

Abstract
Background and Purpose
Investigators have suggested that the chemokine receptor CCR1 plays a role in multiple myeloma. Studies using antisense and neutralizing antibodies to CCR1 showed that down-regulation of the receptor altered disease progression in a mouse model. More recently, experiments utilizing scid mice injected with human myeloma cells demonstrated that the CCR1 antagonist BX471 reduced osteolytic lesions, while the CCR1 antagonist MLN-3897 prevented myeloma cell adhesion to osteoclasts. However, information is limited regarding the pharmacology of CCR1 antagonists in myeloma cells.
Experimental Approach
We compared several well-studied CCR1 antagonists including AZD4818, BX471, CCX354, CP-481715, MLN-3897 and PS899877 for their ability to inhibit binding of [125I]-CCL3 in vitro using membranes prepared from RPMI 8226 cells, a human multiple myeloma cell line that endogenously expresses CCR1. In addition, antagonists were assessed for their ability to modulate CCL3-mediated internalization of CCR1 and CCL3-mediated cell migration using RPMI 8226 cells. As many GPCRs signal through β–arrestin-dependent pathways that are separate and distinct from those driven by G-proteins, we also evaluated the compounds for their ability to alter β-arrestin translocation.
Key Results
There were clear differences between the CCR1 antagonists in their ability to inhibit CCL3 binding to myeloma cells, as well as in their ability to inhibit G–protein-dependent and -independent functional responses.
Conclusions and Implications
Our studies demonstrate that tissue phenotype seems to be relevant with regards to CCR1. Moreover, it appears that for CCR1 antagonists, inhibition of β-arrestin translocation is not necessarily linked to chemotaxis or receptor internalization.
Table of Links
| TARGETS | LIGANDS |
|---|---|
| CCR1 | BX471 |
| CCR4 | CCL3 |
| CCR5 | CCL15 |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Since it was first cloned in 1993 (Neote et al., 1993), the chemokine receptor CCR1 has been the target of intensive research. CCR1 is endogenously expressed on a broad range of immunological cell types, including monocytes, lymphocytes, basophils, eosinophils, neutrophils and mast cells. As a result, CCR1 has been associated with diverse pathologies related to the immune response, and identifying antagonists for CCR1 has been an active area of research for many pharmaceutical companies. Further information on chemokine receptors including CCR1 can be found at the International Union of Basic and Clinical Pharmacology (IUPHAR)/BPS website (http://www.guidetopharmacology.org/). Scientists from Berlex (now Bayer HealthCare Pharmaceuticals) provided the first small molecule antagonist for CCR1 in 1998 (Hesselgesser et al., 1998), and since then, numerous inhibitors have been identified (Gladue et al., 2003; Vallet et al., 2007; Merritt et al., 2009; 2010; Kerstjens et al., 2010; Dairaghi et al., 2011; 2012; Gardner et al., 2013; Pennell et al., 2013; Hossain et al., 2014). To date, six compounds targeting CCR1 have undergone clinical trials (Table 1) for multiple sclerosis, rheumatoid arthritis and chronic obstructive pulmonary disease (Gladue et al., 2010; Karash and Gilchrist, 2011). While no CCR1 antagonists have received approval from the US Food and Drug Administration, two compounds, namely Bristol Myers Squibb's BMS817399 and Chemocentrix's CCX354, are currently in phase II and phase III trials respectively.
Table 1.
Structures of the CCR1 antagonists utilized in these studies
| Compound | Structure | Reported affinity (Ki) | References |
|---|---|---|---|
| AZD4818 |
*
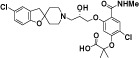
|
Not reported | Gladue et al., 2010 |
| CP-481715 | 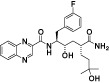 |
CCL3 IC50 = 74 Nm; used HEK_CCR1; fluorescently labelled CCL3 IC50 = 9.1 nM | Gladue et al., 2003; Vallet et al., 2007 |
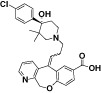
| BX-471 | 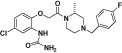 |
CCL3 IC50 = 1.0 nM; used HEK_CCR1 | Anders et al., 2002; Horuk, 2005) |
| MLN-3897 |
*
|
CCL3 IC50 = 2.3 nM; used THP-1 membranes | Carson and Harriman, 2004 |
| CCX354 |
*
|
CCL15IC50 = 1.5 nM; used human monocytes | Dairaghi et al., 2011 |
| PS899877 |  |
CCL3 IC50 = 69 nM; used HEK_CCR1 | Merritt et al., 2010 |
With the exception of CP481715, the compounds were synthesized by Dr Merritt at Kean University based on the referenced publication. CP481715 was provided by Pfizer.
Denotes a putative structure based on evaluation of patent applications.
There has been some speculation that CCR1 may play a role in the progression of multiple myeloma (MM) (Karash and Gilchrist, 2011; Vallet and Anderson, 2011). MM is the second most common adult haematological malignancy in the USA with approximately 20 000 new cases and 11 000 deaths annually and accounts for approximately 10% of all haematological cancers (Raab et al., 2009). The primary symptoms for MM include bone pain and osteolytic lesions that result from the interaction of malignant plasma cells with bone marrow, resulting in an imbalance between osteoclasts, responsible for bone resorption, and osteoblasts, responsible for bone deposition (Sezer, 2009). Early work by Lentzsch et al. (2003) demonstrated the in vitro chemotactic effect of CCL3 on MM cells. Studies have shown that CCL3 (previously known as macrophage inflammatory protein-1α; MIP-1α), an endogenous ligand for CCR1, is secreted at high concentrations by MM cell lines as well as patient-derived MM cells, and levels of CCL3 are elevated in the bone marrow plasma of most patients with active myeloma (Choi et al., 2000). DNA array studies show CCL3 mRNA is presently at much higher levels in the bone marrow and marrow supernatants from patients with myeloma bone disease than patients with other haematological malignancies or normal control individuals (Magrangeas et al., 2003). Furthermore, levels of CCL3 correlate with the extent of bone disease (Roussou et al., 2009). Studies with CCR1-neutralizing antibodies indicate that the receptor enhances adhesion of myeloma cells to bone marrow stromal cells (Oba et al., 2005). The CCR1 antagonists, BX471 (Oba et al., 2005) and MLN-3897 (Vallet et al., 2007) impair CCL3-mediated osteoclast formation. This is of particular importance given that osteoclast-mediated bone destruction is a frequent complication of MM. BX471 has also been shown to inhibit proliferation of MM cell lines (Lentzsch et al., 2005), and experiments utilizing scid mice injected with human myeloma cells found end-term treatment with BX471 resulted in a significant reduction (40%) of osteolytic lesions (Menu et al., 2006).
We focus our work on six CCR1 antagonists that were initially identified using competitive binding assays with CCL3 or CCL15 and HEK cells overexpressing recombinant human CCR1 (Table 1). Previous work on these allosteric inhibitors includes (i) measurement of calcium transients; (ii) chemotaxis of leukocytes; and (iii) leukocyte up-regulation of CD11b (Gladue et al., 2003; Pease and Horuk, 2009; Merritt et al., 2010). However, there have been limited studies with these CCR1 antagonists using assays that are more relevant to MM (Karash and Gilchrist, 2011). Given the differences between natural and recombinant GPCR systems (Kenakin, 1996), we performed competitive binding assays with membranes prepared from RPMI 8226 and compared the results with those obtained with HEK293-EM4 cells stably transfected with a chimeric G-protein that links Gi-coupled receptors to Gq (An et al., 1999) and human CCR1 (HEK_CCR1). We then initiated studies on CCL3-mediated chemotaxis and CCR1 receptor internalization using the myeloma cell line RPMI 8226. Finally, we examined the ability of the CCR1 antagonists to inhibit CCL3-mediated β-arrestin translocation. When originally identified, translocation of β-arrestin proteins was thought to limit GPCR signalling by physically interceding between receptors and G-proteins and inducing receptor internalization. More recently, researchers have demonstrated that β-arrestin signalling can also play an important role in chemotaxis for some chemokine receptors (Sun et al., 2002). Thus, we felt it was critical to assess the effects of CCR1 antagonists on β-arrestin translocation. Together, our experimental results indicate there are clear differences between the CCR1 antagonists. Variances were observed in their ability to inhibit CCL3 binding, as well as CCL3-mediated functional responses such as receptor internalization, cell migration and β-arrestin translocation.
Methods
Chemokines
Recombinant carrier free hCCL3 used for all experiments was purchased from R&D Systems (catalogue number: 270-LD-050; Minneapolis, MN, USA) and [125I]-CCL3 was obtained from PerkinElmer (product number: NEX298005UC; Specific Activity of 2200Ci (81.4TBq)/mmol; Waltham, MA, USA). Recombinant hCCL3 arrived as a dry powder and was reconstituted as a 100 μM stock solution in PBS, distributed into small aliquots, and stored at −20°C, while the [125I]-CCL3 was made into an 11 nM stock solution with sterile water according to the manufacturer's instructions.
CCR1 antagonists
With the exception of CP481715, all compounds were synthesized by Dr. Merritt at Kean University. CP481715 was provided by Pfizer (Groton, CT, USA). The chemical structures of AZD-4818, MLN-3897 and CCX354 have not been disclosed. The putative structure shown in Table 1 for AZD-4818 was one of two possibilities suggested (Norman, 2009) based on an evaluation of patent applications from AstraZeneca. While it is not known that this compound is AZD-4818, it is clearly an advanced analogue based on patent applications (Hansson et al., 2008; Cooper et al., 2009), and it may be a dual antagonist for both CCR1 and CCR3 (Hemmerling et al., 2009). The putative structure shown in Table 1 for CCX354 was a potent compound identified in a patent application (Pennell et al., 2010), which occurred with high frequency throughout the application. The putative structure for MLN-3897 shown in Table 1 is based on patents and presentations by Millennium (a wholly owned subsidiary of Takeda Pharmaceutical Company), and this compound, with only a few closely related analogues, includes procedures for large scale synthesis (Carson and Harriman, 2004). All compounds were made up as 10 mM stocks in DMSO (Sigma-Aldrich, St. Louis, MO, USA) and distributed into aliquots and stored at −20 or −80°C until needed. Serial dilutions were made immediately before the assay in which they were used.
Cell lines
The MM cell lines RPMI 8226 (catalogue number: CCL-155) and U266 (catalogue number: TIB-196) were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were maintained in RPMI 1640 supplemented with 10% heat-inactivated FBS (Atlantic Biologicals, Miami, FL, USA), 100 U mL−1 penicillin (PEN; Life Technologies, Grand Island, NY, USA), and 100 μg mL−1 streptomycin (STREP; Life Technologies) in an atmosphere of 5% CO2 at 37°C. In agreement with others (Trentin et al., 2007; Badr et al., 2011), we found that most RPMI 8226 cells endogenously express both CCR1 and CCR5 when assessed by flow cytometry (Supporting Information). This was in sharp contrast from U266 cells, which had a population of cells with either CCR1 or CCR5, but few cells containing both receptors (data not shown). A stably transfected cell line HEK293-EM4 (Robbins and Horlick, 1998) expressing hCCR1 and Gαqi5 was maintained in DMEM with 10% FBS and PEN/STREP. For β-arrestin translocation assays, we utilized PathHunter™ hCCR1_CHO cells (DiscoveRx Corporation, Fremont, CA, USA). The hCCR1_CHO cells were maintained in DMEM/F12 medium supplemented with 10% FBS, 1X PEN/STREP, 300 μg mL−1 hygromycin B (Life Technologies), and 800 μg mL−1 geneticin (Life Technologies) in an atmosphere of 5% CO2 at 37°C.
Binding assay
Membranes were prepared from HEK_CCR1 or RPMI 8226 cells as previously described (Gilchrist et al., 1998) and stored in aliquots at −80°C until needed. For the competition assays, membranes were suspended at 10 μg mL−1 in HEM buffer comprised of 50 mM HEPES, pH 7.5, 1 mM EDTA and 5 mM MgCl2. Compounds were serially diluted in HEM buffer to a 10× concentration and then added to the membrane mixture. A final concentration of 2 pM [125I]-CCL3 was added and the tubes were incubated at 37°C, shaking, for 2 h. Initial experiments were performed to determine that equilibrium had been reached at the 2 h time point (data not shown). The bound and free radioligands were separated by filtration through Whatman GF/C filter paper (Brandel, Gaithersburg, MD, USA) soaked in TEM2 buffer comprised of 20 mm Tris-HCl, pH 7.4, 0.5 mM EDTA and 5 mM MgCl2 supplemented with 0.3% polyethyleneimine (Sigma-Aldrich) and 20 mg·mL−1 BSA (EMD Millipore, Billerica, MA, USA) using a tissue harvester (Brandel). Filters were washed twice with ice-cold TEM2 buffer and then counted (1 min per sample) using a Packard Gamma Counter (Perkin-Elmer, Waltham, MA, USA). Binding assays were performed in duplicate, and non-specific binding was determined by adding cold CCL3 (100 nM) at the same time as the radioligand to some samples. Data from binding experiments were analysed by non-linear regression analysis to determine IC50 values (GraphPad Prism version 6.0, GraphPad Software Inc. San Diego, CA). For saturation experiments, we used 10 μg mL−1 of membrane prepared from HEK_CCR1 or RPMI 8226 cells and increasing concentrations of [125I]-CCL3 (0–100 pM) with and without cold CCL3 (100 nM) to define the non-specific binding. Binding experiments were carried out at 37°C for 2 h and the KD and Bmax values were determined using GraphPad Prism version 6.0. Experiments were conducted in duplicate and repeated as indicated in Table 2.
Table 2.
Curve-fitting parameters for [125I]-CCL3 competition binding assays
| AZD4818 | BX471 | CCX354 | CP481715 | MLN-3897 | PS899877 | |
|---|---|---|---|---|---|---|
| pIC50 for HEK_CCR1/Gqi5 membranes | 8.629 ± 0.201 n = 2 | 7.74 ± 0.425 n = 3 | 8.88 ± 0.198 n = 1 | 7.971 ± 0.205 n = 3 | 9.933 ± 0.2217 n = 1 | 6.985 ± 0.2818 n = 1 |
| pIC50 for RPMI 8226 membranes | 6.797 ± 0.3827 n = 3 | 7.518 ± 0.3415 n = 4 | 6.507 ± 0.1991 n = 1 | 7.162 ± 0.2005 n = 6 | 8.839 ± 0.5723 n = 4 | 6.948 ± 0.232 n = 3 |
The table summarizes the pIC50 values of the six CCR1 antagonists analysed using membranes prepared from either HEK_hCCR1 cells or the MM cell line RPMI 8226.
Receptor internalization assay
Receptor internalization experiments were performed using flow cytometry. Staining for surface CCR1 and CCR5 was performed as recommended by the manufacturer of the PE-conjugated anti-CCR1 (Clone: 53504; R&D Systems) and FITC conjugated anti-CCR5 (clone: HEK/1/85a; BioLegend, San Diego, CA, USA). Briefly, 5 × 106 RPMI 8226 cells were suspended in RPMI 1640 media with 1% FBS and incubated with various concentrations of antagonist. After 15 min, the cells were activated with CCL3 (1 nM) and incubated for 2 h at 37°C. After being washed with RPMI 1640 media with 1% FBS, cells were suspended in PBS with 1% FBS and 10 μL of each mAb was added. Thereafter, cells were incubated for 30 min in the dark at 4°C. Following two washes with PBS with 1% FBS, samples were analysed on a BD FACScalibur flow cytometer using Cell Quest software (Becton Dickinson, Franklin Lakes, NJ, USA). At least 10 000 events were acquired. Healthy populations were identified and gated on FITC versus PE plots. For the cell surface expression figure, the percentage of cells in the upper left quadrant (high CCR1/low CCR5) without CCL3 exposure was set to 1.0 and the fold change in fluorescence of the 1 nM CCL3 only (control), and 1 nM CCL3 with increasing concentrations of CCR1 antagonist are shown. To calculate the IC50 values (Table 3), the samples with 1 nM CCL3 (no antagonist) were set to 1.0, and a non-linear regression analysis was run using GraphPad Prism version 6.0. Experiments were repeated as indicated in Table 3.
Table 3.
Curve-fitting parameters for CCL3-mediated receptor internalization, chemotaxis and β-arrestin translocation
| AZD4818 | BX471 | CCX354 | CP481715 | MLN-3897 | PS899877 | |
|---|---|---|---|---|---|---|
| pIC50 for CCL3-mediated CCR1 internalizationa | 6.91 ± 0.24 n = 4 | 6.87 ± 1.34 n = 2 | 6.08 ± 2.39 n = 2 | NC | 6.07 ± 0.55 n = 3 | 6.68 ± 0.20 n = 2 |
| pIC50 for CCL3-mediated chemotaxisb | 8.96 ± 0.71 n = 3 | 8.49 ± 0.36 n = 4 | 9.06 ± 0.51 n = 4 | 7.56 ± 0.14 n = 4 | 9.2 ± 0.10 n = 3 | 8.32 ± 0.32 n = 3 |
| pkB for β-arrestin translocationc | 8.58 ± 0.59 n = 4 | 9.15 ± 0.51 n = 7 | 6.93 ± 0.04 n = 2 | 8.26 ± 0.98 n = 4 | 7.74 ± 0.44 n = 8 | 8.86 ± 0.21 n = 2 |
The table summarizes the curve parameters of the six CCR1 antagonists analysed. For CCR1 internalization and chemotaxis, the results provided are the pIC50 mean ± SE for the noted number of independent experiments. In the case of CCR1 internalization ∼10 0000 events were examined for each experiment. For chemotaxis each point was performed in quadruplicate and the number of cells that migrated spontaneously to the chemotaxis buffer was subtracted. The chemotactic index was then determined by dividing the number of migrated cells in the presence of the antagonist by the number of cells that migrated spontaneously to CCL3. Non-linear regression analysis was performed using GraphPad Prism (version 6.0). For β-arrestin translocation the results provided are the mean equilibrium dissociation constant (pkB) ± SEM (Christopoulos and Kenakin, 2002) for the noted number of independent experiments, each being performed in triplicate. NC indicates a curve could not be generated.
Values obtained with baseline constrained to 1.0 and maximum set to be greater than 2.0.
Values obtained with baseline constrained to 1.0.
Values obtained with no constraints on the baseline or maxima of concentration–response curves.
Chemotaxis assay
MultiScreen®-Migration Invasion and Chemotaxis filter plates (EMD Millipore) with 8 μm pore size were used for all chemotaxis experiments. We utilized the fluorescent dye Calcein AM to label RMPI 8226 cells. The cells were washed once with HBSS supplemented with 10 mM HEPES (HHBSS) and suspended at 4 × 106 cells mL−1. Calcein AM (2 μM) was added, and the cells were incubated for 30 min at 37°C. Cells were then washed twice with HHBSS before being suspended in HHBSS at 2 × 106 cells mL−1. Labelled cells (50 μL) were added to the top chambers along with antagonists (or vehicle controls). The bottom chambers contained 150 μL control media (HHBSS) or chemoattractant (1 nM CCL3 in control media). The plate was placed in the 37°C incubator for 3 h. After 3 h, 50 μL of the cell-containing media were removed from the bottom chamber and transferred to a 96-well black plate with a clear bottom. This plate was read using EX490/EM520 on a DTX800 multimode plate reader. For chemotaxis, each point was performed in quadruplicate and the number of cells that migrated spontaneously to the chemotaxis buffer was subtracted. The chemotactic index was determined by dividing the number of migrated cells in the presence of the antagonist by the number of cells that migrated spontaneously to CCL3. Experiments were performed in quadruplicate and IC50 values calculated using non-linear regression analysis (GraphPad Prism version 6.0). For Table 3, the chemotactic index was averaged for triplicate experiments with each compound.
PathHunter β-arrestin translocation assay
To quantitatively assess β-arrestin translocation, we utilized an enzyme fragment complementation format from DiscoveRx Corporation (Fremont, CA, USA) in which CCR1 is fused to a ProLink™ peptide derived from β-galactosidase, and β-arrestin 2 is fused to an N-terminal deletion mutant of β-galactosidase [enzyme acceptor (EA) ]. Following addition of CCL3, the β-arrestin–EA fusion protein binds activated CCR1-ProLink. PathHunter hCCR1_CHO cells (DiscoveRx Corporation) were plated in 96-well half volume white plates at 1 × 104 cells per well in Optimem (Life Technologies) and allowed to attach overnight. Initial experiments established an EC50 of 200 pM for CCL3. Serial dilutions of the antagonists were made in Optimem and added to triplicate wells. The plates were incubated at 37°C with 5% CO2 for 90 min and then increasing concentrations of CCL3 were added and the plates incubated at 37°C with 5% CO2 for an additional 60 min. Detection reagent was added and the plate incubated for 60 min at room temperature. The plates were read using the luminescent setting on a DTX800 multimode plate reader. The pKB values were determined using the Gaddum/Schild equation on GraphPad Prism version 6.0. Experiments were conducted in triplicate and repeated as indicated in Table 3.
Statistics
The number of experiments for each assay is stated as n in the tables. Unless stated otherwise, data in the figures are expressed as mean ± SEM, as determined by GraphPad Prism software analysis version 6.0. Values of P < 0.05 were accepted as significant and were obtained using Student's t-test.
Results
Binding studies
Saturation experiments using [125I]-CCL3 were performed to determine the binding parameters (KD and Bmax) for HEK_CCR1Gqi5 and RPMI 8226 cell membranes. For RPMI 8226 cells, we determined a KD-value of 3 nM (Log KD −8.6 ± 1.65, n = 2) and a Bmax value of 2.47 ± 0.57 fmol·mg−1. Similar experiments using membranes prepared from HEK_CCR1Gqi5 showed a KD-value of 10 nM (log KD −8.01 ± 2.37, n = 2) and a Bmax value of 2.81 ± 0.26 fmol mg−1. In [125I]-CCL3 competition binding assays, CCL3 was found to have an affinity similar to that previously reported (IC50 = 1.5 nM for HEK_CCR1Gqi5; Tsou et al., 1998). We also examined another MM cell line, U266 and found while the affinity was similar to that of RPMI 8226 cells (2.38 nM), the Bmax was considerably reduced (0.4612 ± 0.120 fmol mg−1; data not shown). This is in agreement with microarray data that indicate CCR1 expression is lower in U266 than in RPMI 8226 cells (GSE6205; Lombardi et al., 2007).
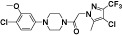
Using membranes from HEK_CCR1Gqi5 cells, we confirmed that AZD-4818, BX471, CCX354, CP481715 and PS899877 are all potent inhibitors of [125I]-CCL3 binding (Figure 1; Table 2). In the cases of BX471, CP481715 and PS899877, our findings are similar to those previously reported with HEK cells (Anders et al., 2002; Vallet et al., 2007; Merritt et al., 2010). We then examined the compounds using membranes from RPMI 8226 cells ( Figure 2; Table 2;). While two compounds were equally potent with membranes from either cell line (BX471, PS899877), the others had a bias for the recombinant HEK_CCR1 system. In the case of AZD4818, CCX354 and CP481715, the affinity is significantly better with membranes from HEK_CCR1 cells than those from RPMI 8226 cells when tested using an unpaired t-test with Welch's correction (P < 0.05). Furthermore, there was a drastic shift in the rank order of potency between the membranes tested. With HEK_CCR1 membranes we found MLN3879 > CCX354 ≥ AZD4818 > CP481715 = BX471 > PS899877 while with membranes from RPMI 8226 cells we found MLN3879 > BX471 > CP481715 ≥ PS899877 > AZD4818 > CCX354.
Figure 1.
Representative competitive binding results of [125I]-CCL3 with CCR1 antagonists. Membranes prepared from RPMI 8226 cells, which endogenously express CCR1 or HEK_CCR1Gqi5 were analysed for their binding to 2 pM [125I]-CCL3 in the presence of increasing concentrations of (A) AXD4818, (B) BX471, (C) CCX354, (D) CP481715, (E) MLN−3897 or (F) PS899877. Non-specific binding was determined in the presence of 100 nM CCL3 and binding shown represents the mean specific binding of replicate samples. Results were normalized such that cpm when no compound was present was set to 1.0 and data were fit using non-linear regression analysis (see Table 2 for summary pIC50 values).
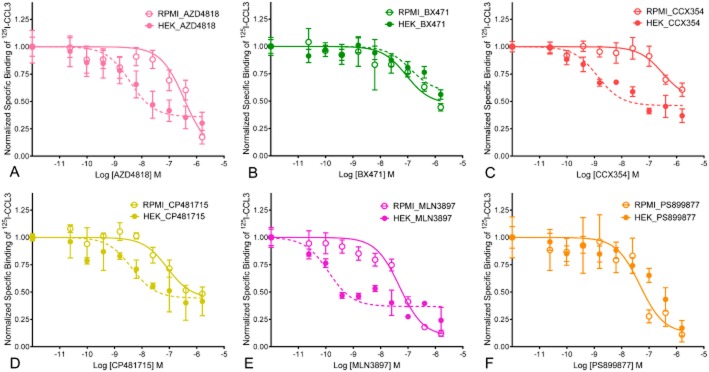
Figure 2.
CCL3 induces CCR1 internalization in MM cells that can be modulated with CCR1 antagonists. Membrane expression of CCR1 in RPMI 8226 cells was determined by flow cytometry analysis using a CCR1-specific mAb, examining ∼100 000 events for each experiment. Results from a single representative experiment for each compound are shown (see Table 3 for summary pIC50 values). Results with and without 1 nM CCL3 show the decrease in CCR1 surface expression after 2 h with the ligand.
CCL3-mediated CCR1 internalization
As noted previously (Trentin et al., 2007; Badr et al., 2011), and confirmed through our binding studies, RPMI 8226 cells endogenously express relatively high levels of CCR1. When treated with CCL3, the receptor is internalized in both a time-dependent (Supporting Information) and dose-dependent (data not shown) fashion. This internalization can be measured using FITC-labelled anti-hCCR1 antibody and flow cytometry to assess the surface expression of the receptor. CCR1 antagonists were tested for their ability to inhibit CCL3-mediated CCR1 internalization using 1 nM CCL3. A 2 h time point was used for all studies. In the presence of CCL3 we observed a decrease in the percentage of cells with surface expression of CCR1 (from 100.0 to 44.6 % ± 2.74, n = 34). Our results indicate that there may be some cross regulation between CCR1 and CCR5 as we found that while CCR1 levels went down with exposure to CCL3 those of CCR5 went up (data not shown). Incubation of cells with AZD-4818, BX471, CCX354, MLN-3897 or PS899877 reduced CCL3-mediated receptor internalization and led to a dose-dependent recovery of surface CCR1 (Table 3; Figure 2) although they all required higher concentrations than what was needed to block binding of 125I-CCL3. In contrast, CP481715 was unable to block CCL3-mediated receptor internalization at any concentration tested.
CCL3-mediated chemotaxis
We then examined the CCR1 antagonists for their ability to inhibit chemotaxis of RPMI 8226 cells in response to CCL3 and found that all compounds inhibited CCL3-mediated chemotaxis of RPMI 8226 cells (Table 3; Figure 3). This result is perhaps not surprising given that most of the compounds had been shown to inhibit cell migration of the monocytic cell line THP-1. However, there was a difference in the rank order (MLN3879 ≥ CCX354 ≥ AZD4818 > BX471 > PS899877 > CP481715) when compared with the ability to block [125I]-CCL3 binding to RMPI membranes. Taken together with the receptor internalization data, the results indicate the compounds have clear differences in their abilities to serve as functional antagonists for CCR1. Some residual chemotaxis is evident, but it is important to point out that RPMI 8226 cells also express CCR5, which is known to respond to CCL3.
Figure 3.

Inhibition of CCL3-mediated chemotaxis of MM cells. RPMI 8226 cells were challenged with CCL3 in a transwell chemotaxis chamber system. CCL3 evoked a concentration-dependent chemotaxis of RPMI 8226 cells. CCR1 antagonists, incubated with the RPMI 8226 cells in the top chambers inhibited chemotaxis to 1 nM CCL3 present in the lower chambers. Data shown are the mean for a single representative experiment performed in quadruplicate (see Table 3 for summary of pIC50 values).
CCL3-mediated β-arrestin translocation
When originally identified, translocation of β-arrestin proteins was thought to play a role in limiting GPCR signalling by (i) physically interceding between the receptor and the G-protein; and (ii) by inducing receptor internalization. More recently, researchers have demonstrated that for a variety of GPCRs, β-arrestin proteins also mediate G–protein-independent signalling, including activation of small GTP-binding proteins and members of the MAPK cascade (DeWire et al., 2007). Notably, β-arrestin has been shown to play an important role in cell migration for some chemokine receptors. In an effort to determine if there were differences between the CCR1 antagonists in their ability to inhibit CCL3-mediated G–protein-independent signalling, we examined their effects using a β-arrestin 2 translocation assay. For this work, we utilized the DiscoveRx Corporation PathHunter assay, which employs enzyme fragment complementation and required that we move to a recombinant system, specifically, an hCCR1_CHO cell line stably transfected with β-arrestin 2–EA and CCR1-ProLink fusion proteins. As has been shown recently by others (Rajagopal et al., 2013), we found there was a dose-dependent increase in β-arrestin translocation in response to CCL3. However, we should note their work utilized a cell line expressing β-arrestin 1.
Cells were pre-exposed to the CCR1 antagonists at 0, 10 or 100 nM. After 90 min, increasing concentrations of CCL3 were added. To quantify the antagonists' potency, pKB values were calculated using the Gaddum/Schild allosteric EC50 shift (GraphPad Prism; Kenakin et al., 2006). Our results (Figure 4; Table 3) demonstrate that the effects of the compounds were varied, with some compounds resulting in a rightward shift of the curve, with no effect on the Emax (PS899877, CP481715), while other compounds showed both a shift to the right as well as a decrease in the maximal response (AZD4818, BX471, MLN-3897). Perhaps most surprising was the complete lack of effect on β-arrestin by CCX354 (Figure 4). Thus, while most of the CCR1 antagonists inhibited CCL3-mediated β-arrestin translocation in a dose-dependent manner, CCX354 had no effect at the concentrations tested.
Figure 4.

Effects of CCR1 antagonists on CCL3-mediated β-arrestin translocation. PathHunter hCCR1_CHO cells (DiscoveRx Corporation) were stimulated with the indicated concentrations of CCL3. Cells were pre-incubated with 0, 1, 10 or 100 nM CCR1 antagonists, and after 90 min increasing concentrations of CCL3 were added. The resulting β-arrestin translocation measurements are given as normalized response. Data shown are the mean from a representative experiment for each antagonist performed in triplicate. For statistical analysis, see Table 3. (A) AZD4818; (B) BX471; (C) MLN−3897; (D) CCX354; (E) CP481715; and (F) PS899877.
Discussion and conclusions
The recent emergence of CCR1 as a potential target for MM coupled with the lack of published information on the molecular mechanisms of action for most CCR1 antagonists has highlighted the need for information to better define their pharmacology in myeloma cells. We examined six compounds, five of which entered into clinical trials (Karash and Gilchrist, 2011), for their ability to inhibit binding of the radiolabelled ligand [125I]-CCL3 to membranes prepared from an MM cell line (RPMI 8226) that endogenously expresses CCR1. While BX-471 (Vaidehi et al., 2006) and CP481715 (Allegretti et al., 2008) are often regarded as allosteric modulators, no study on how any of the compounds tested alter the dissociation rate of an orthosteric ligand has been published. These same six compounds were then compared for their ability to alter CCL3-mediated receptor internalization and chemotaxis using RPMI 8226 cells. Finally, we examined the effects of the compounds on CCL3-mediated β-arrestin translocation using the PathHunter CHO cell line stably transfected with β-arrestin–EA and CCR1-ProLink fusion proteins.
Results from our experiments indicate membranes from myeloma cells had a different rank order of potency for the six antagonists than membranes from HEK_CCR1 cells. Interestingly, BX471 and PS899877 showed similar potency of [125I]-CCL3 binding with both cell types while AZD4818, CCX354, CP481715 and MLN-3897 were better inhibitors when HEK_CCR1 membranes were tested. Similar cell lineage-dependent effects have been noted for other chemokine allosteric inhibitors (Allegretti et al., 2008), highlighting the need to carefully choose the cells to be screened during drug development. Our competition binding studies using the myeloma cell line, which express both CCR1 and CCR5, also suggest that CCX354 may not be completely selective for CCR1.
CCL3 induces a chemotactic response of RPMI 8226 myeloma cells that was inhibited in a dose-dependent manner by all six of the CCR1 antagonists tested. This contrasted with what was observed with other functional responses such as CCL3-mediated receptor internalization, which indicated that CP481715 did not inhibit CCL3-mediated receptor internalization and CCX354 did not alter CCL3-mediated β-arrestin translocation.
We found that for all of the compounds, the amount needed to inhibit binding of [125I]-CCL3 to membrane was different than that needed to inhibit CCL3-mediated chemotaxis and/or CCL3-mediated β-arrestin translocation. For example, the IC50 for PS899877 inhibition of [125I]-CCL3 binding was 105 and 149 nM for HEK and RPMI membranes, respectively, while the IC50 for CCL3-mediated RMPI 8226 chemotaxis was only 4.8 nM. Similarly, the IC50 for BX471 inhibition of [125I]-CCL3 binding was 47 and 58 nM for HEK and RPMI membranes, respectively, while the IC50 for CCL3-mediated RMPI 8226 chemotaxis was only 3 nM. The disconnect between how a compound affects CCL3 binding versus functional responses is not unexpected with allosteric modulators (May et al., 2007) as they may promote conformations of the receptor that alter its ability to interact with the orthosteric ligand or with intracellular partner proteins.
When the CCR1 antagonists were evaluated for their ability to alter β-arrestin translocation, we found that most inhibited CCL-3-mediated β-arrestin translocation in a dose-dependent manner. However, our studies indicate that CCX354 was unable to inhibit CCL3-mediated β-arrestin translocation. It is possible that this antagonist is not as effective at blocking GPCR kinase-mediated phosphorylation of the receptor (Neel et al., 2005). β-arrestin signalling has been shown to be involved in receptor internalization and chemotaxis through other chemokine receptors. Yet our results demonstrating CCX354 did not block arrestin translocation, but was effective at inhibiting CCL3-mediated chemotaxis (IC50 = 0.9 nM) while CP481715 was able to inhibit arrestin translocation (IC50 = 5.5 nM), but not CCR1 internalization suggest that for CCR1 these events may not necessarily be linked. Based on our results, studies comparing the CCR1 antagonists for their ability to inhibit CCL3-mediated MAPK phosphorylation, a pathway often mediated through β-arrestin, are warranted. It is also important to note that recent work by two independent groups suggests CCR1 may constitutively interact with β-arrestin 1 (Rajagopal et al., 2013) and β-arrestin 2 (Gilliland et al., 2013).
In conclusion, we achieved potent inhibition of [125I]-CCL3 binding as well as CCL3-induced chemotaxis, and receptor internalization using an MM cell line expressing endogenous CCR1 with multiple compounds from several different chemotypes. We found that neither HEK nor RPMI 8226 binding assays were consistently predictive of potency in functional responses (Figure 5). There appears to be biased antagonism with some compounds such as BX471 and PS899877, showing preference for reducing myeloma cell migration and β-arrestin translocation over CCR1 internalization or β-arrestin translocation. This type of biased inhibition was recently shown for CCR4 (Ajram et al., 2014). Moreover, our work supports the growing body of evidence demonstrating that allosteric antagonists stabilize unique receptor conformations. Taken together, our findings indicating there are clear differences between the CCR1 antagonists highlights the need to assess compounds using a spectrum of assays. Given that CCR1, like many of the chemokine receptors, can be activated by a plethora of ligands (CCL3, CCL4, CCL5, CCL6, CCL7, CCL9, CCL14, CCL15, CCL16, CCL23), we hope, in the future, to examine whether the six antagonists examined alter responses to additional ligands or if like other allosteric compounds they are probe-dependent. It will also be interesting to determine if CCX354, which was initially identified using CCL15 (Table 1), has a different response pattern (Figure 5) when CCL15 is used as the activating ligand rather than CCL3.
Figure 5.
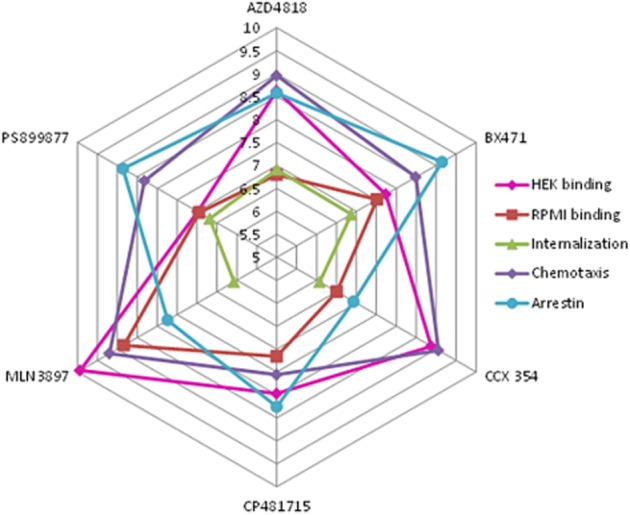
Radar plot for CCR1 antagonists. A radar plot of the six CCR1 antagonists and their pIC50 values for (i) displacement of [125I]-CCL3 binding to HEK membrane; (ii) displacement of 125I-CCL3 binding to RPMI 8226 membrane; (ii) CCL3-mediated internalization of CCR1 in RPMI 8226 cells; (iv) CCL3-mediated chemotaxis of RPMI 8226 cells; and as well as (v) pKB values for β-arrestin translocation. For statistical analysis, see Tables 2 and 3.
Acknowledgments
We would like to thank Brian Zanotti for his assistance with the flow cytometry work. The work described was supported with generous funding provided by the Chicago College of Pharmacy in the form of Research Grants (A. G.) as well as Student Research Awards from the Chicago College of Pharmacy (A. P., D. P., J. A.), the Chicago College of Osteopathic Medicine (KA) and the Kean University Foundation (J. R. M.).
Glossary
- EA
enzyme acceptor
- MM
multiple myeloma
- MIP-1α
macrophage inflammatory protein-1α
- PEN
penicillin
- STREP
streptomycin
Author contributions
A. G. and J. R. M. participated in research design. J. A., K. M. A., A. F., T. D. G., A.G., S. J. H., D. S. P., E. S. and A. W. conducted experiments. K. A. B., J. R. M. and J. L. Y. contributed new reagents or analytic tools. A. G. and M. R. M. performed data analysis. A. G., M. R. M. and J. R. M. wrote or contributed to the writing of the payer.
Conflicts of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12835
Figure S1 An example of the raw results from flow cytometry to measure CCL3-mediated receptor internalization for RPMI 8226 cells. Shown are the four quadrants with the upper left being those cells with high CCR1 expression and CCR5 expression; the lower right being those cells with high CCR5 expression and low CCR1 expression; the upper right being those cells with high CCR1 and high CCR5 expression; and the lower left being those cells that are not expressing CCR1 or CCR5. Panel A is untreated RPMI 8226 cells and panel B is RPMI 8226 cells exposed to 1 nM CCL3, a chemokine capable of activating both CCR1 and CCR5, for 2 h.
Figure S2 CCL3-mediated internalization is time-dependent. The percentage of cells expressing high levels of CCR1 (upper left quadrant) decreases following exposure to 1 nM CCL3.
References
- Ajram L, Begg M, Slack R, Cryan J, Hall D, Hodgson S, et al. Internalization of the chemokine receptor CCR4 can be evoked by orthosteric and allosteric receptor antagonists. Eur J Pharmacol. 2014;729C:75–85. doi: 10.1016/j.ejphar.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegretti M, Bertini R, Bizzarri C, Beccari A, Mantovani A, Locati M. Allosteric inhibitors of chemoattractant receptors: opportunities and pitfalls. Trends Pharmacol Sci. 2008;29:280–286. doi: 10.1016/j.tips.2008.03.005. [DOI] [PubMed] [Google Scholar]
- An S, Bleu T, Zheng Y. Transduction of intracellular calcium signals through G protein-mediated activation of phospholipase C by recombinant sphingosine 1-phosphate receptors. Mol Pharmacol. 1999;55:787–794. [PubMed] [Google Scholar]
- Anders H, Vielhauer V, Frink M, Linde Y, Cohen C, Blattner S, et al. A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J Clin Invest. 2002;109:251–259. doi: 10.1172/JCI14040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badr G, Lefevre E, Mohany M. Thymoquinone inhibits the CXCL12-induced chemotaxis of multiple myeloma cells and increases their susceptibility to Fas-mediated apoptosis. PLoS ONE. 2011;6:e23741. doi: 10.1371/journal.pone.0023741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson KG, Harriman GC. CCR1 Antagonists for The Treatment of I.A. Demyelinating Inflammatory Disease, PCT. Patent no. WO/2004/043965: Millennium Pharmaceuticals, Inc; 2004. [Google Scholar]
- Choi S, Cruz J, Craig F, Chung H, Devlin R, Roodman G, et al. Macrophage inflammatory protein 1-alpha is a potential osteoclast stimulatory factor in multiple myeloma. Blood. 2000;96:671–675. [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Cooper M, Elkins B, Gibson S, Gu E, Hassall I, Hemmerling M, et al. 2009. A process for the preparation of intermediates and their use in the the synthesis of spiropiperidine compounds. Patent no. WO/2009/011653.
- Dairaghi D, Zhang P, Wang Y, Seitz L, Johnson D, Miao S, et al. Pharmacokinetic and pharmacodynamic evaluation of the novel CCR1 antagonist CCX354 in healthy human subjects: implications for selection of clinical dose. Clin Pharmacol Ther. 2011;89:726–734. doi: 10.1038/clpt.2011.33. [DOI] [PubMed] [Google Scholar]
- Dairaghi D, Oyajobi B, Gupta A, McCluskey B, Miao S, Powers J, et al. CCR1 blockade reduces tumor burden and osteolysis in vivo in a mouse model of myeloma bone disease. Blood. 2012;120:1449–1457. doi: 10.1182/blood-2011-10-384784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire S, Ahn S, Lefkowitz R, Shenoy S. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Gardner D, Santella JB, Jr, Duncia J, Carter P, Dhar T, Wu H, et al. The discovery of BMS-457, a potent and selective CCR1 antagonist. Bioorg Med Chem Lett. 2013;23:3833–3840. doi: 10.1016/j.bmcl.2013.04.079. [DOI] [PubMed] [Google Scholar]
- Gilchrist A, Mazzoni M, Dineen B, Dice A, Linden J, Dunwiddie T, et al. Antagonists of the receptor-G protein interface block Gi-coupled signal transduction. J Biol Chem. 1998;273:14912–14919. doi: 10.1074/jbc.273.24.14912. [DOI] [PubMed] [Google Scholar]
- Gilliland C, Salanga C, Kawamura T, Trejo J, Handel T. The chemokine receptor CCR1 is constitutively active, which leads to G protein-independent, β-arrestin-mediated internalization. J Biol Chem. 2013;288:32194–32210. doi: 10.1074/jbc.M113.503797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladue R, Tylaska L, Brissette W, Lira P, Kath J, Poss C, et al. CP-481 715, a potent and selective CCR1 antagonist with potential therapeutic implications for inflammatory diseases. J Biol Chem. 2003;278:40473–40480. doi: 10.1074/jbc.M306875200. [DOI] [PubMed] [Google Scholar]
- Gladue R, Brown M, Zwillich S. CCR1 antagonists: what have we learned from clinical trials. Curr Top Med Chem. 2010;10:1268–1277. doi: 10.2174/156802610791561237. [DOI] [PubMed] [Google Scholar]
- Hansson J, Eriksson T, Mensonides-Harsema M, Mo J. 2008. Novel combination of compounds to be used in the treatment of airway diseases, especially chronic obstructive pulmonary disease (COPD) and asthma. : AstraZeneca. Patent no. WO/2008/103126.
- Hemmerling M, Ivanova S, Mensonides-Harsema M, Schulz H. 2009. Spiropiperidine compounds, a process of their preparation, pharmaceutical compositions containing them, and their use in the treatment of airway diseases, inflammatory diseases, COPD or asthma.: AstraZeneca. Patent no. WO/2009/011655.
- Hesselgesser J, Ng H, Liang M, Zheng W, May K, Bauman J, et al. Identification and characterization of small molecule functional antagonists of the CCR1 chemokine receptor. J Biol Chem. 1998;273:15687–15692. doi: 10.1074/jbc.273.25.15687. [DOI] [PubMed] [Google Scholar]
- Horuk R. BX471: a CCR1 antagonist with anti-inflammatory activity in man. Mini Rev Med Chem. 2005;5:791–804. doi: 10.2174/1389557054867057. [DOI] [PubMed] [Google Scholar]
- Hossain N, Mensonides-Harsema M, Cooper M, Eriksson T, Ivanova S, Bergström L. Structure activity relationships of fused bicyclic and urea derivatives of spirocyclic compounds as potent CCR1 antagonists. Bioorg Med Chem Lett. 2014;24:108–112. doi: 10.1016/j.bmcl.2013.11.062. [DOI] [PubMed] [Google Scholar]
- Karash A, Gilchrist A. Therapeutic potential of CCR1 antagonists for multiple myeloma. Future Med Chem. 2011;3:1889–1908. doi: 10.4155/fmc.11.144. [DOI] [PubMed] [Google Scholar]
- Kenakin T. The classification of seven transmembrane receptors in recombinant expression systems. Pharmacol Rev. 1996;48:413–463. [PubMed] [Google Scholar]
- Kenakin T, Jenkinson S, Watson C. Determining the potency and molecular mechanism of action of insurmountable antagonists. J Pharmacol Exp Ther. 2006;319:710–723. doi: 10.1124/jpet.106.107375. [DOI] [PubMed] [Google Scholar]
- Kerstjens H, Bjermer L, Eriksson L, Dahlström K, Vestbo J. Tolerability and efficacy of inhaled AZD4818, a CCR1 antagonist, in moderate to severe COPD patients. Respir Med. 2010;104:1297–1303. doi: 10.1016/j.rmed.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Lentzsch S, Gries M, Janz M, Bargou R, Dörken B, Mapara M. Macrophage inflammatory protein 1-alpha (MIP-1 alpha ) triggers migration and signaling cascades mediating survival and proliferation in multiple myeloma (MM) cells. Blood. 2003;101:3568–3573. doi: 10.1182/blood-2002-08-2383. [DOI] [PubMed] [Google Scholar]
- Lentzsch S, Anderson G, Cuiling L, Horuk R, Mapara M, Ghobrial I, et al. ASH annual meeting abstracts. Blood. 2005;106:Abstract 3383. [Google Scholar]
- Lombardi L, Poretti G, Mattioli M, Fabris S, Agnelli L, Bicciato S, et al. Molecular characterization of human multiple myeloma cell lines by integrative genomics: insights into the biology of the disease. Genes Chromosomes Cancer. 2007;46:226–238. doi: 10.1002/gcc.20404. [DOI] [PubMed] [Google Scholar]
- Magrangeas F, Nasser V, Avet-Loiseau H, Loriod B, Decaux O, Granjeaud S, et al. Gene expression profiling of multiple myeloma reveals molecular portraits in relation to the pathogenesis of the disease. Blood. 2003;101:4998–5006. doi: 10.1182/blood-2002-11-3385. [DOI] [PubMed] [Google Scholar]
- May L, Leach K, Sexton P, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- Menu E, De Leenheer E, De Raeve H, Coulton L, Imanishi T, Miyashita K, et al. Role of CCR1 and CCR5 in homing and growth of multiple myeloma and in the development of osteolytic lesions: a study in the 5TMM model. Clin Exp Metastasis. 2006;23:291–300. doi: 10.1007/s10585-006-9038-6. [DOI] [PubMed] [Google Scholar]
- Merritt J, Liu J, Quadros E, Morris M, Liu R, Zhang R, et al. Novel pyrrolidine ureas as C-C chemokine receptor 1 (CCR1) antagonists. J Med Chem. 2009;52:1295–1301. doi: 10.1021/jm801416q. [DOI] [PubMed] [Google Scholar]
- Merritt J, James R, Paradkar V, Zhang C, Liu R, Liu J, et al. Novel pyrrolidine heterocycles as CCR1 antagonists. Bioorg Med Chem Lett. 2010;20:5477–5479. doi: 10.1016/j.bmcl.2010.07.082. [DOI] [PubMed] [Google Scholar]
- Neel N, Schutyser E, Sai J, Fan G, Richmond A. Chemokine receptor internalization and intracellular trafficking. Cytokine Growth Factor Rev. 2005;16:637–658. doi: 10.1016/j.cytogfr.2005.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neote K, DiGregorio D, Mak J, Horuk R, Schall T. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell. 1993;72:415–425. doi: 10.1016/0092-8674(93)90118-a. [DOI] [PubMed] [Google Scholar]
- Norman P. AZD-4818, a chemokine CCR1 antagonist: WO2008103126 and WO2009011653. Expert Opin Ther Pat. 2009;19:1629–1633. doi: 10.1517/13543770903118996. [DOI] [PubMed] [Google Scholar]
- Oba Y, Lee J, Ehrlich L, Chung H, Jelinek D, Callander N, et al. MIP-1alpha utilizes both CCR1 and CCR5 to induce osteoclast formation and increase adhesion of myeloma cells to marrow stromal cells. Exp Hematol. 2005;33:272–278. doi: 10.1016/j.exphem.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease J, Horuk R. Chemokine receptor antagonists: part 1. Expert Opin Ther Pat. 2009;19:39–58. doi: 10.1517/13543770802641346. [DOI] [PubMed] [Google Scholar]
- Pennell A, Aggen J, Wright J, Sen S, McMaster B, Dairaghi D, et al. 2010. Substituted piperazines: Chemocentryx, Inc. Patent no. US2004/0162282.
- Pennell A, Aggen J, Sen S, Chen W, Xu Y, Sullivan E, et al. 1-(4-Phenylpiperazin-1-yl)-2-(1H-pyrazol-1-yl)ethanones as novel CCR1 antagonists. Bioorg Med Chem Lett. 2013;23:1228–1231. doi: 10.1016/j.bmcl.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Raab M, Podar K, Breitkreutz I, Richardson P, Anderson K. Multiple myeloma. Lancet. 2009;374:324–339. doi: 10.1016/S0140-6736(09)60221-X. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Bassoni D, Campbell J, Gerard N, Gerard C, Wehrman T. Biased agonism as a mechanism for differential signaling by chemokine receptors. J Biol Chem. 2013;288:35039–35048. doi: 10.1074/jbc.M113.479113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins A, Horlick R. Macrophage scavenger receptor confers an adherent phenotype to cells in culture. Biotechniques. 1998;25:240–244. doi: 10.2144/98252st04. [DOI] [PubMed] [Google Scholar]
- Roussou M, Tasidou A, Dimopoulos M, Kastritis E, Migkou M, Christoulas D, et al. Increased expression of macrophage inflammatory protein-1alpha on trephine biopsies correlates with extensive bone disease, increased angiogenesis and advanced stage in newly diagnosed patients with multiple myeloma. Leukemia. 2009;23:2177–2181. doi: 10.1038/leu.2009.130. [DOI] [PubMed] [Google Scholar]
- Sezer O. Myeloma bone disease: recent advances in biology, diagnosis, and treatment. Oncologist. 2009;14:276–283. doi: 10.1634/theoncologist.2009-0003. [DOI] [PubMed] [Google Scholar]
- Sun Y, Cheng Z, Ma L, Pei G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem. 2002;277:49212–49219. doi: 10.1074/jbc.M207294200. [DOI] [PubMed] [Google Scholar]
- Trentin L, Miorin M, Facco M, Baesso I, Carraro S, Cabrelle A, et al. Multiple myeloma plasma cells show different chemokine receptor profiles at sites of disease activity. Br J Haematol. 2007;138:594–602. doi: 10.1111/j.1365-2141.2007.06686.x. [DOI] [PubMed] [Google Scholar]
- Tsou C, Gladue R, Carroll L, Paradis T, Boyd J, Nelson R, et al. Identification of C-C chemokine receptor 1 (CCR1) as the monocyte hemofiltrate C-C chemokine (HCC)-1 receptor. J Exp Med. 1998;188:603–608. doi: 10.1084/jem.188.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidehi N, Schlyer S, Trabanino R, Floriano W, Abrol R, Sharma S, et al. Predictions of CCR1 chemokine receptor structure and BX 471 antagonist binding followed by experimental validation. J Biol Chem. 2006;281:27613–27620. doi: 10.1074/jbc.M601389200. [DOI] [PubMed] [Google Scholar]
- Vallet S, Anderson K. CCR1 as a target for multiple myeloma. Expert Opin Ther Targets. 2011;15:1037–1047. doi: 10.1517/14728222.2011.586634. [DOI] [PubMed] [Google Scholar]
- Vallet S, Raje N, Ishitsuka K, Hideshima T, Podar K, Chhetri S, et al. MLN-3897, a novel CCR1 inhibitor, impairs osteoclastogenesis and inhibits the interaction of multiple myeloma cells and osteoclasts. Blood. 2007;110:3744–3752. doi: 10.1182/blood-2007-05-093294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 An example of the raw results from flow cytometry to measure CCL3-mediated receptor internalization for RPMI 8226 cells. Shown are the four quadrants with the upper left being those cells with high CCR1 expression and CCR5 expression; the lower right being those cells with high CCR5 expression and low CCR1 expression; the upper right being those cells with high CCR1 and high CCR5 expression; and the lower left being those cells that are not expressing CCR1 or CCR5. Panel A is untreated RPMI 8226 cells and panel B is RPMI 8226 cells exposed to 1 nM CCL3, a chemokine capable of activating both CCR1 and CCR5, for 2 h.
Figure S2 CCL3-mediated internalization is time-dependent. The percentage of cells expressing high levels of CCR1 (upper left quadrant) decreases following exposure to 1 nM CCL3.