

Abstract
Glucocorticoid (GC) is often given when preterm delivery is expected. This treatment is successful in stimulating the development of the fetal lung. However, reports and related research regarding the prolonged effects of prenatal GC on the development of immunity are very limited. Some data, derived from infants whose mothers were given immunosuppressants during pregnancy for the treatment of autoimmune disorders, suggest that prenatal exposure to GC may have only a limited effect on the development of the immune system. What is unknown is whether the immune modulation effects of prenatal GC might appear at a later childhood stage and beyond. Here we evaluated the immune programming influenced by prenatal GC. Pregnant Sprague-Dawley rats received dexamethasone (DEX; 0·1 mg/kg/day) or saline at gestational days 14–20. Male offspring were killed at day 7 or day 120 after birth. Spleens were collected for immune study. Of the inflammation mediators, matrix metalloproteinase-9, tumour necrosis factor-α (TNF-α) and granulocyte–macrophage colony-stimulating factor mRNAs decreased in the prenatal DEX group at an early stage after birth. Upon concanavalin A stimulation, prenatal DEX treatment reduced TNF-α production, but not interferon-γ production, by splenocytes at day 120 after birth compared with the vehicle group. Decreased levels of active chromatin signs (acetylation of histone H3 lysines, H3K4me1/3, and H3K36me3) in TNF-α promoter were compatible with the expressions of TNF-α. Our results suggest that prenatal DEX has a profound and lasting impact on the developing immune system even to the adult stage. Epigenetic histone modifications regulate TNF-α expression following prenatal DEX in rats.
Keywords: epigenetic, prenatal glucocorticoid, tumour necrosis factor-α
Introduction
During the course of human pregnancy, glucocorticoid (GC) treatment is often given when preterm delivery is expected. This treatment is successful in maturing the fetal lung and decreasing the occurrence of respiratory distress syndrome. However, in animal studies, some side-effects with regards to prenatal GC treatment have been described. The immune system is sensitive to GC treatment. In humans, the effects of prenatal GC treatment on the development of the immune system have only been evaluated in small series. Kavelaars et al.1 reported enhanced natural killer cell activity and decreased T-cell proliferation in cord blood of preterm infants born after prenatal GC. However, the programming effects of prenatal GC on immunity have not been well studied. There have only been a limited number of studies showing the immune modulatory effects of prenatal GC in infants; these were observed in infants whose mothers were given immunosuppressants during pregnancy for the treatment of autoimmune disorders. Leucocyte count, sub-populations and antibody production were not different between the exposed babies and the babies that were not exposed.2,3 Studies also showed that the ability of infant leucocytes to produce cytokines [interleukin-2 (IL-2), interferon-γ (IFN-γ)] is not altered with prenatal GC treatment.2,4 Prenatal GC was also shown to not influence the lymphocyte apoptosis in preterm neonates.5 Hence prenatal exposure to GC seems to only have a limited effect on the developing immune system of humans. However, what remains unknown is whether these effects may appear at a later childhood stage and even beyond. To address these questions, we attempted to evaluate the immune programming influenced by prenatal GC.
Materials and methods
This study was carried out in strict accordance with the recommendations outlined in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the Kaohsiung Chang Gung Memorial Hospital. Virgin Sprague-Dawley (SD) rats (12–16 weeks old) were obtained (BioLASCO Taiwan Co., Ltd., Taipei, Taiwan), and then housed and maintained in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. Male SD rats were caged with individual females until mating was confirmed. Pregnant SD rats received intraperitoneal dexamethasone (DEX) (0·1 mg/kg body weight) or vehicle (normal saline) daily from gestational days 14 to 20 to conduct a prenatal DEX exposure model. Male offspring were assigned to two groups (n = 6 to n = 8/group): vehicle and DEX. Rats were killed at 7 or 120 days after birth to assess the short-term and long-term effects of prenatal DEX at the early child and adult periods, respectively. Both bodyweight and spleen weights were assessed after death. The spleens were then used for further studies.
Histological examination
Immediately after the rats were killed, the spleen were harvested, stored in saline on ice, and dissected from the surrounding tissues. Then the spleen was fixed in 10% formalin neutral buffer solution, pH 7·4 (Wako Junyaku, Osaka, Japan). Four-micrometer-thick sections were made and stained with haematoxylin and eosin for morphometric analysis. Images were captured with a mounted digital camera under a 10 × objective of a Nikon Eclipse E600 microscope (Nikon, NY).
Cell culture and drug treatment
Splenocytes were separated from the whole spleen as follows in brief: the spleen was washed with PBS and pressed with a syringe plunger through 70μm nylon mesh (BD Biosciences, San jose, CA). Erythrocytes were lysed by exposure to distilled water for 15 seconds, and the remaining splenocytes were washed with PBS. All spleen cells were counted and 2 × 106 cells/ml were plated in 24-well plates in enriched medium (RPMI-1640 medium supplemented with 1% non-essential amino acids, 1% pyruvate, 10% heat-inactivated fetal bovine serum and antibiotics). Cultured splenocytes were then stimulated with or without 5 μg/ml of concanavalin A (Con A). The cell pellets were collected at the indicated times and processed for mRNA detection or protein analysis and culture supernatants were analysed for secretory TNF-α/IFN-γ detection with ELISA (Biolegend, San Diego, CA).
Western blot
PRO-PREP kit (iNtRON Biotechnology, Seoul, Korea) was used to extract total proteins from frozen tissue samples. Fifty-milligram tissue samples were homogenized in 600 μl of PRO-PREP solution and were incubated for 30 min on ice for cell lysis. Homogenized solution was centrifuged at 23000 g for 5 min at 4° and the supernatant was transferred to a fresh 1·5-ml tube. Protein concentrations were determined by using a Bio-Rad Protein Assay kit (Bio-Rad, Hercules, CA). For Western blotting assay, protein samples were incubated with SDS loading buffer (10 mm Tris–HCl, pH 6·8, 1% SDS, 25% glycerol, 0·1 mm β-mercaptoethanol, and 0·03% bromophenol blue), boiled for 5 min, and subjected to 10% (weight/volume) SDS–PAGE. After being transferred to a PVDF membrane (NEF1002001PK; Perkin Elmer, Waltham, MA) and blocked with Tris-buffered saline Tween 20 (T-TBS) containing 5% dry milk, the membranes were then incubated overnight with the following anti-rat antibodies: p-p38, total p38 (Santa Cruz Biotechnology, Santa Cruz, CA), nuclear factor-κB (NF-κB), c-Jun, phosphorylated Jun N-terminal kinase (p-JNK), phosphorylated extracellular signal-regulated kinase (p-ERK) and total ERK (Abcam, Cambridge, UK), protein kinase Cα (PKCα; BD Biosciences PharMingen, San Diego, CA); and PKCβ (BD Biosciences) diluted 1 : 200 in TBS containing 1% skimmed milk. After five washes with 0·1% T-TBS, the membranes were incubated for 2 hr with peroxidase-labelled secondary antibody diluted 1 : 1000 in T-TBS. After washes with T-TBS, the membranes were developed using an enhanced chemiluminescence Plus kit (NEL105001EA; PerkinElmer) for Chemi Doc system (Bio-Rad) exposure and images were scanned. Images were analysed using Quantity One software (Bio-Rad). All Western blotting was performed for at least six separate experiments.
Reverse transcription- PCR
In brief, total RNA was extracted from the spleen tissue with a Trizol Reagent (#15596-018; Invitrogen, Carlsbad, CA). A 5-μg sample of total RNA was reversed-transcribed with 200 U of Moloney murine leukaemia virus reverse transcriptase (#28025-021; Invitrogen) in 20 μl of total reaction volume containing reverse transcriptase buffer, random primer, dNTP and RNase inhibitor. PCR was performed in 20 μl of total reaction volume containing 2 μg of cDNA, primers specific for IL-1, IL-8, matrix metalloproteinase 9 (MMP-9), tumour necrosis factor-α (TNF-α), granulocyte–macrophage colony-stimulating factor (GM-CSF) and β-actin; 2·5 mm MgCl2: and Maxima SYBR Green/Fluorescein qPCR Master Mix (2×) (no. K0242; Thermo Scientific, San Jose, CA). The cycling protocol consisted of one cycle of 10 min at 95° followed by 45 cycles of denaturation for 10 seconds at 95°, annealing for 20 seconds at 55°, and extension for 20 seconds at 72°. The primers were as follows:
TNF-α: 5′-GGCTGCCCCGACTACGT-3′ (sense) and 5′-AGGGCAAGGGCTCTTGATG-3′ (antisense).
IL-1: 5′-GCACCTTCTTTTCCTTCATCTTTG-3′ (sense) and 5′-TGCAGCTGTCTAATGGGAACAT-3′ (antisense).
IL-8: 5′-TGCACCCAAACCGAAGTCATAGCC-3′ (sense) and 5′-GCGTTCACCAGACAGACGCCA-3′ (antisense).
MMP-9: 5′-TCACGGAGGAAGCCAATTG-3′ (sense) and 5′-CTTGGACTGCGGATCCTCAA-3′ (antisense).
GM-CSF: 5′-ATGGCGCCTTGACCATGATA-3′ (sense) and 5′-ATGAAATCCTCAAAGGTGGTGACT-3′ (antisense).
β-actin: 5′- TACTGCCCTGGCTCCTA-3′ (sense) and 5′- GGGCCGGACTCATCGTA-3′ (antisense).
Serial dilutions of the standard cDNA were also used for parallel amplifications. The threshold cycles (Ct) were calculated with LightCycler software (ver. 1.5.0; Roche, Basel, Switzerland). Standard curves are plotted with Ct-versus-log cDNA quantities, and the quantities of samples were determined from the standard curves. For the relative quantification of gene expression, the comparative threshold cycle (CT) method was employed. The averaged CT was subtracted from the corresponding averaged β-actin value for each sample, resulting in ΔCT. ΔΔCT was achieved by subtracting the average control ΔCT value from the average experimental ΔCT. The fold increase was established by calculating 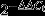 for experimental versus control samples as previously described.6
for experimental versus control samples as previously described.6
Histone deacetylase/histone acetyltransferase activity determination
For nuclear fractions, protein lysates were extracted from spleen tissues using the EpiQuik Nuclear Extraction kit (Epigentek, Brooklyn, NY), quantified, and then normalized to the same concentration. The measurement of histone deacetylase (HDAC) and histone acetyltransferase (HAT) activities were performed using HDAC or HAT (Epigentek) activity assay kits based on the manufacturer’s instructions.
Chromatin immunoprecipitation
Fifty milligrams of spleen tissue was cut into small pieces, fixed by addition of 900 μl of warmed 1% formaldehyde (37°) and then incubated for 10 min at room temperature. The cross-linking was stopped by addition of 100 μl of 125 mm glycine for 5 min at room temperature, followed by centrifugation and washing the pellet twice with ice-cold PBS. Chromatin immunoprecipitation assay (ChIP) was performed using the EZ-Magna ChIP™ A kit (Cat# 17-408; Millipore, Billerica, MA) according to the manufacturer’s directions. DNA was sheared by sonication to an average length of 200–1000 bp and 5 μl of the supernatant was removed as ‘Input’ and saved at 4°. The supernatant (containing 50 μl chromatin DNA, 450 μl dilution buffer/protease inhibitor and 20 μl protein A magnetic beads) was incubated overnight at 4° with 5 μg of the antibodies indicated in the immunoprecipitations. Antibodies used in the immunoprecipitations were specific for acetyl histone H3 (#06–599), acetyl histone H3 lysine 4/lysine 9 (Cell Signaling, Danvers, MA, #9677), monomethyl histone H3 lysine 4 (Abcam, ab8895), trimethyl-histone H3 lysine 4 (Abcam, ab8580), trimethyl-histone H3 lysine 36 (Abcam, ab9050). The immunoprecipitated DNA was then eluted in a total volume of 50 μl and was amplified and quantified by real-time PCR, which was performed with an annealing temperature of 57° for a total of 45 cycles. The primer of Rat TNF-α promoter-1: Forward 5′-CAGAAGCTTCGTGGAAAACTCA-3′ and Reverse 5′-GAATTCACGGACCCCACAAG-3′; Rat TNF-α promoter-2: Forward 5′-CGTTGAGTCTCCCCCCTTATG-3′ and Reverse 5′-TGTCCGTTCCCCTTTGATTTC-3′; Rat TNF-α promoter-3: Forward 5′-GGTGAGGACGGAGAGGAGATT-3′ and Reverse 5′-TGGGAGTTAGTACCAGGGTGTTC-3′. Results were expressed as fold over control or percentage of control.
Statistics
Data were expressed as mean ± standard error of the mean. Mann–Whitney U-test was used when two groups were analysed. Results with a P-value < 0·05 were considered to be statistically significant. All statistical tests were performed using spss 15.0 for Windows XP (SPSS, Inc., Chicago, IL).
Results
Prenatal DEX treatment induced relative splenomegaly
As shown in Table 1, the litter size and male pup mortality rate were not different between these two groups. The DEX group had a lower birth bodyweight (BW) than the vehicle group (5·92 ± 0·12 versus 7·97 ± 0·15 g, P = 0·001). But there was no difference regarding BW at day 7 and day 120 between these two groups. The average spleen weight of pup of DEX group was more than the vehicle group both at day 7 (0·11 ± 0·01 versus 0·08 ± 0·00 g, P = 0·001) and day 120 (0·73 ± 0·01 versus 0·91 ± 0·04 g, P = 0·003). After correcting for BW, the DEX group still had higher spleen weight-to-BW ratios than the vehicle group both at day 7 (0·86 ± 0·04 versus 0·53 ± 0·03, P = 0·001) and day 120 (0·17 ± 0·01 versus 0·14 ± 0·00, P = 0·002).
Table 1.
Pregnancy outcome following fetal exposure to dexamethasone (DEX; mean ± SE)
| Vehicle | Prenatal DEX | P | |
|---|---|---|---|
| Gestation length (days) | 22·95 ± 0·16 | 22·63 ± 0·14 | NS |
| Litter size | 12·38 ± 0·93 | 10·79 ± 0·62 | NS |
| Stillbirth/litter | 0·11 ± 0·08 | 0·17 ± 0·11 | NS |
| Sex ratio (F/M) | 1·28 ± 0·16 | 1·17 ± 0·13 | NS |
| Pup body weight at birth (g) | 7·97 ± 0·15 | 5·92 ± 0·12 | 0·001 |
| Pup spleen weight at day 7 (g) | 0·08 ± 0·00 | 0·11 ± 0·01 | 0·001 |
| Pup body weight at day 7 (g) | 14·59 ± 0·28 | 13·58 ± 0·38 | NS |
| Pup spleen weight/body weight at day 7 | 0·53 ± 0·03 | 0·86 ± 0·04 | 0·001 |
| Pup spleen weight at day 120 (g) | 0·73 ± 0·01 | 0·91 ± 0·04 | 0·003 |
| Pup body weight at day 120 (g) | 512·05 ± 10·82 | 540·75 ± 19·77 | NS |
| Pup spleen weight/body weight at day 120 | 0·14 ± 0·00 | 0·17 ± 0·01 | 0·002 |
Prenatal DEX treatment induced impaired spleen follicle development
Regarding immunohistochemistry stain, there were fewer developed follicles in the DEX group than in the vehicle group at day 120 (Fig. 1).
Figure 1.
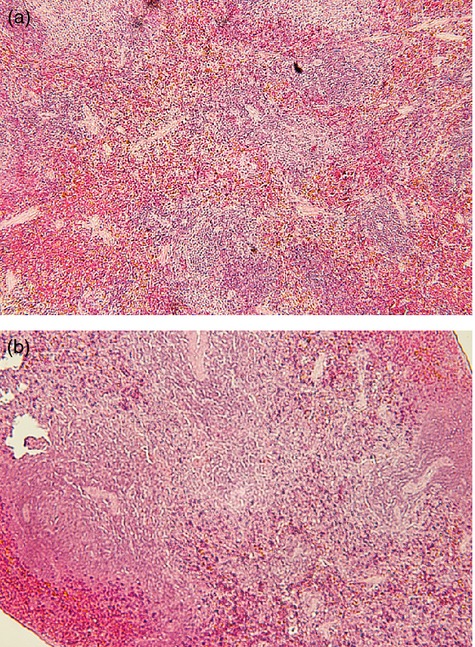
The haematoxylin & eosin staining of rat spleens. Representative blot of (a) vehicle, (b) prenatal dexamethasone (DEX) group of spleen of day 120.
Prenatal DEX treatment decreased several splenic pro-inflammatory mediator mRNA expressions even at long-term stage
At first, we compared the pro-inflammatory mediator (IL-1, IL-8, MMP-9, TNF-α and GM-CSF) mRNA expressions between vehicle and DEX groups both at short-term and long-term stages. Of the inflammation mediators, the MMP-9, TNF-α and GM-CSF mRNA was decreased in the DEX group at the early stage (Fig. 2a,b,d). At the long-term stage, only MMP-9 mRNA was decreased in DEX group (Fig. 3d).
Figure 2.
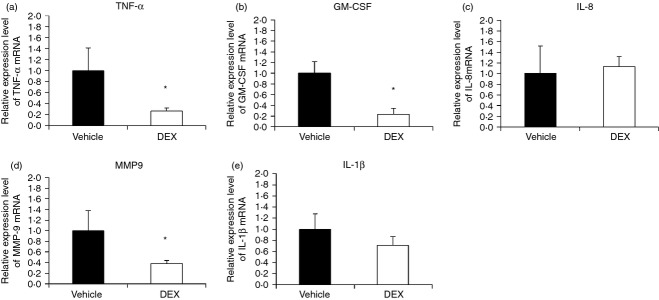
Quantitative RT-PCR analysis of expression of pro-inflammatory mediator mRNA in the spleens of vehicle and dexamethasone (DEX) groups in the short-term. Representative RT-PCR analysis of (a) tumour necrosis factor-α (TNF-α), (b) granulocyte–macrophage colony-stimulating factor (GM-CSF), (c) interleukin-8 (IL-8), (d) matrix metalloproteinase 9 (MMP9), (e) IL-1β mRNA levels of the spleens of vehicle and DEX groups at day 7. Data presented were calculated from eight replicate experiments. *vehicle vs DEX, P < 0·05.
Figure 3.

Quantitative RT-PCR analysis of expression of pro-inflammatory mediators mRNA in the spleens of vehicle and dexamethasone (DEX) groups in the long-term. Representative RT-PCR analysis of (a) tumour necrosis factor-α (TNF-α), (b) granulocyte–macrophage colony-stimulating factor (GM-CSF), (c) interleukin-8 (IL-8), (d) matrix metalloproteinase 9 (MMP9), (e) IL-1β mRNA levels of the spleens of vehicle and DEX groups at day 120. Data presented were calculated from eight replicate experiments. *vehicle vs DEX, P < 0·05.
Prenatal DEX decreased TNF-α production of cultured splenocytes at long-term stage
Then we isolated and stimulated the splenocytes with Con A for 72 hr. The culture supernatants were then collected and indicated for TNF-α production. As shown in Fig. 4(a), rat splenocytes at day 7 had only limited TNF-α production ability. There were no significant differences regarding TNF-α production between the vehicle and DEX groups at day 7 (vehicle versus prenatal DEX: 28·4 ± 2·3 versus 25·3 ± 4·1 pg/ml; P = 0·103). The TNF-α production ability of splenocyte was fully matured at day 120. When compared with vehicle, the DEX group showed depressed TNF-α production at the long-term stage (vehicle versus prenatal DEX: 140·3 ± 14·4 versus 83·1 ± 10·6 pg/ml; P = 0·012) (Fig. 4b). When TNF-α mRNA expressions of cultured splenocytes were investigated, the DEX group also showed a greater decrease in TNF-α mRNA expression than the vehicle group upon Con A stimulation (vehicle versus DEX: 1·0 ± 0·2 versus 0·6 ± 0·1 fold; P = 0·028) (Fig. 4c). Hence, the suppressed TNF-α production in the DEX group was partly through transcriptional regulation.
Figure 4.
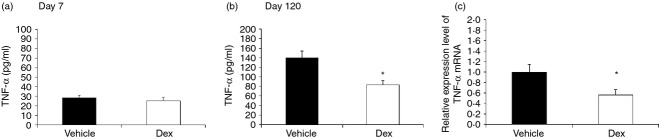
Effects of prenatal dexamethasone (DEX) treatment on tumour necrosis factor-α (TNF-α) production of cultured splenocytes. Isolated splenocytes from the vehicle and DEX groups at (a) day 7 or (b) day 120 were suspended to 2 × 106/ml in 24-well plates then treated with 5 μg/ml of concanavalin A (Con A). The culture supernatants were collected at 72 hr and then indicated for TNF-α detection with ELISA. The scales of the y-axes were different between day 7 and day 120. (c) The cell pellets were collected for TNF-α mRNA expression with RT-PCR. The data shown are from six replicated experiments. *vehicle vs DEX, P < 0·05.
Prenatal DEX treatment did not decrease IFN-γ production of cultured splenocytes
Pro-inflammatory mediators (IL-1β, IL-8, MMP-9, TNF-α and GM-CSF) represent the innate immune response, so IFN-γ was determined to represent adaptive immune manifestation. After splenocytes were stimulated with Con A for 72 hr, the culture supernatants were indicated for IFN-γ production. As shown in Fig. 5, the DEX group showed comparable IFN-γ production to the vehicle group both at early stage (vehicle versus DEX: 63·1 ± 7·6 versus 98·2 ± 15·7; P = 0·18) and long-term stage (vehicle versus DEX: 2234·5 ± 696·8 versus 2061·1 ± 261·8; P = 0·94) (Fig. 5).
Figure 5.
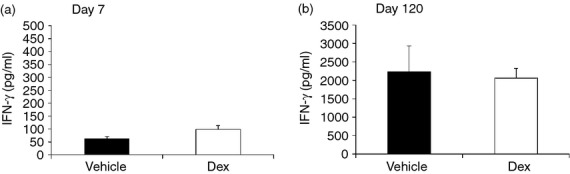
Effects of prenatal dexamethasone (DEX) treatment on interferon-γ (IFN-γ) production of cultured splenocytes. Isolated splenocytes from the vehicle and DEX groups at (a) day 7 or (b) day 120 were suspended to 2 × 106/ml in 24-well plates then treated with 5 μg/ml of Concanavalin A (Con A). The culture supernatants were collected at 72 hr and then indicated for IFN-γ detection with ELISA. The scales of the y-axes were different between day 7 and day 120. The data shown are from six replicate experiments.
PKCα and PKCβ were increased in the short-term but decreased in the long-term with prenatal DEX treatment
Since TNF-α mRNA was suppressed with prenatal DEX treatment, upstream signal molecules of TNF-α mRNA were investigated in further experiments. With Western blot, we found that the p-p38, NF-κB, c-Jun, p-JNK and p-ERK protein expressions were no different between the vehicle and DEX groups (Fig. 6). However, the total ERK protein was more abundant in the DEX group at an early stage. The PKCα and PKCβ proteins were increased in the short-term but decreased in the long-term with prenatal DEX treatment (Fig. 6).
Figure 6.

The tumour necrosis factor-α (TNF-α) -associated signal molecules expression with prenatal dexamethasone (DEX) treatment. (a) Splenic tissue lysates from vehicle and DEX groups were analysed by Western blotting with the indicated antibodies. The results were representative of six replicate experiments. (b) Plots illustrate the TNF-α-associated signal molecule expressions of spleen from vehicle and DEX groups at day 7 and day 120. Data presented were calculated from six replicate experiments. *P < 0·05.
Prenatal DEX treatment decreased HDAC but not HAT activity of rat spleen
Histone acetylation and deacetylation enhances and represses gene transcription, respectively. Acetylation and deacetylation of histone is through the enzymatic activity of HAT and HDAC. Then we investigated the effects of prenatal DEX on the HAT and HDAC activity of rat spleen. Prenatal DEX treatment had limited effects on both HAT and HDAC activity compared with vehicle in the short-term (data not shown). But prenatal DEX treatment decreased HDAC but not HAT activity of rat spleen in the long-term (relative folds of HDAC activity; vehicle versus prenatal DEX: 1 ± 0·04 versus 0·9 ± 0·02; P = 0·04) (Fig. 7).
Figure 7.
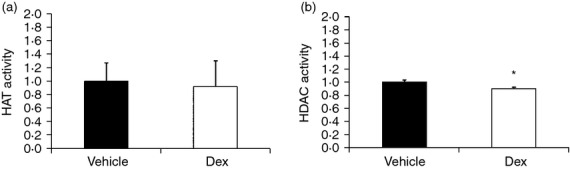
Prenatal dexamethasone (DEX) treatment decreased histone deacetylase (HDAC) but not histone acetyltransferase (HAT) activity of rat spleen at the long-term stage. Bar graphs showing (a) HAT activity and (b) HDAC activity of prenatal DEX group relative to vehicle. Results were expressed as fold difference over vehicle (mean ± SEM; *P < 0·05; n = 6).
Prenatal DEX-induced TNF-α decrease is associated with histone H3 modifications
Spleen tissue treated with/without prenatal DEX
Chromatin immunoprecipitation assays were performed on the nuclear extracts by using antibodies directed against acetyl-histone H3 (total Lys sites) and acetyl-histone H3 (Lys 9/Lys 14). Quantitative real-time PCR analysis was used to determine the percentage of TNF-α promoter input DNA that was bound to these proteins. We designed three pairs of TNF-α promoter primers for the ChIP study. Because prenatal DEX caused a TNF-α transcript difference from vehicle in the long-term, spleen tissues from the long-term stage were chosen for the ChIP study. We found, among the three TNF-α promoters, only the TNF-α promoter 3 (within 240 bp upstream of exon) was responsible for a significant acetylation change of histone H3 by prenatal DEX (Fig. 8a). Prenatal DEX treatment resulted in a total histone H3 acetylation decrease for TNF-α promoter 3 (Fig. 8a). Prenatal DEX treatment also resulted in a decrease in H3K9/K14 acetylation (Fig. 8b).
Figure 8.

Histone H3 lysine acetylation levels at the tumour necrosis factor-α (TNF-α) promoter of spleen with/without prenatal dexamethasone (DEX) treatment. Bar graphs showing (a) the total H3 lysine acetylation levels at different TNF-α promoters. (b) H3K9/K14 acetylation level of prenatal DEX group relative to vehicle at the TNF-α promoter 3. Chromatin immunoprecipitation assays were performed with H3K4/K9 acetylation antibody. Results were expressed as fold difference over vehicle (mean ± SEM; *P < 0·05; n = 6)
Then we attempted to investigate whether promoter histone H3 lysine methylation plays a role in prenatal DEX-related transcriptional decrease of TNF-α in rat spleens. The methylated histone H3K4 correlates with transcriptionally competent chromatin and is associated with active genes.7,8 H3K4me levels of TNF-α promoter were assessed by ChIP assays with H3K4me1, H3K4me3 or H3K36me3 antibodies. Similarly, among the three TNF-α promoters, only the TNF-α promoter 3 was responsible for significant methylation change of histone H3 by prenatal DEX. H3K4me1/3 and H3K36me3 levels in the TNF-α promoter were decreased in rat spleens from prenatal DEX treatment (Fig. 9). These decreases in promoter H3K4me1/3 and H3K36me3 levels correlated with the decreased expression of TNF-α.
Figure 9.

Histone H3 lysine methylation levels at the tumour necrosis factor-α (TNF-α) promoter of spleen with/without prenatal dexamethasone (DEX) treatment. Bar graphs showing the (a) H3K4me1, (b) H3K4me3 and (c) H3K36me3 levels of prenatal DEX group relative to vehicle at the TNF-α promoter. Chromatin immunoprecipitation assays were performed with H3K4me1, H3K4me3 and H3K36me3 antibodies. Results were expressed as fold difference over vehicle (mean ± SEM; *P < 0·05; n = 6).
Discussion
Here we evaluated the immune programming influenced by prenatal DEX treatment. The mRNA expression of pro-inflammation mediators was influenced by prenatal DEX treatment both at the early and long-term stages. Upon Con A stimulation, prenatal GC treatment reduced the TNF-α but not IFN-γ production of splenocytes at the long-term stage. This suggests that prenatal DEX treatment has a profound impact on immune programming, especially for innate immunity.
Wistar rats and Sprague-Dawley rats are now the most commonly used laboratory animals worldwide. This rat model is widely used for toxicology, teratology, experimental oncology, experimental gerontology, cardiovascular research and immunology studies.9,10 Since there are many differences in anatomy, physiology and development between different species, verification of the phase in days of the animal and its correlation with age in years of humans is necessary. Rats have a brief and accelerated childhood compared with humans and become sexually mature at about 6 weeks of age. When considering the different phases of a rat’s life, we chose day 7 and day 120 of the rat to represent the infant and adult stages, respectively.9
Prenatal exposure to GC seems to only have a limited effect on the developing immune system of humans.2–5 However, these scant data are only limited to infants. We did not know whether the immune modulation effects of prenatal GC would appear at a later childhood stage and perhaps even beyond. Here we evaluated how immune programming was influenced by prenatal DEX treatment. Our study had two important findings. First, although the TNF-α production between vehicle and DEX group was not different in the short-term, the mRNA of several pro-inflammatory mediators (TNF-α, GM-CSF, MMP-9) was suppressed by prenatal DEX treatment. The cytokine production difference between the vehicle and DEX groups might be masked by immature immunity at the short-term stage. We found that the splenocytes of the DEX group had impaired TNF-α production compared with vehicle at the long-term stage. This suggested that prenatal DEX treatment has a profound and lasting impact on the developing immune system even to adulthood. This unique finding is first reported in the related literature and worthy of our attention. Second, innate immunity seems more susceptible to prenatal DEX treatment than adaptive immunity. The immune system is composed of innate and adaptive immunity. In general, innate immunity refers to relatively non-specific defence mechanisms that are able to respond immediately or within hours to the chemical properties of an antigen.11 Many pro-inflammatory cytokines are released by cells of the innate immune system upon stimulation such as IL-1β, IL-8, TNF-α and GM-CSF.12–15 In contrast, adaptive immunity refers to the antigen-specific immune response, including the development of immunological memory. Adaptive immune responses are generated by clonal selection of lymphocytes and need a longer period of time to learn.16 Interleukin-2 and IFN-γ are generally considered to be characteristic of the adaptive immune response of T helper type 1.17–19 The T helper type 1 immune response activates the macrophages and induces B cells to make corresponding antibodies, and this leads to cell-mediated immunity.16 GC was known to have a direct inhibitory effect both on innate and adaptive immunity. The effects of GC on the innate immune system include phagocyte function impairment and decreased production of pro-inflammatory mediators.20,21 For adaptive immunity, GC impaired a variety of T-cell functions, cytokine production (such as IL-2, IFN-γ), and induced T-cell apoptosis.22 However, study about the effects of prenatal GC on developing immunity is limited. From our data, the TNF-α production but not IFN-γ was diminished in the DEX group in the long-term. This suggests that innate immunity is more susceptible to prenatal DEX than adaptive immunity. The impact of prenatal GC might leave offspring more vulnerable to pathogenic invasion even in adult life.
GC was reported to inhibit inflammatory cytokines secretion both by affecting gene transcription and post-translational events.23,24 In our study, the rat spleen mRNA of TNF-α was diminished in the presence of prenatal DEX treatment. This suggests that prenatal DEX treatment has, at least, transcriptional regulatory effects on offspring.
Histone acetylation and deacetylation enhances and represses gene transcription, respectively. In general, acetylation of histone H3 lysines (H3KAc) is associated with active gene transcription. H3KAc is mediated by HATs. Inhibition of HAT and recruitment of HDAC2 to sites of inflammatory gene transcription has been proposed to explain the anti-inflammatory activity of steroids.25,26 However, it is not known whether or not promoter histone H3 lysine modification plays a role in prenatal DEX-induced transcriptional change of TNF-α in the spleen. Here we showed that prenatal DEX treatment led to the decrease of H3 lysine acetylation and to diminishing of H3K4me1/3, and H3K36me3 signs at the TNF-α promoter in rat spleen. This suggests that a loss of the active acetylation of the H3 lysines, H3K4me1/3 and H3K36me3, at least in part, contribute to a decrease in the expression of TNF-α with prenatal DEX treatment.
Multiple functions have been ascribed to PKC, including transcription regulation, immune responses, cellular growth regulation and roles in learning and memory. DEX regulated the PKC protein expression and activity variously depended on administration dose, timing and cell types.27–29 In our study, we found early enhancement and late attenuation of PKCα/PKCβ proteins of the spleen by prenatal DEX treatment. In fact, PKC have dual effects on TNF-α regulation.30–33 Hence, late attenuation of PKCα/PKCβ proteins of the spleen by prenatal DEX exposure might contribute the depressed pro-inflammatory mediators. The early enhancement of PKCα/PKCβ proteins of the spleen by prenatal DEX treatment may be just a compensation manifestation. The exact mechanism regarding the regulation of the PKC protein clearly requires further investigation.
During pregnancy, GC is often given when preterm delivery is expected. This treatment is successful in stimulating the development of the fetal lung. However, reports about the prolonged effects of prenatal GC on the development of the immune system itself are rather limited. Our results suggest that prenatal GC has a profound effect on the innate immune system, more so than the adaptive immune system. The impact of prenatal GC might leave offspring more vulnerable to pathogenic invasion even in adult life. However, this study was an animal model in rats and different doses of GC account for different impacts on the respective immune systems. Hence further clinical evaluation and observation of the individuals who had received prenatal DEX treatment in the real world are necessary to verify our study results.
Acknowledgments
This study was supported in part by grants CMRPG8B0141, CMRPG8B0142 (H. R. Yu), CMRPG8C0171 (C.H. Kang) and NSC 102-2314-B-182A-042-MY3 (H. R. Yu) from the National Science Council, Taiwan.
Disclosures
The authors declare no commercial or financial conflict of interest.
References
- Kavelaars A, van der Pompe G, Bakker JM, van Hasselt PM, Cats B, Visser GH, Heijnen CJ. Altered immune function in human newborns after prenatal administration of betamethasone: enhanced natural killer cell activity and decreased T cell proliferation in cord blood. Pediatr Res. 1999;45:306–12. doi: 10.1203/00006450-199903000-00003. [DOI] [PubMed] [Google Scholar]
- Biggioggero M, Borghi MO, Gerosa M, Trespidi L, Cimaz R, Meroni PI. Immune function in children born to mothers with autoimmune diseases and exposed in utero to immunosuppressants. Lupus. 2007;16:651–6. doi: 10.1177/0961203307079569. [DOI] [PubMed] [Google Scholar]
- Motta M, Ciardelli L, Marconi M, Tincani A, Gasparoni A, Lojacono A, Chirico G. Immune system development in infants born to mothers with autoimmune disease, exposed in utero to immunosuppressive agents. Am J Perinatol. 2007;24:441–7. doi: 10.1055/s-2007-986679. [DOI] [PubMed] [Google Scholar]
- Cimaz R, Meregalli E, Biggioggero M, Borghi O, Tincani A, Motta M, Airò P, Meroni PL. Alterations in the immune system of children from mothers treated with immunosuppressive agents during pregnancy. Toxicol Lett. 2004;149:155–62. doi: 10.1016/j.toxlet.2003.12.030. [DOI] [PubMed] [Google Scholar]
- Agakidis C, Sarafidis K, Tzimouli V, et al. Antenatal betamethasone does not influence lymphocyte apoptosis in preterm neonates. Am J Perinatol. 2009;26:485–90. doi: 10.1055/s-0029-1214249. [DOI] [PubMed] [Google Scholar]
- Yu HR, Chang JC, Chen RF, Chuang H, Hong KC, Wang L, Yang KD. Different antigens trigger different Th1/Th2 reactions in neonatal mononuclear cells (MNCs) relating to T-bet/GATA-3 expression. J Leukoc Biol. 2003;74:952–8. doi: 10.1189/jlb.0902474. [DOI] [PubMed] [Google Scholar]
- Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–69. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- Kimura H. Histone modifications for human epigenome analysis. J Hum Genet. 2013;58:439–45. doi: 10.1038/jhg.2013.66. [DOI] [PubMed] [Google Scholar]
- Sengupta P. The laboratory rat: relating its age with human’s. Int J Prev Med. 2013;4:624–30. [PMC free article] [PubMed] [Google Scholar]
- Quinn R. Comparing rat’s to human’s age: how old is my rat in people years? Nutrition. 2005;21:775–7. doi: 10.1016/j.nut.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Travis J. Origins. On the origin of the immune system. Science. 2009;324:580–2. doi: 10.1126/science.324_580. [DOI] [PubMed] [Google Scholar]
- Eder C. Mechanisms of interleukin-1β release. Immunobiology. 2009;214:543–53. doi: 10.1016/j.imbio.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Lacy P, Stow JL. Cytokine release from innate immune cells: association with diverse membrane trafficking pathways. Blood. 2011;118:9–18. doi: 10.1182/blood-2010-08-265892. [DOI] [PubMed] [Google Scholar]
- Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996;334:1717–25. doi: 10.1056/NEJM199606273342607. [DOI] [PubMed] [Google Scholar]
- Ferrero MC, Fossati CA, Baldi PC. Direct and monocyte-induced innate immune response of human lung epithelial cells to Brucella abortus infection. Microbes Infect. 2010;12:736–47. doi: 10.1016/j.micinf.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–5. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardelli F, Ferrantini M. Cytokines as a link between innate and adaptive antitumor immunity. Trends Immunol. 2002;23:201–8. doi: 10.1016/s1471-4906(02)02195-6. [DOI] [PubMed] [Google Scholar]
- Viallard JF, Pellegrin JL, Ranchin V, et al. Th1 (IL-2, interferon-γ (IFN-γ)) and Th2 (IL-10, IL-4) cytokine production by peripheral blood mononuclear cells (PBMC) from patients with systemic lupus erythematosus (SLE) Clin Exp Immunol. 1999;115:189–95. doi: 10.1046/j.1365-2249.1999.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, Libby P. Interferon-γ, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res. 2008;103:467–76. doi: 10.1161/CIRCRESAHA.108.177105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumpas DT, Chrousos GP, Wilder RL, Cupps TR, Balow JE. Glucocorticoid therapy for immune-mediated diseases: basic and clinical correlates. Ann Intern Med. 1993;119:1198–208. doi: 10.7326/0003-4819-119-12-199312150-00007. [DOI] [PubMed] [Google Scholar]
- Fauci AS, Dale DC, Balow JE. Glucocorticosteroid therapy: mechanisms of action and clinical considerations. Ann Intern Med. 1976;84:304–15. doi: 10.7326/0003-4819-84-3-304. [DOI] [PubMed] [Google Scholar]
- Franchimont D, Louis E, Dewe W, Martens H, Vrindts-Gevaert Y, De Groote D, Belaiche J, Geenen V. Effects of dexamethasone on the profile of cytokine secretion in human whole blood cell cultures. Regul Pept. 1998;73:59–65. doi: 10.1016/s0167-0115(97)01063-x. [DOI] [PubMed] [Google Scholar]
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N Engl J Med. 2005;353:1711–23. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- Tobler A, Meier R, Seitz M, Dewald B, Baggiolini M, Fey MF. Glucocorticoids downregulate gene expression of GM-CSF, NAP-1/IL-8, and IL-6, but not of M-CSF in human fibroblasts. Blood. 1992;79:45–51. [PubMed] [Google Scholar]
- Ito K, Caramori G, Lim S, Oates T, Chung KF, Barnes PJ, Adcock IM. Expression and activity of histone deacetylases in human asthmatic airways. Am J Respir Crit Care Med. 2002;166:392–6. doi: 10.1164/rccm.2110060. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Histone deacetylase-2 and airway disease. Ther Adv Respir Dis. 2009;3:235–43. doi: 10.1177/1753465809348648. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Pandey GN. Administration of dexamethasone up-regulates protein kinase C activity and the expression of γ and ε protein kinase C isozymes in the rat brain. J Neurochem. 1999;72:380–7. doi: 10.1046/j.1471-4159.1999.0720380.x. [DOI] [PubMed] [Google Scholar]
- Xu H, Song J, Gao X, Xu Z, Xu X, Xia Y, Dai Y. Paeoniflorin attenuates lipopolysaccharide-induced permeability of endothelial cells: involvements of F-actin expression and phosphorylations of PI3K/Akt and PKC. Inflammation. 2013;36:216–25. doi: 10.1007/s10753-012-9537-3. [DOI] [PubMed] [Google Scholar]
- Tian L, Philp JA, Shipston MJ. Glucocorticoid block of protein kinase C signalling in mouse pituitary corticotroph AtT20 D16:16 cells. J Physiol. 1999;516(Pt 3):757–68. doi: 10.1111/j.1469-7793.1999.0757u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine SJ, Logun C, Chopra DP, Rhim JS, Shelhamer JH. Protein kinase C, interleukin-1β, and corticosteroids regulate shedding of the type I, 55 kDa TNF receptor from human airway epithelial cells. Am J Respir Cell Mol Biol. 1996;14:254–61. doi: 10.1165/ajrcmb.14.3.8845176. [DOI] [PubMed] [Google Scholar]
- Jialal I, Kaur H, Devaraj S. Human C-reactive protein accentuates macrophage activity in biobreeding diabetic rats. J Diabetes Complications. 2013;27:23–8. doi: 10.1016/j.jdiacomp.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jialal I, Devaraj S. Antisense to protein kinase C-α and p47phox attenuates the pro-inflammatory effects of human C-reactive protein in macrophages of biobreeding diabetic rats. Diab Vasc Dis Res. 2012;9:315–9. doi: 10.1177/1479164112452165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Lee SM, Suk K, Lee WH. A novel pathway responsible for lipopolysaccharide-induced translational regulation of TNF-α and IL-6 expression involves protein kinase C and fascin. J Immunol. 2011;187:6327–34. doi: 10.4049/jimmunol.1100612. [DOI] [PubMed] [Google Scholar]