

Abstract
Herpes simplex virus type 1 (HSV-1), a member of the herpes virus family, is characterized by a short replication cycle, high cytopathogenicity and distinct neurotropism. Primary infection and reactivation may cause severe diseases in immunocompetent and immunosuppressed individuals. This study investigated the role of human plasmacytoid dendritic cells (pDC) in the activation of natural killer (NK) cells for the control of herpesviral infections. Within peripheral blood mononuclear cells, UV-inactivated HSV-1 and CpG-A induced CD69 up-regulation on NK cells, whereas infectious HSV-1 was particularly active in inducing NK cell effector functions interferon-γ (IFN-γ) secretion and degranulation. The pDC-derived IFN-α significantly contributed to NK cell activation, as evident from neutralization and cell depletion experiments. In addition, monocyte-derived tumour necrosis factor-α (TNF-α) induced after exposure to infectious HSV-1 was found to stimulate IFN-γ secretion. A minority of monocytes was shown to be non-productively infected in experiments using fluorescently labelled viruses and quantitative PCR analyses. HSV-1-exposed monocytes up-regulated classical HLA-ABC and non-classical HLA-E molecules at the cell surface in an IFN-α-dependent manner, whereas stress molecules MICA/B were not induced. Notably, depletion of monocytes reduced NK cell effector functions induced by infectious HSV-1 (P < 0·05). Altogether, our data suggest a model in which HSV-1-stimulated pDC and monocytes activate NK cells via secretion of IFN-α and TNF-α. In addition, infection of monocytes induces NK cell effector functions via TNF-α-dependent and TNF-α-independent mechanisms. Hence, pDC and monocytes, which are among the first cells infiltrating herpetic lesions, appear to have important bystander functions for NK cells to control these viral infections.
Keywords: cytokines, dendritic cells, human, natural killer cells, viral
Introduction
Herpes simplex virus type 1 (HSV-1) is a member of the α-herpes virus subfamily with a seroprevalence of 70–80%.1 It is characterized by a short replication cycle, high cytopathogenicity and distinct neurotropism.2 Primary infections cause lytic lesions at oral or genital mucocutaneous sites, followed by transport of the virus to trigeminal or dorsal root ganglia, where lifelong latency is established. Reactivations occur frequently, usually causing self-limiting oral or genital lesions. Under conditions of immunosuppression, but also in immunocompetent individuals, primary infection and reactivation may cause severe sequelae such as encephalitis, acute retinal necrosis or systemic disease.
Studies in children suffering from severe herpes simplex infections revealed the central role of type I interferon (IFN) production and signalling for the innate immune control of these viruses.3 Early studies by Fitzgerald-Bocarsly identified the ‘interferon-producing cells’ as an important accessory cell population for the cytolytic killing of HSV-infected fibroblasts.4 These cells were subsequently characterized as plasmacytoid dendritic cells (pDC), the major producers of type I interferons in the blood.5,6 The role of pDC in the immune control of HSV infections was confirmed in murine models. Local footpad and corneal HSV-1 infections in MyD88−/− and Toll-like receptor 9−/− mice resulted in decreased IFN-α production, but mice were still able to control the infection.7 Increased pathogenesis in genital HSV-2 infections was observed after antibody-dependent pDC depletion8 and in IFN-α receptor knockout mice.9 Recently, specific depletion of pDC in CLEC4C-DTR transgenic mice corroborated the crucial role of these cells in IFN-α production, secretion of pro-inflammatory cytokines, and survival in systemic, but not local, HSV infections.10
Along with pDC, natural killer (NK) cells are important in inducing innate anti-HSV responses.11,12 NK cells were first identified as killing tumour cells without previous activation.13 In follow-up studies, it became clear that tumour cells, virus-infected and allogeneic cells induced NK cell effector functions via the ‘missing-self’ HLA repertoire at the cell surface.14 NK cells comprise a CD16+ CD56dim subset, which accounts for the majority of blood NK cells, migrates to the site of infection and is mostly cytolytic. The minor CD16(+) CD56bright subset migrates to lymphatic tissue and mostly secretes cytokines, in particular IFN-γ.15 A murine model of ocular HSV-1 infection showed anterior-to-posterior spread of HSV-1 after NK cell depletion.16 Interleukin-15 deficient (IL-15−/−) mice lacking NK cells were found to be 100-fold more susceptible to genital HSV-2 infection, while mice lacking IFN-γ were only 10-fold more susceptible than control mice.17 NK cell depletion resulted in increased HSV-1 titres in the lung after intranasal inoculation of mice.18
Follow-up studies addressed the interplay of pDC and NK cells, with continuing discussions about the role of soluble and cell-associated factors. Human NK cell activation and cytolytic functions were reported to be induced by pDC-derived type I IFN upon stimulation with influenza virus, CpG and poly (I:C).19 Other studies described how pDC-derived IFN-α and tumour necrosis factor-α (TNF-α) were responsible for CpG-induced NK cell activation and IFN-γ secretion,20 whereas NK cell degranulation and cytotoxicity required direct contact with pDC.21 In recurrent human HSV-2 lesions, infiltrating pDC were detected in close proximity to activated T lymphocytes and NK cells.22 Murine models confirmed that NK cell activation required type I IFN signalling as IFN-α receptor knockout mice lacked IFN-γ production in vaginal HSV-2 infections.9 In systemic HSV infections of CLEC4C-DTR mice, pDC were shown to be important for NK cell activation, IFN-γ production, and degranulation.10
Studies addressing the cross-talk between NK cells and pDC in human HSV infections are still limited. Therefore, we used sucrose gradient-purified HSV-1 to analyse in detail the induction of NK cell activation by ultraviolet-inactivated (HSVUV) and infectious (HSVINF) virus within the peripheral blood mononuclear cell (PBMC) context. Plasmacytoid DC and monocytes were involved in HSV-1-induced NK cell activation, but infection of monocytes additionally induced NK cell effector functions. In these processes, IFN-α and TNF-α were determined as crucial cytokines. Our findings appear to be important for the control of herpes virus infections as monocytes, NK cells and pDC are among the first cells infiltrating herpetic lesions.22
Material and methods
Isolation and cultivation of cells
The PBMC were isolated from EDTA-anticoagulated blood of healthy volunteers using standard Biocoll density gradient centrifugation (Biochrom AG, Berlin, Germany). A total of 21 different donors (13 female, eight male) were included in the study (age range 25–55 years). Plasmacytoid DC were purified or depleted from PBMC using the CD304 MicroBead Kit with MS/LS columns (Miltenyi Biotec, Bergisch-Gladbach, Germany), as previously described.23,24 Monocytes were purified or depleted from PBMC by positive selection using CD14 MicroBeads, and NK cells by negative selection using the NK Cell Isolation Kit (both Miltenyi Biotec) according to the manufacturer’s recommendations. The purity of isolated NK cells was regularly above 93%. Cell viability was analysed by trypan blue staining. Cells were cultivated in RPMI-1640 (Invitrogen, Darmstadt, Germany), supplemented with 10% heat-inactivated (56°, 60 min) fetal calf serum (FCS; Sigma-Aldrich, Munich, Germany), 0·3 mg/ml glutamine, 200 U/ml penicillin and 90 U/ml streptomycin. The study was approved by the Ethical Committee of the Medical Faculty, Friedrich-Alexander-Universität Erlangen-Nürnberg (No. 3299).
Generation of viral stocks
Vero cells deficient for IFN-α and IFN-β1 genes25 were infected with a clinical HSV-1 isolate26 for 2 hr, washed and incubated in Dulbecco’s modified Eagle’s medium (Invitrogen) with 10% FCS and supplements as described above. After 3–4 days of infection, the cell culture was subjected to two freeze–thaw cycles. Lysates were cleared by centrifugation at 440 g for 5 min, and the resulting supernatants were centrifuged at 50 000 g at 4° for 90 min. Viral pellets were incubated in the residual liquid overnight at 4°, resuspended, dounced 20 times and then loaded onto a continuous gradient (30% to 15% sucrose in virus standard buffer, 0·05 m Tris–HCl, 0·012 m KCl, 0·005 m EDTA, 0·1% BSA). After centrifugation at 50 000 g for 30 min, the visible viral layer was harvested and centrifuged at 78 000 g for 90 min. Virus pellets were filtered through 0·22-μm pores and stored at −80° (HSVINF). Aliquots of the viral stocks were completely inactivated (HSVUV) by application of 1 Joule/cm2 using the Bio-Link 254 UV cross-linker (Vilber Lourmat, Eberhardzell, Germany). The autofluorescing HSV-1 166v isolate, which expresses a green fluorescent protein (GFP) -fused VP22 protein,27 was propagated on Vero cells, and the HSV-1d106S isolate,28 which expresses GFP under the control of the CMV promoter, was propagated on complementing E11 cells. Stocks of 166v were only filtered through 0·22-μm pores, whereas supernatants containing HSV-1d106S were sucrose-purified as described above. The 50% tissue culture infective dose was determined for all viral stocks.
Generation of pDC supernatants
A total of 5 × 105 pDC were exposed to HSV-1INF (multiplicity of infection 1) at 37° for 3 hr, washed with Dulbecco’s PBS and incubated with trypsin EDTA at 37° for 15 min. Cells were washed and cultivated in RPMI-1640 media with supplements including 20 ng/ml IL-3 (R&D Systems, Wiesbaden-Nordenstadt, Germany) for 18 hr before removal of supernatants.
Stimulation of cells
Either PBMC or respectively depleted cells were plated at a density of 1 × 106 cells/500 μl in 24-well flat-bottom plates (Greiner, Frickenhausen, Germany). Cells were stimulated using the endotoxin-free CpG-A oligodeoxynucleotide (ODN) 6016 (5′-T*C-G-A-C-G-T-C-G-T-G-G*G*G*G-3′, where * represents phosphorothioate and - represents a phosphodiester bond) at 0·75 μm, provided by Coley Pharmaceutical GmbH – A Pfizer Company (Düsseldorf, Germany); HSV-1INF at a multiplicity of infection of 0·5 or a corresponding volume of HSVUV. Recombinant human IL-2 (Roche-Pharma, Grenzach-Wyhlen, Germany) was used as a positive control stimulus at a concentration of 100 U/ml in a separate culture condition. Supernatants were harvested at the indicated time periods after stimulation and stored at −20°, whereas cells were immediately processed for FACS analysis. NK cells were plated at a density of 2·5 × 105 cells/500 μl and stimulated with increasing concentrations of recombinant human IFN-α2b (hrIFN-α) (Miltenyi Biotec) or pDC supernatants containing corresponding levels of IFN-α2a/2b for 3–18 hr.
Neutralization experiments
Neutralization experiments were performed using murine IgG1 antibodies to IL-1β (clone 8516) and TNF-α (clone 28 401) together with the mouse IgG1 isotype control (clone 11 711) (all R&D Systems); and the murine IgG2a antibody to the human IFN-α receptor (clone MMHAR-2) together with the mouse IgG2a isotype control (clone PPV-04) (both Acris, Herford, Germany). The concentration of antibodies used for neutralization was 15 μg/ml throughout the study.
Infection experiments
A total of 5 × 105 monocytes were exposed to HSV-1INF, HSV-1UV and HSV-1 166v (multiplicity of infection 1) for 24 and 48 hr, and subsequently analysed for the expression of GFP, CD14 (clone 61D3; AbD Serotec, Düsseldorf, Germany), CD33 (clone WM53), CD64 (clone 10·1), HLA-ABC (clone W6/32), HLA-E (clone 3D12), and stress-induced molecules MICA/MICB (clone 6D4; all Biolegend, London, UK). Cellular contaminations within the monocyte preparation were identified using CD1c (clone L161; Biolegend), CD3 (clone UCHT; Biolegend or AbD Serotec, Düsseldorf, Germany), CD19 (clone HIB19; Biolegend), CD56 (clone HCD56; Biolegend), and CD123 (clone AC145; Miltenyi Biotec).
FACS analysis
Cells were incubated on ice for 10 min and harvested by thorough pipetting. They were washed once with FACS buffer (Dulbecco’s PBS plus 1% FCS and 1 mm EDTA) and incubated with FcR blocking reagent (Miltenyi Biotec) at 4° for 10 min. Then, cells were stained with antibodies against specific cell surface markers at 4° for 20 min, washed, and fixed using a 4% paraformaldehyde solution. In all experiments, NK cells were identified as positive for CD56 and negative for CD3 and CD14. Within PBMC, pDC were identified as positive for CD303 (Miltenyi Biotec) and negative for CD3 and CD14; monocytes as positive for CD14; T cells as positive for CD3, negative for CD14, and positive for CD4 (clone RPA-T4; Biolegend), CD8 (clone RPA-T8; Biolegend, or clone MEM-31; Immunotools, Friesoythe, Germany), or TCR-γ/δ (clone 11F2; Miltenyi Biotec); B cells as positive for CD19 and negative for CD3 and CD14. Respective isotype antibodies were used as controls. Activation of cells was evaluated via FITC- or Alexa Fluor 700-labelling of CD69 (clone FN50 obtained from Miltenyi Biotec or Biolegend, respectively). Cellular degranulation was investigated by adding 5 μl of the Alexa Fluor 488-labelled CD107a antibody (clone eBioH4A3; eBioscience, Frankfurt, Germany) to the cell culture 1·5 hr before cell harvest. Live and dead cell staining was performed using a Fixable Violet Dead Cell Stain Kit (Invitrogen). Cells were collected using the multiparameter LSR-II flow cytometer together with the facsdiva software for automatic compensation and measurement of samples (BD Biosciences, Heidelberg, Germany). Data were analysed using the fcs express 3 Software (De Novo Software, Los Angeles, CA).
Quantification of cytokines
IFN-α2a/2b levels in the cell culture supernatants were measured using an ELISA module set (eBioscience); samples with values above the linear range were diluted as appropriate. Other cytokines were quantified using the Th1/Th2 11 plex RTU FlowCytomix Multiplex assay and flowcytomixpro software according to the manufacturer’s recommendations (eBioscience). IFN-γ and TNF-α production by different cells was determined using the respective Secretion Assay Detection Kits (Miltenyi Biotec). In brief, harvested cells were washed with cold cytokine buffer, resuspended in cold media with supplements, and incubated on ice for 5 min after addition of the cytokine catch reagent antibody. Thereafter, cells were incubated in warm media with supplements in a microtube shaker for 45 min. The reaction was stopped by adding cold cytokine buffer, followed by FACS labelling.
Statistics
All experiments were performed using cells of different donors. The Student’s t-test was used for comparisons of two datasets, the Tukey HSD test for three or more datasets to account for multiple comparisons. Two-sided P-values ≤ 0·05 were considered significant.
Results
Stimulation of PBMC with HSV-1 leads to NK cell activation
To analyse the potential of HSV-1 to induce NK cell activation and effector functions within the PBMC context, PBMC were exposed to HSVUV and HSVINF. IL-2 and CpG-A served as control stimuli, representing direct29 and pDC-dependent30 NK cell activation, respectively. As evident from flow cytometry analyses, all stimuli significantly up-regulated CD69 expression on NK cells compared with mock at 12 hr post stimulation (P < 0·01) (Fig. 1a). In these experiments, HSVINF was significantly more active than HSVUV on CD56dim NK cells (P < 0·01). At 18 hr post stimulation, all stimuli were similarly active on CD56dim NK cells, whereas CD56bright NK cells were more pronouncedly activated by HSVINF compared with HSVUV (P < 0·05) (data not shown). This indicates faster and more potent NK cell activation by HSVINF versus HSVUV. In contrast, the ratio of CD56bright to CD56dim NK cells and hence NK cell differentiation was not affected by any of the stimuli used (data not shown).
Figure 1.
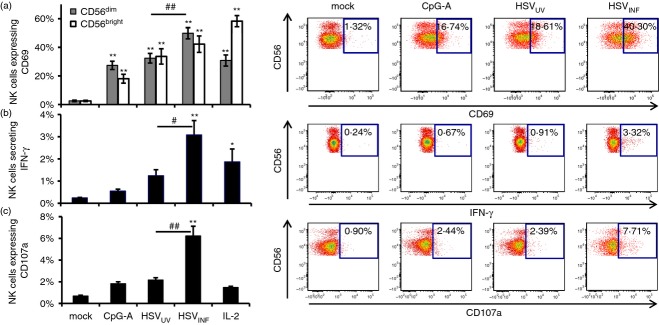
Stimulation of peripheral blood mononuclear cells (PBMC) with herpes simplex virus type 1 (HSV-1) induces natural killer (NK) cell activation. A total of 1 × 106 PBMC were exposed to CpG-A (ODN 6016), UV-inactivated and infectious HSV-1 (HSVUV and HSVINF), and interleukin-2 (IL-2). At 12 hr post stimulation, NK cell activation, cytokine secretion and degranulation were determined via CD69 expression, interferon-γ (IFN-γ)secretion, and CD107a surface expression, respectively. The percentages of NK cells expressing these markers are given as mean and standard error of 15 independent experiments. *P < 0·05, **P < 0·01 versus mock; #< 0·05, ##P < 0·01 HSVUV versus HSVINF (Tukey HSD). A representative FACS plot is shown on the right.
To find out whether CD69 up-regulation reflected induction of NK cell effector functions, we simultaneously evaluated IFN-γ secretion and surface expression of CD107a. This lysosomal-associated membrane protein 1 is only expressed at the cell surface after degranulation. The correlation between CD107a surface expression and cytokine secretion as well as cytotoxicity was demonstrated for NK cells.31 At 12 hr post stimulation, CpG-A and HSVUV were not significantly different from the mock control in inducing IFN-γ secretion or degranulation (Fig. 1b,c). Interleukin-2 induced IFN-γ secretion (P < 0·05) but no degranulation, while HSVINF induced significant IFN-γ secretion and degranulation compared with mock and HSVUV (P < 0·05) (Fig. 1b,c). These data underline the importance of viral infectivity for the induction of NK cell effector functions.
HSV-1 activates NK cells in part via IFN-α induction
IFN-α was reported as a major stimulus for NK cell activation.4 Therefore, we investigated whether HSV-1 activates NK cells within PBMC via induction of IFN-α production. CpG-A, HSVUV and HSVINF induced significant IFN-α production at 12 and 18 hr post stimulation (P < 0·05) (Fig. 2a). HSVINF was more active than HSVUV at 12 hr post stimulation (P < 0·05), which was no longer significant at 18 hr post stimulation. The effect of IFN-α on purified NK cells was evaluated in more detail performing time–course experiments. Supernatants of HSVINF-stimulated pDC containing IFN-α2a/2b concentrations as low as 20 and 40 pg/ml significantly up-regulated CD69 compared with mock at 12 and 3 hr post stimulation, respectively (P < 0·05) (Fig. 2b). To compare the effect of pDC supernatants to human recombinant IFN-α2b (hrIFN-α), both solutions were adjusted to similar concentrations of IFN-α2a/2b. Serial dilutions confirmed IFN-α as a potent stimulus for NK cell activation and revealed a more pronounced activity of pDC supernatants (Fig. 2c). This activity was significantly reduced by neutralizing the IFN-α receptor (P < 0·05) (Fig. 2d), indicating that IFN-α and other type I IFNs are the main soluble factors in pDC-induced NK cell activation.
Figure 2.
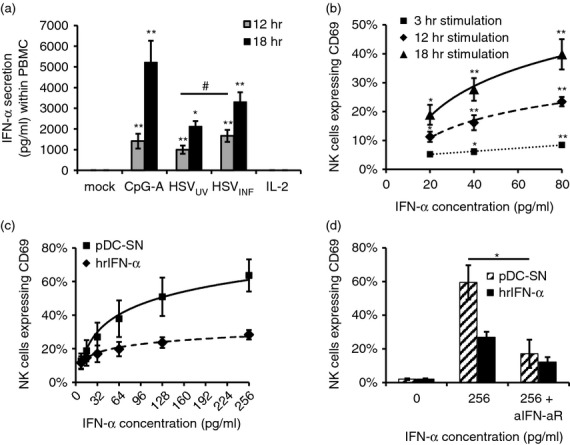
Herpes simplex virus type 1 (HSV-1) activates natural killer (NK) cells in part via interferon-α (IFN-α). (a) IFN-α production after stimulation of peripheral blood mononuclear cells with CpG-A, UV-inactivated and infectious HSV-1 (HSVUV and HSVINF), and interleukin-2 (IL-2) for 12 and 18 hr. Results obtained by ELISA represent mean and standard error of 15 and 7 independent experiments, respectively. *P < 0·05, **P < 0·01 versus mock; #P < 0·05 HSVUV versus HSVINF (Tukey HSD). (b) Time- and concentration-dependent up-regulation of CD69 on purified NK cells after exposure to supernatants obtained from HSVINF-stimulated plasmacytoid dendritic cells (pDC-SN). Data obtained by flow cytometry represent mean and standard error of three independent experiments. *P < 0·05, **P < 0·01 versus mock (Tukey HSD). (c) Comparison of CD69 up-regulation on purified NK cells after exposure to pDC-SN and human recombinant interferon-α2b (hrIFN-α) for 18 hr. (d) Effect of a neutralizing antibody to the IFN-α receptor (aIFN-aR) on activation of purified NK cells. Data represent mean and standard error of four independent experiments. *P < 0·05, mock versus aIFNa-R (Student’s t-test).
TNF-α plays a major role in HSV-1-induced NK cell activation
To find out whether other cytokines besides type I IFNs are involved in NK cell activation, we performed a Th1/Th2 multiplex cytokine bead array on supernatants of PBMC stimulated with CpG-A, HSVUV and HSVINF. Neither stimulus induced significant secretion of IL-2, IL-4, IL-5, IL-10, IL-12p70, IFN-γ or TNF-β, whereas CpG-A up-regulated IL-6 (P < 0·05) (data not shown). Interleukin-8 was secreted in all samples including the mock control, and two cytokines, namely IL-1β and TNF-α, were significantly increased in HSVINF-stimulated PBMC compared with mock and HSVUV (P < 0·05) (Fig. 3a). To investigate the role of these cytokines in HSVINF-induced NK cell activation, PBMC were stimulated in the presence of neutralizing antibodies against IL-1β, TNF-α or the isotype control. Neutralization of TNF-α significantly reduced CD69 up-regulation on NK cells induced by CpG-A and HSV-1 at 12 hr post stimulation (P < 0·01) (Fig. 3b). TNF-α was also significantly involved in HSVINF-induced IFN-γ secretion by NK cells (P < 0·05), whereas it had no effect on NK cell degranulation (Fig. 3b). These findings evidence a crucial role for TNF-α in HSVINF-induced NK cell activation and IFN-γ secretion, while neutralization of IL-1β had no effect.
Figure 3.
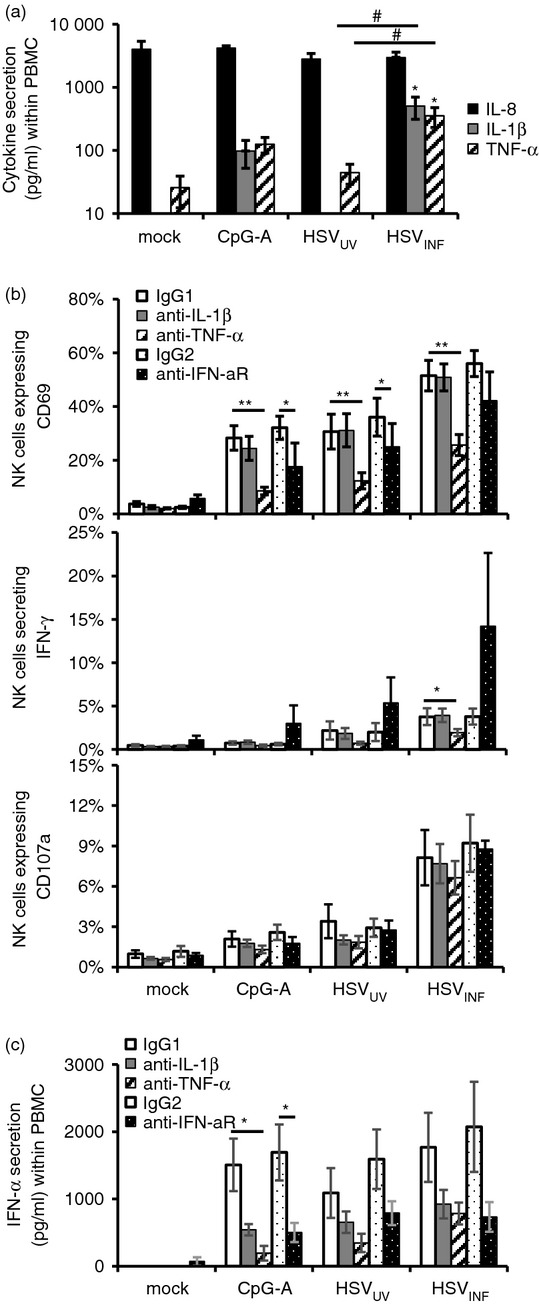
Herpes simplex virus type 1 (HSV-1) activates natural killer (NK) cells in part via tumour necrosis factor-α (TNF-α). (a) Secretion of interleukin-8 (IL-8), IL-1β and TNF-α in peripheral blood mononuclear cells (PBMC) stimulated with CpG-A, UV-inactivated and infectious HSV-1 (HSVUV and HSVINF) for 18 hr. Data were obtained using a T helper type 1 (Th1)/Th2 11 multiplex cytokine bead array. Only data with significant differences between HSVINF and HSVUV are shown. Results represent mean and standard error of seven independent experiments. *P < 0·05 versus mock; #P < 0·05 HSVUV versus HSVINF (Tukey HSD). (b) Percentage of NK cells expressing CD69, interferon-γ (IFN-γ), and CD107a, and (c) IFN-α production after stimulation of PBMC in the presence of neutralizing antibodies to IL-1β, TNF-α and IFN-α receptor (anti-IFN-aR). Results represent mean and standard error of five independent experiments. *P < 0·05, **P < 0·01 neutralizing antibody versus respective isotype control (Student’s t-test).
To study the contribution of pDC-derived IFN-α in HSVINF-induced NK cell activation, PBMC were stimulated in the presence of a neutralizing antibody against the IFN-α receptor. This blocking diminished CpG-A-induced and HSVUV-induced CD69 up-regulation (P = 0·05 and P < 0·01, respectively), but had no effect on NK cell effector functions (Fig. 3b). Neutralization of the IFN-α receptor decreased IFN-α secretion within PBMC (Fig. 3c), which was consistent with the known autocrine loop.32 A similar effect was observed with neutralization of IL-1β and TNF-α, suggesting that all three cytokines are involved in the secretion of large amounts of IFN-α upon stimulation with CpG-A and HSV-1.
Monocytes contribute to HSV-1-induced TNF-α production
Since TNF-α appeared to be a key effector molecule in the HSVINF-induced NK cell activation, we analysed the secretion of this respective cytokine upon stimulation of PBMC using flow cytometry analyses. Seven individual cell populations, namely pDC, monocytes, B cells, NK cells, γδ T cells, CD4+ and CD8+ T cells, responded to HSVINF exposure with significant TNF-α secretion compared with mock (P < 0·05) (Fig. 4a). Plasmacytoid DC and monocytes also responded to stimulation with CpG-A and HSVUV (P < 0·05), and B cells to CpG-A (P < 0·01). A significant difference between HSVUV and HSVINF stimulation was only observed for monocytes (P < 0·01).
Figure 4.
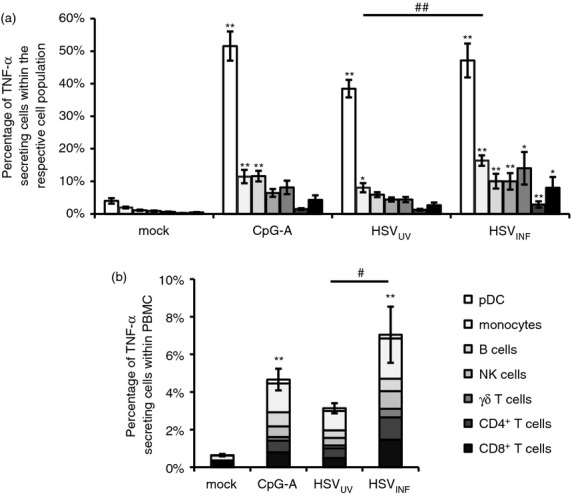
Plasmacytoid dendritic cells (pDC) and monocytes are major sources of tumour necrosis factor-α (TNF-α) upon exposure to herpes simplex virus type 1 (HSV-1). (a) Percentages of pDC, monocytes, B cells, natural killer (NK) cells, γδ T cells, CD4+ and CD8+ T cells secreting TNF-α at 18 hr post stimulation, analysed by flow cytometry for the individual cell populations. (b) Overall TNF-α secretion within peripheral blood mononuclear cells (PBMC), considering the frequency of each cell type within the PBMC population. Data represent mean and standard error of five independent experiments. *P < 0·05, **P < 0·01 versus mock; #P < 0·05, ##P < 0·01 HSVUV versus HSVINF (Tukey HSD). The figure legend applies to both figures.
The percentage of cells secreting TNF-α was highest among pDC. However, they are a rare cell population within PBMC. To adjust for the different frequencies of individual cell populations within PBMC, we multiplied the percentage of TNF-α-secreting cells within an individual cell population by the frequency of this cell population within PBMC (Fig. 4b). When the percentages of all TNF-α secreting cell populations were assembled, significant TNF-α secretion was only observed after stimulation with CpG-A and HSVINF (P < 0·01). HSVINF induced significantly more TNF-α than HSVUV (P < 0·05), which confirmed the data obtained in the bead array. In this analysis, monocytes were identified as most numerous TNF-α producing cell population upon stimulation with HSVINF.
Monocytes can be infected by HSV-1
As TNF-α secretion by monocytes was significantly affected by HSV-1 infectivity (Fig. 4a), and HSV-1 may infect monocytes as reported in early studies,33 we decided to investigate monocytes as potential target cells for HSV-1 infection. Monocytes were isolated from PBMC and exposed to HSVUV, HSVINF and HSVGFP expressing a GFP-VP22 fusion protein.27 The percentage of monocytes expressing GFP in flow cytometry analyses was significantly higher after exposure to HSVGFP than to mock, HSVUV and HSVINF at 24 hr (P < 0·01) and 48 hr (P < 0·01 for mock and HSVUV, n.s. for HSVINF) (Fig. 5a). GFP expression was already observed at 12 hr post infection (data not shown). The percentage of infected monocytes declined from 24 to 48 hr, indicating abortive rather than productive infection, in concordance with early observations by other groups.33,34 To investigate whether productive infection occurred, supernatants of HSV-1-infected monocytes were analysed for HSV-1 DNA. Quantitative PCR analyses showed replication of HSVGFP in control fibroblasts, while HSV-1 DNA dropped sharply in supernatants of these cells after exposure to infectious, but non-replicative HSVd106S.28 In monocytes, replication kinetics of HSVGFP and HSVd106S were similar, confirming non-productive infection in these cells (Fig. 5b). Since VP22 is a tegument protein present within viral particles, the observed fluorescence might have resulted from viruses sticking to the outside of exposed monocytes. Therefore, experiments were repeated using HSVd106S, which expresses GFP under the control of the cytomegalovirus promoter. Cells exposed to this virus only fluoresce if they have been infected and express GFP. Infection rates with HSVd106S were similar to HSVGFP (Fig. 5c), confirming actual infection of a minority of monocytes by HSV-1.
Figure 5.
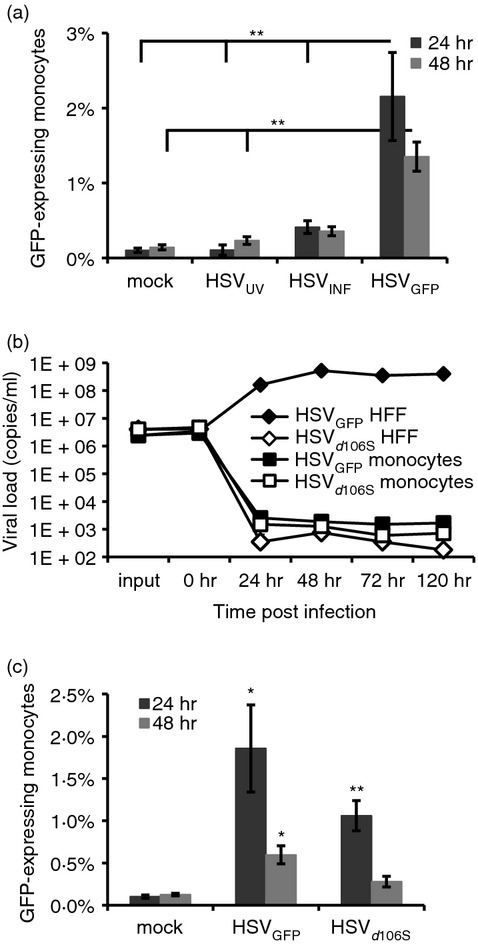
Herpes simplex virus type 1 (HSV-1) infects a minority of monocytes non-productively. (a) Percentages of purified monocytes expressing green fluorescent protein (GFP) as analysed by flow cytometry at 24 and 48 hr post infection with HSVGFP, which codes for a GFP-fused VP22 protein. UV-inactivated and infectious HSV-1 (HSVUV and HSVINF) were included as controls. Data represent mean and standard error of 11 independent experiments. **P < 0·01 as indicated (Tukey HSD). (b) Quantitative PCR analyses of HSV-1 DNA in supernatants of HSVGFP- and HSVd106S-infected human foreskin fibroblasts (HFF) and monocytes at indicated time-points post infection. Results were reproduced with monocytes of three donors. (c) Purified monocytes were infected in parallel with HSVGFP and HSVd106S, which expresses GFP under the control of the cytomegalovirus promoter. Data represent mean and standard error of six independent experiments. *P < 0·05, **P < 0·01 versus mock (Student’s t-test).
Monocytes up-regulate MHC-I, not MICA/B, upon exposure to infectious HSV-1
Down-regulation of MHC-I molecules by HSV-1 might provide an explanation for the activation of NK cells by HSV-1-infected monocytes. Therefore, we investigated the expression of classical HLA-ABC and non-classical HLA-E molecules on monocytes. HLA-ABC molecules were up-regulated on monocytes upon stimulation with HSVINF and HSVGFP compared with HSVUV at 24 and 48 hr post stimulation (P < 0·01), with rising kinetics from 24 to 48 hr (Fig. 6a). Similar data were obtained using HSVd106S (data not shown) and investigating HLA-E up-regulation (Fig. 6b). These findings indicate that MHC-I molecules are up-regulated and not down-regulated on monocytes upon exposure to infectious HSV-1. Since IFN-α was reported to induce up-regulation of MHC-I molecules on cells,35 monocyte supernatants were analysed for IFN-α production. Reproducible secretion was observed only after stimulation with HSVINF and HSVGFP, but not HSVUV (Fig. 6c). To test the hypothesis that IFN-α was responsible for HLA-ABC and HLA-E up-regulation, we conducted neutralization experiments infecting monocytes with HSVGFP in the presence of the IFN-α receptor antibody or the respective isotype. Neutralization of the IFN-α receptor significantly prevented up-regulation of HLA-ABC (Fig. 6d) and HLA-E (data not shown) (all P < 0·05) and increased monocyte infection at 48 hr post infection (Fig. 6e). Consequently, IFN-α restricts HSV-1 infection in exposed monocytes and up-regulates MHC-I molecules on the cell surface. We also analysed expression of stress molecules MICA/MICB, because they are known to trigger NK cell activation.14 Monocytes did not express MICA/MICB upon exposure to HSV-1 (Fig. 6f). Hence, HSV-stimulated monocytes do not contribute to NK cell activation via MHC-I down-regulation or expression of MICA/MICB.
Figure 6.
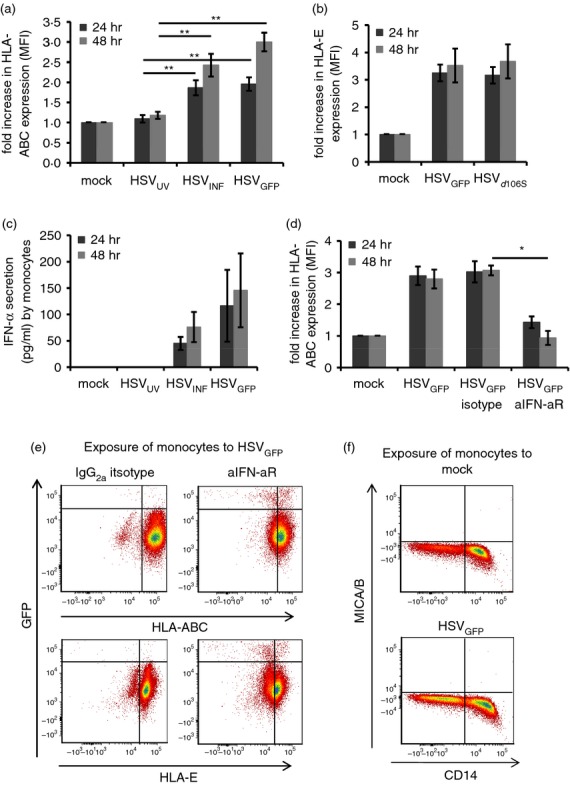
Herpes simplex virus type 1 (HSV-1) induces up-regulation of MHC-I, but not stress molecules MICA/B. (a) Fold increase in HLA-ABC or (b) HLA-E expression as analysed by flow cytometry after exposure of purified monocytes to HSVUV, HSVINF, HSVGFP, and HSVd106S for 24 and 48 hr. **P < 0·01 compared with HSVUV (Tukey HSD). Results are representative of eleven (HLA-ABC) and three (HLA-E) independent experiments. (c) Interferon-α (IFN-α) production after exposure of purified monocytes to HSVUV, HSVINF, and HSVGFP for 24 and 48 hr. Data represent eight independent experiments. (d) HLA-ABC expression on monocytes exposed to HSVGFP in the presence of a neutralizing IFN-α receptor antibody (aIFN-aR). Data represent three independent experiments. *P < 0·05, **P < 0·01 compared with the isotype control (Student’s t-test). All merged data are presented as mean and standard error. (e) Representative example of GFP and HLA-ABC- and HLA-E expression in monocytes exposed to HSVGFP in the presence of aIFN-aR or IgG2a isotype. (f) Representative example for MICA/B expression on isolated monocytes 24 hr post stimulation.
Monocytes mediate NK cell effector functions upon HSV-1 infection
To quantify the actual contribution of monocytes in comparison to pDC to HSVINF-induced NK cell activation, we conducted cell depletion experiments. Depletion of monocytes and pDC significantly reduced CD69 up-regulation induced by HSVUV and HSVINF (P < 0·05) (Fig. 7a). For HSVINF (not HSVUV), depletion of monocytes had a much stronger effect than depletion of pDC (P < 0·01), confirming the important role of pDC and particularly monocytes in HSVINF-induced NK cell activation. Further, monocytes (not pDC) were responsible for inducing IFN-γ secretion (P < 0·05) (Fig. 7b) and degranulation (P < 0·01) (Fig. 7c) after exposure of PBMC to HSVINF, indicating that monocytes are indispensable for the induction of NK cell effector functions by HSVINF. Depletion of pDC and monocytes significantly reduced IFN-α production (P < 0·05) (Fig. 7d), pinpointing both cell populations as major contributors to IFN-α secretion. Altogether, our data reveal that pDC-triggered NK cell activation by HSVINF depends on the infection of monocytes, which eventually induces NK cell effector functions via TNF-α-dependent and TNF-α-independent mechanisms.
Figure 7.

Herpes simplex virus type 1 (HSV-1)-exposed monocytes induce natural killer (NK) cell effector functions. NK cell expression of (a) CD69, (b) interferon-γ (IFN-γ), and (c) CD107a, as well as (d) IFN-α production after stimulation of peripheral blood mononuclear cells (PBMC) with CpG-A (ODN 6016), UV-inactivated and infectious HSV-1 (HSVUV and HSVINF). PBMC were either used as isolated or after depletion (delta) of monocytes or plasmacytoid dendritic cells (pDC). Data represent mean and standard error of eight independent experiments. *P < 0·05, **P < 0·01 as indicated (Tukey HSD).
Discussion
Our systematic analysis of cytokine induction and NK cell activation by different stimuli identified HSV-1 as a potent and fast inducer of NK cell activation in the context of human PBMC. Interestingly, we observed differences in the stimulating potential between HSVUV and HSVINF, concerning NK cell activation and cytokine secretion. HSVINF induced faster and stronger CD69 up-regulation than HSVUV and, in contrast to HSVUV, also caused IFN-γ secretion and degranulation (Fig. 1). This stands in contrast to a study, where NK cell activity of HSV-1-stimulated PBMC was similar for infectious and UV-inactivated virus,36 which may be explained by different methodical approaches: Ahmad et al. studied the increase of basic lytic activity against NK target cells K562, whereas we investigated degranulation of NK cells in the absence of cytotoxicity-inducing target cells. In our experiments, only a minority of NK cells produced IFN-γ and/or degranulated upon HSVINF stimulation. A similar low responsiveness of peripheral blood NK cells to classical NK cell stimulation was reported by others.14,37 An explanation may be that we did not use stimulating cytokines (e.g. IL-2) in addition to HSVINF. In conditions of inflammation, however, NK cell effector functions may be further augmented in vivo. Notably, we identified IFN-γ production and degranulation in CD56dim and CD56bright NK cells. The CD56dim NK cell subset was originally identified to be mostly cytolytic.15 Recent data, however, describe these cells as rapid producers of IFN-γ upon antibody-mediated stimulation of natural killer receptors.38 Our data show, for the first time, that CD56dim NK cells are also a source of IFN-γ upon HSVINF stimulation.
Interferon-α was described by others as the main cytokine in the induction of NK cell activation after stimulation of human pDC with influenza virus, CpG and poly (I:C).19–21 Our data identified HSV-1 as potent inducer of IFN-α production within human PBMC, with HSVINF being faster and more potent than HSVUV (Fig. 2a). Supernatants of HSVINF-stimulated pDC induced time- and dose-dependent NK cell activation, which was significantly reduced after neutralization of the IFN-α receptor (Fig. 2d). Sucrose-purified HSV-1 was not able to activate isolated NK cells (data not shown), which emphasizes the crucial role of pDC-derived IFN-α in the induction of NK cell activation by HSV-1.
The induction of IFN-α production was able to explain some but not all effects observed after stimulation of PBMC with HSVINF, in particular the induction of NK cell effector functions. Using a cytokine bead array, we identified TNF-α and IL-1β as further cytokines, which were significantly induced by HSVINF compared with HSVUV (Fig. 3a). Other groups have shown an important role of TNF-α in the NK cell activation induced by CpG-activated human pDC.19,20 Studies with TNF-α knockout mice showed decreased survival rates in acute corneal HSV-1 infections and increased reactivation rates after UV light stimulation,39 and lethal encephalitis after intranasal HSV-1 infection.40 Our neutralization experiments support the role of this cytokine in the control of herpes viral infections, as TNF-α turned out to contribute significantly to HSV-1-induced NK cell CD69 up-regulation and IFN-γ secretion (Fig. 3b).
Interleukin-1β costimulated IFN-γ production of CD56bright NK cells together with IL-12 or, in particular, IL-15.41 A mouse model of lethal encephalitis after intranasal HSV-1 infection confirmed its role in vivo.40 Our neutralization studies did not reveal a direct impact of IL-1β on NK cell activation and effector functions, but rather an indirect effect via enhanced IFN-α production (Fig. 3c). Strikingly, all three cytokines affected IFN-α production, suggesting a positive feedback loop for IFN-α secretion, in which, besides IFN-α itself,32 IL-1β and TNF-α are involved. Induction of high IFN-α levels by HSV-1 may imply a cross-talk between the involved cell populations through secretion of these cytokines. Other cytokines, which appear to contribute to NK cell activation, are IL-15 and IL-18, which were found to be of relevance in murine models of vaginal HSV-2 and intravenous HSV-1 infections.17,42 Our experiments also showed that not all cytokines induced each other: neutralization of the IFN-α receptor increased the HSVINF-induced IFN-γ production (Fig. 3b). These findings suggest that IFN-α inhibits production of IFN-γ, which was similarly described in another study of murine cytomegalovirus-induced type I IFN production.43 In this process, signal transducer and activator of transcription 1 (STAT1) was described to be crucially involved.44 In contrast, in lymphocytic choriomeningitis infection, type I IFNs activate STAT4 and thereby induce IFN-γ production.45 These findings suggest a plasticity of NK cell innate immune responses in different infection models.46 To further investigate this process, we addressed the impact of combined blockade of TNF-α and IFN-α receptor on NK cell functions. Combination of blocking antibodies did not have additive effects on NK cell CD69 up-regulation compared with blockade with single antibodies, whereas IFN-γ production after combined blockade was comparable to the isotype control (data not shown). These findings suggest opposing roles for TNF-α and IFN-α in HSVINF-induced IFN-γ secretion.
Our study identified pDC and monocytes as potent TNF-α producers in response to HSVINF stimulation (Fig. 4). The significant difference in TNF-α secretion between HSVUV-exposed and HSVINF-exposed monocytes could in part explain the inability of HSVUV (in contrast to HSVINF) to induce significant IFN-γ secretion, because neutralization of TNF-α significantly diminished HSVINF-induced IFN-γ secretion. These observations suggest a crucial role for viral infectivity in the induction of pro-inflammatory cytokines and NK cell effector functions. Analyses of two different GFP-expressing HSV-1 isolates revealed infection of a minority of monocytes, yet without inducing a productive replication cycle (Fig. 5), in concordance with previous studies of monocyte infection by HSV-1.33,47 The infected cells were indeed monocytes, as confirmed by staining of CD14 (data not shown). Low infection rates of monocytes were in part due to IFN-α, as blocking of the IFN-α receptor increased infection rates (Fig. 6e), which confirms previous studies reporting that type I interferons suppress HSV-1 replication in vitro and in vivo.48,49 Another explanation for the low infection rates may be the lack of pre-stimulation, which has similarly been reported for T lymphocytes, whose infectability was enhanced after phytohaemagglutinin stimulation.50 It was also recently reported that SAMHD1 restricts HSV-1 DNA replication in macrophages.51 It should be noted, however, that a frequency of 1–2% infected monocytes in our experiments contrasted with roughly 20% of monocytes secreting TNF-α upon exposure to HSVINF. Hence, infection of monocytes appears to stimulate a number of uninfected bystander cells to secrete TNF-α.
Depletion experiments confirmed pDC as potent mediators of HSV-induced NK cell activation via IFN-α production (Fig. 7a,d). More importantly, these studies showed the relevance of monocytes in both NK cell activation and IFN-α secretion. Monocytes may contribute to high IFN-α levels via secretion of IFN-α itself, as observed for infected monocytes (Fig. 6c), and/or via secretion of IL-1β and TNF-α, thereby stimulating IFN-α secretion by pDC, as suggested by cytokine neutralization (Fig. 3c). A new aspect in our study is that monocytes were identified as an indispensable cell population in the induction of NK cell effector functions by HSVINF within the PBMC context (Fig. 7b,c). We could furthermore determine TNF-α secretion as an important mechanism in the induction of IFN-γ, but the exact process, in which degranulation was induced, remained elusive. Most likely, infected monocytes are directly recognized by NK cells as target cells and induce NK cell activation and effector functions through various possible mechanisms.
Induction of NK cell cytotoxicity via down-regulation of HLA molecules on infected cells was demonstrated for HSV-1 and HSV-2.52,53 In our studies, we observed IFN-α-dependent overall up-regulation of HLA-ABC and HLA-E on monocytes. Unfortunately, we were not able to investigate HLA-A, HLA-B and HLA-C separately, because no specific antibodies are available. HLA up-regulation may protect monocytes from NK cell killing. In turn, monocytes may produce TNF-α to enhance NK cell killing of HSV-infected cells in inflamed tissue.54 MICA, which was up-regulated on Toll-like receptor-stimulated monocytes,55 was not induced by HSV-1, consistent with a study in which MICA in infected cells was down-regulated by late HSV-1 gene products.56 Immediate early gene expression was found to be sufficient for NK cell-mediated lysis of HSV-infected fibroblasts.54 Follow-up studies found that the HSV-1 immediate early protein ICP0 induced lysis of HSV-infected cells via the natural cytotoxicity receptors.57 Yet, the induced molecules on target cells were not identified. Possible candidates may be CD244 (2B4, SLAMF4), which is up-regulated on NK cells and interacts with CD48 on macrophages;58 the glucocorticoid-induced TNF receptor-ligand (GITRL), involved in the induction of NK cell cytotoxicity by CpG-stimulated pDC;59 or B7-H6, a cell surface ligand for NKp30, induced on pro-inflammatory monocytes upon stimulation with IL-1β and TNF-α.60
Altogether, our data propose a model in which the cross-talk between NK cells, pDC, and monocytes is mediated in part by IFN-α and TNF-α, eventually causing activation of effector NK cells secreting IFN-γ and exhibiting degranulation. In this process, both HSV-1-stimulated pDC and monocytes initially activate NK cells, but only monocytes exposed to HSVINF induce effector functions via TNF-α-dependent and TNF-α-independent mechanisms. These data may stimulate further studies investigating the role of cell contact-dependent molecules in the induction of NK cell effector functions. Plasmacytoid DC accumulate in herpes virus-induced lesions in the skin61,62 and mucosa,8 as well as in draining lymph nodes,63 where they encounter dendritic cells, NK and T cells.22,59,64 Analysing the interactions between pDC, NK cells, and monocytes is important because these cells are among the first to infiltrate herpetic lesions,22 and thereby may contribute to the efficient control of primary and recurrent HSV infections.
Acknowledgments
We thank Bernhard Fleckenstein, Erlangen, and André Gessner, Regensburg, for continuous support. Ingrid Müller-Fleckenstein, Institute of Clinical and Molecular Virology, Erlangen, is acknowledged for help in preparing virus stocks, and Anette Rohrhofer, Diagnostic Services of the Institute of Microbiology and Hygiene, Regensburg, for excellent technical assistance in performing HSV-1 real-time analyses. The HSV-1 isolate 166v was generously contributed by Gillian Elliot and Peter O’Hare, Section of Virology, Faculty of Medicine, Imperial College London, London, UK. HSV-1d106S and complementing E11 cells were kindly provided by David Knipe, Department of Microbiology and Molecular Genetics, Harvard Medical School, Boston, MA, and Neal DeLuca, Department of Microbiology and Molecular Genetics, University of Pittsburgh School of Medicine, Pittsburgh, PA. CpG-A ODN 6016 was contributed by Jörg Vollmer, Pfizer Oligonucleotide Therapeutics Unit – Coley Pharmaceutical GmbH, Düsseldorf, Germany. This study was supported by the German Research Foundation (SCHM1702/3-1), the graduate colleges 1071 (‘Viruses of the immune system’; to K.T.) and 1660 (‘Key signals of adaptive immune response’; to S.T.), and the ‘Akademie der Wissenschaften und Literatur zu Mainz’.
Glossary
- FCS
fetal calf serum
- HSV
herpes simplex virus
- IFN
interferon
- IL
interleukin
- INF
infectious
- NK
natural killer
- ODN
oligodeoxynucleotide
- PBMC
peripheral blood mononuclear cells
- pDC
plasmacytoid dendritic cells
- TNF
tumour necrosis factor
- UV
UV-inactivated
Disclosures
The authors declare no financial or commercial conflict of interest.
References
- Cunningham AL, Taylor R, Taylor J, Marks C, Shaw J, Mindel A. Prevalence of infection with herpes simplex virus types 1 and 2 in Australia: a nationwide population based survey. Sex Transm Infect. 2006;82:164–8. doi: 10.1136/sti.2005.016899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley RJ. Herpesviruses (Human). eLS 2011.
- Sancho-Shimizu V, Perez de Diego R, Jouanguy E, Zhang SY, Casanova JL. Inborn errors of anti-viral interferon immunity in humans. Curr Opin Virol. 2011;1:487–96. doi: 10.1016/j.coviro.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman M, Howell D, Fitzgerald-Bocarsly P. Interferon-α-dependent and -independent participation of accessory cells in natural killer cell-mediated lysis of HSV-1-infected fibroblasts. J Leukoc Biol. 1992;52:473–82. doi: 10.1002/jlb.52.5.473. [DOI] [PubMed] [Google Scholar]
- Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5:919–23. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–7. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004;103:1433–7. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- Lund JM, Linehan MM, Iijima N, Iwasaki A. Cutting edge: plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. J Immunol. 2006;177:7510–4. doi: 10.4049/jimmunol.177.11.7510. [DOI] [PubMed] [Google Scholar]
- Gill N, Chenoweth MJ, Verdu EF, Ashkar AA. NK cells require type I IFN receptor for antiviral responses during genital HSV-2 infection. Cell Immunol. 2011;269:29–37. doi: 10.1016/j.cellimm.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Swiecki M, Wang Y, Gilfillan S, Colonna M. Plasmacytoid dendritic cells contribute to systemic but not local antiviral responses to HSV infections. PLoS Pathog. 2013;9:e1003728. doi: 10.1371/journal.ppat.1003728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew T, Taylor KE, Mossman KL. Innate and adaptive immune responses to herpes simplex virus. Viruses. 2009;1:979–1002. doi: 10.3390/v1030979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster P, Boscheinen JB, Tennert K, Schmidt B. The role of plasmacytoid dendritic cells in innate and adaptive immune responses against α herpes virus infections. Adv Virol. 2011;2011:679271. doi: 10.1155/2011/679271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E. What is natural in natural killer cells? Immunol Lett. 2006;107:1–7. doi: 10.1016/j.imlet.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–40. doi: 10.1016/s1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- Tanigawa M, Bigger JE, Kanter MY, Atherton SS. Natural killer cells prevent direct anterior-to-posterior spread of herpes simplex virus type 1 in the eye. Invest Ophthalmol Vis Sci. 2000;41:132–7. [PubMed] [Google Scholar]
- Ashkar AA, Rosenthal KL. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol. 2003;77:10168–71. doi: 10.1128/JVI.77.18.10168-10171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading PC, Whitney PG, Barr DP, Smyth MJ, Brooks AG. NK cells contribute to the early clearance of HSV-1 from the lung but cannot control replication in the central nervous system following intranasal infection. Eur J Immunol. 2006;36:897–905. doi: 10.1002/eji.200535710. [DOI] [PubMed] [Google Scholar]
- Gerosa F, Gobbi A, Zorzi P, Burg S, Briere F, Carra G, Trinchieri G. The reciprocal interaction of NK cells with plasmacytoid or myeloid dendritic cells profoundly affects innate resistance functions. J Immunol. 2005;174:727–34. doi: 10.4049/jimmunol.174.2.727. [DOI] [PubMed] [Google Scholar]
- Marshall JD, Heeke DS, Abbate C, Yee P, Van Nest G. Induction of interferon-γ from natural killer cells by immunostimulatory CpG DNA is mediated through plasmacytoid-dendritic-cell-produced interferon-α and tumour necrosis factor-α. Immunology. 2006;117:38–46. doi: 10.1111/j.1365-2567.2005.02261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benlahrech A, Donaghy H, Rozis G, Goodier M, Klavinskis L, Gotch F, Patterson S. Human NK cell up-regulation of CD69, HLA-DR, interferon γ secretion and cytotoxic activity by plasmacytoid dendritic cells is regulated through overlapping but different pathways. Sensors (Basel) 2009;9:386–403. doi: 10.3390/s90100386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaghy H, Bosnjak L, Harman AN, Marsden V, Tyring SK, Meng TC, Cunningham AL. Role for plasmacytoid dendritic cells in the immune control of recurrent human herpes simplex virus infection. J Virol. 2009;83:1952–61. doi: 10.1128/JVI.01578-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritschet K, Donhauser N, Schuster P, et al. CD4- and dynamin-dependent endocytosis of HIV-1 into plasmacytoid dendritic cells. Virology. 2012;423:152–64. doi: 10.1016/j.virol.2011.11.026. [DOI] [PubMed] [Google Scholar]
- Schuster P, Donhauser N, Pritschet K, Ries M, Haupt S, Kittan NA, Korn K, Schmidt B. Co-ordinated regulation of plasmacytoid dendritic cell surface receptors upon stimulation with herpes simplex virus type 1. Immunology. 2010;129:234–47. doi: 10.1111/j.1365-2567.2009.03176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz MO, Ziemin S, Le Beau MM, Pitha P, Smith SD, Chilcote RR, Rowley JD. Homozygous deletion of the α- and β1-interferon genes in human leukemia and derived cell lines. Proc Natl Acad Sci USA. 1988;85:5259–63. doi: 10.1073/pnas.85.14.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittan NA, Bergua A, Haupt S, Donhauser N, Schuster P, Korn K, Harrer T, Schmidt B. Impaired plasmacytoid dendritic cell innate immune responses in patients with herpes virus-associated acute retinal necrosis. J Immunol. 2007;179:4219–30. doi: 10.4049/jimmunol.179.6.4219. [DOI] [PubMed] [Google Scholar]
- Elliott G, O’Hare P. Live-cell analysis of a green fluorescent protein-tagged herpes simplex virus infection. J Virol. 1999;73:4110–9. doi: 10.1128/jvi.73.5.4110-4119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Broberg E, Watanabe D, Dudek T, Deluca N, Knipe DM. Genetic engineering of a modified herpes simplex virus 1 vaccine vector. Vaccine. 2009;27:2760–7. doi: 10.1016/j.vaccine.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G, Matsumoto-Kobayashi M, Clark SC, Seehra J, London L, Perussia B. Response of resting human peripheral blood natural killer cells to interleukin 2. J Exp Med. 1984;160:1147–69. doi: 10.1084/jem.160.4.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-α genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–9. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels CA, Kleinerman ES, Snyderman R. Abortive and productive infections of human mononuclear phagocytes by type I herpes simplex virus. Am J Pathol. 1978;91:119–36. [PMC free article] [PubMed] [Google Scholar]
- Albers I, Kirchner H, Domke-Opitz I. Resistance of human blood monocytes to infection with herpes simplex virus. Virology. 1989;169:466–9. doi: 10.1016/0042-6822(89)90174-8. [DOI] [PubMed] [Google Scholar]
- Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad A, Sharif-Askari E, Fawaz L, Menezes J. Innate immune response of the human host to exposure with herpes simplex virus type 1: in vitro control of the virus infection by enhanced natural killer activity via interleukin-15 induction. J Virol. 2000;74:7196–203. doi: 10.1128/jvi.74.16.7196-7203.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfossi N, Andre P, Guia S, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–42. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- De Maria A, Bozzano F, Cantoni C, Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56dim CD16+ NK cells as rapid producers of abundant IFN-γ on activation. Proc Natl Acad Sci USA. 2011;108:728–32. doi: 10.1073/pnas.1012356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami M, Kita M, Yan XQ, Yamamoto T, Iida T, Sekikawa K, Iwakura Y, Imanishi J. Role of IFN-γ and tumor necrosis factor-α in herpes simplex virus type 1 infection. J Interferon Cytokine Res. 2002;22:671–6. doi: 10.1089/10799900260100150. [DOI] [PubMed] [Google Scholar]
- Sergerie Y, Rivest S, Boivin G. Tumor necrosis factor-α and interleukin-1β play a critical role in the resistance against lethal herpes simplex virus encephalitis. J Infect Dis. 2007;196:853–60. doi: 10.1086/520094. [DOI] [PubMed] [Google Scholar]
- Cooper MA, Fehniger TA, Ponnappan A, Mehta V, Wewers MD, Caligiuri MA. Interleukin-1β costimulates interferon-γ production by human natural killer cells. Eur J Immunol. 2001;31:792–801. doi: 10.1002/1521-4141(200103)31:3<792::aid-immu792>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Barr DP, Belz GT, Reading PC, Wojtasiak M, Whitney PG, Heath WR, Carbone FR, Brooks AG. A role for plasmacytoid dendritic cells in the rapid IL-18-dependent activation of NK cells following HSV-1 infection. Eur J Immunol. 2007;37:1334–42. doi: 10.1002/eji.200636362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousens LP, Orange JS, Su HC, Biron CA. Interferon-αβ inhibition of interleukin 12 and interferon-γ production in vitro and endogenously during viral infection. Proc Natl Acad Sci USA. 1997;94:634–9. doi: 10.1073/pnas.94.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KB, Cousens LP, Doughty LA, Pien GC, Durbin JE, Biron CA. Interferon αβ-mediated inhibition and promotion of interferon γ: STAT1 resolves a paradox. Nat Immunol. 2000;1:70–6. doi: 10.1038/76940. [DOI] [PubMed] [Google Scholar]
- Mack EA, Kallal LE, Demers DA, Biron CA. Type 1 interferon induction of natural killer cell γ interferon production for defense during lymphocytic choriomeningitis virus infection. MBio. 2011;2:pii:e00169–11. doi: 10.1128/mBio.00169-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A, Biron CA. Type 1 interferons and the virus–host relationship: a lesson in detente. Science. 2006;312:879–82. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- Bruun T, Kristoffersen AK, Rollag H, Degre M. Interaction of herpes simplex virus with mononuclear phagocytes is dependent on the differentiation stage of the cells. APMIS. 1998;106:305–14. doi: 10.1111/j.1699-0463.1998.tb01351.x. [DOI] [PubMed] [Google Scholar]
- Harle P, Noisakran S, Carr DJ. The application of a plasmid DNA encoding IFN-α1 postinfection enhances cumulative survival of herpes simplex virus type 2 vaginally infected mice. J Immunol. 2001;166:1803–12. doi: 10.4049/jimmunol.166.3.1803. [DOI] [PubMed] [Google Scholar]
- Noisakran S, Campbell IL, Carr DJ. IFN-α1 plasmid construct affords protection against HSV-1 infection in transfected L929 fibroblasts. J Interferon Cytokine Res. 2000;20:107–15. doi: 10.1089/107999000312784. [DOI] [PubMed] [Google Scholar]
- Maccario R, Revello MG, Comoli P, Gerna G. Herpes simplex virus-1-specific human cytotoxic T lymphocytes are induced in vitro by autologous virus-infected mononuclear cells. Viral Immunol. 1992;5:93–103. doi: 10.1089/vim.1992.5.93. [DOI] [PubMed] [Google Scholar]
- Kim ET, White TE, Brandariz-Nunez A, Diaz-Griffero F, Weitzman MD. SAMHD1 restricts herpes simplex virus 1 in macrophages by limiting DNA replication. J Virol. 2013;87:12949–56. doi: 10.1128/JVI.02291-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elboim M, Grodzovski I, Djian E, Wolf DG, Mandelboim O. HSV-2 specifically down regulates HLA-C expression to render HSV-2-infected DCs susceptible to NK cell killing. PLoS Pathog. 2013;9:e1003226. doi: 10.1371/journal.ppat.1003226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard B, Fruh K. A role for MHC class I down-regulation in NK cell lysis of herpes virus-infected cells. Eur J Immunol. 2000;30:509–15. doi: 10.1002/1521-4141(200002)30:2<509::AID-IMMU509>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Fitzgerald-Bocarsly P, Howell DM, Pettera L, Tehrani S, Lopez C. Immediate-early gene expression is sufficient for induction of natural killer cell-mediated lysis of herpes simplex virus type 1-infected fibroblasts. J Virol. 1991;65:3151–60. doi: 10.1128/jvi.65.6.3151-3160.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss M, Decker P, Baltz KM, et al. Interaction of monocytes with NK cells upon Toll-like receptor-induced expression of the NKG2D ligand MICA. J Immunol. 2008;181:6711–9. doi: 10.4049/jimmunol.181.10.6711. [DOI] [PubMed] [Google Scholar]
- Schepis D, D’Amato M, Studahl M, Bergstrom T, Karre K, Berg L. Herpes simplex virus infection downmodulates NKG2D ligand expression. Scand J Immunol. 2009;69:429–36. doi: 10.1111/j.1365-3083.2009.02241.x. [DOI] [PubMed] [Google Scholar]
- Chisholm SE, Howard K, Gomez MV, Reyburn HT. Expression of ICP0 is sufficient to trigger natural killer cell recognition of herpes simplex virus-infected cells by natural cytotoxicity receptors. J Infect Dis. 2007;195:1160–8. doi: 10.1086/512862. [DOI] [PubMed] [Google Scholar]
- Nedvetzki S, Sowinski S, Eagle RA, et al. Reciprocal regulation of human natural killer cells and macrophages associated with distinct immune synapses. Blood. 2007;109:3776–85. doi: 10.1182/blood-2006-10-052977. [DOI] [PubMed] [Google Scholar]
- Hanabuchi S, Watanabe N, Wang YH, et al. Human plasmacytoid predendritic cells activate NK cells through glucocorticoid-induced tumor necrosis factor receptor-ligand (GITRL) Blood. 2006;107:3617–23. doi: 10.1182/blood-2005-08-3419. [DOI] [PubMed] [Google Scholar]
- Matta J, Baratin M, Chiche L, et al. Induction of B7-H6, a ligand for the natural killer cell-activating receptor NKp30, in inflammatory conditions. Blood. 2013;122:394–404. doi: 10.1182/blood-2013-01-481705. [DOI] [PubMed] [Google Scholar]
- Gerlini G, Mariotti G, Bianchi B, Pimpinelli N. Massive recruitment of type I interferon producing plasmacytoid dendritic cells in varicella skin lesions. J Invest Dermatol. 2006;126:507–9. doi: 10.1038/sj.jid.5700052. [DOI] [PubMed] [Google Scholar]
- Kohrgruber N, Groger M, Meraner P, et al. Plasmacytoid dendritic cell recruitment by immobilized CXCR3 ligands. J Immunol. 2004;173:6592–602. doi: 10.4049/jimmunol.173.11.6592. [DOI] [PubMed] [Google Scholar]
- Yoneyama H, Matsuno K, Toda E, et al. Plasmacytoid DCs help lymph node DCs to induce anti-HSV CTLs. J Exp Med. 2005;202:425–35. doi: 10.1084/jem.20041961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huch JH, Cunningham AL, Arvin AM, Nasr N, Santegoets SJ, Slobedman E, Slobedman B, Abendroth A. Impact of varicella-zoster virus on dendritic cell subsets in human skin during natural infection. J Virol. 2010;84:4060–72. doi: 10.1128/JVI.01450-09. [DOI] [PMC free article] [PubMed] [Google Scholar]