

Abstract
Hypoxic exposure is associated with impaired cardiac energetics in humans and altered mitochondrial function, with suppressed complex I-supported respiration, in rat heart. This response might limit reactive oxygen species generation, but at the cost of impaired electron transport chain (ETC) activity. Dietary nitrate supplementation improves mitochondrial efficiency and can promote tissue oxygenation by enhancing blood flow. We therefore hypothesised that ETC dysfunction, impaired energetics and oxidative damage in the hearts of rats exposed to chronic hypoxia could be alleviated by sustained administration of a moderate dose of dietary nitrate. Male Wistar rats (n = 40) were given water supplemented with 0.7 mmol l−1 NaCl (as control) or 0.7 mmol l−1 NaNO3, elevating plasma nitrate levels by 80%, and were exposed to 13% O2 (hypoxia) or normoxia (n = 10 per group) for 14 days. Respiration rates, ETC protein levels, mitochondrial density, ATP content and protein carbonylation were measured in cardiac muscle. Complex I respiration rates and protein levels were 33% lower in hypoxic/NaCl rats compared with normoxic/NaCl controls. Protein carbonylation was 65% higher in hearts of hypoxic rats compared with controls, indicating increased oxidative stress, whilst ATP levels were 62% lower. Respiration rates, complex I protein and activity, protein carbonylation and ATP levels were all fully protected in the hearts of nitrate-supplemented hypoxic rats. Both in normoxia and hypoxia, dietary nitrate suppressed cardiac arginase expression and activity and markedly elevated cardiac l-arginine concentrations, unmasking a novel mechanism of action by which nitrate enhances tissue NO bioavailability. Dietary nitrate therefore alleviates metabolic abnormalities in the hypoxic heart, improving myocardial energetics.
Introduction
Human exposure to sustained hypobaric hypoxia at high altitude (Holloway et al. 2011b) or normobaric hypoxia in a chamber (Holloway et al. 2011a) results in impaired cardiac energetics upon the subject's return to normoxia. The underlying mechanisms remain unresolved, but lowered electron transport chain (ETC) and Krebs cycle enzyme activities have been reported in the hypoxic rat heart (Heather et al. 2012), suggesting altered mitochondrial function. Similarly, sustained high altitude exposure results in altered human skeletal muscle energetics (Edwards et al. 2010) associated with a loss of mitochondrial density (Murray, 2009; Levett et al. 2012), downregulation of ETC complexes I (NADH dehydrogenase) and IV (cytochrome c oxidase; COX) (Levett et al. 2012) and suppressed mitochondrial respiratory function (Jacobs et al. 2012). Such metabolic modifications are likely to be mediated by the hypoxia-inducible factor (HIF) family of transcription factors, which are stabilised under conditions of low cellular oxygen (Semenza, 2007) and by reactive oxygen species (ROS; Guzy & Schumacker, 2006) and act to suppress oxidative metabolism, decreasing tissue oxygen demand in line with the decreased oxygen supply.
It is not immediately clear, however, why tissue oxygenation might fall in humans at high altitude, as, with appropriate acclimatisation, a robust erythropoietic response ensures that blood oxygen content remains constant up to ∼7000 m above sea level, despite the fall in barometric pressure and oxyhaemoglobin saturation (Grocott et al. 2009). It is possible that microcirculatory dysfunction restricts local oxygen delivery; indeed, impaired blood flow through the sublingual microcirculation has been reported at high altitude (Martin et al. 2009), and this may in turn reflect a deficiency of the biological messenger and vasodilator nitric oxide (NO). In support of this, microcirculatory blood flow was found to correlate inversely with plasma levels of nitrite in humans at altitude, suggesting that the availability of bioactive nitrogen oxides (NOx) influences human acclimatisation to hypoxia (Levett et al. 2011). Moreover, Tibetan natives displayed higher forearm blood flows than lowlanders at high altitude alongside elevated circulating levels of NO products such as nitrate (Erzurum et al. 2007).
Dietary nitrate supplementation has been shown to improve mitochondrial efficiency in humans (Larsen et al. 2010), decreasing the oxygen cost of exercise (Larsen et al. 2009). Meanwhile nitrite, a product of either NO oxidation or nitrate reduction, has been found to protect mitochondria during ischaemia/reperfusion, ameliorating the rise in ROS production and cytochrome c release, whilst preserving ATP synthesis (Shiva et al. 2007). Although nitrite may exert NO-like bioactivity in its own right (Bryan et al. 2005), it has recently been suggested that the hypoxia-driven reduction of nitrite to NO during ischaemia might preserve high-energy phosphate levels via an interaction with COX that preserves the efficiency of proton pumping (Clerc et al. 2007).
We postulated that an enhanced systemic availability of NOx, achieved via sustained administration of a moderate dose of dietary inorganic nitrate, might improve tissue oxygenation during chronic exposure to environmental hypoxia, preventing oxidative stress and mitochondrial dysfunction and thereby improving cardiac energetics. To investigate this, we studied rats exposed to atmospheric hypoxia (13% O2) or normoxia (21% O2) for 2 weeks and supplemented animals with either 0.7 mmol l−1 NaCl (as control) or 0.7 mmol l−1 NaNO3 in drinking water. In pilot studies this dose was found to result in a similar nitrate intake per unit body mass to that previously shown to alter mitochondrial function in humans (Larsen et al. 2010) and to elevate plasma nitrogen oxide levels. Mitochondrial function was assessed in cardiac muscle fibres alongside ETC protein levels and mitochondrial density. Protein carbonyls were analysed as a marker of oxidative stress, whilst high-energy phosphates reflected tissue energetics. Additionally, to investigate a potential mechanism, expression of arginase I and II and tissue arginase activity were measured in cardiac extracts alongside the expression of HIF-1 targets to indicate possible changes in arginine availability/utilisation and tissue oxygenation status. We found that dietary nitrate fully prevents the metabolic and energetic perturbations that arise in the myocardium following sustained exposure to environmental hypoxia.
Methods
Ethical approval
All experiments conformed to UK Home Office guidelines under the Animals in Scientific Procedures Act and were reviewed by the University of Cambridge Animal Welfare and Ethical Review Committee.
Animals
Male Wistar rats (273 ± 2 g; n = 40) were obtained from a commercial breeder (Charles River, Margate, UK), pair-housed in conventional cages with a normal 12 h/12 h light/dark photoperiod and maintained on distilled water and a standardized quality-controlled rodent chow to normalise micronutrient levels (RM1(E) SQC, Special Diets Services, UK). After 12 days, animals received either 0.7 mmol l−1 NaNO3 (nitrate group) or 0.7 mmol l−1 NaCl (control group matched for salinity and sodium intake; both ultra-pure, Sigma, St Louis, MO, USA) in distilled water (n = 20 per group); food and water intakes were measured daily to assess nitrate intakes. After 4 days, half of each group was transferred to hypoxia chambers (PFI Systems, Milton Keynes, UK) maintained at 13% O2 with 20 air changes per hour (n = 10 per group), whilst the remaining animals were housed in 21% O2. All animals remained in these atmospheric conditions, with access to food and NaCl/NaNO3-supplemented water ad libitum for a further 14 days. After 14 days, animals were anaesthetised with a 500 mg kg−1 i.p. injection of sodium pentobarbital (Vétoquinol, Buckingham, UK). Following cessation of peripheral nervous function, the chest cavity was opened and blood samples were drawn from the left ventricle by cardiac puncture, treated with EDTA and centrifuged at 5000 g for 5 min at 4°C. The heart was then immediately excised, rapidly washed in ice-cold PBS, blotted and weighed. A portion of the left ventricle was placed in ice-cold biopsy preservation solution (BIOPS; 10 mmol l−1 Ca-EGTA buffer, 0.1 μmol l−1 free calcium, 20 mmol l−1 imidazole, 20 mmol l−1 taurine, 50 mmol l−1 K-Mes, 0.5 mmol l−1 dithiothreitol, 6.56 mmol l−1 MgCl2, 5.77 mmol l−1 ATP and 15 mmol l−1 phosphocreatine, pH 7.1) whilst another portion of the left ventricle was snap-frozen for immunoblotting and metabolite analysis.
Measurement of nitrate, nitrite and nitroso-compounds
Nitrate and nitrite concentrations in plasma and cardiac tissue were quantified by ion chromatography using a dedicated HPLC system (ENO-20 Analyser, Eicom, Japan) with sequential on-line reduction of nitrate to nitrite by a cadmium column and post-column derivatisation with a modified Griess reagent, as described (Bryan et al. 2004). Frozen samples were thawed in the presence of 5 mmol l−1 N-ethylmaleimide (NEM) and 10 mmol l−1 EDTA, and tissues homogenised in NEM/EDTA-supplemented PBS at a dilution factor of 1:6 (w/v). After a further 10 min of incubation at room temperature samples were vortexed, deproteinated by addition of ice-cold methanol (1:1, v/v) and centrifuged. Thereafter, supernatants were subjected to immediate analysis. The concentrations of total nitroso-compounds (i.e. the sum of S-nitrosothiols and N-nitrosamines) in plasma were quantified by gas phase chemiluminescence using the triiodide method, as described (Bryan et al. 2004).
Respirometry
Cardiac muscle fibre bundles were dissected from fresh left ventricle in ice-cold BIOPS and incubated with gentle rocking for 20 min at 4°C in 1.5 ml BIOPS supplemented with 20 μl of 5 mg mL−1 saponin to selectively permeabilise the cell membrane, leaving mitochondrial membranes intact (Kuznetsov et al. 2008). After permeabilisation, fibres were washed with fresh respiration medium (0.5 mmol l−1 EGTA, 3 mmol l−1 MgCl2.6H2O, 20 mmol l−1 taurine, 10 mmol l−1 KH2PO4, 20 mmol l−1 HEPES, 1 g l−1 BSA, 60 mmol l−1 potassium lactobionate, 110 mmol l−1 mannitol and 0.3 mmol l−1 dithiothreitol, adjusted to pH 7.1 with 5 m KOH) three times for 5 min at 4°C with gentle rocking. For analysis, fibres were added to 500 μl atmospheric oxygen-equilibrated respiratory medium maintained at 37°C in a water-jacketed Clark-type oxygen electrode chamber (Strathkelvin Instruments, Motherwell, UK). Respiration rates were analysed by substrate/inhibitor-titration (see inset to Fig.3), essentially as described previously (Kuznetsov et al. 2008). Respiration was first measured in the presence of complex I substrates (10 mmol l−1 glutamate, 5 mmol l−1 malate), initially in the absence of ADP (State 2) and thereafter following the addition of 2 mmol l−1 ADP to stimulate ATP synthesis (State 3). A respiratory inhibitor was then added to prevent electron flow from complex I (0.5 μmol l−1 rotenone), before respiration rates were measured in the presence of a complex II substrate (10 mmol l−1 succinate). In a second assay, respiration was measured in the presence of palmitoyl-carnitine (0.04 mmol l−1) and malate (5 mmol l−1), in the presence and absence of 2 mmol l−1 ADP. Muscle fibres were recovered from electrode chambers and dried for 48 h at 80°C, such that respiration rates could later be normalized to tissue dry mass. Respiratory control ratios (RCRs), an indicator of the coupling of substrate oxidation to ADP phosphorylation, were calculated as State 3/State 2.
Figure 3. Mitochondrial density and biogenesis markers, and palmitoyl-carnitine respiration.
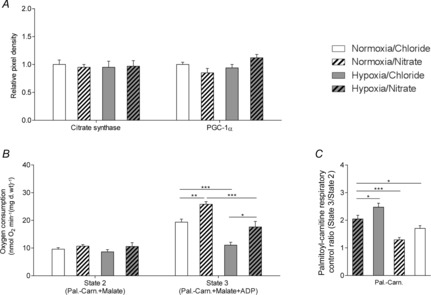
A, levels of citrate synthase and PGC-1α in the hearts of normoxic and hypoxic rats with/without nitrate supplementation. B and C, respiration rates (B) and respiratory control ratio (C) with palmitoyl-carnitine and malate substrates in permeabilised cardiac muscle fibres from normoxic and hypoxic rats with/without nitrate supplementation. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Complex I spectrophotometric assay
NADH/ubiquinone oxidoreductase activity was measured spectrophotometrically in freeze-thawed tissue according to published protocols (Pon et al. 2007).
Immunoblotting
Protein levels were measured in whole heart homogenates, using SDS-PAGE and immunoblotting, as described (Heather et al. 2006). Evenness of protein loading and quality of transfer were confirmed by Ponceau staining and homogeneity between gels ensured by loading multiple control samples on every gel. Bands were quantified using UN-SCAN-It software (Silk Scientific, Orem, UT, USA). The Total OXPHOS Detection Kit (Mitosciences, Eugene, OR, USA) is a five-antibody cocktail which determines protein levels of the ETC complexes. Antibodies to the 20 kDa subunit of complex I (ND6), the 30 kDa iron–sulphur subunit of complex II (Fe-S), the core-2 subunit of complex III (47 kDa), Subunit 1 of complex IV (39 kDa) and the F1α subunit of ATP-synthase (53 kDa) provide information on levels of these complexes in their active form, as these subunits are labile when unassembled. Citrate synthase and PGC-1α protein levels were detected using antibodies purchased from Abcam (Cambridge, UK), respectively ab96600 (52 kDa) and ab54481 (105 kDa).
Detection of protein carbonylation
Protein carbonylation was measured using an OxyBlot Protein Oxidation Detection Kit (Millipore, Billerica, MA, USA) according to the manufacturer's instructions. The extent of modification of proteins by ROS and other free radical species was determined after derivatization of the carbonyl groups with 2,4-dinitrophenylhydrazine. SDS-PAGE and immunoblotting were then performed using standard procedures with a primary antibody against the derivatised carbonyl groups. Evenness of protein loading and homogeneity between gels were confirmed using Ponceau staining, as above.
Measurement of adenylates and l-arginine
Metabolites were extracted from heart tissue using an established water/chloroform/methanol procedure (Le Belle et al. 2002). To approximately 100 mg crushed tissue, 600 μl 2:1 methanol/chloroform was added before processing in a tissue lyser (Qiagen, UK) for 20 min at 25 Hz. After homogenisation, 200 μl water and 200 μl chloroform were added, before thorough vortexing and centrifugation at 13,000 g for 15 min. The aqueous and organic fractions were removed and aliquoted into separate 2 ml tubes and kept on ice. A further 600 μl 2:1 methanol/chloroform was added to protein pellet and lysate, and the extraction process repeated as above. After centrifugation, the two layers were removed, and combined with the other aqueous and organic fractions kept on ice, and dried overnight in a stream of nitrogen under a fume hood.
Adenosine mono-, di- and triphosphate (AMP, ADP and ATP, respectively) and l-arginine were measured using hydrophilic interaction liquid chromatography-mass spectrometry (HILIC-MS). Samples were reconstituted in 70:30 acetonitrile/water and analysed on a Quattro Premier XE triple quadrupole-mass spectrometer (Waters, Elstree, UK). Multiple reaction monitoring in positive ion mode was used with the following optimised mass transitions: AMP 348.03 > 135.90; ADP 427 > 135.90; ATP 507.91 > 135.90; l-arginine 231.20 > 70.10. AMP, ADP, ATP and l-arginine had separately optimised cone voltages and collision energies: 95.0 V, 18 eV; 30.0 V, 30.0 eV; 30.0 V, 30.0 eV; and 35.0 V, 15.0 eV; respectively. Other optimisation parameters common to each species were as follows: capillary voltage 3.5 kV; source temperature 110°C for adenylates and 150°C for l-arginine with desolvation temperature of 350°C for all metabolites. Chromatographic separation was achieved using a 100 mm × 2.1 mm × 5 μm ZIC-HILIC column (Sequant, Sweden) with isocratic 70:30 acetonitrile/100 mm ammonium acetate at a flow rate of 400 μl min−1 for 8 min. Owing to the necessity of preparing muscle fibres for mitochondrial respiratory analysis, it was not possible to instantly snap-freeze heart tissue for mass spectrometry, and hence the levels of ATP, ADP, AMP and l-arginine are presented as relative levels rather than true in vivo concentrations, although the levels are legitimately comparable within and between experimental groups.
RNA purification
Total RNA was purified from crushed, frozen heart using an RNeasy Mini Kit (QIAgen, Valencia, CA, USA) according to the manufacturer's specifications. Approximately 25 mg tissue was resuspended in 350 μl working lysis buffer, before centrifugation at 14,000 g for 3 min in a QIAshredder spin column (QIAgen). The lysate supernatant was then digested with proteinase K (QIAgen) for 15 min at 55°C to remove fibrous proteins that would otherwise decrease RNA yield. The solution was centrifuged at 14,000 g for 1 min, transferred to a new tube and 450 μl 100% ethanol added. After mixing, the solution was transferred to an RNeasy spin column and centrifuged at 8000 g for 15 s to bind RNA to the membrane. Flow-through was discarded and 350 μl wash buffer added before centrifugation at 8000 g for 15 s to wash the membrane. To remove DNA from the sample, 80 μl working DNase I (QIAgen) solution was added directly to the spin column membrane and samples were incubated at room temperature for 15 min. After addition of a further 350 μl wash buffer, the spin column was centrifuged at 8000 g for 15 s and flow-through was discarded. To wash the membrane, 500 μl working ethanol buffer was added before centrifugation at 8000 g for 15 s, after which flow-through was discarded. A further 500 μl working ethanol buffer was added before centrifugation at 8000 g for 2 min. The spin column was placed in a new collection tube and centrifuged at 14,000 g for 1 min to prevent carryover of ethanol. To elute pure RNA, 50 μl RNase-free water was added directly to the membrane and the column placed in a new 1.5 ml collection tube before centrifugation at 8000 g for 1 min. RNA concentration was quantified at 260 nm using a SmartSpec Plus spectrophotometer (Bio-Rad, Hercules, CA, USA).
Reverse transcription and RT-qPCR
Primer annealing was performed in a 10 μl reaction volume using 1 nmol l−1 dNTPs, 50 ng random hexamers and 1 μg total RNA, at 65°C for 5 min in a Veriti 96-well Thermocycler (Applied Biosystems, Foster City, CA, USA). Complimentary DNA was produced from the primer annealing reaction using 200 U SuperScript III (reverse transcriptase), 40 U RNaseOUT (recombinant RNase inhibitor), 20 nmol l−1 dithiothreitol, 20 nmol l−1 MgCl2 and 1× reverse transcriptase buffer (Life Technologies, Foster City, CA, USA) in a 20 μl final reaction volume. Thermocycler parameters were as follows: initial incubation, 10 min at 25°C; cDNA synthesis, 50 min at 50°C; and reaction termination, 5 min at 85°C. For analysis of steady-state mRNA levels, relative abundance of transcripts of interest was assessed by quantitative PCR in SYBR Green FastStart Universal Master Mix (Applied Biosystems) with a StepOnePlus detection system (Applied Biosciences). QuantiTect primer assays for Vegfa, Pgk1, Slc1a2, Ca9, Nos2, Arg1 and Arg2 were obtained from QIAgen. Thermocycler parameters were as follows: initial incubation, 95°C for 10 min; with 40 cycles of both elongation, 15 s at 95°C; and cooling, 1 min at 60°C. Expression levels were normalised to Actb using the ΔCT method, and subsequently to normoxia/control to give fold-changes.
Arginase activity
Arginase activity was determined using an arginase assay kit (Abnova, Taipei City, Taiwan) according to the manufacturer's specifications. The method utilises a chromogen that complexes with urea produced by the arginase reaction to produce a colour that is directly proportional to arginase activity in the sample.
Statistics
Results are expressed as means ± SEM. Data were collated in Excel before one- or two-way ANOVA was used to determine significant differences across experimental groups (Graphpad, Instat). Bonferroni post hoc analysis allowed for multiple analysis of selected groups where appropriate. Differences were considered statistically significant when P ≤ 0.05.
Results
Morphology
Neither exposure to environmental hypoxia (13% O2) nor dietary nitrate supplementation for 2 weeks, alone or in combination, caused a significant difference in final body weights or whole heart weights in rats (Table1), although trends towards lower body weights and increased heart weights led to 12% greater heart weight/body weight ratios in both groups of hypoxic rats (P ≤ 0.01) when compared with normoxic rats. Hypoxic rats also showed ventricular remodelling, having lower left ventricle weights and higher right ventricle weights than their normoxic counterparts, and this was not affected by dietary nitrate supplementation (Table1). Lung wet weight/dry weight ratios were the same in all animals, and therefore acclimatisation of these rats to hypoxia did not cause pulmonary oedema. Epididymal fat pad mass, measured as an index of whole body adiposity, and tibia length, used as a surrogate measure of body size, were the same in all animals. No differences in food or water intake were observed across the treatment groups.
Table 1.
Morphological characteristics
| Normoxic Control (n = 10) | Normoxic Nitrate (n = 10) | Hypoxic Control (n = 9) | Hypoxic Nitrate (n = 9) | |
|---|---|---|---|---|
| Morphological data | ||||
| Starting body weight (g) | 268 ± 5 | 268 ± 6 | 276 ± 2 | 280 ± 4 |
| End body weight (g) | 429 ± 8 | 431 ± 9 | 416 ± 14 | 412 ± 12 |
| Heart weight (g) | 1.2 ± 0.0 | 1.3 ± 0.0 | 1.3 ± 0.0 | 1.3 ± 0.1 |
| Heart/body mass × 1000 | 2.85 ± 0.05 | 2.92 ± 0.05 | 3.18 ± 0.06*** | 3.19 ± 0.10** |
| Left ventricle mass (g) | 0.86 ± 0.02 | 0.84 ± 0.02 | 0.72 ± 0.03** | 0.77 ± 0.03* |
| Right ventricle mass (g) | 0.21 ± 0.01 | 0.22 ± 0.02 | 0.39 ± 0.02*** | 0.39 ± 0.02*** |
| Epididymal fat pad (g) | 3.5 ± 0.3 | 3.2 ± 0.2 | 4.4 ± 0.31 | 3.5 ± 0.34 |
| Tibia length (mm) | 47.0 ± 0.2 | 47.0 ± 0.2 | 47.0 ± 0.4 | 47.0 ± 0.4 |
| Lung wet/dry mass | 4.70 ± 0.07 | 4.58 ± 0.08 | 4.68 ± 0.05 | 4.71 ± 0.03 |
| Food/water intakes | ||||
| Food intake (g day−1) | 32.1 ± 2.1 | 32.9 ± 1.3 | 29.0 ± 0.8 | 27.1 ± 0.8 |
| Water intake (ml day−1) | 51.4 ± 6.0 | 51.1 ± 2.2 | 40.9 ± 4.8 | 35.5 ± 2.5 |
Rats were housed in normoxia (21% O2) and hypoxia (13% O2) supplemented with either 0.7 mmol l−1 NaCl (Control) or NaNO3 (Nitrate), and corresponding food/water intakes during the treatment phase. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 vs. normoxic control.
Nitrate intake and resulting nitrate and nitrite levels
Nitrate intake was calculated from the amount of food and water consumed each day and measured nitrate concentrations in both. No differences were observed in food intake between any of the groups and thus there was no difference in nitrate intake from food. Due to consumption of nitrate-supplemented water, normoxic nitrate-supplemented animals had a nitrate intake over seven times greater than normoxic controls (P ≤ 0.001), whilst hypoxic nitrate-supplemented animals had nitrate intakes over five times greater (P ≤ 0.001) than normoxic controls and six times greater than non-supplemented hypoxic rats (Fig.1A).
Figure 1. Nitrate intake, plasma nitrogen oxide (NOx) levels and cardiac NOx levels.
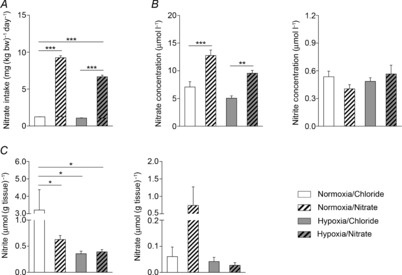
A, total dietary nitrate intake from food and water in normoxic and hypoxic rats with/without nitrate supplementation. B, plasma levels of nitrate and nitrite. C, cardiac levels of nitrate and nitrite. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
To determine how nitrate intake altered circulating nitrite and nitrate levels, their concentrations were measured in plasma. In normoxic rats, nitrate supplementation increased circulating nitrate concentrations by 80% (P ≤ 0.001) compared with normoxic controls, whilst nitrite concentrations were unchanged. In hypoxic rats, plasma nitrate levels tended to be lower than in normoxic counterparts, although this did not reach statistical significance. In nitrate-supplemented hypoxic rats, nitrate concentrations were 89% higher than in non-supplemented hypoxic rats, again, with no change in nitrite concentration (Fig.1B).
Next we determined tissue levels of nitrate and its putative reduction product, nitrite, in the heart. Here, hypoxia resulted in cardiac nitrate levels that were around 5- to 7-fold lower than in normoxic controls (P ≤ 0.05; Fig.1C), and these levels did not change with nitrate supplementation. Unexpectedly, cardiac nitrate content was also significantly decreased in the normoxic nitrate supplementation group compared with controls. Cardiac nitrite concentrations were not significantly different across all groups, but tended to be higher in nitrate-supplemented, normoxic animals compared with the other three groups.
Cardiac mitochondrial function and protein expression
Mitochondrial respiration was measured in permeabilised cardiac muscle fibre bundles using a substrate/inhibitor titration to indicate the capacity for electron flow through different complexes of the ETC. Complex I-supported state 2 respiration rates (in the presence of glutamate/malate) were similar in all groups, whilst ADP-stimulated, state 3 rates were 33% lower (P ≤ 0.05) in hypoxic rats when compared with normoxic controls. Nitrate supplementation did not alter complex I respiration in normoxic rats, but restored normal complex I respiration in the hearts of hypoxic rats (Fig.2A). RCR (state 3/state 2) was 35% lower in the hearts of hypoxic rats when compared with those of normoxic controls (Fig.2B). RCR was not altered by nitrate supplementation in either group, but was not significantly different from normal levels in nitrate-supplemented hypoxic rats. Complex II-supported state 3 rates (succinate/rotenone) were similar across all groups (Fig.2A).
Figure 2. Cardiac mitochondrial function, protein levels and enzyme activity.
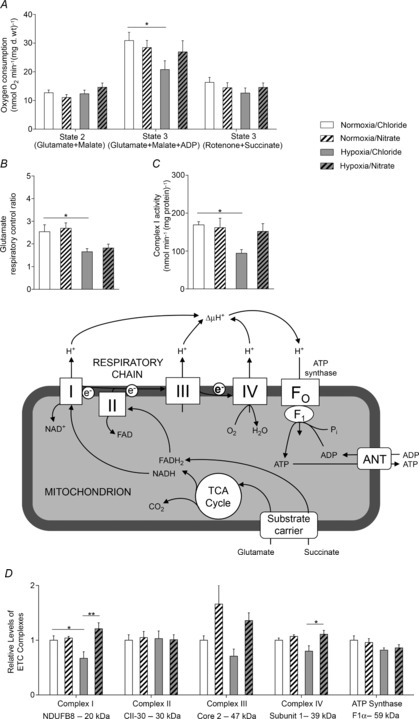
A and B, respiration rates (A) and respiratory control ratio (B) with complex I (glutamate/malate) and complex II (rotenone/succinate) substrates, acquired in permeabilised cardiac muscle fibres from normoxic and hypoxic rats with/without nitrate supplementation using a substrate inhibitor titration (see inset, adapted from Cole et al. 2011). C and D, complex I enzyme activity (C) and levels of representative ETC complex subunits (D), in the hearts of rats. *P ≤ 0.05, **P ≤ 0.01.
Complex I activity levels, measured by a spectrophotometric assay, were 44% lower in the hearts of hypoxic rats than in those of normoxic rats (P ≤ 0.05), and were restored to normal levels by nitrate supplementation (Fig.2C). Protein expression of complex I reflected oxygen consumption rates and enzyme activity levels at this complex, being 33% lower in hypoxic hearts than in normoxic controls (P ≤ 0.05), but restored to normoxic levels in nitrate-supplemented hypoxic rats. Cardiac levels of complex IV were lower in hypoxic rats than in normoxic controls, although this did not reach significance. Nitrate alone did not alter complex IV levels, but this was 39% higher in nitrate-supplemented hypoxic rats compared with non-supplemented hypoxic rats (P ≤ 0.05). Cardiac complex III levels were not significantly altered across groups, but tended to be lower in hypoxic than in normoxic rats, and higher in nitrate-supplemented rats than in non-supplemented animals. Cardiac levels of complex II and ATP-synthase (Fig.2D) were the same in all rats as were levels of citrate synthase and PGC-1α (Fig.3A), suggesting no changes in cardiac mitochondrial density or mitochondrial biogenesis as a result of either hypoxic exposure or nitrate supplementation.
Cardiac state 2 respiration rates in the presence of the more physiological substrate, palmitoyl-carnitine plus malate, were the same in the hearts of all groups, but state 3 respiration rates were 43% lower in the hearts of hypoxic non-supplemented rats compared with normoxic controls (P ≤ 0.001; Fig.3B). State 3 rates, however, were the same in the hearts of nitrate-supplemented hypoxic rats and normoxic controls, whilst nitrate-supplemented normoxic rat hearts had 25% higher state 3 rates than normoxic controls (P ≤ 0.01; Fig.3B). With palmitoyl-carnitine/malate, normoxia/nitrate rats had a 21% higher RCR than normoxic controls (P ≤ 0.05), whilst hypoxia/chloride and hypoxia/nitrate rats had 37% (P ≤ 0.001) and 17% (P ≤ 0.05) lower RCRs, respectively, than normoxic controls (Fig.3C).
Energetics and oxidative stress
Cardiac ATP levels were 62% lower in hypoxic rats than in normoxic controls (P ≤ 0.001) but were maintained at normoxic levels in nitrate-supplemented hypoxic rats (Fig.4A). ADP levels were 48% lower in the hearts of hypoxic rats compared with the hearts of normoxic controls (P ≤ 0.001), but were 28% higher in the hearts of nitrate-supplemented normoxic rats (P ≤ 0.05) and maintained at normoxic levels in the hearts of nitrate-supplemented hypoxic rats (Fig.4A). Cardiac AMP levels were unaltered by nitrate supplementation alone, but were 57% higher (P ≤ 0.05) in non-supplemented hypoxic animals and maintained at normoxic levels following nitrate supplementation (Fig.4A).
Figure 4. Cardiac energetics and oxidative stress, and plasma nitroso-compounds.
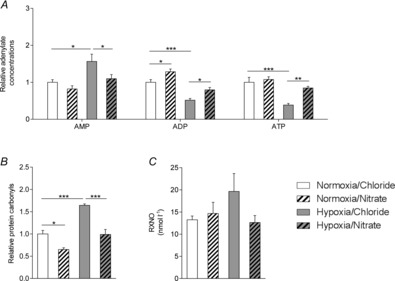
A and B, adenosine monophosphate, diphosphate and triphosphate levels (A) and protein carbonyls (B) in the hearts of normoxic and hypoxic rats with/without nitrate supplementation. C, plasma nitroso-compounds from the same rats. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Nitrate supplementation lowered protein carbonyl levels in hearts of both normoxic and hypoxic rats. Compared with normoxic controls, levels of protein carbonyls were 29% lower in nitrate-supplemented normoxic rats (P ≤ 0.05). Whilst protein carbonyl levels were 31% higher in non-supplemented hypoxic rats than normoxic controls (P ≤ 0.001), indicating greater oxidative stress, protein carbonyl content was normal in nitrate-supplemented hypoxic rats, suggesting that nitrate supplementation ameliorates the hypoxia-driven increase in cardiac oxidative stress (Fig.4B). Similarly, hypoxic exposure resulted in 48% higher total plasma nitroso-compound levels (although this did not reach statistical significance), consistent with enhanced systemic oxidative stress, whilst nitrate supplementation prevented this (Fig.4C).
Arginase expression and activity
Expression of the HIF-2 target Arg1 was 29% higher in the hearts of hypoxic rats compared with normoxic/controls (P ≤ 0.05), but was 54% lower in hearts of nitrate-supplemented normoxic rats (P ≤ 0.05) and 71% lower in hearts of nitrate-supplemented hypoxic rats (P ≤ 0.01) than in those of normoxic controls (Fig.5A). Expression of Arg2, a putative target of both HIF-1 and HIF-2, followed a similar profile to that of Arg1, although its expression was not elevated following 2 weeks of hypoxic exposure. Nitrate supplementation, however, resulted in 41% lower Arg2 expression in the hearts of nitrate-supplemented normoxic rats (P ≤ 0.05) and 37% lower Arg2 expression in the hearts of nitrate-supplemented hypoxic rats (P ≤ 0.05) compared with those of normoxic controls (Fig.5A). Total cardiac arginase activity reflected these expression patterns, being 51% higher in hypoxic than normoxic rats (P ≤ 0.01). There was no elevation in arginase activity in the hearts of hypoxic rats when supplemented with nitrate; indeed, in these hearts activity was 61% lower than in those of untreated hypoxic rats (P ≤ 0.001) and 41% lower than in those of untreated normoxic controls (P ≤ 0.05) (Fig.5B). Correspondingly, in the normoxic nitrate-supplemented rats cardiac l-arginine levels were 2.3-fold higher than in normoxic control rats (P ≤ 0.05), whilst l-arginine levels in the hearts of hypoxic nitrate-supplemented rats were 3.3-fold higher than in those of hypoxic controls (P ≤ 0.05). Cardiac l-arginine levels were not, however, altered by hypoxia alone (Fig.5C). Taken together, these results indicate that constitutive and hypoxia-induced arginase activity is a major factor that limits the availability of l-arginine for other processes (such as NO production via NOS) in the heart.
Figure 5. Expression and activity of cardiac arginase.
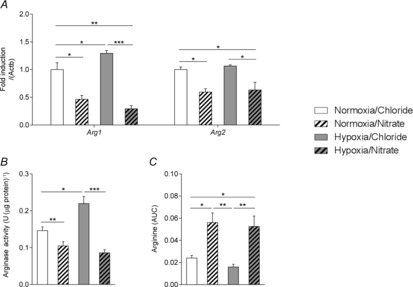
Expression of arginase I and arginase II (A), total arginase activity (B) and l-arginine levels (C) in the hearts of normoxic and hypoxic rats with/without nitrate supplementation. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Expression of HIF-1 targets
After 2 weeks of exposure to hypoxia there were no changes in expression of any HIF-1 targets in the heart. The expression of the HIF-1 targets Vegfa and Pgk1 were 73% (P ≤ 0.01) and 71% lower (P ≤ 0.05) in the hearts of nitrate-supplemented normoxic rats compared with non-supplemented controls (Fig.6A), whilst there were no nitrate-induced changes in the expression of Slc1a2 (Fig.6A), Ca9 or Nos2 (Fig.6B). In nitrate-supplemented hypoxic rats, however, Vegfa was 44% lower (P ≤ 0.05; Fig.6A), Ca9 54% lower (P ≤ 0.05) and Nos2 52% lower (P ≤ 0.01) than in non-supplemented hypoxic rats (Fig.6B).
Figure 6. HIF-1 targets.
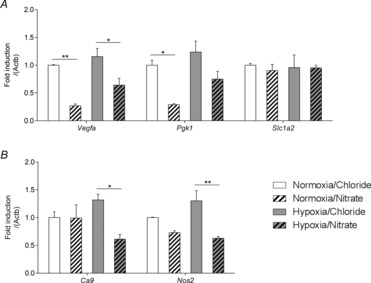
Relative expression of HIF-1 targets (A: Vegfa, Pgk1 and Slc1a2; B: Ca9, Nos2), in the hearts of normoxic and hypoxic rats with/without nitrate supplementation. *P ≤ 0.05, **P ≤ 0.01.
Discussion
Sustained exposure to environmental hypoxia is associated with altered cardiac energetics (Holloway et al. 2011b) and mitochondrial function (Heather et al. 2012). Here we investigated whether a moderate dietary intake of nitrate, a potential precursor of NO under hypoxic conditions (Lundberg & Govoni, 2004), can ameliorate these energetic abnormalities in hypoxic rats. As expected, hypoxic exposure increased oxidative stress and caused an energetic impairment in the heart. Hypoxia elicited a specific loss of complex I protein levels and enzyme activity in heart mitochondria, with no changes in either mitochondrial density or biogenesis, whilst fatty acid oxidation capacity decreased. Dietary nitrate supplementation enhanced circulating plasma nitrate levels and fully prevented the hypoxia-induced loss of complex I, decreased fatty acid oxidation, impaired energetics and oxidative stress. In addition, our study hints towards a novel mechanism by which nitrate administration might enhance NO bioavailability: via the suppression of tissue arginase activity, redirecting l-arginine flux from ornithine/urea production to NO/citrulline formation under both hypoxic and normoxic conditions (Fig.7). Whilst we did not measure cardiac NO production in this study, in a separate study from our laboratory, which used the same dose of dietary nitrate in normoxic rats alone, cardiac cGMP levels were 38% higher in the hearts of the nitrate-supplemented group (P ≤ 0.01; data not shown). cGMP is a marker of NO bioactivity, secondary to the activation of soluble guanylate cyclase by NO. This novel action of nitrate may thus increase the proportion of NO production from endogenous sources by shifting substrate (l-arginine) utilisation from arginase to NO-synthase, therefore preventing mitochondrial dysfunction and combating oxidative stress. It is possible that the same mechanism accounts for some of the effects of inorganic nitrate in humans, including the blood pressure lowering (Webb et al. 2008) and oxygen-sparing effects (Larsen et al. 2010) reported by others.
Figure 7. Representation of the effects of hypoxia on mitochondrial ATP synthesis, ROS production and arginase activity (right).
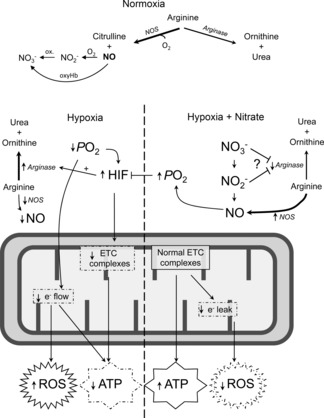
Possible mechanism by which nitrate supplementation may elicit observed effects, resulting in improved redox homeostasis and ATP production via inhibition of arginase and increased bioavailability of NO (right), restoring tissue 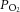 . Variations in arrow thickness under normoxic and hypoxic conditions (with and without nitrate supplementation) reflect differences in relative flux rates through major l-arginine consuming pathways.
. Variations in arrow thickness under normoxic and hypoxic conditions (with and without nitrate supplementation) reflect differences in relative flux rates through major l-arginine consuming pathways.
A major strength of our study lies in the tightly controlled intake of nitrate within experimental groups, achieved via provision of a quality-controlled diet and nitrate supplementation via drinking water, with the matching of salinity and sodium intake in control animals. Whilst an additional non-NaCl-supplemented control group was not used, validation studies from our laboratory have shown that the dose of NaCl used has no effect on any of the study outcomes or any other outcome we have measured. An additional strength is the moderate dose of nitrate employed. As such, the circulating nitrate concentrations measured in this study can readily be achieved in humans via a slight modification of the diet. NO production rates (Siervo et al. 2011) and circulating nitrate and nitrite concentrations (Pannala et al. 2003) are very similar in rats and humans, and indeed the nitrate dose used in our study matches that used in human studies (Larsen et al. 2010). In addition, the study involved the use of a wide range of experimental techniques to understand the hypoxia and nitrate-induced changes in tissue oxygen metabolism and energetics, whilst observing an excellent agreement in findings across an array of platforms. Our study also has some limitations. Due to technical constraints, we can only present relative levels of adenosine phosphates here, but the levels are legitimately comparable within and between experimental groups. Another possible limitation of the current study relates to the lack of data on changes in cardiac function and/or haemodynamics in response to hypoxia and nitrate supplementation. Whilst it is now important to establish the functional consequences of our findings in rigorously controlled studies, it would clearly be a mistake to report here a single measure of cardiovascular function that was altered by hypoxia but reversed by nitrate, and thereby to conclude that this was due to the changes we report in mitochondrial function, rather than a consequence of any of the other systemic effects of hypoxia and/or nitrate.
The degree of hypoxic exposure experienced by rats in this study represented an inspired 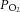 of 12.1 kPa (91 mmHg) and is approximately the same as that experienced at 4000 m above sea level, an altitude at which millions of people worldwide live and work (West, 2004). Exposure to this level of environmental hypoxia for 2 weeks was sufficient to elicit changes in the expression of HIF target genes in the heart, with the HIF-2 target (Arg1) showing the greatest upregulation but no apparent upregulation of HIF-1 targets at this time point. This might suggest a relatively advanced state of hypoxic acclimatisation, as HIF-1 stabilization is thought to dominate during acute exposure whilst HIF-2 is stabilized over longer durations of hypoxia (Koh & Powis, 2012). Unfortunately, due to the need to prepare muscle fibre bundles from these hearts, we were unable to snap-freeze myocardium sufficiently rapidly enough to accurately measure HIF protein levels. In humans at high altitude, we have reported altered heart and skeletal muscle energetics (Edwards et al. 2010; Holloway et al. 2011b), and in skeletal muscle this is associated with a loss of mitochondrial density (Levett et al. 2012) and the downregulation of the transcriptional co-activator, PGC-1α, a master regulator of mitochondrial biogenesis (Levett et al. 2012). In contrast, in the hypoxic rat heart we found that there was no loss of citrate synthase or PGC-1α, which appears to corroborate the findings of one of our co-authors who reported no difference in the yield of either interfibrillar or subsarcolemmal mitochondria when isolated from the hypoxic rat heart (Heather et al. 2012).
of 12.1 kPa (91 mmHg) and is approximately the same as that experienced at 4000 m above sea level, an altitude at which millions of people worldwide live and work (West, 2004). Exposure to this level of environmental hypoxia for 2 weeks was sufficient to elicit changes in the expression of HIF target genes in the heart, with the HIF-2 target (Arg1) showing the greatest upregulation but no apparent upregulation of HIF-1 targets at this time point. This might suggest a relatively advanced state of hypoxic acclimatisation, as HIF-1 stabilization is thought to dominate during acute exposure whilst HIF-2 is stabilized over longer durations of hypoxia (Koh & Powis, 2012). Unfortunately, due to the need to prepare muscle fibre bundles from these hearts, we were unable to snap-freeze myocardium sufficiently rapidly enough to accurately measure HIF protein levels. In humans at high altitude, we have reported altered heart and skeletal muscle energetics (Edwards et al. 2010; Holloway et al. 2011b), and in skeletal muscle this is associated with a loss of mitochondrial density (Levett et al. 2012) and the downregulation of the transcriptional co-activator, PGC-1α, a master regulator of mitochondrial biogenesis (Levett et al. 2012). In contrast, in the hypoxic rat heart we found that there was no loss of citrate synthase or PGC-1α, which appears to corroborate the findings of one of our co-authors who reported no difference in the yield of either interfibrillar or subsarcolemmal mitochondria when isolated from the hypoxic rat heart (Heather et al. 2012).
Following hypoxic exposure we did, however, find decreased respiration rates, enzyme activity and protein levels of ETC complex I in the heart. We have previously found a loss of complex I protein in human skeletal muscle (Levett et al. 2012) after prolonged hypoxia and a loss of complex I-supported respiration in hypoxic human fibroblasts (Colleoni et al. 2013), suggesting a specific effect of hypoxia on this complex. Interestingly, Heather et al. (2012) reported complex I-related respiratory changes in the hypoxic rat heart that were confined to the subsarcolemmal population of mitochondria, perhaps suggesting that in heart, as in skeletal muscle (Levett et al. 2012), this subpopulation is more susceptible to hypoxia-driven impairments. The specific loss of complex I might serve to decrease superoxide generation via reverse electron transport through this complex (Turrens & Boveris, 1980), but at the cost of decreased ATP synthesis. Whilst we did not measure ATP synthesis rates directly, we can infer that the capacity for mitochondrial ATP synthesis is decreased in these hearts as the decreased RCRs indicate worsened coupling of oxygen consumption to ATP synthesis, alongside decreased overall capacity for oxidation of both complex I substrates and the more physiological substrate, palmitoyl-carnitine. Thus, the mitochondrial changes measured here probably underlie the observed depression in cardiac energetics.
In the hypoxic heart, a moderate dose of dietary nitrate prevented the loss of complex I, and whilst this might result from improved tissue oxygenation alone, the reversible formation of S-nitrosothiols is known to alter the structure and function of proteins, including complex I subunits (Clementi et al. 1998; Piantadosi, 2012). Indeed, reversible nitrosation of complex I is known to inactivate it in several ways (Galkin & Moncada, 2007) and during ischaemia/reperfusion this modification is known to be cardioprotective (Larsen et al. 2012; Chouchani et al. 2013). Future studies might aim to investigate whether enhanced NO bioavailability via dietary nitrate supplementation results in such protective modifications, although it should be noted that complex I activity was preserved in the hearts of normoxic rats with dietary nitrate supplementation in our study.
Nitrate supplementation therefore protected cardiac ATP levels and also prevented the hypoxia-induced increase in protein carbonyls in heart, but notably also lowered protein carbonyls in the normoxic heart. Our findings suggest that nitrate improves myocardial oxygenation in hypoxia, although it remains unclear whether this is due to enhanced oxygen delivery via improvements in blood flow, or local effects to decrease oxygen utilization, for example by improving mitochondrial efficiency. Moreover, expression of the HIF-1 targets Vegfa and Pgk1 was downregulated in the hearts of nitrate-supplemented normoxic rats, possibly reflecting the elevated tissue nitrate levels we observed in these hearts, which may improve tissue oxygenation even in the absence of an environmental hypoxic stimulus; however, this raises a further question about the mechanism by which nitrate might elicit this protective effect. Nitrate supplementation also prevented the hypoxic rise in total plasma nitroso-compounds (nitrosothiols and nitrosamines, products the formation of which is known to be linked to cellular redox status) (Bryan et al. 2004), providing further evidence for reduced systemic oxidative stress following nitrate treatment.
Nitrate is widely believed to be a precursor of NO under hypoxic conditions via sequential reduction, first to nitrite and then to NO (Lundberg et al. 2008). The latter step involves multiple enzyme systems and is potently inhibited by oxygen (Feelisch et al. 2008), but despite its conceptual appeal as an NOS-independent route of NO production it is not yet clear what role, if any, the purported nitrate–nitrite–NO reduction pathway plays in physiology. While the mechanism by which nitrate administration gives rise to NO under normoxic conditions remains similarly unresolved (Tsikas et al. 2013), our findings suggest that nitrate improves tissue oxygenation even in the normoxic heart. A novel observation arising from our investigation is the suppression of cardiac arginase expression and activity by nitrate. This action could conceivably shift l-arginine utilisation away from arginase (and the production of ornithine and urea) towards NO synthase to enhance NO bioavailability under both normoxic and hypoxic conditions, with increased NO production preventing oxidative stress and the loss of complex I function (Fig.7). Of note, tissue arginase is elevated in hypoxia (Pernow & Jung, 2013), and increased activity has been reported in numerous cardiovascular pathologies, including congestive heart failure, myocardial ischaemia and pulmonary arterial hypertension (Pernow & Jung, 2013), whilst arginase II over-expression in the mouse endothelium induces endothelial dysfunction, hypertension and artherosclerosis (Vaisman et al. 2012). As arginase appears to exert its effects primarily through the phenomenon of ‘arginine steal’, arginase inhibition has been proposed as a therapeutic strategy in such pathologies to enhance NO bioavailability and limit oxidative stress. Our results suggest that this might be readily achievable via a moderate dose of dietary nitrate. Whilst the underlying mechanism remains incompletely resolved at this stage, nitrate administration decreases the expression of both arginase I and II, which are recognised targets of HIF-2; thus, the possible interaction between nitrate and the HIF-signalling pathway appears to warrant further investigation. Surprisingly little is known about the transport of nitrate across cell membranes and how intra- and extracellular concentrations are sensed and regulated. The seemingly paradoxical decrease in cardiac nitrate levels observed on nitrate supplementation under normoxic conditions suggests that measuring plasma levels alone may be inadequate to assess bodily NO/nitrate status and hints at a richer level of regulation than is commonly assumed. Our results are consistent with the notion that nitrate may be part of a negative feedback loop that serves to integrate dietary nitrate intake and NO production from NOS (where nitrate represents the final oxidation product). Indeed, a similar feedback mechanism has been proposed to exist for nitrite whereby low doses inhibit endothelial NOS to increase blood pressure (Bryan et al. 2005). Irrespective of the mechanistic details, the thought that the ubiquitous dietary constituent nitrate might contribute to bodily NO production in humans is intriguing.
The functional consequences of the changes to complex I expression and activity exerted by hypoxia deserve further consideration, as indeed do the protective effects of nitrate. Mice with a cardiac-specific complex I deficiency were recently shown to have normal cardiac function in vivo, but a predisposition to accelerated heart failure upon pressure overload stress or with repeated pregnancy (Karamanlidis et al. 2013). Thus, whilst complex I loss might exert protection under short-term hypoxic conditions or during ischaemia/reperfusion, the longer-term repercussions of ETC suppression and energetic impairment might include a predisposition to cardiac failure; indeed, poor cardiac energetics predicts mortality in patients with chronic heart failure (Neubauer, 2007).
The finding that nitrate supplementation protects cardiac mitochondrial function and energetics in sustained hypoxia sheds further light on the adaptations of high altitude native populations to life in chronic environmental hypoxia. Tibetans have elevated circulating levels of NO products associated with improved blood flow (Erzurum et al. 2007) and the source of nitrogen oxides in their case is not dietary. Instead, genomic studies aiming to elucidate the genetic basis of human high altitude adaptation have highlighted a region of the NOS2A gene on chromosome 17 as being distinctive in both Andeans and Tibetans compared with lowlanders (Bigham et al. 2009, 2010). NOS2A encodes inducible nitric oxide synthase (iNOS) and is a vital source of NO. Little is currently known about the energy metabolism or cardiac energetics of Tibetans, or whether enhanced nitrogen oxide availability improves the acclimatisation of lowland natives to high altitude, but these are likely to be fruitful avenues for further study. In addition, the potential of moderate dietary nitrate supplementation for the treatment of hypoxia-related metabolic diseases deserves further attention. For instance, heart failure is characterized by poor cardiac energetics, even in patients receiving optimal therapy, and the degree of energetic impairment predicts prognosis (Neubauer, 2007). Myocardial hypoxia (Willam et al. 2006), energetic impairment (Murray et al. 2006) and mitochondrial dysfunction (Murray et al. 2007, 2008) are all features of the non-necrotic, hypertrophied regions of the chronically infarcted rat heart, and this model of heart failure would be an interesting one in which to investigate the therapeutic and cardioprotective potential of dietary nitrate. Moreover, arginase levels are increased in the failing human heart and correlate with severity, whilst arginase inhibition improves microcirculatory function in these patients by increasing NO availability (Quitter et al. 2012). It would be interesting to investigate whether arginase was similarly protective in the pure hypoxia setting. Additionally, chronic obstructive pulmonary disease (COPD) shares many features with chronic hypoxic exposure at altitude, including similar patterns of muscle wasting, weight loss and altered cellular metabolism (Raguso et al. 2004) characterised by a decline in skeletal muscle mitochondrial function (Meyer et al. 2013). Meanwhile, in critically ill patients who experience hypoxaemia and tissue hypoxia, survival is strongly associated with the early induction of mitochondrial biogenesis (Carre et al. 2010), and nitrate supplementation might augment this process.
Conclusion
We report hypoxia-induced changes in mitochondrial function in the rat heart, which might underlie the metabolic perturbations observed in the hearts of healthy humans at high altitude. A moderate dose of dietary nitrate alleviated these abnormalities, preventing the hypoxia-driven downregulation of mitochondrial complex I, impaired energetics and oxidative stress, potentially via inhibition of arginase activity. Our observation that arginase is suppressed by nitrate, even in normoxia, holds promise for novel dietary interventions aimed at improving NO production under a variety of conditions unrelated to hypoxia.
Acknowledgments
The authors gratefully acknowledge the technical assistance of Mr Steven Murfitt.
Glossary
- BIOPS
biopsy-preservation solution
- COX
cytochrome c oxidase
- ETC
electron transport chain
- HIF
hypoxia-inducible factor
- NO
nitric oxide
- NOx
nitrogen oxides
- RCR
respiratory control ratio
- ROS
reactive oxygen species
Key points
Exposure to environmental hypoxia, at high altitude or in a chamber, impairs cardiac energetics and alters mitochondrial function.
Inorganic nitrate, a ubiquitous dietary constituent, improves mitochondrial efficiency, lowering the oxygen cost of exercise, whilst elevated circulating nitrogen oxide levels in high-altitude natives enhances blood flow.
Here we report that dietary nitrate supplementation prevents hypoxia-induced changes in cardiac mitochondrial function and energetics, whilst ameliorating oxidative stress, suggesting improved tissue oxygenation.
Furthermore, nitrate supplementation suppresses cardiac arginase expression and increases tissue l-arginine levels under both hypoxic and normoxic conditions, underpinning a novel mechanism to enhance the availability of nitric oxide.
Nitrate supplementation may thus be of benefit to individuals exposed to hypobaric hypoxia at altitude or in patients with diseases characterised by tissue hypoxia and energetic impairment, such as heart failure and chronic obstructive pulmonary disease, or in the critically ill.
Additional information
Competing interests
The authors confirm that there are no conflicts of interest.
Author contributions
The experiments in this study were primarily performed in the Department of Physiology, Development and Neuroscience, University of Cambridge, with further analysis of samples taking place in the Department of Biochemistry, University of Cambridge; Department of Physiology, Anatomy and Genetics, University of Oxford; and University Hospital Southampton. T.A., B.O.F., M.F. and A.J.M. conceived and designed the experiments. T.A., B.O.F., C.B-P., J.A.W., A.S.C., L.C.H., J.L.G., R.S.J., M.F. and A.J.M. collected, analysed and interpreted the experimental data. T.A., B.O.F., M.F. and A.J.M. drafted the article and revised it critically for important intellectual content. All authors have read and approved the manuscript.
Funding
This work was primarily funded by a British Heart Foundation PhD Studentship to T.A. (FS/09/050). The authors are also supported by a Research Councils UK Academic Fellowship to A.J.M., a Diabetes UK RD Lawrence Fellowship (11/0004175) to L.C.H., the EU Framework 7 Inheritance project to J.L.G., a Wellcome Trust Principal Research Fellowship to R.S.J. and funds from the Faculty of Medicine, University of Southampton to M.F.
References
- Bigham A, Bauchet M, Pinto D, Mao X, Akey JM, Mei R, Scherer SW, Julian CG, Wilson MJ, Lopez Herraez D, Brutsaert T, Parra EJ, Moore LG. Shriver MD. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 2010;6:e1001116. doi: 10.1371/journal.pgen.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigham AW, Mao X, Mei R, Brutsaert T, Wilson MJ, Julian CG, Parra EJ, Akey JM, Moore LG. Shriver MD. Identifying positive selection candidate loci for high-altitude adaptation in Andean populations. Hum Genomics. 2009;4:79–90. doi: 10.1186/1479-7364-4-2-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan NS, Fernandez BO, Bauer SM, Garcia-Saura MF, Milsom AB, Rassaf T, Maloney RE, Bharti A, Rodriguez J. Feelisch M. Nitrite is a signaling molecule and regulator of gene expression in mammalian tissues. Nat Chem Biol. 2005;1:290–297. doi: 10.1038/nchembio734. [DOI] [PubMed] [Google Scholar]
- Bryan NS, Rassaf T, Maloney RE, Rodriguez CM, Saijo F, Rodriguez JR. Feelisch M. Cellular targets and mechanisms of nitros(yl)ation: an insight into their nature and kinetics in vivo. Proc Natl Acad Sci USA. 2004;101:4308–4313. doi: 10.1073/pnas.0306706101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carre JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P, Stotz M. Singer M. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med. 2010;182:745–751. doi: 10.1164/rccm.201003-0326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cocheme HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RA, Krieg T, Brookes PS. Murphy MP. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi E, Brown GC, Feelisch M. Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc P, Rigoulet M, Leverve X. Fontaine E. Nitric oxide increases oxidative phosphorylation efficiency. J Bioenerg Biomembr. 2007;39:158–166. doi: 10.1007/s10863-007-9074-1. [DOI] [PubMed] [Google Scholar]
- Cole MA, Murray AJ, Cochlin LE, Heather LC, McAleese S, Knight NS, Sutton E, Jamil AA, Parassol N. Clarke K. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol. 2011;106:447–457. doi: 10.1007/s00395-011-0156-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colleoni F, Padmanabhan N, Yung HW, Watson ED, Cetin I, Tissot van Patot MC, Burton GJ. Murray AJ. Suppression of mitochondrial electron transport chain function in the hypoxic human placenta: a role for miRNA-210 and protein synthesis inhibition. PLoS One. 2013;8:e55194. doi: 10.1371/journal.pone.0055194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LM, Murray AJ, Tyler DJ, Kemp GJ, Holloway CJ, Robbins PA, Neubauer S, Levett D, Montgomery HE, Grocott MP. Clarke K. The effect of high-altitude on human skeletal muscle energetics: P-MRS results from the Caudwell Xtreme Everest expedition. PLoS One. 2010;5:e10681. doi: 10.1371/journal.pone.0010681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzurum SC, Ghosh S, Janocha AJ, Xu W, Bauer S, Bryan NS, Tejero J, Hemann C, Hille R, Stuehr DJ, Feelisch M. Beall CM. Higher blood flow and circulating NO products offset high-altitude hypoxia among Tibetans. Proc Natl Acad Sci U S A. 2007;104:17593–17598. doi: 10.1073/pnas.0707462104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feelisch M, Fernandez BO, Bryan NS, Garcia-Saura MF, Bauer S, Whitlock DR, Ford PC, Janero DR, Rodriguez J. Ashrafian H. Tissue processing of nitrite in hypoxia: an intricate interplay of nitric oxide-generating and -scavenging systems. J Biol Chem. 2008;283:33927–33934. doi: 10.1074/jbc.M806654200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin A. Moncada S. S-nitrosation of mitochondrial complex I depends on its structural conformation. J Biol Chem. 2007;282:37448–37453. doi: 10.1074/jbc.M707543200. [DOI] [PubMed] [Google Scholar]
- Grocott MP, Martin DS, Levett DZ, McMorrow R, Windsor J. Montgomery HE. Arterial blood gases and oxygen content in climbers on Mount Everest. N Engl J Med. 2009;360:140–149. doi: 10.1056/NEJMoa0801581. [DOI] [PubMed] [Google Scholar]
- Guzy RD. Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006;91:807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- Heather LC, Cole MA, Lygate CA, Evans RD, Stuckey DJ, Murray AJ, Neubauer S. Clarke K. Fatty acid transporter levels and palmitate oxidation rate correlate with ejection fraction in the infarcted rat heart. Cardiovasc Res. 2006;72:430–437. doi: 10.1016/j.cardiores.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Heather LC, Cole MA, Tan JJ, Ambrose LJ, Pope S, Abd-Jamil AH, Carter EE, Dodd MS, Yeoh KK, Schofield CJ. Clarke K. Metabolic adaptation to chronic hypoxia in cardiac mitochondria. Basic Res Cardiol. 2012;107:268. doi: 10.1007/s00395-012-0268-2. [DOI] [PubMed] [Google Scholar]
- Holloway C, Cochlin L, Codreanu I, Bloch E, Fatemian M, Szmigielski C, Atherton H, Heather L, Francis J, Neubauer S, Robbins P, Montgomery H. Clarke K. Normobaric hypoxia impairs human cardiac energetics. FASEB J. 2011a;25:3130–3135. doi: 10.1096/fj.11-183426. [DOI] [PubMed] [Google Scholar]
- Holloway CJ, Montgomery HE, Murray AJ, Cochlin LE, Codreanu I, Hopwood N, Johnson AW, Rider OJ, Levett DZ, Tyler DJ, Francis JM, Neubauer S, Grocott MP. Clarke K. Cardiac response to hypobaric hypoxia: persistent changes in cardiac mass, function, and energy metabolism after a trek to Mt Everest Base Camp. FASEB J. 2011b;25:792–796. doi: 10.1096/fj.10-172999. [DOI] [PubMed] [Google Scholar]
- Jacobs RA, Siebenmann C, Hug M, Toigo M, Meinild AK. Lundby C. Twenty-eight days at 3454-m altitude diminishes respiratory capacity but enhances efficiency in human skeletal muscle mitochondria. FASEB J. 2012;26:5192–5200. doi: 10.1096/fj.12-218206. [DOI] [PubMed] [Google Scholar]
- Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC, Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W. Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013;18:239–250. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MY. Powis G. Passing the baton: the HIF switch. Trends Biochem Sci. 2012;37:364–372. doi: 10.1016/j.tibs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R. Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008;3:965–976. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]
- Larsen FJ, Schiffer TA, Borniquel S, Sahlin K, Ekblom B, Lundberg JO. Weitzberg E. Dietary inorganic nitrate improves mitochondrial efficiency in humans. Cell Metab. 2010;13:149–159. doi: 10.1016/j.cmet.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Larsen FJ, Schiffer TA, Weitzberg E. Lundberg JO. Regulation of mitochondrial function and energetics by reactive nitrogen oxides. Free Radic Biol Med. 2012;53:1919–1928. doi: 10.1016/j.freeradbiomed.2012.08.580. [DOI] [PubMed] [Google Scholar]
- Larsen FJ, Weitzberg E, Lundberg JO. Ekblom B. Dietary nitrate reduces maximal oxygen consumption while maintaining work performance in maximal exercise. Free Radic Biol Med. 2009;48:342–347. doi: 10.1016/j.freeradbiomed.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Le Belle JE, Harris NG, Williams SR. Bhakoo KK. A comparison of cell and tissue extraction techniques using high-resolution 1H-NMR spectroscopy. NMR Biomed. 2002;15:37–44. doi: 10.1002/nbm.740. [DOI] [PubMed] [Google Scholar]
- Levett DZ, Fernandez BO, Riley HL, Martin DS, Mitchell K, Leckstrom CA, Ince C, Whipp BJ, Mythen MG, Montgomery HE, Grocott MP. Feelisch M. The role of nitrogen oxides in human adaptation to hypoxia. Sci Rep. 2011;1:109. doi: 10.1038/srep00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levett DZ, Radford EJ, Menassa DA, Graber EF, Morash AJ, Hoppeler H, Clarke K, Martin DS, Ferguson-Smith AC, Montgomery HE, Grocott MP. Murray AJ. Acclimatization of skeletal muscle mitochondria to high-altitude hypoxia during an ascent of Everest. FASEB J. 2012;26:1431–1441. doi: 10.1096/fj.11-197772. [DOI] [PubMed] [Google Scholar]
- Lundberg JO. Govoni M. Inorganic nitrate is a possible source for systemic generation of nitric oxide. Free Radic Biol Med. 2004;37:395–400. doi: 10.1016/j.freeradbiomed.2004.04.027. [DOI] [PubMed] [Google Scholar]
- Lundberg JO, Weitzberg E. Gladwin MT. The nitrate–nitrite–nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- Martin DS, Ince C, Goedhart P, Levett DZ. Grocott MP. Abnormal blood flow in the sublingual microcirculation at high altitude. Eur J Appl Physiol. 2009;106:473–478. doi: 10.1007/s00421-009-1023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A, Zoll J, Charles AL, Charloux A, de Blay F, Diemunsch P, Sibilia J, Piquard F. Geny B. Skeletal muscle mitochondrial dysfunction during chronic obstructive pulmonary disease: central actor and therapeutic target. Exp Physiol. 2013;98:1063–1078. doi: 10.1113/expphysiol.2012.069468. [DOI] [PubMed] [Google Scholar]
- Murray AJ. Metabolic adaptation of skeletal muscle to high altitude hypoxia: how new technologies could resolve the controversies. Genome Med. 2009;1:117. doi: 10.1186/gm117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ, Cole MA, Lygate CA, Carr CA, Stuckey DJ, Little SE, Neubauer S. Clarke K. Increased mitochondrial uncoupling proteins, respiratory uncoupling and decreased efficiency in the chronically infarcted rat heart. J Mol Cell Cardiol. 2008;44:694–700. doi: 10.1016/j.yjmcc.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Edwards LM. Clarke K. Mitochondria and heart failure. Curr Opin Clin Nutr Metab Care. 2007;10:704–711. doi: 10.1097/MCO.0b013e3282f0ecbe. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Lygate CA, Cole MA, Carr CA, Radda GK, Neubauer S. Clarke K. Insulin resistance, abnormal energy metabolism and increased ischemic damage in the chronically infarcted rat heart. Cardiovasc Res. 2006;71:149–157. doi: 10.1016/j.cardiores.2006.02.031. [DOI] [PubMed] [Google Scholar]
- Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- Pannala AS, Mani AR, Spencer JP, Skinner V, Bruckdorfer KR, Moore KP. Rice-Evans CA. The effect of dietary nitrate on salivary, plasma, and urinary nitrate metabolism in humans. Free Radic Biol Med. 2003;34:576–584. doi: 10.1016/s0891-5849(02)01353-9. [DOI] [PubMed] [Google Scholar]
- Pernow J. Jung C. Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovasc Res. 2013;98:334–343. doi: 10.1093/cvr/cvt036. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim Biophys Acta. 2012;1820:712–721. doi: 10.1016/j.bbagen.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pon LA, Schon EA American Society for Cell Biology. Mitochondria. San Diego: Academic Press; 2007. [Google Scholar]
- Quitter F, Figulla HR, Ferrari M, Pernow J. Jung C. Increased arginase levels in heart failure represent a therapeutic target to rescue microvascular perfusion. Clin Hemorheol Microcirc. 2012;54:75–85. doi: 10.3233/CH-2012-1617. [DOI] [PubMed] [Google Scholar]
- Raguso CA, Guinot SL, Janssens JP, Kayser B. Pichard C. Chronic hypoxia: common traits between chronic obstructive pulmonary disease and altitude. Curr Opin Clin Nutr Metab Care. 2004;7:411–417. doi: 10.1097/01.mco.0000134372.78438.09. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007;2007:cm8. doi: 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, Wang X, MacArthur PH, Shoja A, Raghavachari N, Calvert JW, Brookes PS, Lefer DJ. Gladwin MT. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–2102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siervo M, Stephan BC, Feelisch M. Bluck LJ. Measurement of in vivo nitric oxide synthesis in humans using stable isotopic methods: a systematic review. Free Radic Biol Med. 2011;51:795–804. doi: 10.1016/j.freeradbiomed.2011.05.032. [DOI] [PubMed] [Google Scholar]
- Tsikas D, Schneider JY. Frolich JC. Antihypertensive inorganic nitrate and nitrite: what is the underlying mechanism? Hypertension. 2013;62:e6. doi: 10.1161/HYPERTENSIONAHA.113.01646. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisman BL, Andrews KL, Khong SM, Wood KC, Moore XL, Fu Y, Kepka-Lenhart DM, Morris SM, Jr, Remaley AT. Chin-Dusting JP. Selective endothelial overexpression of arginase II induces endothelial dysfunction and hypertension and enhances atherosclerosis in mice. PLoS One. 2012;7:e39487. doi: 10.1371/journal.pone.0039487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AJ, Patel N, Loukogeorgakis S, Okorie M, Aboud Z, Misra S, Rashid R, Miall P, Deanfield J, Benjamin N, MacAllister R, Hobbs AJ. Ahluwalia A. Acute blood pressure lowering, vasoprotective, and antiplatelet properties of dietary nitrate via bioconversion to nitrite. Hypertension. 2008;51:784–790. doi: 10.1161/HYPERTENSIONAHA.107.103523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JB. The physiologic basis of high-altitude diseases. Ann Intern Med. 2004;141:789–800. doi: 10.7326/0003-4819-141-10-200411160-00010. [DOI] [PubMed] [Google Scholar]
- Willam C, Maxwell PH, Nichols L, Lygate C, Tian YM, Bernhardt W, Wiesener M, Ratcliffe PJ, Eckardt KU. Pugh CW. HIF prolyl hydroxylases in the rat; organ distribution and changes in expression following hypoxia and coronary artery ligation. J Mol Cell Cardiol. 2006;41:68–77. doi: 10.1016/j.yjmcc.2006.04.009. [DOI] [PubMed] [Google Scholar]