytic regulation of TGF-beta signaling. Cell Res. 2009;19:58–70. doi: 10.1038/cr.2008.315. [DOI] [PubMed] [Google Scholar]
- Clark JM, Diehl AM. Nonalcoholic fatty liver disease: an underrecognized cause of cryptogenic cirrhosis. JAMA. 2003;289:3000–3004. doi: 10.1001/jama.289.22.3000. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- Dooley S, Delvoux B, Streckert M, Bonzel L, Stopa M, ten Dijke P, et al. Transforming growth factor beta signal transduction in hepatic stellate cells via Smad2/3 phosphorylation, a pathway that is abrogated during in vitro progression to myofibroblasts. TGFbeta signal transduction during transdifferentiation of hepatic stellate cells. FEBS Lett. 2001;502:4–10. doi: 10.1016/s0014-5793(01)02656-4. [DOI] [PubMed] [Google Scholar]
- Dooley S, Hamzavi J, Breitkopf K, Wiercinska E, Said HM, Lorenzen J, et al. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology. 2003;125:178–191. doi: 10.1016/s0016-5085(03)00666-8. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Mencarelli A, Palazzetti B, Del Soldato P, Morelli A, Ignarro LJ. An NO derivative of ursodeoxycholic acid protects against Fas-mediated liver injury by inhibiting caspase activity. Proc Natl Acad Sci U S A. 2001;98:2652–2657. doi: 10.1073/pnas.041603898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003;38:879–889. doi: 10.1053/jhep.2003.50384. [DOI] [PubMed] [Google Scholar]
- George J, Pera N, Phung N, Leclercq I, Yun Hou J, Farrell G. Lipid peroxidation, stellate cell activation and hepatic fibrogenesis in a rat model of chronic steatohepatitis. J Hepatol. 2003;39:756–764. doi: 10.1016/s0168-8278(03)00376-3. [DOI] [PubMed] [Google Scholar]
- Goto M, Okamoto Y, Yamamoto M, Aki H. Anti-inflammatory effects of 5-aminosalicylic acid conjugates with chenodeoxycholic acid and ursodeoxycholic acid on carrageenan-induced colitis in guinea-pigs. J Pharm Pharmacol. 2001;53:1711–1720. doi: 10.1211/0022357011778115. [DOI] [PubMed] [Google Scholar]
- Gressner OA, Lahme B, Rehbein K, Siluschek M, Weiskirchen R, Gressner AM. Pharmacological application of caffeine inhibits TGF-beta-stimulated connective tissue growth factor expression in hepatocytes via PPARgamma and SMAD2/3-dependent pathways. J Hepatol. 2008;49:758–767. doi: 10.1016/j.jhep.2008.03.029. [DOI] [PubMed] [Google Scholar]
- Inagaki Y, Mamura M, Kanamaru Y, Greenwel P, Nemoto T, Takehara K, et al. Constitutive phosphorylation and nuclear localization of Smad3 are correlated with increased collagen gene transcription in activated hepatic stellate cells. J Cell Physiol. 2001;187:117–123. doi: 10.1002/1097-4652(2001)9999:9999<00::AID-JCP1059>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. 2007;282:22089–22101. doi: 10.1074/jbc.M700998200. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R, Pan X, Malladi P, Wainwright MS, Whitington PF. Mitochondrial reactive oxygen species signal hepatocyte steatosis by regulating the phosphatidylinositol 3-kinase cell survival pathway. J Biol Chem. 2007;282:21327–21336. doi: 10.1074/jbc.M701759200. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Massague J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature. 1997;389:618–622. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J Cell Sci. 2006;119((Pt 23)):4803–4810. doi: 10.1242/jcs.03270. [DOI] [PubMed] [Google Scholar]
- Li F, Zeng B, Chai Y, Cai P, Fan C, Cheng T. The linker region of Smad2 mediates TGF-beta-dependent ERK2-induced collagen synthesis. Biochem Biophys Res Commun. 2009;386:289–293. doi: 10.1016/j.bbrc.2009.05.084. [DOI] [PubMed] [Google Scholar]
- Li J, Kim CI, Leo MA, Mak KM, Rojkind M, Lieber CS. Polyunsaturated lecithin prevents acetaldehyde-mediated hepatic collagen accumulation by stimulating collagenase activity in cultured lipocytes. Hepatology. 1992;15:373–381. doi: 10.1002/hep.1840150303. [DOI] [PubMed] [Google Scholar]
- Lieber CS, Robins SJ, Li J, DeCarli LM, Mak KM, Fasulo JM, et al. Phosphatidylcholine protects against fibrosis and cirrhosis in the baboon. Gastroenterology. 1994;106:152–159. doi: 10.1016/s0016-5085(94)95023-7. [DOI] [PubMed] [Google Scholar]
- Lo RS, Massague J. Ubiquitin-dependent degradation of TGF-beta-activated smad2. Nat Cell Biol. 1999;1:472–478. doi: 10.1038/70258. [DOI] [PubMed] [Google Scholar]
- Ma X, Zhao J, Lieber CS. Polyenylphosphatidylcholine attenuates non-alcoholic hepatic fibrosis and accelerates its regression. J Hepatol. 1996;24:604–613. doi: 10.1016/s0168-8278(96)80147-4. [DOI] [PubMed] [Google Scholar]
- Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology. 2008;134:1641–1654. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S, Matsuzaki K, Yoshida K, Furukawa F, Tahashi Y, Yamagata H, et al. TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene. 2004;23:7416–7429. doi: 10.1038/sj.onc.1207981. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114((Pt 24)):4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- Oneta CM, Mak KM, Lieber CS. Dilinoleoylphosphatidylcholine selectively modulates lipopolysaccharide-induced Kupffer cell activation. J Lab Clin Med. 1999;134:466–470. doi: 10.1016/s0022-2143(99)90167-1. [DOI] [PubMed] [Google Scholar]
- Pathil A, Mueller J, Warth A, Chamulitrat W, Stremmel W. Ursodeoxycholyl lysophosphatidylethanolamide improves steatosis and inflammation in murine models of nonalcoholic fatty liver disease. Hepatology. 2012;55:1369–1378. doi: 10.1002/hep.25531. [DOI] [PubMed] [Google Scholar]
- Pathil A, Warth A, Chamulitrat W, Stremmel W. The synthetic bile acid–phospholipid conjugate ursodeoxycholyl lysophosphatidylethanolamide suppresses TNFalpha-induced liver injury. J Hepatol. 2011;54:674–684. doi: 10.1016/j.jhep.2010.07.028. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reunanen N, Foschi M, Han J, Kahari VM. Activation of extracellular signal-regulated kinase 1/2 inhibits type I collagen expression by human skin fibroblasts. J Biol Chem. 2000;275:34634–34639. doi: 10.1074/jbc.C000175200. [DOI] [PubMed] [Google Scholar]
- Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res. 2008;49:1068–1076. doi: 10.1194/jlr.M800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota G, Knockaert M, Alarcon C, Montalvo E, Brivanlou AH, Massague J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J Biol Chem. 2006;281:40412–40419. doi: 10.1074/jbc.M610172200. [DOI] [PubMed] [Google Scholar]
- Schnabl B, Kweon YO, Frederick JP, Wang XF, Rippe RA, Brenner DA. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology. 2001;34:89–100. doi: 10.1053/jhep.2001.25349. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Treede I, Braun A, Sparla R, Kuhnel M, Giese T, Turner JR, et al. Anti-inflammatory effects of phosphatidylcholine. J Biol Chem. 2007;282:27155–27164. doi: 10.1074/jbc.M704408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- Wrighton KH, Lin X, Feng XH. Phospho-control of TGF-beta superfamily signaling. Cell Res. 2009;19:8–20. doi: 10.1038/cr.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, Blaner WS, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R. Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci U S A. 2001;98:974–979. doi: 10.1073/pnas.98.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 UDCA-LPE decreases ethanol-induced pSmad3 in hepatic stellate cells. (A) Representative Western blot analysis of pSmad3 and Smad3 in LX2 hepatic stellate cells cultured for 4 h with conditioned medium from CL48 liver cells exposed to control medium, ethanol 50 mM (Eth) or ethanol + 90 μM UDCA-LPE for 24 h. (B) Graphical presentation of relative protein levels of pSmad3 and pSmad3/Smad3 ratio as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3. ###P < 0.001 versus Con; ***P < 0.001 versus Eth.
Figure S2 Reduction of pSmad3 by UDCA-LPE was greated than that after UDCA, LPE or PC separately. (A) Representative Western blot analysis of pSmad3 and Smad3 in CL48 liver cells incubated with control medium, TGF-β1 (4 ng·mL−1), TGF-β1 + 90 μM UDCA, TGF-β1 + 90 μM LPE, TGF-β1 + 90 μM PC or TGF-β1 + 90 μM UDCA-LPE for 1 h. GAPDH was used as control for equal protein loading. (B) Graphical presentation of relative protein levels of pSmad3 and pSmad3/Smad3 ratio as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3. ###P < 0.001 versus Con; ***P < 0.001 versus TGF-β1.
Figure S3 Smurf2 protein levels are not changed by UDCA-LPE treatment. (A,B) Representative Western blot analysis of Smurf2 in (A) LX2 hepatic stellate cells and (B) CL48 liver cells incubated with control medium, TGF-β1 (4 ng·mL−1) or TGF-β1 + 90 μM UDCA-LPE for 1 h. GAPDH was used as control for equal protein loading. (C,D) Graphical presentation of relative Smurf2 protein levels in (C) CL48 cells and (D) LX2 cells as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3.
Figure S4 Reduction of pSmad3 in livers of MCD mice by UDCA-LPE. (A) Representative Western blot analysis of pSmad3 in liver tissue lysates of C57BL6 mice fed a MCD diet for 11 weeks followed by 30 mg·kg−1 UDCA-LPE 3× per week for the last 2.5 weeks on diet. GAPDH was used as control for equal protein loading. (B) Graphical presentation of relative protein levels of pSmad3 and pSmad3/Smad3 ratio as estimated by densitometric analysis, values were relative to GAPDH and normalized to untreated controls, N = 3. ###P < 0.001 versus Con; ***P < 0.001 versus MCD.